

Abstract
Background
Huntington disease (HD) is a neurodegenerative disorder characterized by motor dysfunction, cognitive deterioration, and psychiatric symptoms, with progressive motor impairments being a prominent feature. The primary objectives of this study are to delineate the disease course of motor function in HD, to provide estimates of the onset of motor impairments and motor diagnosis, and to examine the effects of genetic and demographic variables on the progression of motor impairments.
Methods
Data from an international multisite, longitudinal observational study of 905 prodromal HD participants with cytosine-adenine-guanine (CAG) repeats of at least 36 and with at least two visits during the followup period from 2001 to 2012 was examined for changes in the diagnostic confidence level from the Unified Huntington's Disease Rating Scale.
Results
HD progression from unimpaired to impaired motor function, as well as the progression from motor impairment to diagnosis, were associated with the linear effect of age and CAG repeat length. Specifically, for every 1-year increase in age, the risk of transition in diagnostic confidence level increased by 11% (95% CI: 7-15%) and for one repeat length increase in CAG, the risk of transition in diagnostic confidence level increased by 47% (95% CI: 27-69%).
Conclusions
Findings show that CAG repeat length and age increased the likelihood of the first onset of motor impairment as well as the age at diagnosis. Results suggest that more accurate estimates of HD onset age can be obtained by incorporating the current status of diagnostic confidence level into predictive models.
Keywords: Huntington disease, hidden Markov model, diagnostic confidence level, prediction, diagnosis, onset
Introduction
Huntington disease (HD) is an autosomal dominant neurodegenerative disease caused by a trinucleotide expansion cytosine-adenine-guanine (CAG) in exon 1 of the HTT gene [1]. Individuals who develop HD have at least 36 CAG repeats. HD is associated with severe motor, cognitive, and psychiatric impairments that typically develop in adulthood [2]. The typical age of diagnosis is between 30 and 50 years, with a range of 2 to 85 years. The mean duration of the disease following diagnosis is between 17 and 20 years [3].
Progression of motor dysfunction is a characteristic of the disease course of HD. For a typical individual with CAG expansion, his or her motor function remains unimpaired from birth until a certain age at which subtle signs and symptoms of motor impairment start to appear, such as a change in handwriting or occurrence of slight involuntary movements. With continued worsening of motor function, further characteristic motor signs of HD emerge, such as chorea, dystonia, bradykinesia, and oculomotor impairments. At a certain point in the disease course the individual receives an HD diagnosis, based on the presence of motor signs and symptoms. Once an individual is diagnosed with HD, the condition slowly progresses toward death, since there are no disease-modifying treatments.
Motor function in HD has most typically been evaluated according to the standardized 15-item motor exam in the Unified Huntington's Disease Rating Scale (UHDRS) [4]. After completing the exam, an experienced and certified motor rater assigns a score according to the diagnostic confidence level (DCL; UHDRS item 17), which asks whether the participant “meets the operational definition of the unequivocal presence of an otherwise unexplained extrapyramidal movement disorder in a subject at risk for HD” (99% confidence). The DCL ranges from 0 to 4 – i.e., from unimpaired (DCL = 0), to non-specific motor impairments (less than 50% confidence) (DCL = 1), up to motor impairments that are unequivocal signs of HD (99% confidence) (DCL = 4). From the perspective of tracking the disease course of motor function, the first receipt of DCL score of 1 represents the onset of motor impairments and that of 4 indicates the onset of HD diagnosis.
The presentation of motor signs and symptoms in HD is variable among individuals. Although chorea is a classic sign of HD, a wide spectrum of other motor signs and symptoms can also occur, such as bradykinesia, dystonia, rigidity, and impairments of eye movements and gait [2, 5-11]. Not all of these impairments manifest in every individual with HD, nor do they follow the same progressive patterns [2, 5-11]. Most notably, some HD gene mutation carriers never develop chorea [2, 10, 11]. Therefore, it is inaccurate to use a single motor sign or symptom as pathognomonic of HD to characterize the disease course for all individuals.
No matter how differently the motor signs and symptoms manifest themselves among different individuals or at different time points, the overall progressive pattern of motor dysfunction is a common feature in HD. In other words, every HD individual should, in principle, experience a period of no motor impairments, followed by a period of subtle motor impairments but without a rating of 4 on the DCL, followed by motor impairments warranting HD diagnosis (DCL = 4). The duration of these periods is unknown and likely varies among individuals. Careful consideration of the age at which motor impairments begin and the age at which a manifest motor diagnosis is given may provide useful information on disease progression. The rate and pattern of motor disease progression have received little attention, despite extensive study of HD age of onset [5, 11-15].
In all existing publications on the modeling of age of HD onset [5, 11-18], predicted age of onset is modeled based only on a participant's baseline information, such as CAG repeat length, age at study entry, and other demographic and neurological measures. There are several limitations to this approach. First, these models consider only HD diagnosis as the outcome. For participants who are not diagnosed during study followup, their outcomes are broadly categorized as censored (not yet diagnosed), and the information on their final statuses of motor function is discarded. Second, none of the models have included the status of motor function at study entry. As HD is a progressive disease, when participants enter a study, their motor function typically shows varying degrees of impairment. In studying age of HD onset, it is preferable to include such information in the model. Finally, none of the existing models can utilize the rich information on status of motor function collected over individual study visits. In summary, the existing models are static in nature and not well-equipped to evaluate the dynamic process of disease progression. In this study, we aim to address these issues by employing a novel statistical framework, under which both the HD diagnosis and intermediate status of motor function can be modelled, and the dynamic process of disease progression can be evaluated. To the best of our knowledge, our study is the first of its kind and fills a gap in research on dynamic modeling of disease progression in HD.
The primary objectives of this article are to provide models for the motor progression of HD from unimpaired to impaired motor function, and from impaired motor function to motor diagnosis. Such models may provide novel and significant contributions toward a better understanding of the natural evolution of motor impairments in HD.
Methods
Participants
Participants were 905 persons with the gene mutation for HD (≥36 CAG) from the PREDICT-HD study [19-22]. Data in PREDICT-HD were collected at 33 sites in the United States, Canada, Australia, Germany, Spain, and the United Kingdom from 2001 to 2014. All participants underwent comprehensive motor, cognitive, psychiatric, and functional evaluations at study entry, and annually thereafter. At study enrollment, participants were required to be 18 years of age or older, have a family history of HD, and have completed independent genetic testing for the HD CAG expansion prior to entry into PREDICT-HD. Confirmatory DNA testing was conducted at the baseline PREDICT-HD visit using a polymerase chain-reaction method to determine CAG-repeat length [23].
For this study, participants were included if they had at least two visits, because our research focuses on the modeling of changes in motor function from one visit to another. If a participant had only one data point, no change could be modelled. Descriptive statistics for the sample are presented in Table 1, stratified by the baseline DCL levels. As can be seen from the table, the gender composition, CAG repeat length and number of visits were very similar across baseline DCL levels.
Table 1.
Descriptive statistics of study participants.
| Baseline DCL | DCL = 0 | DCL = 1 | DCL =2 | DCL = 3 |
|---|---|---|---|---|
| Number (percentage) of participants | 351 (38.8%) | 395 (43.6%) | 119 (13.1%) | 40 (4.4%) |
| Number (percentage) of females | 211 (60.1%) | 267 (67.6%) | 74 (62.2%) | 25 (62.5%) |
| Mean (standard deviation) of age | 39.3 (9.8) | 40.5 (9.8) | 43.0 (10.8) | 45.5 (10.7) |
| Median (range) of CAG repeat length | 42 (38-50) | 42 (38-52) | 42 (38-51) | 43 (39-51) |
| Median (range) of number of visits | 5 (2-10) | 5 (2-10) | 6 (2-10) | 5 (2-10) |
DCL diagnostic confidence level, CAG cytosine-adenine-guanine
Statistical Analysis Methods
We argue the diagnostic confidence levels of 0 and 4 represent two clinical extremes of motor function in disease progression, but the intermediate levels of 1, 2, and 3 are less distinct from one another. The intermediate levels reflect varying degrees of confidence a motor rater has on a participant's motor dysfunction. To prevent the over-interpretation of the clinical meaning of each of the intermediate confidence levels, we think that it makes practical sense to group them together into one category representing broader motor impairment below the threshold of a motor diagnosis. Therefore, DCL data from each participant was categorized into three states: the level of 0 corresponds to the “unimpaired” state, the levels of 1, 2 and 3 collectively represent the impaired state, and the level of 4 represents HD diagnosis. In the remainder of the article, the newly created variable with three states will be the outcome variable in all analyses.
Although in principle, motor dysfunction should progress from unimpaired to impaired to HD diagnosis, the observed data did not always follow such a progressive pattern, and deviations frequently occur. This is illustrated in the left panel of Figure 1 using the observed data from two participants in the PREDICT-HD study. For example, the disease course of participant A in general follows the overall pattern of unimpaired to impaired to diagnosis. However, the participant received a diagnosis at the fourth visit, and the diagnosis was reversed in the next four visits (back to impaired). Based on the observed disease trajectory, it is natural to hypothesize that participant A′s diagnosis at the fourth visit could be just a random deviation and the participant should not be declared as HD diagnosed in light of his/her motor performance at the next four visits. For participant B, the disease course also shows a progressive trend. However, there are variations in disease status at the last three visits (fluctuating between impaired and HD diagnosis). There is a possibility that the impaired state that patient B received at the fifth visit could in fact be HD diagnosis.
Fig 1.
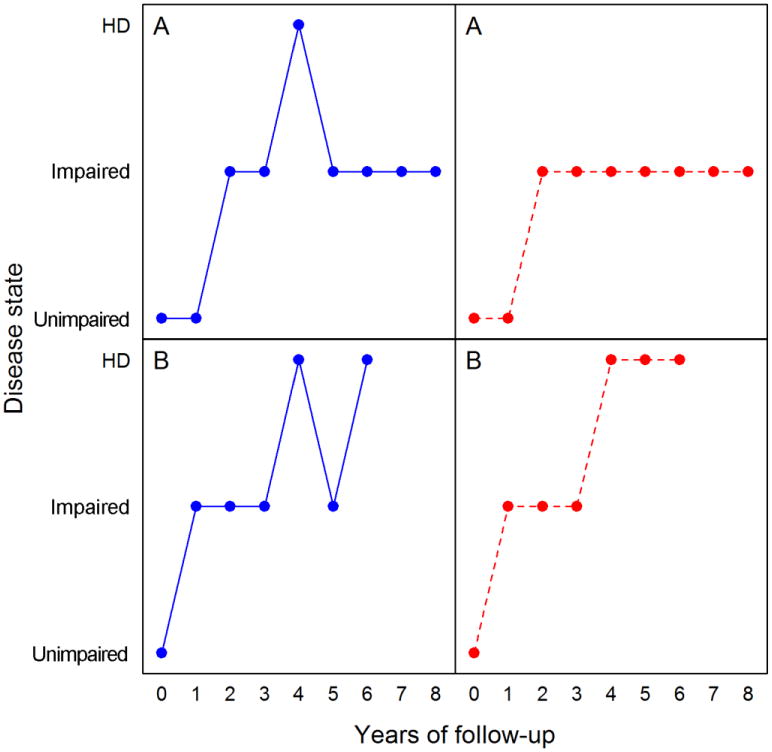
Examples of observed data from two participants in the PREDICT-HD study. HD Huntington disease
There are many other participants whose disease trajectories show similar deviations during followup. Table 2 represents the frequency of transitions between each possible pair of disease states based on the PREDICT-HD data. As can be seen from the table, most transitions show a progressive pattern, such as from unimpaired to impaired, or from impaired to HD diagnosis. However, many reverse transitions also occur, such as from impaired to unimpaired, or from HD diagnosis to impaired. Since the disease is relentless and there is currently no treatment, these reverse transitions can be treated as random deviations or measurement noise. The actual causes of the deviations in the observed disease trajectories are not clear. Some possible reasons include changes of performance of motor assessors, temporary effects of some drugs taken by the participants, or psychological variations over the participants' clinical visits.
Table 2.
A summary of the number of transitions between different states of motor impairment over followup in the PREDICT-HD study.
| To: Unimpaired | Impaired | HD | |
|---|---|---|---|
| From: Unimpaired | 757 | 449 | 6 |
| Impaired | 317 | 1768 | 236 |
| HD | 0 | 57 | 261 |
The examples in Figure 1 and the summary statistics in Table 2 indicate the observed data may not always show a progressive pattern [24, 25]. A disease course showing a strict progressive pattern can be considered as an idealized disease process. Such a process makes it possible to define HD diagnosis or duration in a disease state in an unequivocal and unique way. An idealized disease process can be derived based on the observed data, and this is one of the primary objectives of this study.
To model the idealized disease course of motor function based on the observed data in the context of expected measurement noise, we employ the hidden Markov model (HMM) [26, 27] for the statistical analysis. The HMM is a statistical model for a categorical response variable, such as disease status, that is measured repeatedly over time, where each outcome is subject to measurement noise. Each level of the categorical variable is called a state. A distinction is made between the noisy observation, which is called an observed state, and its noise-free counterpart, which is called the hidden state (or idealized state). Using the HMM model, the noise-free disease states can be derived from the noisy observations, and the transitions between idealized disease states, as well as factors affecting these transitions, can be systematically evaluated. When modeling the progressive nature of the disease course of motor function in HD, we assume that the idealized disease states cannot transition backward from HD diagnosis to impaired or from impaired to unimpaired. Under this assumption, each HD gene mutation carrier has a unique and unequivocally defined onset age of motor impairments and onset age of HD diagnosis.
Three individual-level covariates were considered in all analyses: CAG repeat length, age at each visit, and sex. All covariates entered the model in a linear fashion. Age was a time-varying covariate indexing the fact that transitions between disease states are age dependent. Both CAG and age were centered at their sample means, and the interaction between age and CAG was also included. The effects of these covariates were assumed to be different for distinct transitions. Estimation of covariate effects and prediction of years of onset were performed using the msm package [28] in the R computing program.
Results
Effects of CAG repeat length, sex and age on disease progression
The estimated covariate effects and their 95% confidence intervals (CIs) are summarized in Table 3. The statistical significance of a covariate effect is based on whether its CI contains 0 or not: if it contains 0, then it is not statistically significant. For the transition from the unimpaired to impaired state, the main effects of age and CAG are statistically significant, but their interaction effect is not. To be specific, for every one-year increase in age, the risk of transition to the next state (i.e., unimpaired to impaired) increases by 11%; whereas for one repeat length increase in CAG, the risk increases by 47%. For the transition from impaired state to HD diagnosis, the main effects of age and CAG as well as their interaction are all significant. Like the transition from unimpaired to impaired, the risk of transition increases with older age and longer CAG repeat length, but the amount of increase in the risk depends on individual age and CAG. For example, when CAG = 43, a one-year increase in age increases the risk of transition by 11%, and when CAG = 45, a one-year increase in age increases the risk of transition by 13%. The effect of sex is not significant for either transition.
Table 3.
Estimated covariate effects (95% CI) in transitions between disease states.
| Variable | Transition from unimpaired to impaired | Transition from impaired to HD |
|---|---|---|
| Age | 0.102 (0.067, 0.136) | 0.099 (0.074, 0.124) |
| CAG | 0.382 (0.241, 0.523) | 0.552 (0.450, 0.653) |
| Gender (Male) | -0.429 (-0.866, 0.008) | -0.216 (-0.539, 0.107) |
| Age*CAG | 0.003 (-0.007, 0.013) | 0.010 (0.002, 0.017) |
HD Huntington disease, CAG cytosine-adenine-guanine
Estimated probabilities of transitions
The probabilities of transition from one disease state to another are shown in the supplementary material. For illustrative purposes, we extracted the estimated 3-year, 5-year and 10-year transition probabilities for an individual at age 40 with CAG repeat length of 43. The 3-year benchmark was chosen to represent a feasible length of a clinical trial, whereas the 10-year benchmark was chosen because it is the longest follow up time of our sample (5 years is a convenient middle value). If the individual's motor function is currently unimpaired, the probabilities for this individual to be diagnosed in 3, 5, and 10 years are 4%, 10%, and 27%, respectively. On the other hand, if the individual already has symptoms and signs of motor impairments, the risk of being diagnosed in 3, 5, and 10 years increases to 19%, 30%, and 51%, respectively.
Estimated years to onset of motor impairments and HD diagnosis
The average number of years to first onset of motor impairments and to HD diagnosis for any specific individual can be estimated. Estimates are shown in the supplementary material for an individual at age 40 with CAG repeat length 43, and another individual at age 45 and CAG repeat length 45. When the individual with CAG = 43 and age = 40 currently has unimpaired motor function, his or her estimated average years to onset of motor impairments and HD diagnosis are approximately 6 and 21, respectively. If the individual already shows motor impairments, the estimated years to HD diagnosis are reduced to 14. For the individual with CAG = 45 and age = 45, if the individual currently has unimpaired motor function, his or her estimated average years to onset of motor impairments and HD diagnosis are approximately 2 and 5, respectively. If the individual already shows motor impairments, the estimated years to HD diagnosis is 3.
Discussion
Using data from the largest and longest-followed cohort of premanifest HD, we examined progression of motor impairment so that the onset of motor impairments and that of HD diagnosis can be defined and evaluated. Estimates of years to first onset of motor impairments and years to HD diagnosis are provided for a broad range of CAG repeat lengths and ages. Transitions from unimpaired to impaired motor functioning and from impaired motor signs to a traditional diagnosis of manifest HD are delineated.
As expected, findings confirmed that CAG plays a critical role in determining the onset of HD diagnosis, as reported in numerous previous studies. Although age and CAG have a significant interaction effect on HD diagnosis (as based on the DCL of 4 from the UHDRS), the interaction is not significant on the onset of “prediagnosis” motor impairments. The lack of significant interaction suggests that age and CAG impact the onset of motor impairments prior to a formal diagnosis in an independent way rather than dependent on each other. One reason could be that some motor signs and symptoms are age-related and increase with age irrespective of being at risk for HD. Another reason may be that since the onset of motor impairments represents the first appearance of impaired motor function, which usually occurs much earlier than HD diagnosis, the toxicity effect of CAG at that point has not accumulated enough such that its impact can be magnified by age.
Our investigation made novel and significant contributions to the study of motor progression in HD. In the literature, when predicting age of HD diagnosis, the current disease state of motor function is not considered [5, 12-18]. However, it is clear that for two individuals with identical demographic and genetic information, if one individual currently has unimpaired motor function while the other already shows motor impairments, it is very likely that the second individual will receive an HD diagnosis sooner than the first person. Current disease state summarizes the dynamic and most updated information on the disease process. Therefore, the incorporation of current disease state should provide a more accurate estimate of years to HD diagnosis. Findings in this paper provide benchmark data that may be informative for designing interventional clinical trials, such as the estimated years to onset of HD diagnosis and that of motor impairments. One potential use of our results is for identifying patients who are in a critical period that is amenable to therapeutic intervention. Another use is to assess the efficacy of an intervention in terms of extending the unimpaired period in motor function or delaying the onset of motor impairments or HD diagnosis.
In this study, we divide the disease course of motor function in the premanifest and prodromal phases into an unimpaired and impaired period based on the longitudinal DCL data. There are at least three advantages to this division. The first is that the presentation of motor signs and symptoms varies significantly among individuals, and no single sign or symptom manifests in all individuals affected by HD. Therefore, it is advantageous to have an unimpaired/impaired division so that the progressive states of motor dysfunction can be summarized and meaningfully compared among HD individuals. The second advantage is that the onset of motor impairments represents an important landmark event in disease progression. Before the occurrence of this landmark event, an HD individual has unimpaired motor function, just like other non-HD individuals; after the onset of this event, the individual starts to develop motor impairments. The duration of the unimpaired period and factors affecting the transition from unimpaired to the impaired period may provide crucial information on disease progression. These questions have not been extensively studied and the “unimpaired/impaired” division makes it possible to address these issues. The third advantage is that the division yields higher reliability of the data compared to using the raw DCL scores. As shown in the study of Hogarth et al. [25], the DCL levels of 1, 2 and 3 have relatively low reliability, and collapsing DCL levels between 0 and 4 clearly improves the reliability of the data.
Our study has two unique strengths. The first is that it has a large sample size, long followup time, and extensive followup visits for each participant. The second is that we account for measurement error in the data, so the disease progression of the motor phenotype can be appropriately reconciled with biological indicators of neurodegeneration. There are also limitations to our study. First, in the model we only considered linear terms of all variables, and nonlinear effects are certainly possible. Second, the range of CAG repeat length in our sample (38-52) is somewhat narrow, and findings might vary with different ranges. Third, some studies have indicated CAG repeat lengths at or above 40 are fully penetrant, whereas penetrance for lengths in the range of 36-39 (both inclusive) might be incomplete and are associated with later onset [29, 30]. We did not examine this issue in our study. Fourth, in the analysis we have not studied variables in other domains, such as cognitive, psychiatric and functional. As HD is associated with impairment in all these domains, it will be interesting to examine the relationships between progression of motor impairments and other measures of clinical and biologic phenotype.
In summary, by taking advantage of the data from one of the largest and longest followup studies in premanifest and prodromal HD, we systematically examined the following unique aspects of disease progression of motor dysfunction: the onset of motor impairments; the onset of HD diagnosis; the transition from the unimpaired period to impaired period of motor function and that from the impaired period to HD diagnosis; and the impact of age, sex and CAG on these transitions.
Supplementary Material
Acknowledgments
This research is supported by the National Institutes of Health, National Institute of Neurological Disorders and Stroke (5R01NS040068) awarded to Jane Paulsen; CHDI Foundation, Inc (A6266; A2015) awarded to Jane Paulsen; and Cognitive and Functional Brain Changes in Preclinical Huntington's Disease (HD) (5R01NS054893) awarded to Jane Paulsen.
We thank the PREDICT-HD sites, the study participants, the National Research Roster for Huntington Disease Patients and Families, the Huntington's Disease Society of America and the Huntington Study Group. This publication was supported by the National Center for Advancing Translational Sciences, and the National Institutes of Health (NIH), through Grant 2 UL1 TR000442-06. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
J. Paulsen is the principal investigator for PREDICT-HD and has therefore received grant funding from the National Institutes of Health/National Institute of Neurological Disorders and Stroke as detailed above. J. Paulsen has also served on an advisory board for Lundbeck, LLC and has a consulting agreement with ProPhase, LLC. J. Long has a consulting agreement with NeuroPhage, LLC.
Conflict of interest: All other authors declare no conflicts of interest.
Ethical standards: PREDICT-HD was approved by the institutional review boards at the University of Iowa and each participating site, and has therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. Written informed consent was obtained from each participant.
PREDICT-HD Investigators, Coordinators, Motor Raters, Cognitive Raters: Isabella De Soriano, Courtney Shadrick, and Amanda Miller (University of Iowa, Iowa City, Iowa, USA);
Edmond Chiu, Joy Preston, Anita Goh, Stephanie Antonopoulos, and Samantha Loi (St. Vincent's Hospital, The University of Melbourne, Kew, Victoria, Australia);
Phyllis Chua and Angela Komiti (The University of Melbourne, Royal Melbourne Hospital, Melbourne, Victoria, Australia);
Lynn Raymond, Joji Decolongon, Mannie Fan, and Allison Coleman (University of British Columbia, Vancouver, British Columbia, Canada);
Christopher A. Ross, Mark Varvaris, Maryjane Ong, and Nadine Yoritomo (Johns Hopkins University, Baltimore, Maryland, USA);
William M. Mallonee and Greg Suter (Hereditary Neurological Disease Centre, Wichita, Kansas, USA);
Ali Samii, Emily P. Freney, and Alma Macaraeg (University of Washington and VA Puget Sound Health Care System, Seattle, Washington, USA);
Randi Jones, Cathy Wood-Siverio, and Stewart A. Factor (Emory University School of Medicine, Atlanta, Georgia, USA);
Roger A. Barker, Sarah Mason, and Natalie Valle Guzman (John van Geest Centre for Brain Repair, Cambridge, UK);
Elizabeth McCusker, Jane Griffith, Clement Loy, Jillian McMillan, and David Gunn (Westmead Hospital, Sydney, New South Wales, Australia);
Michael Orth, Sigurd Süβmuth, Katrin Barth, Sonja Trautmann, Daniela Schwenk, and Carolin Eschenbach (University of Ulm, Ulm, Germany);
Kimberly Quaid, Melissa Wesson, and Joanne Wojcieszek (Indiana University School of Medicine, Indianapolis, Indiana, USA);
Mark Guttman, Alanna Sheinberg, Albie Law, and Irita Karmalkar (Centre for Addiction and Mental Health, University of Toronto, Markham, Ontario, Canada);
Susan Perlman and Brian Clemente (UCLA Medical Center, Los Angeles, California, USA);
Michael D. Geschwind, Sharon Sha, Joseph Winer, and Gabriela Satris (University of California, San Francisco, San Francisco, California, USA);
Tom Warner and Maggie Burrows (National Hospital for Neurology and Neurosurgery, London, UK);
Anne Rosser, Kathy Price, and Sarah Hunt (Cardiff University, Cardiff, Wales, UK);
Frederick Marshall, Amy Chesire, Mary Wodarski, and Charlyne Hickey (University of Rochester, Rochester, New York, USA);
Peter Panegyres, Joseph Lee, Maria Tedesco, and Brenton Maxwell (Neurosciences Unit, Graylands, Selby-Lemnos & Special Care Health Services, Perth, Western Australia, Australia);
Joel Perlmutter, Stacey Barton, and Shineeka Smith (Washington University, St. Louis, Missouri, USA);
Zosia Miedzybrodzka, Daniela Rae, Vivien Vaughan, and Mariella D'Alessandro (Clinical Genetics Centre, Aberdeen, Scotland, UK);
David Craufurd, Judith Bek, and Elizabeth Howard (University of Manchester, Manchester, UK);
Pietro Mazzoni, Karen Marder, and Paula Wasserman (Columbia University Medical Center, New York, New York, USA);
Rajeev Kumar, Diane Erickson, Christina Reeves, and Breanna Nickels (Colorado Neurological Institute, Englewood, Colorado, USA);
Vicki Wheelock, Lisa Kjer, Amanda Martin, and Sarah Farias (University of California, Davis, Sacramento, California, USA);
Wayne Martin, Oksana Suchowersky, Pamela King, Marguerite Wieler, and Satwinder Sran (University of Alberta, Edmonton, Alberta, Canada);
Anwar Ahmed, Stephen Rao, Christine Reece, Alex Bura, and Lyla Mourany (Cleveland Clinic Foundation, Cleveland, Ohio, USA);
Executive Committee: Principal Investigator Jane S. Paulsen, Jeffrey D. Long, Hans J. Johnson, Thomas Brashers-Krug, Phil Danzer, Amanda Miller, H. Jeremy Bockholt, and Kelsey Montross.
Scientific Consultants: Deborah Harrington (University of California, San Diego);
Holly Westervelt (Rhode Island Hospital/Alpert Medical School of Brown University);
Elizabeth Aylward (Seattle Children's Research Institute);
Stephen Rao (Cleveland Clinic);
David J. Moser, Janet Williams, Nancy Downing, Vincent A. Magnotta, Hans J. Johnson, Thomas Brashers-Krug, Jatin Vaidya, Daniel O'Leary, and Eun Young Kim (University of Iowa).
Core Sections: Biostatistics: Jeffrey D. Long, Ji-In Kim, Spencer Lourens (University of Iowa);
Ying Zhang and Wenjing Lu (University of Indiana). Ethics: Cheryl Erwin (Texas Tech University Health Sciences Center);
Thomas Brashers-Krug, Janet Williams (University of Iowa); and Martha Nance (University of Minnesota).
Biomedical Informatics: H. Jeremy Bockholt, Jason Evans, and Roland Zschiegner (University of Iowa).
References
- 1.Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-E. [DOI] [PubMed] [Google Scholar]
- 2.Walker FO. Huntington's disease. Lancet. 2007;369:218–228. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- 3.Roos RA. Huntington's disease: a clinical review. Orphanet J Rare Dis. 2010;5:40. doi: 10.1186/1750-1172-5-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huntington Study Group. Unified Huntington's Disease Rating Scale: reliability and consistency. Mov Disord. 1996;11:136–142. doi: 10.1002/mds.870110204. [DOI] [PubMed] [Google Scholar]
- 5.Penney JB, Jr, Young AB, Shoulson I, et al. Huntington's disease in Venezuela: 7 years of follow-up on symptomatic and asymptomatic individuals. Mov Disord. 1990;5:93–99. doi: 10.1002/mds.870050202. [DOI] [PubMed] [Google Scholar]
- 6.Louis ED, Lee P, Quinn L, Marder K. Dystonia in Huntington's disease: prevalence and clinical characteristics. Mov Disord. 1999;14:95–101. doi: 10.1002/1531-8257(199901)14:1<95::aid-mds1016>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 7.Kirkwood SC, Siemers E, Bond C, Conneally PM, Christian JC, Foroud T. Confirmation of subtle motor changes among presymptomatic carriers of the Huntington disease gene. Arch Neurol. 2000;57:1040–1044. doi: 10.1001/archneur.57.7.1040. [DOI] [PubMed] [Google Scholar]
- 8.Garcia Ruiz PJ, Hernandez J, Cantarero S, Bartolome M, Sanchez Bernardos V, Garcia de Yebenez J. Bradykinesia in Huntington's disease. A prospective, follow-up study. J Neurol. 2002;249:437–440. doi: 10.1007/s004150200035. [DOI] [PubMed] [Google Scholar]
- 9.Blekher TM, Yee RD, Kirkwood SC, Hake AM, Stout JC, Weaver MR, Foroud TM. Oculomotor control in asymptomatic and recently diagnosed individuals with the genetic marker for Huntington's disease. Vision Res. 2004;44:2729–2736. doi: 10.1016/j.visres.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 10.Biglan KM, Zhang Y, Long JD, Geschwind M, Kang GA, Killoran A, Lu W, McCusker E, Mills JA, Raymond LA, Testa C, Wojcieszek J, Paulsen JS PREDICT-HD Investigators of the Huntington Study Group. Refining the diagnosis of Huntington disease: the PREDICT-HD study. Front Aging Neurosci. 2013;5:12. doi: 10.3389/fnagi.2013.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biglan KM, Ross CA, Langbehn DR, Aylward EH, Stout JC, Queller S, Carlozzi NE, Duff K, Beglinger LJ, Paulsen JS PREDICT-HD Investigators of the Huntington Study Group. Motor abnormalities in premanifest persons with Huntington's disease: the PREDICT-HD study. Mov Disord. 2009;24:1763–1772. doi: 10.1002/mds.22601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andrew SE, Goldberg YP, Kremer B, Telenius H, Theilmann J, Adam S, Starr E, Squitieri F, Lin B, Kalchman MA, Graham RK, Hayden MR. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease. Nat Genet. 1993;4:398–403. doi: 10.1038/ng0893-398. [DOI] [PubMed] [Google Scholar]
- 13.Stine OC, Pleasant N, Franz ML, Abbott MH, Folstein SE, Ross CA. Correlation between the onset age of Huntington's disease and length of the trinucleotide repeat in IT-15. Hum Mol Genet. 1993;2:1547–1549. doi: 10.1093/hmg/2.10.1547. [DOI] [PubMed] [Google Scholar]
- 14.Lucotte G, Turpin JC, Riess O, Epplen JT, Siedlaczk I, Loirat F, Hazout S. Confidence intervals for predicted age of onset, given the size of (CAG)n repeat, in Huntington's disease. Hum Genet. 1995;95:231–232. doi: 10.1007/BF00209410. [DOI] [PubMed] [Google Scholar]
- 15.Foroud T, Gray J, Ivashina J, Conneally PM. Differences in duration of Huntington's disease based on age at onset. J Neurol Neurosurg Psychiatry. 1999;66:52–56. doi: 10.1136/jnnp.66.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Squitieri F, Sabbadini G, Mandich P, Gellera C, Di Maria E, Bellone E, Castellotti B, Nargi E, de Grazia U, Frontali M, Novelletto A. Family and molecular data for a fine analysis of age at onset in Huntington disease. Am J Med Genet. 2000;95:366–373. doi: 10.1002/1096-8628(20001211)95:4<366::aid-ajmg13>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 17.Langbehn DR, Hayden MR, Paulsen JS PREDICT-HD Investigators. CAG-repeat length and the age of onset in Huntington disease (HD): A review and validation study of statistical approaches. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:397–408. doi: 10.1002/ajmg.b.30992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andresen JM, Gayan J, Cherny SS, Brocklebank D, Alkorta-Aranburu G, Addis EA, Cardon LR, Housman DE, Wexler NS. Replication of twelve association studies for Huntington's disease residual age of onset in large Venezuelan kindreds. J Med Genet. 2007;44:44–50. doi: 10.1136/jmg.2006.045153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paulsen JS, Magnotta VA, Mikos AE, Paulson HL, Penziner E, Andreasen NC, Nopoulos PC. Brain structure in preclinical Huntington's disease. Biol Psychiatry. 2006;59:57–63. doi: 10.1016/j.biopsych.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 20.Paulsen JS, Langbehn DR, Stout JC, Aylward E, Ross CA, Nance M, Guttman M, Johnson S, MacDonald M, Beglinger LJ, Duff K, Kayson E, Biglan K, Shoulson I, Oakes D, Hayden M PREDICT-HD Investigators and Coordinators of the Huntington Study Group. Detection of Huntington's disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry. 2008;79:874–880. doi: 10.1136/jnnp.2007.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paulsen JS, Long JD, Johnson HJ, Aylward EH, Ross CA, Williams JK, Nance MA, Erwin CJ, Westervelt HJ, Harrington DL, Bockholt HJ, Zhang Y, McCusker EA, Chiu EM, Panegyres PK PREDICT-HD Investigators Coordinators of the Huntington Study Group. Clinical and Biomarker Changes in Premanifest Huntington Disease Show Trial Feasibility: A Decade of the PREDICT-HD Study. Front Aging Neurosci. 2014;6:78. doi: 10.3389/fnagi.2014.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paulsen JS, Hayden M, Stout JC, Langbehn DR, Aylward E, Ross CA, Guttman M, Nance M, Kieburtz K, Oakes D, Shoulson I, Kayson E, Johnson S, Penziner E Predict-HD Investigators of the Huntington Study Group. Preparing for preventive clinical trials: the Predict-HD study. Arch Neurol. 2006;63:883–890. doi: 10.1001/archneur.63.6.883. [DOI] [PubMed] [Google Scholar]
- 23.Warner JP, Barron LH, Brock DJ. A new polymerase chain reaction (PCR) assay for the trinucleotide repeat that is unstable and expanded on Huntington's disease chromosomes. Mol Cell Probes. 1993;7:235–239. doi: 10.1006/mcpr.1993.1034. [DOI] [PubMed] [Google Scholar]
- 24.Witjes-Ane MN, Mertens B, van Vugt JP, Bachoud-Levi AC, van Ommen GJ, Roos RA. Longitudinal evaluation of “presymptomatic”; carriers of Huntington's disease. J Neuropsychiatry Clin Neurosci. 2007;19:310–317. doi: 10.1176/appi.neuropsych.19.3.310. [DOI] [PubMed] [Google Scholar]
- 25.Hogarth P, Kayson E, Kieburtz K, Marder K, Oakes D, Rosas D, Shoulson I, Wexler NS, Young AB, Zhao H. Interrater agreement in the assessment of motor manifestations of Huntington's disease. Mov Disord. 2005;20:293–297. doi: 10.1002/mds.20332. [DOI] [PubMed] [Google Scholar]
- 26.Rabiner LR. A tutorial on hidden Markov models and selected applications in speech recognition. Proceedings of the IEEE. 1989;77:257–286. doi: 10.1109/5.18626. [DOI] [Google Scholar]
- 27.Zucchini W, MacDonald IL. Hidden Markov models for time series : an introduction using R. CRC Press; Boca Raton: 2009. [Google Scholar]
- 28.Jackson CH. Multi-State Models for Panel Data: The msm Package for R. Journal of Statistical Software. 2011;38:1–28. [Google Scholar]
- 29.Rubinsztein DC, Leggo J, Coles R, et al. Phenotypic characterization of individuals with 30-40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36-39 repeats. Am J Hum Genet. 1996;59:16–22. [PMC free article] [PubMed] [Google Scholar]
- 30.Sequeiros J, Ramos EM, Cerqueira J, Costa MC, Sousa A, Pinto-Basto J, Alonso I. Large normal and reduced penetrance alleles in Huntington disease: instability in families and frequency at the laboratory, at the clinic and in the population. Clin Genet. 2010;78:381–387. doi: 10.1111/j.1399-0004.2010.01388.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.