

The crystal structures described here demonstrate the conformational flexibility of the active-site cavity of the plasmepsins. The interactions of the different moieties in the P1′ position of the inhibitors have to be taken into account in the design of new potent plasmepsin inhibitors.
Keywords: malaria, Plasmodium falciparum, plasmepsin, hydroxyethylamine, inhibitor
Abstract
Plasmepsin II (PMII) is one of the ten plasmepsins (PMs) identified in the genome of Plasmodium falciparum, the causative agent of the most severe and deadliest form of malaria. Owing to the emergence of P. falciparum strains that are resistant to current antimalarial agents such as chloroquine and sulfadoxine/pyrimethamine, there is a constant pressure to find new and lasting chemotherapeutic drug therapies. Previously, the crystal structure of PMII in complex with NU655, a potent antimalarial hydroxyethylamine-based inhibitor, and the design of new compounds based on it have been reported. In the current study, two of these newly designed hydroxyethylamine-based inhibitors, PG418 and PG394, were cocrystallized with PMII and their structures were solved, analyzed and compared with that of the PMII–NU655 complex. Structural analysis of the PMII–PG418 complex revealed that the flap loop can adopt a fully closed conformation, stabilized by interactions with the inhibitor, and a fully open conformation, causing an overall expansion in the active-site cavity, which in turn causes unstable binding of the inhibitor. PG418 also stabilizes the flexible loop Gln275–Met286 of another monomer in the asymmetric unit of PMII, which is disordered in the PMII–NU655 complex structure. The crystal structure of PMII in complex with the inhibitor PG418 demonstrates the conformational flexibility of the active-site cavity of the plasmepsins. The interactions of the different moieties in the P1′ position of PG418 and PG394 with Thr217 have to be taken into account in the design of new potent plasmepsin inhibitors.
1. Introduction
Malaria is one of the most severe infectious diseases in the world, with approximately 198 million cases and 598 000 attributed deaths reported globally in 2013 (World Health Organization, 2013 ▸). Human infection can be caused by four major species of the malaria parasite, i.e. Plasmodium falciparum, P. vivax, P. malariae and P. ovale, of which P. falciparum is responsible for more than 95% of malaria-related morbidity and mortality (Breman et al., 2001 ▸). P. falciparum, the most lethal of the four parasites infecting humans, is becoming increasingly resistant to many of the current frontline antimalarial drugs, including chloroquine, quinine, artemisinin and sulfadoxime/pyrimethamine (Egan & Kaschula, 2007 ▸; White, 1998 ▸). This highlights the urgent need for new efficacious drugs to combat this disease.
Ten plasmepsins have been identified in the genome of the malaria parasite P. falciparum (Coombs et al., 2001 ▸). Four of them, called the digestive plasmepsins, PMI, PMII, PMIV and histo-aspartic protease (HAP), reside in the acidic food vacuole and are involved in haemoglobin degradation (Banerjee et al., 2002 ▸; Liu et al., 2005 ▸). The functional role of nondigestive plasmepsins is an active field of research and it has recently been shown that PMV, PMIX and PMX are essential for survival of the parasite (Bonilla et al., 2007 ▸). There is evidence that haemoglobin digestion by plasmepsins has redundant mechanisms and that the in vivo effect of plasmepsin inhibitors might be exerted through their action on nondigestive plasmepsins (Bonilla et al., 2007 ▸; Meyers & Goldberg, 2012 ▸). Owing to their important role in providing nutrients for the growing parasites, plasmepsins have been identified as promising targets for the development of novel antimalarial drugs (Banerjee et al., 2002 ▸). Indeed, inhibitors of aspartic proteases have been shown to exhibit potent antiparasitic activity (Francis et al., 1994 ▸; Carroll et al., 1998 ▸; Miura et al., 2010 ▸), although the majority of them do not display nanomolar activity in cell-based models.
Of the plasmepsins, PMII is relatively easy to obtain in milligram quanitities for protein–ligand investigations. It has 65–73% sequence identity to other plasmepsins, but only 35% identity to the closest human aspartic protease relative, cathepsin D (Francis et al., 1994 ▸). This makes PMII a good target for structure-based drug design of new inhibitors for the other digestive and nondigestive plasmepsins. PMII is made up of 329 amino acids folded into two topologically similar N- and C-terminal domains (Fig. 1 ▸). The binding cleft contains Asp34 and Asp214, which together represent the catalytic dyad.
Figure 1.

Ribbon-diagram representation of PMII cocrystallized with three hydroxyethylamine-based inhibitors. Superposition of PMII in complex with PG418 (green), PG394 (brown) and NU655 (cyan). Inhibitors and catalytic Asp34 and Asp214 side chains are shown as stick models. The flap loop, flexible loop (residues 236–245), the loop comprising residues 10–16 and the loop comprising residues 275–286 are shown in darker colours.
To date, one of the most active groups of aspartic protease inhibitor compounds identified against intra-erythrocytic P. falciparum cell growth is based on the hydroxyethylamine moiety (Table 1 ▸; Jaudzems et al., 2014 ▸; Rathi et al., 2013 ▸; Ciana et al., 2013 ▸). These inhibitors are characterized by a hydroxyl group which replaces the catalytic water molecule in the active site, forming hydrogen bonds to the catalytically active aspartates Asp34 and Asp214. They have also been used as transition-state mimics in the design of inhibitors of other aspartic proteases, including HIV protease (Tucker et al., 1992 ▸; Petroková et al., 2004 ▸) and β-secretase (Sandgren et al., 2012 ▸; Rueeger et al., 2013 ▸).
Table 1. Inhibition of P. falciparum PMII, PMI and PMIV and human cathepsin D by the studied inhibitors (Jaudzems et al., 2014 ▸).
| IC50 (µM) | |||||
|---|---|---|---|---|---|
| Compound | Formula | PMII | PMI | PMIV | Cathepsin D |
| PG418 |
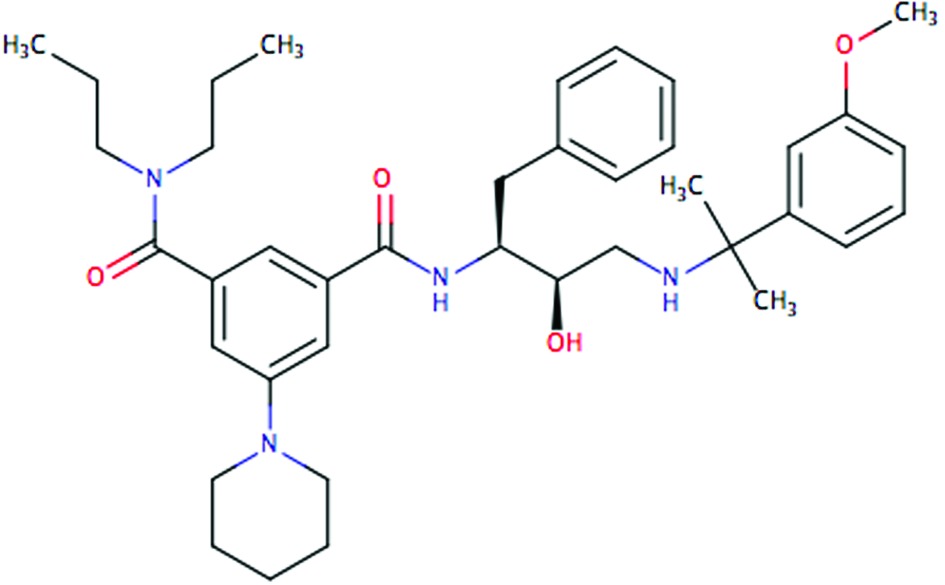
|
0.070 | 0.30 | 0.024 | 0.042 |
| PG394 |
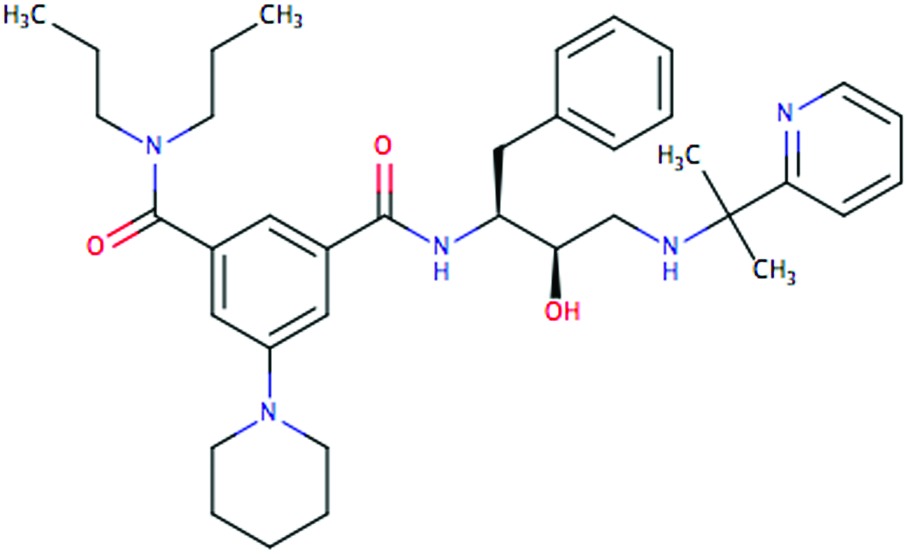
|
0.8 | 4.1 | 0.25 | 0.35 |
| NU655 |
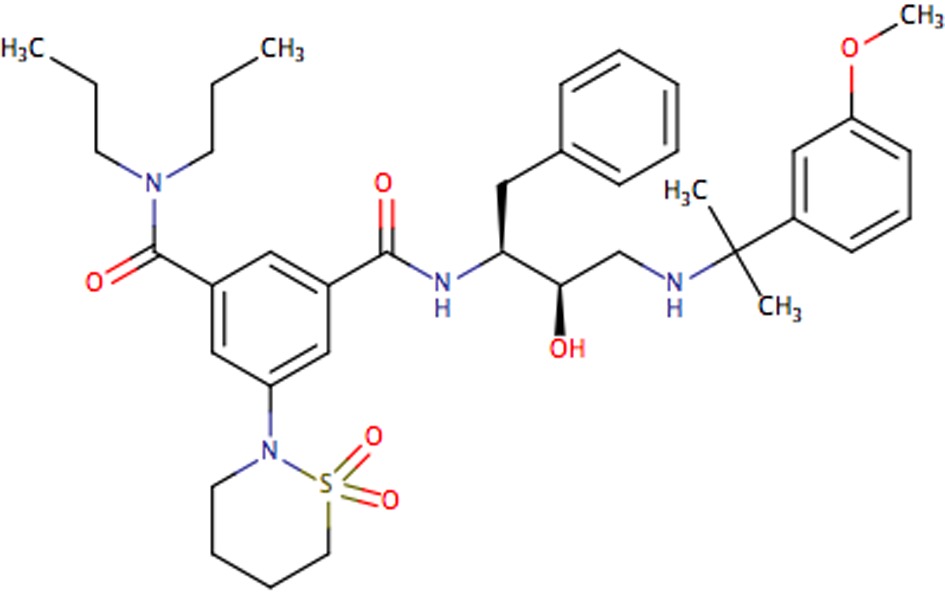
|
0.15 | 0.70 | 0.029 | 0.043 |
Previously, we determined the structure of PMII in complex with NU655 (Gamo et al., 2010 ▸) and used it as a guide for the design of new plasmepsin inhibitors (Jaudzems et al., 2014 ▸). Subsequent attempts to model the newly synthesized hydroxyethylamine inhibitors into the active site of the available structures of PMII or PMIV were unable to explain the structure–activity relationships; therefore, we performed structural studies of some of these optimized inhibitors in complex with PMII. In this paper, we present the crystal structures of PMII with two inhibitors: PG418 and PG394 (Table 1 ▸).
2. Materials and methods
2.1. Protein expression and purification
Two different constructs of the proenzyme of PMII, with a pro-segment of 50 residues, were overexpressed using the pET-3a vector (Novagen) in Escherichia coli BL21 (DE3) cells. The construct that was used to form the complex with the PG394 inhibitor had the mutation Met205Ser, which reduces the autolytic activity (Gulnik et al., 2002 ▸). In both cases the protein was isolated from inclusion bodies. Refolding and purification were performed as described in Beyer et al. (2004 ▸).
The purified pro-PMII was autoactivated by the addition of a one-tenth volume of 1 M sodium citrate pH 4.6 followed by incubation at 37°C for 30 min. The enzyme solution was then returned to pH 8.0 by the addition of 1 M Tris–HCl pH 8.0 and was further purified by gel filtration on a Superdex 75 10/300 column (GE Healthcare). The protein was concentrated to 10.8 mg ml−1 in 20 mM Tris–HCl pH 8.0 using a 10 kDa cutoff Amicon concentrator. Stocks of 50 µl of the protein were flash-frozen in liquid nitrogen and stored at −80°C.
The synthesis of the two inhibitors PG418 and PG394 has been described previously (Jaudzems et al., 2014 ▸).
2.2. Crystallization and data collection
PMII was pre-incubated with the ligand PG418, previously dissolved in DMSO at a final concentration of 100 mM, at a molar ratio of 1:20 at room temperature for 2 h and cocrystallized by vapour diffusion using MRC 96-well sitting-drop plates (Molecular Dimensions); 1 µl PMII–ligand complex solution was mixed with 1 µl reservoir solution consisting of 100 mM sodium citrate pH 4.6, 25%(w/v) PEG 3350.
Cocrystallization of the PMII–PG394 complex was also performed by the sitting-drop technique in 96-well MRC plates (Molecular Dimensions). However, in this case 1 µl protein solution was mixed with 1 µl reservoir solution [100 mM sodium citrate pH 6.5, 80%(w/v) ammonium sulfate] and 0.2 µl 100 mM inhibitor in 100%(v/v) DMSO.
The crystals were cryoprotected with mother liquor enriched with 30%(v/v) glycerol and were flash-cooled in liquid nitrogen.
Data for crystals of PMII in complex with PG418 and PG394 were collected on beamline I911-3 at the MAX-lab synchrotron, Lund, Sweden. Images were processed with iMosflm (Leslie et al., 2002 ▸; Powell et al., 2013 ▸) and scaled with SCALA (Evans, 2006 ▸) from the CCP4 suite (Winn et al., 2011 ▸).
2.3. Structure solution and refinement
The orientation and position of PMII was determined using the molecular-replacement program Phaser (McCoy et al., 2007 ▸) from the CCP4 suite (Winn et al., 2011 ▸), using the PMII structure with PDB entry 2bju (Prade et al., 2005 ▸) as a search model. Rigid-body refinement, followed by Cartesian simulated annealing of the protein alone, were initially applied to the structure using PHENIX (Echols et al., 2014 ▸), which resulted in a high-quality electron-density map for manual rebuilding of the structure in Coot (Emsley & Cowtan, 2004 ▸). The PG418 and PG394 inhibitor parameter files were generated with the ligand sketcher LIDIA (Debreczeni & Emsley, 2012 ▸) and restraints were generated with PRODRG in Coot (Debreczeni & Emsley, 2012 ▸).
The model of plasmepsin II in complex with PG418 was improved by iterative cycles of manual rebuilding with Coot and automated refinement with PHENIX, applying TLS and noncrystallographic symmetry (NCS) restraints owing to the presence of three monomers in the asymmetric unit.
The structure of PMII–PG394 was improved by iterative cycles of manual rebuilding with Coot and automated refinement with PHENIX, applying TLS and noncrystallographic symmetry (NCS) restraints owing to the presence of four monomers in the asymmetric unit. While PMII–PG418 was refined in real space, refinement of PMII–PG394 was performed in reciprocal space owing to the lower resolution (3.3 Å; Headd et al., 2012 ▸). The electron-density maps were improved using thermal B-factor sharpening, which increases the detail of side-chain conformations (Brunger et al., 2009 ▸).
Statistics for data collection, final refinement and validation by MolProbity (Chen et al., 2010 ▸) of both structures are summarized in Table 2 ▸. The superposition of models and r.m.s. deviations were calculated using LSQKAB (Kabsch et al., 1976 ▸) from the CCP4 suite (Winn et al., 2011 ▸). Buried surfaces and residues at the intermolecular contacts in the crystals were identified with the PISA server (http://www.ebi.ac.uk/pdbe/prot_int/pistart.html; Krissinel & Henrick, 2007 ▸). The majority of the figures were generated with PyMOL (v.1.5.0.1; Schrödinger).
Table 2. Data-collection and refinement statistics.
| PMII–PG418 | PMII–PG394 | |
|---|---|---|
| Data processing | ||
| Space group | P212121 | P213 |
| Unit-cell parameters (Å) | a = 81.22, b = 104.60, c = 111.68 | a = b = c = 177.79 |
| Wavelength (Å) | 0.972 | 0.972 |
| Resolution (Å) | 77–2.27 | 56.22–3.30 |
| R sym or R merge (%) | 15 (71) | 12 (68) |
| 〈I/σ(I)〉 | 11.6 (2.1) | 7.5 (2.2) |
| Completeness (%) | 100 (99.9) | 96.9 (90.8) |
| Wilson B factor (Å2) | 25.3 | 79.9 |
| CC1/2 | 0.995 (0.596) | 0.995 (0.616) |
| Refinement | ||
| Resolution (Å) | 64–2.3 | 56–3.3 |
| No. of unique reflections | 45010 (6470) | 27554 |
| R work/R free (%) | 17.2 (25.9)/22.7 (30.4) | 18.8 (27.9)/21.1 (31.3) |
| No. of atoms | ||
| Protein | 7815 | 10414 |
| Ligands | 177 | 198 |
| Water | 383 | 3 |
| Average B factors (Å2) | ||
| Protein | 31.6 | 84.9 |
| Ligands | 51.2 | 82.2 |
| Water | 32.5 | 50.5 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.009 | 0.008 |
| Bond angles (°) | 1.24 | 0.90 |
| Ramachandran plot | ||
| Most favoured (%) | 96.9 | 95.6 |
| Outliers (%) | 0.0 | 0.2 |
| MolProbity overall score | 1.49 | 1.60 |
| MolProbity clashcore | 5.55 | 5.37 |
Coordinates and structure factors were deposited in the Protein Data Bank as entries 4y6m (PMII–PG418 complex) and 4ya8 (PMII–PG394 complex).
3. Results and discussion
3.1. Overall structure
The crystal structures of PMII in complex with PG418 and PG394 were solved by the molecular-replacement method and refined to resolutions of 2.3 and 3.3 Å, respectively. The two complexes crystallized in different space groups. The PMII–PG418 complex crystallized in space group P212121 with three molecules in the crystallographic asymmetric unit. The PMII–PG394 complex crystallized in space group P213 with four molecules in the crystallographic asymmetric unit. Simulated-annealing mF o − DF c OMIT maps for the ligand structures of PG418 and PG394 are shown in Fig. 2 ▸. The real-space correlation coefficients (RSCCs) for the PG418 ligand are 0.93 for monomers A and B and and 0.75 for monomer C (Fig. 6b). The corresponding RSCCs for the PG394 ligand are around 0.94 for monomers A, C and D and 0.92 for monomer B.
Figure 2.

Simulated-annealing mF o − DF c OMIT maps of ligands in complex with PMII. The simulated-annealing mF o − DF c ligand-omit maps are shown as a grey mesh at 3σ. (a) Two conformers of the PG418 ligand are shown with C atoms of monomer A (green) and of monomer B (lilac); (b) PG394 ligand corresponding to monomer D.
The overall structures of PMII–PG418 (with the exception of monomer C), PMII–PG394 and PMII–NU655 are very similar (Fig. 1 ▸). The flap loop, which gates access to the active site, is in a closed conformation in all three PMII complex structures. The major differences are found in the Gln275–Met286 loop, which is disordered in the structure of PMII in complex with NU655 (PDB entry 4cku; Jaudzems et al., 2014 ▸) and in the loop comprising residues Asp10–Phe16. The latter loop forms the substrate (or inhibitor) binding pocket S3 and presents similar, more closed conformations in the structures of PMII cocrystallized with PG418 and PG394 compared with the structure of PMII in complex with NU655, in which the pocket is wider by approximately 2.4 Å.
We also observed differences in the flexible areas of PMII when we compared the monomers in the crystallographic asymmetric unit. In the PMII–PG394 structure the only significant difference among the four molecules in the crystallographic asymmetric unit lies in the Gln275–Met286 loop (r.m.s.d. of 3.6 Å for Cα atoms between loops in monomers A and B). This difference is owing to different crystallographic contacts (data not shown). The monomers of the PMII–PG418 complex structure (Fig. 3 ▸ a) assigned as A and B superpose very well. However, when the A and B monomers are compared with the C monomer, the r.m.s.d. for Cα atoms of 329 residues is around 1.3 Å. The main differences between monomers A/B and C in the asymmetric unit are found in the Met75–Val82 loop, known as the flap loop (r.m.s.d. of 7.9 Å between monomers A and C) and in the Val236–Tyr245 loop, known as the flexible loop (r.m.s.d. of 3.2 Å between monomers A and C).
Figure 3.

Comparison of monomers in the asymmetric unit of the PMII–PG418 crystal structure. (a) Superposition of Cα atoms of the three monomers of the asymmetric unit of the PMII–PG418 complex structure. Molecule A is in green, molecule B is in lilac and molecule C is in magenta. The flap loop is encircled with a black ellipse. PG418 of monomer A is shown in spheres with carbon in white, oxygen in red and nitrogen in blue. (b) Close-up view showing the conformational differences of the flap region in monomers A and C of the complex of PMII with PG418. Tyr77 and Trp41 side chains of both monomers and the inhibitor PG418 of monomer A are shown as sticks.
The PMII–PG418 structure shows two different conformations of the flap loop. It has a flap-closed conformation in monomers A and B and a flap-open conformation in monomer C. The angle between the flap loop of monomer A and the flap loop of a superimposed monomer C is about 32.5° (Fig. 3 ▸). In the flap-open conformation of monomer C the opening of the flap loop moves the conserved Tyr77 away, abolishing a hydrogen-bond contact with the indole NH of the highly conserved Trp41 (Fig. 3 ▸ b). The interaction between these two side chains has typically been observed in structures of aspartic proteinases inhibited by peptidomimetic inhibitors (e.g. pepstatin; PDB entry 1sme; Silva et al., 1996 ▸). The lack of this interaction facilitates the flexibility of Trp41, which displays two conformations in monomer C (Fig. 3 ▸ b). The flap loop is an important element of the catalytic machinery. In the presence of inhibitors it is usually in a closed conformation. However, there are some cases where the flap-open conformation has been reported: HAP in complex with KNI-10006 (Bhaumik et al., 2011 ▸) and PMII in complex with an achiral inhibitor (Prade et al., 2005 ▸; Bhaumik et al., 2011 ▸). These conformational differences present between monomers of the asymmetric unit demonstrate an intrinsic interdomain flexibility of PMII and are related to its catalytic activity. In fact, similar flexibility has also been detected in HIV-1 protease (Yedidi et al., 2012 ▸).
3.2. Gln275–Met286 loop and the dimeric interface of the PMII–PG418 complex structure
While the Gln275–Met286 loop is disordered in the structure of the PMII–NU655 complex (PDB entry 4cku), in the structure of the PMII–PG418 complex it is stabilized by packing of another monomer of the asymmetric unit close to the active-site cavity (Fig. 4 ▸), and the loop is in close proximity to the inhibitor PG418. Fig. 4 ▸ shows the noncrystallographic dimer of monomers A and C in the crystallographic asymmetric unit. The buried surface area between the two monomers is 2820 Å2. We have not observed this packing in the complex structure of PMII with either PG394 or NU655. However, dimer formation of two PMII monomers also occurs in the complex of PMII with pepstatin A and an achiral inhibitor, but in these cases the loop that interacts with the inhibitor is the flexible loop (Fig. 1 ▸), residues Val236–Tyr245 (PDB entry 1xdh, Silva et al., 1996 ▸; PDB entry 2bju, Prade et al., 2005 ▸; PDB entries 1lee and 1lf2, Asojo et al., 2002 ▸). The interaction of the Gln275–Met286 loop with the active site in the PMII–PG418 structure moves away two water molecules that are present in the active site of the PMII–NU655 structure.
Figure 4.

Dimerization of the PMII–PG418 complex in the asymmetric unit. Molecule A and molecule C are coloured green and magenta, respectively. PG418 of both monomers is coloured by atom type: carbon in white, oxygen in red and nitrogen in blue. The loop comprising residues 275–286 of monomer C interacts with the active site and inhibitor of monomer A.
3.3. Active-site interactions
Since the structure of the PMII–PG418 complex was obtained at better resolution than the structure of the PMII–PG394 complex, the interactions of PG418 with PMII are described in more detail.
The structure of the PMII–NU655 complex (Jaudzems et al., 2014 ▸) showed that there are three sites of potentially repulsive interactions between the inhibitor and PMII active-site residues (Fig. 5 ▸ a). The compounds in the present work were designed with appropriate chemical modifications at these positions in order to improve intermolecular interactions (Table 1 ▸). The modified compounds lacked the sulfonyl group of the 2-(1,2-thiazinane-1,1-dioxide) moiety that is involved in unfavourable interactions between the inhibitor and residue Val78 of the flap loop. The 3-methoxyphenyl group in the crystal structure of the PMII–PG418 complex is very close to the polar side chains of Thr217 and Asp214 (Fig. 5 ▸ b). In PG394 the 3-methoxyphenyl group is substituted with a 2-pyridyl group in position P1′. The N atom in this ring was proposed in order to introduce a hydrogen-bond acceptor for the Thr217 side-chain hydroxyl (Fig. 5 ▸ c).
Figure 5.

Protein–ligand interactions across the active sites of plasmepsin II in complexes with hydroxyethylamine-based inhibitors. (a) PMII–NU655, (b) PMII–PG418, (c) PMII–PG394. (d) Wall-eyed stereoview of the superposition of the PMII active site with the three inhibitors.
The electron density of the inhibitor PG418 (Fig. 2 ▸ a) is very similar in monomers A and B, and it is good for the central part of the inhibitor but weaker or lacking for aliphatic propyl groups and for the methoxy group of the isopropyl-2-(3-methoxyphenyl) moiety that occupies the S1′ and part of the S2 pockets. The methoxy group of the isopropyl-2-(3-methoxyphenyl) moiety of PG418 is present in two orientations, making different interactions as described below. Owing to the fully open conformation of the flap loop (Figs. 3 ▸ b and 6 ▸ d), the inhibitor PG418 in monomer C is substantially more disordered with poor electron density (Fig. 6 ▸ b), presumably caused by the unstable binding of the inhibitor that is not involved in interactions with the catalytic dyad residues Asp34 or Asp214 (data not shown). A closer examination of the binding cavity of the PMII–PG418 complex in monomers A and C reveals that the inhibitor is more solvent-exposed in the active site of monomer C (Figs. 6 ▸ c and 6 ▸ d).
Figure 6.

Differences in the active-site conformation of the PMII–PG418 complex between monomers A and C. (a) and (b) show mF o − DF c OMIT maps of monomers A and C, respectively, contoured at 3σ around PG418. (c) and (d) show the electrostatic surface within 7 Å around PG418 in monomer A and monomer C, respectively. Diagrams were drawn with CCP4mg (McNicholas et al., 2011 ▸).
The interactions of PG418 and PG394 with the active-site residues of PMII are in general very similar to those previously described for the inhibitor NU655 (Fig. 5 ▸ a). As in the PMII–NU655 complex, the hydroxyl groups of PG418 and PG394 are not within the characteristic hydrogen-bond distances of both carboxylic acids, Asp34 and Asp214 (Asojo et al., 2003 ▸; Silva et al., 1996 ▸). In the PMII–PG418 complex (Fig. 5 ▸ b) the hydroxyl group is only at a hydrogen-bonding distance from Asp34 (O–O distance of 2.73 Å), the carbonyl group of Gly36 (3.11 Å) and a water molecule. However, Asp214 is hydrogen-bonded to the secondary amino group (N–O distance of 2.83 Å). Other hydrogen-bond interactions present in the studied complex structures are formed with the more central groups of the inhibitor, mimicking the interactions of the substrate backbone, i.e. between the benzamide and Gly216 and the N,N-dipropylamide and Ser218 (Fig. 5 ▸ d). In contrast to PG394 and NU655, there are neither polar nor van der Waals interactions between PG418 and the flap loop (Fig. 5 ▸ b). A water molecule bridges the carbonyl group of the inhibitor and the N atom of Val78. The 3-methoxyphenyl moiety occupies the S1′ and part of the S2 pockets. In one of the conformations described for monomer A (Fig. 5 ▸ b), the O atom is interacting with the hydroxyl group of Thr217, while in the other orientation the 3-methoxyphenyl group is solvent-exposed. Additionally, in this second orientation there are repulsive interactions (not shown) of the ring with the hydrophilic S2 pocket of the active site, as described for NU655 (Fig. 5 ▸ a). In Fig. 5 ▸(d) the superposition of the active site of PMII in complex with PG418 (monomer A), PG394 and NU655 is shown and the two orientations of the 3-methoxyphenyl group are demonstrated. These repulsive interactions made us to consider replacing the 3-methoxyphenyl group with 2-pyridyl, 3-pyridyl and 4-pyridyl groups, respectively, in the hope that the N atom might act as a hydrogen-bond acceptor. The pyridyl N atom of PG394 indeed establishes a hydrogen-bond interaction with the hydroxyl group of Thr217 (N–O distance of 3.04 Å) and a nonconstructive interaction with the aspartate group of Asp214 (N–O distance of 3.20 Å). Besides this, from Fig. 5 ▸(d) it can be seen that the flap loop in the PMII–PG394 complex has a more closed conformation than with PG418 and that the benzamide carbonyl of PG394 establishes a hydrogen-bond interaction with the N atom of Ser79. However, the biochemical experiments performed to measure the activity of PG394 (Jaudzems et al., 2014 ▸) showed a decrease in potency by an order of magnitude against all aspartic proteases (Table 1 ▸). This discrepancy could be owing to protonation of the pyridine N atom at the pH of the assay (pH 4.6). As was mentioned in §2, the crystals were obtained at pH 6.5, which could modify the protonation state of the pyridine N atom, and at this pH this N atom may act as a hydrogen-bond acceptor.
The most important differences in the interactions between the three PMII complexes discussed here are at position P3. The 2-(1,2-thiazinane-1,1-dioxide) moiety of NU655 is substituted by a piperidine ring in PG418 and PG394. While the sulfonyl group of the 2-(1,2-thiazinane-1,1-dioxide) moiety is only 3.0 Å from the hydrophobic Val78 side chain, the piperidinyl group of PG418 does not interact with any residue of the flap loop, as mentioned above. This complete absence of hydrogen-bond interactions between the tightly bound inhibitor PG418 and the flap, as well as the diminished hydrophobic interactions, allowed the flap to twist and lift its tip in monomer C (Fig. 2 ▸ b and Fig. 6 ▸ d). The inhibitor PG418 establishes van der Waals interactions with the Glu278–Leu284 loop of another monomer (Fig. 5 ▸ b), and these interactions move away two water molecules that were present in the structure of the PMII–NU655 complex (Figs. 5 ▸ a and 5 ▸ d), in which this loop was disordered. However, the latter interactions are presumably of no biological importance, since they are only made possible by crystal contacts with different monomers in the asymmetric unit.
4. Conclusion
The current paper presents the crystal structure of plasmepsin II in complex with two hydroxyethylamine-based inhibitors designed from a previously studied very potent antimalarial inhibitor, NU655. The crystal structure of plasmepsin II from P. falciparum in complex with the inhibitor PG418 demonstrates the previously reported conformational flexibility of the active-site cavity of the plasmepsins, as it presents snapshots of a fully open and a fully closed conformation of the flap loop. The replacement of the methoxyphenyl group of PG418 with the pyridine group of PG394 allows hydrogen-bond formation to the residue Thr217 at the S1′ pocket. This information can be used in the design of more potent hydroxyethylamine-based inhibitors.
Supplementary Material
PDB reference: PMII–PG418 complex, 4y6m
PDB reference: PMII–PG394 complex, 4ya8
Acknowledgments
Financial support from InnovaBalt project (EU Grant 316149) and FP7 program Biostruct-X proposal 7869. We thank the staff of MAX-lab synchrotron for the support during data collection.
References
- Asojo, O. A., Afonina, E., Gulnik, S. V., Yu, B., Erickson, J. W., Randad, R., Medjahed, D. & Silva, A. M. (2002). Acta Cryst. D58, 2001–2008. [DOI] [PubMed]
- Asojo, O. A., Gulnik, S. V., Afonina, E., Yu, B., Ellman, J. A., Haque, T. S. & Silva, A. M. (2003). J. Mol. Biol. 327, 173–181. [DOI] [PubMed]
- Banerjee, R., Liu, J., Beatty, W., Pelosof, L., Klemba, M. & Goldberg, D. E. (2002). Proc. Natl Acad. Sci. USA, 99, 990–995. [DOI] [PMC free article] [PubMed]
- Beyer, B. B., Goldfarb, N. E. & Dunn, B. M. (2004). Curr. Protoc. Protein Sci., Unit 21.14. 10.1002/0471140864.ps2114s32. [DOI] [PubMed]
- Bhaumik, P., Xiao, H., Hidaka, K., Gustchina, A., Kiso, Y., Yada, R. Y. & Wlodawer, A. (2011). Biochemistry, 50, 8862–8879. [DOI] [PMC free article] [PubMed]
- Bonilla, J. A., Bonilla, T. D., Yowell, C. A., Fujioka, H. & Dame, J. B. (2007). Mol. Microbiol. 65, 64–75. [DOI] [PubMed]
- Breman, J. G., Egan, A. & Keusch, G. T. (2001). Am. J. Trop. Med. Hyg. 64, iv–vii. [DOI] [PubMed]
- Brunger, A. T., DeLaBarre, B., Davies, J. M. & Weis, W. I. (2009). Acta Cryst. D65, 128–133. [DOI] [PMC free article] [PubMed]
- Carroll, C. D., Patel, H., Johnson, T. O., Guo, T., Orlowski, M., He, Z.-M., Cavallaro, C. L., Guo, J., Oksman, A., Gluzman, I. Y., Connelly, J., Chelsky, D., Goldberg, D. E. & Dolle, R. E. (1998). Bioorg. Med. Chem. Lett. 8, 2315–2320. [DOI] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Ciana, C. L., Siegrist, R., Aissaoui, H., Marx, L., Racine, S., Meyer, S., Binkert, C., de Kanter, R., Fischli, C., Wittlin, S. & Boss, C. (2013). Bioorg. Med. Chem. Lett. 23, 658–662. [DOI] [PubMed]
- Coombs, G. H., Goldberg, D. E., Klemba, M., Berry, C., Kay, J. & Mottram, J. C. (2001). Trends Parasitol. 17, 532–537. [DOI] [PubMed]
- Debreczeni, J. É. & Emsley, P. (2012). Acta Cryst. D68, 425–430. [DOI] [PMC free article] [PubMed]
- Echols, N., Moriarty, N. W., Klei, H. E., Afonine, P. V., Bunkóczi, G., Headd, J. J., McCoy, A. J., Oeffner, R. D., Read, R. J., Terwilliger, T. C. & Adams, P. D. (2014). Acta Cryst. D70, 144–154. [DOI] [PMC free article] [PubMed]
- Egan, T. J. & Kaschula, C. H. (2007). Curr. Opin. Infect. Dis. 20, 598–604. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Francis, S. E., Gluzman, I. Y., Oksman, A., Knickerbocker, A., Mueller, R., Bryant, M. L., Sherman, D. R., Russell, D. G. & Goldberg, D. E. (1994). EMBO J. 13, 306–317. [DOI] [PMC free article] [PubMed]
- Gamo, F. J., Sanz, L. M., Vidal, J., de Cozar, C., Alvarez, E., Lavandera, J. L., Vanderwall, D. E., Green, D. V., Kumar, V., Hasan, S., Brown, J. R., Peishoff, C. E., Cardon, L. R. & Garcia-Bustos, J. F. (2010). Nature (London), 465, 305–310. [DOI] [PubMed]
- Gulnik, S. V., Afonina, E. I., Gustchina, E., Yu, B., Silva, A. M., Kim, Y. & Erickson, J. W. (2002). Protein Expr. Purif. 24, 412–419. [DOI] [PubMed]
- Headd, J. J., Echols, N., Afonine, P. V., Grosse-Kunstleve, R. W., Chen, V. B., Moriarty, N. W., Richardson, D. C., Richardson, J. S. & Adams, P. D. (2012). Acta Cryst. D68, 381–390. [DOI] [PMC free article] [PubMed]
- Jaudzems, K., Tars, K., Maurops, G., Ivdra, N., Otikovs, M., Leitans, J., Kanepe-Lapsa, I., Domraceva, I., Mutule, I., Trapencieris, P., Blackman, M. J. & Jirgensons, A. (2014). ACS Med. Chem. Lett. 5, 373–377. [DOI] [PMC free article] [PubMed]
- Kabsch, W., Kabsch, H. & Eisenberg, D. (1976). J. Mol. Biol. 100, 283–291. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Leslie, A. G. W., Powell, H. R., Winter, G., Svensson, O., Spruce, D., McSweeney, S., Love, D., Kinder, S., Duke, E. & Nave, C. (2002). Acta Cryst. D58, 1924–1928. [DOI] [PubMed]
- Liu, J., Gluzman, I. Y., Drew, M. E. & Goldberg, D. E. (2005). J. Biol. Chem. 280, 1432–1437. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- McNicholas, S., Potterton, E., Wilson, K. S. & Noble, M. E. M. (2011). Acta Cryst. D67, 386–394. [DOI] [PMC free article] [PubMed]
- Meyers, M. J. & Goldberg, D. E. (2012). Curr. Top. Med. Chem. 12, 445–455. [DOI] [PMC free article] [PubMed]
- Miura, T., Hidaka, K., Uemura, T., Kashimoto, K., Hori, Y., Kawasaki, Y., Ruben, A. J., Freire, E., Kimura, T. & Kiso, Y. (2010). Bioorg. Med. Chem. Lett. 20, 4836–4839. [DOI] [PubMed]
- Petroková, H., Dusková, J., Dohnálek, J., Skálová, T., Vondrácková-Buchtelová, E., Soucek, M., Konvalinka, J., Brynda, J., Fábry, M., Sedlácek, J. & Hasek, J. (2004). Eur. J. Biochem. 271, 4451–4461. [DOI] [PubMed]
- Powell, H. R., Johnson, O. & Leslie, A. G. W. (2013). Acta Cryst. D69, 1195–1203. [DOI] [PMC free article] [PubMed]
- Prade, L., Jones, A. F., Boss, C., Richard-Bildstein, S., Meyer, S., Binkert, C. & Bur, D. (2005). J. Biol. Chem. 280, 23837–23843. [DOI] [PubMed]
- Rathi, B., Singh, A. K., Kishan, R., Singh, N., Latha, N., Srinivasan, S., Pandey, K. C., Tiwari, H. K. & Singh, B. K. (2013). Bioorg. Med. Chem. 21, 5503–5509. [DOI] [PubMed]
- Rueeger, H., Lueoend, R., Machauer, R., Veenstra, S. J., Jacobson, L. H., Staufenbiel, M., Desrayaud, S., Rondeau, J. M., Möbitz, H. & Neumann, U. (2013). Bioorg. Med. Chem. Lett. 23, 5300–5306. [DOI] [PubMed]
- Sandgren, V., Agback, T., Johansson, P. O., Lindberg, J., Kvarnström, I., Samuelsson, B., Belda, O. & Dahlgren, A. (2012). Bioorg. Med. Chem. 20, 4377–4389. [DOI] [PubMed]
- Silva, A. M., Lee, A. Y., Gulnik, S. V., Maier, P., Collins, J., Bhat, T. N., Collins, P. J., Cachau, R. E., Luker, K. E., Gluzman, I. Y., Francis, S. E., Oksman, A., Goldberg, D. E. & Erickson, J. W. (1996). Proc. Natl Acad. Sci. USA, 93, 10034–10039. [DOI] [PMC free article] [PubMed]
- Tucker, T. J., Lumma, W. C. Jr, Payne, L. S., Wai, J. M., de Solms, S. J., Giuliani, E. A., Darke, P. L., Heimbach, J. C., Zugay, J. A. & Schleif, W. A. (1992). J. Med. Chem. 35, 2525–2533. [DOI] [PubMed]
- White, N. J. (1998). Br. Med. Bull. 54, 703–715. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- World Health Organization. (2013). World Malaria Report 2013. Geneva: World Health Organization. http://www.who.int/malaria/publications/world_malaria_report_2013/report/en/.
- Yedidi, R. S., Liu, Z., Wang, Y., Brunzelle, J. S., Kovari, I. A., Woster, P. M., Kovari, L. C. & Gupta, D. (2012). Biochem. Biophys. Res. Commun. 421, 413–417. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: PMII–PG418 complex, 4y6m
PDB reference: PMII–PG394 complex, 4ya8