

Abstract
Phagocytes are key cellular participants determining important aspects of host exposure to nanomaterials, initiating clearance, biodistribution and the tenuous balance between host tolerance and adverse nanotoxicity. Macrophages in particular are believed to be among the first and primary cell types that process nanoparticles, mediating host inflammatory and immunological biological responses. These processes occur ubiquitously throughout tissues where nanomaterials are present, including the host mononuclear phagocytic system (MPS) residents in dedicated host filtration organs (i.e., liver, kidney spleen, and lung). Thus, to understand nanomaterials exposure risks it is critical to understand how nanomaterials are recognized, internalized, trafficked and distributed within diverse types of host macrophages and how possible cell-based reactions resulting from nanomaterial exposures further inflammatory host responses in vivo. This review focuses on describing macrophage-based initiation of downstream hallmark immunological and inflammatory processes resulting from phagocyte exposure to and internalization of nanomaterials.
Keywords: macrophage, clearance, circulation, toxicity, biodistribution, imaging, drug delivery
Graphical Abstract
1. Introduction
Nanoparticles in diverse forms and designs have substantial clinical potential; they may eventually be capable of directing specific cell recognition, internal trafficking and processing pathways. Additionally, nanoparticles may be able to overcome traditional drug delivery roadblocks that currently prevent many types of treatments from becoming viable therapies. For example, hydrophobic drugs, nucleic acids and proteins have been encapsulated within nanoparticles to reduce intracellular degradation, increase circulation times and improve therapeutic characteristics[1]. In 2013, 241 companies and institutions had a total of 789 ongoing clinical trials and 103 unique nanomedicine investigational products[2], seeking to exploit unique delivery properties.
However, despite promising potential and sharp increases in clinical trials and investigational products, only 38 products have received Food and Drug Administration (FDA) regulatory approval for patient use across 60 years of investigational research [2]. Nanomedicine translation faces significant challenges related to more efficient clinical development when compared to the 32 new but traditional small molecular weight therapeutics receiving FDA approval in 2012 alone. Nanomaterials, unlike small molecular weight, soluble therapeutics, approximate the same size scale as biologics, do not permeate epithelial membranes (gut, skin, eye) efficiently, and often are decorated with diverse non-specifically adsorbed host proteins, increasing their non-specific cellular interactions. Both size and physical properties (e.g., materials chemistry, interfacial properties, particle transport dynamics) are often used to explain limitations in clinical translation, related to their observed increased cellular recognition that prompts host recognition, nanoparticle clearance and associated inflammatory effects [3, 4]. Issues plaguing nanomaterials circulation and targeting in humans can be attributed to rapid vascular filtration and clearance of therapeutics and diagnostics, and to induction of host inflammatory responses due to non-specific recognition and uptake of nanoconstructs by macrophages in vivo [5–11]. Rapid blood clearance limits nanomaterial accumulation at target delivery sites; nanoparticle accumulation in macrophages within clearance organs initiates inflammatory responses, inducing toxicity [12–22]. A historical review of published literature indicates that approximately 95% of systemized nanomaterials doses are sequestered by filtration organs and never reach their intended targets [23–25]. This outcome is generally non-distinguishable from biodistributions of circulating microparticles studied in vivo for decades, and importantly, clinically insignificant for many drug classes in their therapeutic value [26].
Because mammals have been environmentally and continuously exposed to a dizzying array of nanomaterials for millennia (e.g., air-borne, water-borne, food-borne nanomaterials of many metallic siliceous, and carbonaceous forms) without significant apparent overt toxicity, mammalian immunological surveillance systems must have evolved mechanisms to tolerate or eliminate adventitious, ambient assaults from daily particle burdens [27–30]. Simple combinations of environmentally ubiquitous nanomaterials and abundant microbes would also suggest that nanoparticles carrying fragments of microbial organisms (e.g., antigens, nucleic acids and membrane chemistry) known to be highly immune-provocative to mammals (e.g., eDNA, dsRNA, endotoxins, exotoxins) would be subject to host immune processing and neutralization as a routine survival function. Therefore, host mechanisms for particle processing are, at some level, highly evolved and difficult to by-pass, despite the best efforts of materials engineering [27–30].
Nanoparticle association with the host highly evolved mononuclear phagocytic system (MPS) is a function of particle opsonization upon contact with blood and rapid recognition of these opsonins via the MPS [31, 32]. This is particularly observed in structurally distinct fenestrated vasculature via liver Kupffer cells and splenic macrophages [33, 34]. If these macrophagic cells are indeed responsible for high particle clearance rates, disappointing imaging and therapeutic efficacy due to poor delivery efficiencies to specific targets and increased clearance organ accumulation are anticipated. Nanoparticle delivery vehicles designed to either avoid or specifically harness this host recognition system could improve payload delivery, reduce inflammatory effects and improve imaging and drug efficacy. However, to rationally design these improved systems, better understanding is needed of nanoparticle-macrophage interactions both at cellular and system-wide levels in physiological conditions.
Macrophages recognize opsonized proteins, specific surface chemistries, and other surface and biological characteristics that mark these nanoparticles, similar to analogous microparticle precedents, for clearance and/or toxicological fates[2, 29, 35–39]. Particle physicochemical characteristics can influence these interactions and may also potentiate toxicological mechanisms [2, 28, 38, 40–48]. What is not understood is how nanoparticle surfaces interact with the complex biological environment to influence phagocytic recognition, clearance, cellular processing and toxicological fates. Developing correlations between nanoparticle physicochemical characteristics and nanoparticle uptake, processing and clearance mechanisms in macrophages would provide a basis to overcome decades of frustration in particle systemic delivery and targeting, and facilitate design of new, more efficacious and safer nanomaterial platforms.
Mesothelioma, pneumoconiosis, and silicosis are clinically relevant well known disease states that occur after post-environmental particulate exposure. These conditions share common features of morbidity, i.e. initiation of inflammation and presentation many years after initial exposure [49–51]. Development of inflammatory-mediated and damage from unresolved oxidative stress mechanisms is a chronic issue, distinct from acute effects in exposure, response, and cumulative pathology. While particles and their associated disease etiologies might be very different from engineered nanomaterials introduced more recently, that the initial phases of these well-studied diseases follow similar patterns to what is reported for acute toxicity studies of engineered nanoparticles is concerning. Recent evidence suggests that long-term silica residence within MPS/RES clearance organs, including the lung, liver and spleen, initiate fibrotic-like lesions via infiltration and microgranulation of hepatocytes (in the liver) and long-term inflammatory responses and recruitment of macrophages/leukocytes [8, 51, 52]. Nanoparticles in circulation share many clearance mechanisms and fates of their microparticle analogs. Inhalation of nanoparticles has also initiated fibrotic-like lesions within lung tissue [50, 53]. Interestingly, fibrotic lesion production can be mitigated with particle surface modification (i.e., hydrophobicity and charge). For example, lung fibrosis was a hallmark for cationic silica nanoparticles, while those with polar or anionic surfaces tended to migrate to the mediastinal lymph nodes [54]. Nonetheless, chronic inhaled exposure to nanomaterials is shown to elicit deleterious lung effects from on-going oxidative stress, enhancing pro-inflammatory effects in airways of chronic obstructive pulmonary disease (COPD) patients[55]. Additionally, detrimental cardiovascular effects from inhaled nanomaterials exposure are observed in epidemiological studies, attributed to particle translocation across the respiratory epithelium into circulation and subsequent toxicity to vascular endothelium, disruption of normal blood coagulation, and changes in autonomic nervous functions that eventually alter cardiac frequency and function[55]. This suggests that the body has developed a common local response to isolate these foreign particulate materials from host biological environments. Clearly, controlling biological fate for engineered nanomaterials requires focus on specific systemic processing mechanisms and their chronic consequences in either storage or elimination. Unanswered issues are how host local particle responses consequent to materials clearance, isolation, and elimination (i.e., oxidative stress, inflammatory, immune responses long term) are affected by nanomaterials chemistry, their morphologies or their biological conditioning, and how any of these downstream events initiate adverse chronic problems. Even more concerning are local particle clearance responses that might promote cancer and immunological disorders. Ultimately, rational design criteria for safe, effective nanomaterials with minimal human exposure risks, perhaps degradable inorganic or polymer systems, that reliably avoid adverse processing and long-term pathological responses are critical.
In this review, we argue that much nanoparticle drug delivery and imaging literature neglects defining the precise and defining relationships between host macrophages and in vivo nanoparticle processing, resulting in continued deficiencies in nanoparticle designs that target or circulate widely, and therefore also limit clinical reliability and translation. Additionally, the current lack of understanding of risks of chronic toxicity from on-going or repeated nanoparticle exposures to host filtration organs and macrophages could have serious consequences and impede clinical advancement. Key mechanistic details that initiate potentially toxic outcomes remain unknown. Long-term nanomaterial fates in clearance organs and possible risks for patients in both acute and chronic exposures are generally unexplored. In vitro and preclinical studies have not yet effectively recapitulated these exposures. This review identifies both nanomaterials failures and successes as a function of macrophage-nanomaterial interactions and resulting potential inflammatory effects, with the goal of improving therapeutic translational capacity.
2. What is a phagocyte/macrophage?
Macrophages and other phagocytes are leukocytic cells capable of phagocytizing or taking up bacteria, cellular debris, and particles through energy-consuming membrane-engulfing as a characteristic phenotype [56–58]. Their primary role is early response to foreign material contamination and its clearance. Macrophages have been known to uptake foreign materials within a matter of minutes, increasing their rates of phagocytosis for positively charged and bacteria-specific proteins [59, 60]. Understanding how macrophages/phagocytes identify and rapidly clear nanoparticles in vivo requires insights into macrophage biology and key cellular characteristics used to identify and ingest foreign materials. Interestingly, the macrophage is a widely classified cell type, encompassing specialized tissue resident macrophages that maintain biological hemostatic detox (i.e. red pulp and Kupffer macrophages lining the fenestrated endothelium of the spleen and liver) and neurological function (microglia of the brain), and the chameleon-like hematopoietic monocytes that differentiate into macrophages and take up residence within most tissues with a multitude of functions[61]. What is not well known is how these cellular features and phenotypes influence cellular-nanoparticle interactions, at least in sufficient detail required for informing nanomaterials design. Ideally, reliable correlations should be drawn between nanoparticle-macrophage interactions and in vivo toxicity and delivery fates. This current evidence is either largely empirical in vivo data, or derived from simplistic model in vitro studies involving monocultured monocyte lines in simplified media on plastic dishes. Relevant context and conditions are largely missing. More informative data are required for controlling nanomaterial circulation and safety.
Macrophages are key in vivo participants in normal inflammatory and immunological processes [62, 63]. As active phagocytes, they display a spectrum of phenotypes, spanning pro-inflammatory to prohealing, and appear capable of reversible transformations between different distinct functional forms [64]. Certain macrophage forms are essential for the destruction and removal of hazardous materials, pathogens, and damaged or abnormal tissues [65, 66]; these native roles are also likely involved in nanoparticle processing. Macrophages also play an essential role in normal wound healing, prompting local angiogenesis and tissue neogenesis. Known also to play a primary role in the macroscale foreign body response to engineered biomaterial implants, [67] macrophages initiate local fibrosis and unresolved chronic inflammation around implants that is not readily eliminated. Evidence suggests that local microenvironmental factors and cues drastically alter macrophage phenotype and differentiation states [68]. This includes altered macrophage morphology, surface receptor expression and function [62, 69], that ultimately affect material recognition and uptake patterns necessary for nanoparticle interactions[70] and nanomaterials processing in vivo.
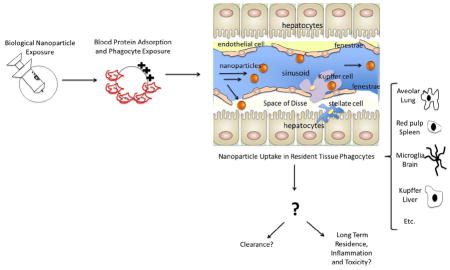
Mature macrophages are terminally differentiated forms of circulating hematopoietic premature precursor monocytes [71] or derive from the tissue precursors in which they reside [72]. Both blood-derived and tissue-resident macrophages participate in macrophage-nanoparticle interactions. Multiple phagocytic model systems from a variety of tissue types (Figure 1) are required to understand macrophage-nanoparticle interactions mechanistically. Discussion of functional roles of tissue-specific macrophages is beyond the scope of this review; readers are directed to excellent, more specific reviews on macrophage populations and their distinct roles [73, 74].
Figure 1.

(Left) Cartoon depicting examples of the body’s resident tissue macrophages responsible for nanoparticle tissue site clearance and inflammatory activation; (Right) Images of silica nanoparticles internalized within macrophages in the liver and spleen (reprinted with permission from [70]).
2.1 Macrophages and their extracellular environment
Macrophage responses to local environmental stimuli impact nanomaterials fates in vivo; yet, altering how macrophages engage nanomaterials requires some improved insights into recognition and processing mechanisms in the context of their local environment. Recent studies illustrate the need to better understand macrophage states in vivo and how macrophage responses to unrelated factors may potentially drastically alter their quiescence, activation and phagocytosis rates. Very efficient nanoparticle uptake and scavenging as a natural MPS function has been consistently shown for decades to efficiently clear nearly all systemically dosed particles (i.e., up to 95% clearance regardless of chemistry [75]), drastically undermining claims for particle “targeting” and nanoparticle therapeutic efficacies [3, 4, 76]. For example, knocking out macrophage-specific scavenger receptors has shown reduced nanoparticle uptake and even reduced proinflammatory signal release [18]. This suggests that particle filtration may also potentiate local inflammatory effects [12, 20, 22]. Local activation of normally quiescent tissue macrophages may provoke rearrangements of their surface receptors, making them more sensitive to nanoparticle recognition, and leading to abnormally increased internalization rates in phagosomes — cellular vacuoles containing phagocytosed material. Increased clearance rates were observed in animals treated with bacterial polymer and known activating agent, zymosan, to activate local macrophages, mimicking natural systemic inflammation. This appears to be opsonization-independent and is enhanced compared to control particle treatments [77]. A similar study evaluated the priming effect of proinflammatory cytokine, interferon(INF)-gamma, released in systemic inflammation, on increased uptake of chitin particles in an alveolar macrophage model [78]. This priming enhanced uptake within the macrophages of clearance organs and likely polarized them to a more phagocytic phenotype. This heightened phagocytic activity accounts for the observed increase in particle uptake. However, others have also shown that reduced nanoparticle phagocytosis occurs in senescent macrophages, suggesting that native terminally differentiated tissue cells may not illicit these responses [79]. These studies illustrate how systemic inflammation and macrophage state might alter particle biodistribution, clearance and resulting efficacies. Hence, specific macrophage phenotypes in the target disease state or particle uptake location should be carefully assessed as influenced by the global state of the host.
2.2 Macrophage phenotype as influenced by environmental factors
Macrophage differentiation, polarization states, and resulting activation drastically alter nanoparticle uptake and biological responses [19, 63, 68]. Understanding this cause-effect can improve particle biodistribution, clearance, targeting and delivery. The cell’s polarization state has also been shown to influence local inflammation and may potentiate local adverse events [64, 80].
Macrophage phenotypes are heterogeneous in vivo. Ongoing debate exists on how best to categorize this heterogeneity [81]. Many researchers utilize the Th1/Th2 paradigm: that macrophages reside within a spectrum defined on one end by a classically activated state (M1) and on the other end by the alternatively activated state (M2). Though the Th1/Th2 model system has been increasingly asserted to be over-simplified [81], we have chosen to use this generalization as one commonly applied to simplify discussion. M1 macrophages are induced via cytokine INF-gamma, representing traditional immune-activating phagocytic janitorial cells[64]. In contrast, M2 macrophages are induced via cytokine interleukin four/thirteen (IL-4/IL-13) exposure and activate angiogenesis, immune suppression and tissue break down/remodeling[63] (Figure 2). Th1/Th2 classification system can be used to study how environmental factors influence nanoparticle cellular processing. For example, in vitro monocultures of M1 macrophages provide the most robust and drastic response to titanium particulate exposure compared to M2 cultures [19]. M1 macrophages also increase inflammatory cytokine and chemokine production, while M2 cells, despite internalizing particles, show a broadly suppressed inflammatory response. This difference suggests that microenvironmental factors capable of inducing phenotypic changes can also influence how cells, particularly macrophages, might respond locally to systemic particulate exposure [19]. Use of multiple co-culture macrophage phenotypes in vitro might ensure that relevant correlative in vivo information is derived. Variations in particle surface characteristics and/or adsorbed proteins may alter how nanoparticles interact with each polarized macrophage phenotype, and the spectrum in between.
Figure 2.

A) Macrophage polarization (M1 versus M2 traditional dichotomy) plays an important role in the uptake of nanoparticles [70, 82]. Traditional polarization of macrophages to an M1 phenotype enhances nanoparticle uptake, while M2 polarization appears to reduce particle uptake. B) This cell uptake variation may be explained by variations in functions of these macrophage polarization states outlined briefly in this cartoon (adapted with permission from [70]).
3. Recognition of nanomaterials by phagocytes
In vivo, the host particle surveillance and clearance systems (i.e., MPS or tissue-resident phagocytes) do not encounter bare nanomaterials. The immediate host biological conditioning produces protein adsorption to the biomaterial surface upon blood or tissue contact [83, 84].
3.1 Recognition of nanomaterials via protein adsorption
The adsorbed protein coating, referred to as “corona” in the nanomaterials literature and also “opsonins” in the drug delivery literature, matures over time in vivo to an equilibrium state largely unknown for nanomaterials in blood. This time-dependent blood protein adsorption process, and what characteristics of nanoparticles initiate desirable and adverse effects, as well as how presence and conformation of adsorbed proteins influence the presentation of nanoparticles to phagocytes will be increasingly important to understand as a determinant of their clearance [27, 28, 35]. Protein opsonization is rapid, and has been well-known to “prime” particles for MPS recognition and clearance [85, 86]. Controlling this ubiquitous protein recognition will facilitate design of new nanoparticles that might not be cleared too rapidly or induce adverse toxicological effects. Both adsorbed protein composition and conformation on the particle surface influence how nanoparticles interact with macrophage surface receptors and mediate phagocytic recognition [27, 28, 35]. For example, the positive collagen domains of macrophages’ surface scavenger receptors (SR AI/II) are known to uptake polyanionic charged materials [87]. Depending on the nature of protein adsorption to the surface, protein conformational changes occur that elicit unnatural protein conformations during opsonization that initiate phagocytosis and potentially inflammatory reactions[27, 28, 35].
Physicochemical interfacial characteristics are known to be important mediators of protein adsorptive and opsonization properties. Adsorbed protein conformational changes can result from surface curvature, topography and surface energy/hydrophobicity (Figure 3) [88, 89]. In order to clearly understand these influences on opsonization, any nanomaterial must first be characterized to assert surface composition and the actual surface chemistry that interacts with biology [90]. While correlations have been made for particles and specific serum protein adsorption behaviors in highly simplified experimental studies, few reports characterize actual whole plasma or whole blood-particle opsonization and its effects on phagocytes as a function of particle size, chemistry and time. Hence, in vivo correlations are largely unsupported by in vitro models.
Figure 3.

Schematic illustration of the ways that particle physicochemical characteristics can influence degrees and conformation of adsorbed proteins. Surface curvature, topography, hydrophilic/hydrophobic chemistry and polymer coating steric barriers on nanoparticle surfaces are shown to alter amounts and conformations of proteins adsorbed to surfaces. Surface-adsorbed proteins (opsonins) influence macrophage recognition and uptake of nanoparticles [28, 32, 91]. Additionally, conformational protein rearrangements on nanoparticle surfaces alters protein epitope exposures to phagocytes [83, 84]. Certain epitopes have the capacity to activate macrophages[31].
Adsorbed protein conformation can be affected by particle surface curvature [92, 93]. For example, Vertegel et al. [92] have found that the small enzyme, lysozyme, adsorbs to silica nanomaterials with diameters as small as 4 nm and as large as 100 nm. They further observed that smaller nanoparticles with higher surface curvatures retained native conformational lysozyme to a greater degree than larger nanoparticles with less surface curvature. Albumin was also observed to retain its native conformation on particles with a higher surface curvature while fibrinogen exhibited the opposite effect [94]. These studies suggest that protein adsorption onto nanoparticle surfaces could potentially be manipulated through controlling surface curvature, although current work is limited to model protein solutions lacking relevance to blood’s complexity. Translation of dilute or single protein adsorption studies to nanomaterials in multicomponent milieu similar to bodily fluids is not well developed or understood.
Protein adsorption and subsequent phagocytic uptake also appear dependent upon particle surface topography [95]. Bovine serum albumin (BSA), for example, adsorbs more heavily to rough platinum surfaces compared to smooth platinum surfaces [96]. Interestingly a separate study claimed that increased BSA adsorption can explain why rough gold nanomaterials showed reduced cell uptake when compared to smooth gold nanorods and spheres in neuronal microglial cell cultures [12].
Changing particle surface energies (e.g., hydrophilicity/hydrophobicity) and imposing immobilized steric barriers (e.g., grafted polymer brush surfaces) may also decrease protein adsorption and subsequent phagocytic recognition of nanomaterials. For example, hydrophilic poly(ethylene glycol) (PEG) has often been immobilized in many forms and approaches to provide a brush-like steric barrier that is shown to reduce protein adsorption and is correlated with increased blood circulation times for some particles [97]. Dextran layers employed on commercial iron oxide MRI agent nanoparticles (i.e., Feridex™) may serve the same role [98].
Nanoparticle curvature, topography and surface energy represent only a few select physicochemical characteristics that can be altered to modify nanomaterials interfacial adsorption processes with proteins that affect their biological interactions [88, 89]. Other characteristics could potentially alter these profiles as well. Nanoparticle protein adsorption is reviewed extensively elsewhere [38, 39, 97, 99]. While protein adsorption conditions the nanoparticle surface appearance to macrophages, macrophages must also recognize these surfaces to facilitate their uptake by phagocytosis. These surface adsorption characteristics are diverse, even in simple model systems, and therefore currently unpredictable in the presence of biological milieu. Hence, presentation of biologically processed nanoparticles to phagocytes yields widely disparate states and profiles, possibly passive, or alternatively, initiating an array of events ranging from possible bio-invisibility to complement activation to oxidative stress to overt toxicity, eliciting a spectrum of possible host responses.
3.2: Recognition of nanoparticles via phagocyte surface receptors
If macrophage particle uptake correlates to nanomaterial in vivo performance, then understanding macrophage recognition of circulating foreign materials may help to elucidate how nanomaterials are cleared or elicit toxicity. Macrophages have evolved distinct pathogenic and foreign material recognition mechanisms [100–102]. These endogenous processes and patterns are likely important to nanomaterial host recognition as well. Host foreign body recognition and pathogenic processing are valuable mechanisms worthy of analysis to inform how nanomaterials are handled by phagocytes. This could provide guidelines for direct manipulation of particle physicochemical characteristics to minimize phagocytic uptake and reduce potential inflammatory-mediated events, increasing technological capabilities of nanomaterials for healthcare applications.
Many nanomaterial uptake and cellular processing mechanisms parallel normal immunological pathogenic processing, suggesting conservation in cellular recognition and pathway regulation. A variety of native surface receptors, called pattern-associated recognition receptors (PRRs), are able to recognize antigenic or epitope presentation patterns from pathogen surfaces or within damaged tissues [103]. Pathogen surface patterns are conserved across a variety of microorganisms, termed pathogen-associated molecular patterns (PAMPs). PAMPs identify injury or cell death patterns, termed damage-associated molecular patterns (DAMPs)[104]. DAMPs usually correspond to host tissues undergoing necrosis and as host-elicited danger signals that initiate local recruitment of immune cells. Because foreign material, pathogens and damaged native tissues present patterns recognized by phagocyte surface receptors, nanoparticles could also potentially present analogous molecular patterns due to their protein adsorption, or associated specifically to the raw material physicochemical properties. Further, these patterns could potentially initiate normal inflammatory events mediated by phagocytic cells. Four specific macrophage surface receptors include: (1) toll-like receptors, (2) mannose receptors, (3) scavenger receptors, and (4) Fc receptors (Table 1 and Figure 4).
Table 1.
Features of primary phagocytic receptors responsible for nanoparticle-mediated cellular uptake.
| Receptor Type | Receptor Subtype | Receptor Description |
|---|---|---|
| Toll-like Receptors | TLR 1,2,4,5,6 | Located on surface of phagocytes [105] |
| TLR 3,7,8, 9 | Found within intracellular compartments [105] | |
| TLR 2,3,7 | Induce cell survival or inflammatory reducing mechanisms such as autophagy [106, 107] | |
| TLR 4 | ||
| Mannose/Lectin Receptors | Recognize complex carbohydrates, detecting mannose, glucose or sugar structures on pathogenic material and glycoproteins [112]. | |
| C-type lectin | Have been engaged by decorating NP surfaces with di-mannose and galactose [113] | |
| Sugar lectin-mannose | Interactions with macrophages can be harnessed with nanoparticulates decorated with large numbers of sugar-like motifs [114, 115] | |
| Mannan | Has been used to coat gelatin nanoparticles to increase specificity of delivery of didanosine to macrophages for the treatment of HIV [116]. These nanoparticles substantially increased the amount of drug delivered to the brain, lymphatics and splenic tissue regions, increasing specificity of delivery and decreasing systemic side effects [117]. |
|
| Mannose (CD206) | ||
| Carbohydrate surface modification |
|
|
| Scavenger receptors |
|
|
| SR-AI/II scavenger receptors | ||
| MARCO | ||
| Fc Receptors (CD64 ) |
Figure 4.

Phagocytic-particle recognition is responsible for nanoparticle clearance and internalization. Macrophage surface receptors are depicted here that could be responsible for nanoparticle recognition. Each receptor that recognizes the nanoparticle will induce a specific internalization mechanism, outlined in the diagram. Downstream inflammatory effects, triggered by receptor engagement, can be induced via these internalization pathways.
3.2.1. Comparative importance of macrophage surface receptors
While all of these cellular receptors can participate in nanoparticle uptake into macrophages, Fc and mannose receptors might play more significant roles than others. Comparative phagocytosis studies have studied rates of uptake between scavenger, mannose and Fc receptors. Nanoparticles targeted to mannose and Fc receptors appear to be internalized rapidly, while scavenger receptors require significantly longer times [131]. This suggests that Fc and mannose receptors are better poised to efficiently internalize nanoparticles. Fc and mannose receptors expression on macrophages is variable and transient, and expression levels for each receptor type vary widely between polarization and activation states of these cells [131]. Additionally, many of these macrophage surface receptors have evolved to detect opsonized materials to increase nanoparticle uptake and clearance; many receptors may work in parallel. Thus, particle targeting via directed particle surface chemistry and protein adsorption may be accomplished by controlled protein adsorption and surface engineering that selectively adsorbs certain proteins in conformationally “appropriate” (non-activating) states selective for specific cell receptor recognition. For example, proteins IgG and C3b induce phagocytosis through the specific phagocyte Fc and complement receptors [131], a strategy that could then be used to target macrophage uptake directly. However, macrophages also uptake nanoparticles in the absence of serum proteins, suggesting these four receptors also intrinsically recognize native nanomaterials [132]. In some cases, adsorbed proteins can also mask materials from surface receptor recognition [133]. An understanding of which proteins and what adsorbed conformations provide this masking are important to elicit nanomaterial “bioinvisibility” in the host.
4. Phagocytic internalization mechanisms
To limit, control or select nanomaterial uptake mechanisms and how they direct intracellular processes and fate, nanoparticle delivery could be directed with higher efficiency to specific cell populations (e.g., disease states) and cellular compartments. This capability would greatly improve drug delivery. Some drugs act only at specific disease sites and within cellular organelles and require internal trafficking efficiently to avoid off-site problems. For example, nucleic acid therapies act primarily through cell nuclear localization. If trafficked instead to the cytosol, to autophagosomes or lysosomes, they will remain therapeutically silent and risk toxicity. Understanding this trafficking process -- from outside to inside and further within cellular targets -- while mitigating adverse events created by nonspecific delivery, will yield important clues for nanomaterials cellular fates. Cellular recognition of foreign materials via pattern recognition receptors prompts material internalization within phagocytes, exploiting active cellular cytoskeletal rearrangements in the cytosol, cell membrane invagination around the foreign material and internal cell vesicle development. These cell internalization mechanisms are broadly classified as either phagocytosis or pinocytosis depending on receptors, membrane mechanisms and intracellular disposition (Figure 5).
Figure 5.

Dissecting the various nanoparticle entry mechanisms into cells. (Top): Confocal images depicting actin polymerization staining within RAW 264.7 cells (an adherent murine monocyte/macrophage line), actin is in red, nanoparticles are in green and the nucleus is depicted in blue. The image on the left helps to visualize clathrin-mediated endocytosis of nanoparticles under 200nm through bowl-like invaginations. The image on the right depicts phagocyte-mediated uptake of nanoparticles larger than 200nm through protrusions; (Middle) transmission electron microscopy image depicting nanoparticle cellular entry, left appears to be through invagination-like mechanisms and right through protrusion-like mechanisms; (Bottom): This image displays the various forms of cellular uptake of nanoparticles. Depicted are (A) phagocytosis, (B) macropinocytosis, (C) clathrin-mediated endocytosis, (D) clathrin- and caveolae-independent endocytosis, (E) caveolae-mediated endocytosis, (F) passive membrane movement (adapted with permission from [134, 135]).
Phagocytosis, a primary mechanism for nanoparticle uptake by macrophages, is broadly used to describe actin rearrangement and pseudopodial envelopment of large bodies into cells [127]. Phagocytosis is usually associated with Fc- and complement mediated (CR)-mediated receptors, enveloping material by cell membrane dynamics in a zipper-like fashion. Only certain classes of cells, usually termed “professional phagocytes,” have this type of cytoskeletal rearrangement capability. These include macrophages, neutrophils, dendritic cells, monocytes, and only in special cases, endothelial and secretory epithelial cells[136, 137]. Cellular pseudopodial vesiculations appear concurrent with internal granule movements and subsequent granule fusion within the cell[127]. Usually following phagocytosis, vesicles containing the foreign material fuse with lysosomal compartments, which then undergo pH reductions capable of destroying pathogens[127]. However, some pathogens are capable of thwarting this host defense mechanism. For example, when M. tuberculosis, S. aureus or Chlamydia are ingested and enter phagosomes, they actively prevent normal phagosomal acidification by down-regulating ATPase and lysosomal-associated membrane protein (LAMP) markers, respectively, thereby avoiding destruction, and even proliferate within the cell [138]. Phagocytosis is limited to particle sizes below 10 microns and more commonly below 6 microns. Rates of phagocytosis vary widely and change depending on cell type, activation state, culture conditions or particle biological conditioning (e.g., endotoxin or protein exposure). Nanomaterials could be designed to utilize similar self-preservation mechanisms to reduce vesicle fusion and alter trafficking and intracellular fate.
Pinocytosis, in contrast, is active in all mammalian cells and in some situations is responsible for nanoparticle uptake. This internalization mechanism can be classified into large volume extracellular internalization (e.g., macropinocytosis and circular dorsal ruffles responsible for 0.5–5 μm internalization) and smaller volume internalization (e.g., clathrin and clathrin-independent mechanisms, capable of 20–500 nm internalization) [139–141]. However, larger volume internalization mechanisms rarely occur without clathrin-mediated internalization. Macropinocytosis and circular dorsal ruffles are characterized by their large membrane protrusions off the cell basic shape, and actin-dependent polymerization mechanisms, similar to phagocytosis [140, 142]. However, phagocytic zipper-like membrane envelopment and uptake into phagocytic vesicles is absent in these processes. Rather, cells simply envelop the foreign substance directly into the cytoplasm. This process is implicated in uptake of viruses and macromolecules and potentially nanomaterials[143]. Clathrin-mediated endocytosis is likely the most well-studied of the internalization mechanisms. Scavenger, mannose and toll like (TL) receptors are known recognition and induction mechanisms for the process[144], involving formation of clathrin pits in the cell membrane that wrap and pull their cargo into the cell. Once enveloping cargo, these pits are cut from membrane invaginations via dynamin scission to form internalized vesicles[139], trafficked to early endosomes, acidified and transformed to late endosomal vesicles and then trafficked to other intracellular destinations including lysosomal compartments, a mechanism that nanoparticles harness[145].
Unlike clathrin-mediated endocytosis, responsible for internalization of size ranges from approximately 100–350 nm, caveolin-mediated endocytosis is responsible for 20–100-nm particle endocytosis [146–148]. Replacing the clathrin bowls are caveolae that generally reside as protein clusters within cell membrane lipid raft domains[146]. Their internalization mechanism; in contrast to membrane wrapping, produces invaginations after completion of a membrane bowl just below the cell membrane surface, ingesting the cargo via membrane dropping[141]. These vesicles are typically transported to the golgi or are excreted from the cell, a mechanism useful for particular nanoparticle delivery scenarios [147]. Cholera toxin utilizes this mechanism to avoid lysosomal internalization, a strategy also valuable for nanomaterial drug delivery[146].
While molecular surface recognition patterns induce cell uptake via interactions with phagocytic receptors, alterations in physicochemical characteristics including geometry and surface chemistry should also be studied for controlling specific particle uptake behaviors with cells (Table 2). These can also be exploited to induce specific intracellular fates and destinations, improving delivery specificity, selectivity, and targeting to avoid toxicity [149–152]. Particle geometry and surface chemistry are discussed further as strategies to distinguish how nanomaterial physicochemical characteristics might be deliberately tailored to engage host biology and better impact their phagocytic recognition, uptake and physiological fate.
Table 2.
Influence of particle geometry, surface charge, and functionalization on macrophage uptake.
| Geometry | Angle < 45 degrees | Microparticles can be completely enveloped and internalized into cellular compartments [153, 154]. |
| Angle > 45 degrees | Microparticles unable to be completely phagocytosed [153, 154]. | |
| Cylindrical (90 degrees at ends) | Microparticles can associate with cellular membrane but membrane wrapping cannot occur due to the energetically unfavorable conformation required to temporarily wrap over the sharp edge [155]. | |
| Rod-like with rounded ends (< 90 degrees) | Are taken up more rapidly than their true cylindrical counterparts [148]. | |
| Oblated ellipsoidal | More efficient phagocytic internalization capability than prolated ellipsoidal particles [21]. | |
| Spherical | ||
| Surface Charge | Positively charged particles | |
| Negatively charged | Generally taken up to a lesser degree than their positively charged counterparts [157, 158]. | |
| Surface Functionality | Polymer coverage | Have decreased protein binding and subsequent phagocytosis compared to bare counterparts [28] |
| Multivalency | Mathematical modeling of multivalency illustrates if ligand density on the surface of the nanoparticles is sufficient, membrane wrapping around the nanoparticles would be optimal when their diameter ranges between 20–35 nm and ligand density is approximately 80% with a minimum coverage of 20% [161]. This model suggests that uptake can be altered significantly with optimization of surface coverage because this can facilitate energetically favorable membrane wrapping. |
5 Phagocytic intracellular fate
Control and manipulation of particle morphological and surface physicochemical properties to interact in predictable ways with physiological components (proteins, cells) would enable exploitation of rational particle engineering strategies to select specific cell types, transport routes, internal cell compartments and more control over dosing, biodistributions, therapeutic action and toxicity. Extracellular particle recognition and processing determines intra-cellular uptake and particle trafficking. Once internalized, three processing events are possible for nanomaterials in phagocytes: (1) cell-autonomous antimicrobial defense mechanisms, (2) native pathogenic or foreign material cellular process mechanisms, and (3) opsonization recognition events due to specific structural surface similarities with pathogens and foreign materials[162]. How nanoparticles are sorted to each pathway is not understood.
5.1 Autophagy as cell response to nanomaterials
Autophagy is an example of an innate defense mechanism that degrades intracellular microbes, dysfunctional cellular organelles, and misfolded proteins. Autophagy is induced in nanoparticle uptake via pattern recognition receptors [163] that then leads to increased activity, leukocyte migration, and inflammatory responses such as specified polarization of immune cells and release of pro-inflammatory cytokines [163–170]. Autophagic dysfunction and vesicle accumulation has been seen following nanoparticle treatment, enhancing cell death through increasing mitochondrial dysregulation[171]. In some cases, however, autophagy is beneficial in providing a cellular coping mechanism for stress instead of apoptosis or necrosis, and reduces the likelihood of toxicity emerging from exposure to nanoparticle systems or massive necrosis. In this stress-coping mechanism, particles are trafficked and compartmentalized into internal autophagosomes, effectively isolated from the surrounding biological environment, reducing associated toxicity and aiding in reducing cellular stress [171]. Traditional cell death mechanisms like necrosis and apoptosis release large volumes of cellular contents into the extracellular environment including reactive oxygen species, reducing local pH and recruiting inflammatory agents, all of which can induce unknown local toxicity[172].
Autophagy is a primary mechanism by which highly positively charged nanoparticles are shown to traffic intracellularly. Positively charged dendrimers, polyplexes, gold nanoparticles, iron oxide and silica nanoparticles have been shown to utilize this mechanism [42, 167, 169, 173, 174]. However, little to no evidence currently exists linking other particle surface properties to autophagic mechanisms or to why this route is apparently selective for cationic charge density. While autophagy may be beneficial for reducing particle toxicity, it may be detrimental to cargo delivery, as trafficking of these positively charged systems to autophagosomes eliminates transport to other target organelles. For example, nucleic acid polyplexes need to be delivered directly to the cell nucleus or mitochondria to work effectively. However, if nucleic acid delivery systems ultimately traffic to autophagic compartments, very little to none of the cargo is delivered to the target site, severely limiting efficacy. Fortunately, approaches for controlling autophagy with surface modifications of nanoparticles have been identified. Modifying multiwall carbon nanotubes (MWCNTs) with organic compounds can help to avoid autophagic responses: a study utilizing a series of microtubule associated protein light chain three (LC3-II) screening assays illustrated that autophagy is highly dependent on particle surface characteristics in astrocyte and human embryonic kidney cell models. This study identified certain immobilized surface organic compounds that more strongly upregulate autophagy, versus other organic compounds that appear to have little to no effect on mechanistic regulation [175]. This suggests that surface chemistry in other systems might also be manipulated to potentially evade this process. Development of such nanoparticles may possibly aid in the design of new materials to modulate downstream inflammatory events. For example, where autophagy facilitates pathogenic removal[176], inducing or exacerbating this cell response using nanoparticles in the presence of infection would represent a promising treatment mechanism. Mycobacterium tuberculosis (M.tb.), a pathogen well-known for its survival and proliferation within phagosomes, inhibits the development of autophagy within macrophages. In alveolar macrophages, PLGA nanoparticles loaded with rapamycine are phagocytized and induce autophagy, which leads to M.tb. elimination [177]. This distinction illustrates the importance of understanding the particle uptake mechanisms, traffic pattern and target site of the material construct in designing new nanoparticle systems. For M.tb, autophagy can be exploited to enhance therapeutic efficacy, while for nucleic acid delivery, autophagy is detrimental to efficacy.
Autophagosome-like compartments that connect to the cell surface, termed surface-connected compartments (SCC), can also process pathogens and foreign materials, and may also prove useful in nano-based targeting and therapy. SCCs are a series of connected intracellular compartments formed when macrophages are subjected to large numbers of highly hydrophobic nanoparticles[178]. These SCC compartments serve as a cellular tool to digest foreign material and quickly release contents back to the extracellular space, potentially facilitating nanomaterials’ degradation while avoiding intracellular contamination. SCC is observed in uptake of low density lipoprotein (LDL), hydrophobic gold and hydroxyapatite particles below 500nm [178–180]. These compartments are mainly formed through actin rearrangement and remain open to the extracellular space while appearing to be connected to the plasma membrane. Autophagic compartmentalization and SCC formation could be a universal phenomenon for macrophage-nanoparticle processing and raises the possibility that these have evolutionarily developed as endogenous host nanoparticle processing mechanisms.
5.2 Directed intracellular delivery
Cell-targeted cargo delivery can be enhanced by methods to reliably select specific intracellular organelles to better predict and enable site-specific action. After foreign materials are taken up into endosomes or phagosomes, they fuse with lysosomal compartments used by cells to neutralize foreign material with isolated, focal heavy enzymatic digestion and reduced pH. This vesicle fusion allows cells abilities to degrade or remove hostile materials and pathogens from their intracellular environment for inactivation. However, these vesicles also encompass recognition motifs (i.e. TLRs, integrins, etc.) that can traffic ingested material to other specific cellular compartments [181]. These motifs, in turn, can be utilized for site-directed delivery. Additionally, nanomaterials might be engineered to escape these compartments through either selected decorated peptide surface motifs or surface compositional alterations, such as increasing particle weak base surface charge density to induce lysosomal burst through the so-called “proton sponge effect” [182]. After release from the lysosomal compartment, carriers can then be trafficked to a specific intracellular compartment (e.g., mitochondria, nucleus, and endosomes) for more efficient delivery of bioactive agents.
Specific nanomaterials engineering strategies guiding both uptake and intracellular trafficking could target nanoparticle systems to the mitochondria or designed to induce apoptosis in cancer, alter potassium dysregulation in heart abnormalities, or reduce oxidative stress effects of aging [183]. A common mitochondrial delivery strategy utilizes intrinsic differences in the local compartmental membrane potential, which is significantly lower than other cellular compartments. Attachment of mitochondriotropic cationic molecules to nanoparticle surfaces (e.g., triphenylphosphonium, TPP), harnesses this membrane potential difference to achieve high mitochondrial localization [184, 185]. Potentially, better nanoparticle strategies to by-pass the MPS, get inside specific cells and to specific sites to control particle intracellular destinies would overall reduce non-specific uptake and potential phagocytic toxicity, improve efficacy and reduce translation failure risks. Producing new, rational and reliable strategies for particle shape, surface chemistry and interactions with biological components would facilitate these improvements in cell-particle processing.
Peptides have been used to target other specific intracellular compartments (e.g., nucleus and endoplasmic reticulum) and could also be advantageous for particle trafficking [186]. The nucleus is a key organelle for delivery of many bioactive agents including most gene therapies. Attachment of nuclear localization signals (NLS) recognized via importin protein family members is shown to mediate particle nuclear transport mechanisms [187]. NLS is also reported to increase doxorubicin-loaded nanoparticle delivery and anti-proliferative effects [188]. These specific targeting strategies to subcellular compartments are reviewed elsewhere but are largely proven only in simplistic cell culture systems in vitro and are largely inefficient in vivo [189, 190].
Directing intracellular fate pathways utilizing engineered nanomaterials platforms must ensure a complete delivery solution in systemic circulation focused on selective opsonization in blood, discrete, reliable cell receptor selection at target tissue sites, uptake mechanisms from delivery vasculature, transport through healthy cells to diseased cells and proper intracellular processing. Nanomaterials have yet to claim these properties in vivo. Particle fabrication costs, intrinsic complexity and difficulties in utilizing intracellular trafficking ligands combined with specific surface chemistry, analytical validation, and particle stability hinders many researchers from incorporating these combined properties into new nanomaterials. However, nanomaterials without directed cellular target specificity nearly always yield indirect non-specific default uptake mechanisms determined by intrinsic physicochemical properties that target the vast majority of any systemized nanomaterial dose to the MPS system and possibly elicit cell toxicity.
5.3 Indirect default intracellular targeting and resulting potential toxicity
Generally, about 95% of every nanomaterial dosing to blood non-specifically targets the MPS filtration organs comprising fenestrated vasculature with high populations of committed phagocytes[2–4]. This occurs independently of any surface-immobilized target motifs (e.g., peptides, ligands, antibodies, etc.). Non-specific targeting of nanomaterials (i.e., scavenging) is the host default processing pathway for any and all systemized nanomaterials. Toxicity associated with high loading of nanomaterials to MPS organs is represented by cell stress biomarkers, specifically subsets of cytokines, chemokines and reactive oxygen species (ROS)[40, 49, 53]. Following the production dynamics of specific cytokine and chemokine markers and possible mechanistic dissection of downstream effects of nanoparticle-induced stress in vivo can be used to follow toxicity responses. Both detailed host processing and ultimate intracellular destinations for nanomaterials must be known in order to allow this information to guide future nanoparticle design improvements that promote more specific, selective cell responses. Toxicological consequences of non-specific cell uptake and acute and chronic tissue burdens must vary both from cell type to cell type and in the context of specific tissue environmental and pathological factors. Yet, few of these details are known. In vitro test systems to monitor cell-nanomaterial toxicities[191] while useful for basic trafficking studies, fall short of predicting actual nanomaterial in vivo responses, particularly systemic toxicities.
Nanoparticles targeting the mitochondria non-specifically can interfere with the mitochondrial membrane potential and its electron transport chain that provides a proton gradient through which ADP is phosphorylated to yield ATP. In this case, cytochrome C is released and apoptosis can be initiated. Nonspecifically targeted iron oxide particles and carbon nanotubes can home to mitochondria, affecting the cellular mechanistic energy machinery, ultimately leading to cell death [192, 193]. Mitochondrial dysfunction has been observed after treatment with silica nanoparticles. In these studies, JC-1 mitochondrial potential assays provided evidence that nanoparticle size and surface modification affected membrane potential changes. Smaller, more positively charged nanoparticles increased induction of these potential changes [194]. Crystalline silica nanoparticles initiate both necrotic and apoptotic cell death mechanisms, a result of mitochondrial damage. Early stages of toxicity with these particles show phagolysosmal escape and cellular damage, resulting in drastic changes in mitochondrial potential [195]. These temporal changes elicited hyperpolarization or depolarization, causing apoptotic or necrotic cell death, respectively. Mitochondrial dysfunction has also been seen in nanoparticle induced lysosomal and autophagic disorders[171]. Such processes are detrimental, facilitate drastic nanoparticle toxicity, and should be avoided in nanoparticle design and development for systemic delivery.
Nanoparticles can generate reactive oxygen species (ROS) and induce mitochondrial damage in macrophages potentially leading to stress-related gene up-regulation and inducing inflammation [45]. Surface hydroxyl groups can interact with iron in biological environments to initiate the Fenton reaction and free radical production [196, 197]. Increased ROS formation initiates cell stress, uncontrolled oxidative chemistry and inflammatory response. Increased oxidative stress markers, mitochondrial dysfunction, and inflammatory gene expression after nanoparticle treatment have been observed during these inflammatory responses [20, 44, 194, 198, 199]. For example, porous silica nanoparticles were reported to increase ROS generation and adversely affect ATP and tumor necrosis factor alpha (TNF-alpha) content in macrophages and other hematopoetic cells. Effects were more pronounced with charged surfaces while hydrophilicity played little to no role [200].
In addition to charge, other nanoparticle surface modifications warrant caution in macrophage interactions. One includes topography. Previous environmental nanotoxicity studies showed that nanoparticle crystal size significantly affected ROS induction [198]. This is due to differences in uptake mechanisms associated with different nanoparticle sizes. Smaller nanoparticles increased cellular toxicity compared to larger nanoparticles, which could be a function of increased specific surface area and increased charged density. This implicates an optimal nanoparticle size range for minimizing toxic effects and improved clinical translation. Porosity also appears to influence inflammatory activation, supporting surface area effects. Investigations of non-porous and porous silica nanomaterials demonstrated a drastic increase in MAPK, TNF-alpha, Il-1beta and NF-kappa beta production when macrophages were subjected to non-porous material, while porous silica yielded no such effect [201]. Future studies should help elucidate which modifications and surface characteristics trigger toxicity and inform nanoparticle design.
6. In vitro macrophage model systems and their correlation in vivo
Little is known about mechanisms through which physicochemical characteristics of nanoparticles induce up-regulation of inflammatory genes or in which stage of nanoparticle-macrophage interaction inflammatory genes are upregulated (i.e., presentation to biological milieu, cellular contact, or after internalization). Better understanding of these mechanisms, activation stages and characteristics would be beneficial when designing nanoparticle systems. More importantly, inflammatory gene upregulation upon nanoparticle insult needs to be confirmed in vivo. Macrophage models may help to decode these understandings if validated, as many macrophage model systems elicit inflammatory mediated events. In vitro macrophage models appear to correlate well with nanoparticle in vivo study results. Primary Kupffer cells exhibit toxicity from quantum dot uptake [202], by upregulating TNF-alpha, IL-1beta and IL-6. These studies predicted an acute inflammatory response in vivo, which is corroborated by results of in vivo quantum dot experiments in rodents [202]. Similar results have been observed with other nanoparticles [202].
Macrophage model systems appear to be capable of predicting correlations between nanoparticle characteristics and inflammatory induction. For example, some nanoparticle preparations induce toxic responses in hemopoietic lineages, while others do not [203]. In one study, nanoparticle-treated primary isolated hemopoeitic cells were compared to traditional monocytic immortalized cell lines. The traditional monocytic cell lines [203] show little to no toxicity towards antimony oxide nanoparticles; however, the nanoparticles did induce significant toxicity within the primary cell cultures, suggesting that primary cell models could improve the predictability of in vivo outcomes. A recently published review suggests that macrophage in vitro assays correlate well with in vivo results [29]. Some reported macrophage phagocytosis assays correlate with their in vivo retention in the MPS system. Release in vitro of TNF-alpha, IL-1beta and IL-8 from macrophage model systems can, for example, accurately predict disseminated intravascular coagulation (DIC) and cytokine storm disorders in vivo. Leukocyte proliferation in vitro correlates with immunostimulation or immunosuppressive effects. Myelosuppressive potential in vivo can be correlated with colony forming unit- granulocyte macrophage assays [29]. The ability of in vitro assays using macrophages to predict in vivo results suggests that some macrophage cultures, if properly validated, can be important model systems for nanoparticle translation. Thus utilizing validated macrophage in vitro assay screening could reduce costs and time-intensive screening compared to in vivo models[204].
While frequently predictive, in vitro cell culture particle toxicity model systems do not always correlate with in vivo results, and until compared to in vivo experiments, results should be used cautiously. In general, cellular model systems are usually more sensitive than in vivo model systems, due to often excessive dosing, poor distributions of particles, effects of aggregation and settling in media, and intrinsic cellular inability to compensate stresses via homeostatic balances. For example, quantum dots are traditionally thought of as highly toxic systems in vitro, but in vivo they tend to be tolerated with little to no observable toxicity [205]. Similar disparities also exist with diverse results reported for silica, gold, and iron oxide, emphasizing the importance of accurate particle characterization and dosimetry in translating in vitro results to an in vivo context.
7. Phagocyte-nanoparticle interactions in vivo
Particle biological conditioning, distributions and macrophage processing in vivo are significantly more complex than in vitro assays can duplicate, altering transport, physical states, tissue residence patterns, and mechanisms of toxicity. Tools to dissect macrophages in organ-specific uptake, induction of toxicity and clearance in vivo are required to understand their current intrinsic lack of targeting, uncontrolled biodistributions, MPS accumulation, reduced circulation times and non-specific versus specific toxicity.
Macrophages as a general class of phagocytes exhibit diverse heterogeneity in temporal functions, tissue-specific phenotypes, and reactivity to insults. This diversity results from environmental conditioning, and could affect nanoparticle processing. For example, Kupffer cells vary phenotypically as a function of their tissue location [206, 207]. Those cells residing within the portal tract region of livers (a structure comprising an artery, vein, bile duct, lymphatic vessel and nerve) exhibit increased levels of scavenger receptors, phagocytosis and lysosomal enzyme activity compared to centrally located cells, which have increased cytokine activities [208]. In contrast to portal region cells in livers, splenic macrophages that reside within the red pulp, white lymphatic pulp and marginal regions vary significantly in levels of endocytosis, with marginal zone macrophages exhibiting significant increases in MARCO receptors [209]. If particle properties could discern between these macrophage phenotypes and target particle recognition mechanisms to specific subpopulations, nanosystems could increase circulation times and deliver to specific tissue sites. The potential to control local macrophage environments by pharmacologically altering or priming these macrophage sites to specific activation states that either present antigens or polarize to specific Th1 or Th2 states has the capacity to increase local wound healing and bactericidal activity. Certain particle physicochemical characteristics including size, shape and surface modification appear to play important roles in vivo affecting nanoparticle tissue accumulation (Table 3)[209, 210].
Table 3.
Influence of nanoparticle geometry, surface charge, and functionalization on in vivo clearance and tissue accumulation.
| Geometry | ||
| Size | Below 15 nm | |
| Above 15 nm | ||
| Above 40 nm | ||
| Shape | Short rod with aspect ratio of 1.5 | Primarily accumulated in the liver [6]. |
| Rod with an aspect ratio of 5 | Resided within the spleen [6]. | |
| Surface Charge | Neutral and Zwitterionic |
|
| Hydrophillic |
|
|
| Hydrophobic |
|
|
| Surface Functionality | Polymer chain length | Longer polymer chain lengths on the surface of nanomaterials have shown faster clearance rates when compared to smaller chain lengths [10]. |
| BSA | Gold nanoparticles surface coated with BSA formed large aggregates and accumulated in the liver and spleen, inducing toxicity within these organ systems [166]. | |
| GSH | GSH-coated nanoparticles were quickly recognized via macrophages in the kidney and showed renal excretion [166]. | |
| Polymers | ||
| Liposomes |
|
|
7.1 Clearance mechanisms and tissue residence
Ideally after payloads have been delivered to their intended site, nanomaterials would be cleared from circulation to avoid inflammatory responses. Some nanoparticles with degradation capabilities or sufficiently small size do show elimination through biliary or renal excretion mechanisms, while others do not (Table 3) [210, 223]. Studies suggest that even after 2 weeks, only limited non-degradable nanomaterial clearance from MPS accumulation occurs, with a majority of particles still residing in clearance organs [7, 224, 225]. The possibility for chronic toxicity as a result of longer-term nanoparticle accumulation and residence, analogous to foreign body responses to more traditional biomaterial implants, remains to be determined for specific nanomaterials in different tissue sites.
7.1.1 Effects of long-term non-degradable nanoparticle tissue residence
Recent evidence suggests that long-term nanoparticle residence within MPS clearance organs such as the liver and spleen, initiates lesions via infiltration and microgranulation of hepatocytes and long-term inflammatory responses [8, 52]. Inhalation of nanoparticles has also initiated lesions within the lung tissue [50, 53]. This suggests that rather than eliminating nanoparticles, the body has developed a local response to isolate these foreign materials from host biological environments. The unanswered question is if this local initial response further triggers other adverse chronic inflammatory or immunological responses. Even more concerning are local responses to nanoparticle accumulations that lead to pre-cancerous conditions and immunological disorders [27–30]. However, lesion formation has been mitigated, at least to some extent, by surface modification addressing hydrophobicity and charge. For example, lung fibrosis occurs for cationic silica nanoparticles residing in lung, while particles with polar or anionic surfaces migrate to the mediastinal lymph nodes, preventing lung fibrosis [54].
The ultimate fate of most nanoparticle systems appears to be primarily accumulation to major (95%) extents within MPS clearance organs such as spleen, liver and kidney. These are specialized organs with fenestrated capillary beds to capture particles, and associated specialized macrophage populations near these vascular beds to sequester particles. Most evidence linking cellular uptake in vivo shows correlations between macrophage uptake and residence within those tissues. Important future studies would include investigations to identify (1) which macrophage subtypes in these tissues are responsible for primary uptake; (2) how macrophage type and reactivity to particle phagocytosis affects the surrounding environment; and (3), if or how nanoparticle secondary processing to produce particle elimination or accumulation occurs. Macrophages may traffic to biliary tracks and dump their contents or become trapped there. Alternatively, harsh intracellular phagosomal conditions within macrophages may manipulate and degrade these ‘nondegradable’ systems to other states that prompt toxicity, oxidative exhaustion, or apoptosis.
Increased particle circulating lifetimes, controlled tissue clearance rates and site selectivity enhanced elimination to excretion organs, and capacity to restore cell homeostasis are critical to nanomaterial safety and toxicity profiles. However, nanomaterial design needs are better focused on more incremental goals of reducing nanoparticle inflammation-related toxicity. Orthopedic implant-centered inflammation in hard tissues represents a prominent clinical example of locally induced nanoparticle inflammation. Implanted metallic bearing surfaces, such as those utilized in total knee or hip replacements, release nanoparticulates as a result of tribological joint function and wear. Released nanoparticles accumulate in the joint space and also migrate in the implant space (i.e., along the hip femoral stem in the endosteal space, producing aseptic osteolysis resulting from local macrophage recruitment and active phagocytosis of particle debris [226]. Continual macrophage exposure to implant wear debris in this bone location eventually produces a local inflammatory response that results in bone resorption around the implant, implant loosening, and in some cases, implant failure [226]. Patient hip tissue samples from cases of implant-centered inflammation have been examined histopathologically; large numbers of CD68-positive foamy macrophage cells were observed in the implant-proximal tissue [226]. The particles have also been counted in the biopsied macrophages and shown to be metallic and/or poly(ethylene). Subsequent gene expression analysis illustrated that foamy macrophages are strongly associated with expression of osteolytic proteins [226]. These findings have been correlated in vitro with macrophage inflammatory genomic profiling as a result of particle wear debris exposure [227]. This suggests a role for macrophages in the induction of aseptic osteolysis as a chronic local inflammatory disorder purely responsive to particulate debris. That this local inflammatory macrophage-induced reaction could reflect a general inflammatory mechanism capable in any tissue with a large accumulation of nanoparticles is the concern. Additionally, that high accumulations of non-degradable particulates in tissue sites both specific (i.e., disease) and non-specific (i.e., MPS organs) might elicit similar responses is also concerning.
7.2 Adverse toxicological effects in vivo
Macrophage-nanoparticle interactions in vivo may initiate global toxicity (i.e. induction of cell stress, release of ROS, cytokine production, etc), as a result of indirect systemic bioactivities and host reactions such as complement activation and thrombolytic events. Complement activation and thrombolytic events are potent responses to chemical and physical insults that initiate powerful zymogen activation and also cytokine and chemokine release that promotes immune and macrophage cellular recruitment, differentiation, and activation. Serious consequences of uncontrolled complement activation include anaphylaxis, and by on-going thrombosis, disseminated coagulopathies. Complement and thrombolytic responses can be mitigated in some particle exposures by variations in surface properties.[228, 229] Nonetheless, dextran-coated superparamagnetic iron oxide nanoparticles clinically used and approved as an iron supplement and magnetic resonance imaging contrast agent have been associated with sufficient complement-related side effects in patients to discontinue them from clinical use (e.g., Feridex™)[98].
7.2.1 Complement activation by nanoparticle-blood contact
Complement activation by nanoparticles initiates several possible reactions. Complement activation related pseudo-allergy (CARPA) has been described in patients exposed to micelles, liposomes and microbubbles. During CARPA reactions, nanoparticles activate complement proteins in blood, generating fragments C3a and C5a as potent stimulants of basophil granulocytes through their C3aR and C5aR receptors. Upon basophil activation, histamine and proteases are excreted from basophil granules, and lipid mediators (leukotrienes, prostaglandins, thromboxanes, PAF) are released from their cell membranes. These potent activation products are responsible for anaphylactic symptoms, causing bronchoconstriction, influencing blood vessel permeability and blood pressure. IgE-mediated allergic responses produce the same mediators from activation of eosinophil granulocytes and mast cells through their FcεR receptors. The alternative complement pathway produces Factor H, a potent pulmonary bronchorestrictor. Other inflammatory reactions are also observed. For example, liposomal formulations of doxorubicin have in some cases caused severe hypersensitivity due to initiation of inflammatory cascades [230]. Hydroxyl surface modifications initiate complement activation and subsequent inflammation, due to interaction with C-reactive protein. Additionally, iron oxide nanoparticles surface-activate C3 and initiate inflammatory mechanisms [231]. Activation of iC3b by nanoparticles might lead to erythrocyte binding and splenic clearance [232, 233]. Macrophages have complement receptors that initiate uptake and are required for pathogen removal [234]. These examples illustrate the need to understand particle surface modifications and relationships to complement activation with its potent adverse effects.
Surface modifications can cause significant differences in the degree of complement activation [231]. Polysaccharide-based dextrans have been used as excipients for decades to reduce protein adsorption and complement/phagocytic activity, resulting in increased circulating half-life of several nanoparticles [235, 236]. Similar methods have been utilized with poly(ethylene glycol) (PEG) [28] and other water-soluble polymers. While this has been met with some success, protein adsorption from plasma (70mg/ml proteins) is inevitable [236, 237]. With non-specific protein adsorption inevitable and pro-coagulant and complement activation observed, particles require scrutiny for complement activation in relevant systems. Heat-treated sera used in cell culture for example lack competent complement systems. Systems similar to these might also potentiate dysopsonin binding in vivo, reducing recognition by the immune system [77, 91, 238]. More information on how nanoparticles interact with the complement system can be reviewed elsewhere [239].
7.2.2 Thrombotic nanoparticle activation
Particle-induced thrombotic activation is another known consequence of nanoparticle-blood contact. Modulation of surface charge and density can trigger different clotting mechanisms [240]. Specifically, negatively charged nonporous silica nanoparticles induce platelet aggregation, but when the same particles were surface-modified with amines this response was reduced [240]; similar results have also been shown with carbon nanoparticles [241]. Interestingly, increased platelet aggregation has been correlated with an increase in particle phagocytosis, suggesting that macrophages might react to platelet-produced reaction pro-coagulant products or ingest the nanoparticle-centered coagulant material and initiate downstream responses [242].
Aggregation and thrombotic initiation has also been observed with cationic dendrimers both in vitro and in vivo [243–245] with effects increasing with increasing cationic charge density. Lower generation dendrimers with less amine charge density attenuated these thrombotic effects, but G7 (highest charge density) dendrimer systems prompt rapid, intense and fatal disseminated intravascular coagulopathies in rodents. This was not observed with hydroxyl- or carboxyl-terminated dendrimer systems. This suggests that appropriate controlled surface modifications might mitigate potent pro-coagulant triggers. Extensive review of nanoparticle systems and their blood compatibility are summarized elsewhere [246]. Delineation of mechanistic details by which surface properties initiate thrombogenesis and complement activation, and whether procoagulant properties observed with cationic dendrimers extend to other nanoparticle structures such as larger nanoparticles, remain to be investigated. A key question is how particle surface properties influence these complex blood protease cascade phenomena that often produce catastrophic systemic consequences, and if nanomaterial engineering could control these triggers sufficiently.
7.2.3 Toxicological implications of nanoparticle-mediated inflammatory processes in vivo
Systemic inflammatory effects, attributed to nanoparticle exposure, such as fibrosis and inflammation, may be directly related to thrombogenesis and complement initiation; however, particles may still induce host inflammatory response without contribution from these blood-borne systems. Important mechanistic details that forebode potentially toxic outcomes remain unknown. For example, two iron oxide particles with similar cores but different surface chemistries initiated very different levels of inflammatory cytokines IL-1beta, IL-8 and TNF-alpha in vivo. One formulation displayed little to no cytokine increase, while the other showed significant increases correlated with an induced in vivo cytokine storm, resulting in splenic congestion[29]. Several studies have shown that when nanoparticles are incubated in vitro with peripheral blood mononuclear cells (PBMCs) and mono mac-6 cells (MM-6) (monocytic and macrophage lines), increased cytokine activation directly correlates with observed toxicity markers in vivo, including disseminated intravascular coagulopathy (DIC) and cytokine storms [29]. IL-8 is also upregulated by 4-nm gold nanoparticles [247]. A study comparing gold and silver nanoparticles found that short-term silver nanoparticle blood exposure induced ROS and NF-kappa B signaling, leading to direct increases in cytokines TNF-alpha and IL-6; however, gold nanoparticles did not exhibit similar profiles over the same time period. When gold particle exposure time was increased, significant inflammatory response was observed [17]. Additionally, osteolysis and potential systemic organ damage might be initiated by NF-kappa B activation nanoparticle exposure as the subsequent induction of IL-8 helps induce TNF-alpha and IL-6 [22]. These examples indicate that nanoparticles are capable of widespread inflammatory responses resulting from prolonged exposure, independent of nanoparticle composition. These outcomes correlate with general coagulation and complement activation, independent of particle composition as well.
Numerous studies have investigated relationships between particle size and cytokine indicators of inflammation. For example, 5-nm silver nanoparticles exhibited higher toxicity than 100-nm silver nanoparticles in macrophage cultures [248]. An often-confounding issue with metallic nanoparticle toxicity assessment is their oxidative stability in milieu, and undetermined presence of oxidized metal ion leaching products that might produce toxicity, not the metal itself. However, this is rarely assessed. Given the abundant evidence for general surface adsorption and activation processes from nanoparticles, and general internalization by phagocytes, nanoparticle toxicity appears to be more a function of surface area exposed to macrophages rather than size [249].
Acute inflammatory reactions to nanoparticles can have profound effects. For example chronic silica exposure is associated with autoimmune diseases and chronic inflammatory states [249]. Toxicological studies provide evidence that microcrystalline forms of silica powerfully activate inflammatory and oxidative stress agents, such as cytokines, chemokines, reactive oxygen species (ROS), reactive nitrogen species (RNS) and nitric oxide [40, 53, 250–252]. Most findings were associated with silicosis and lung cancer in mine workers [13, 53, 253–255], both inflammatory results of chronic exposure to crystalline silica exclusively via inhalation. However, little information exists for long-term biological effects of amorphous silica. The rate of particle dissolution, degradation and elimination of amorphous silica are not well studied. Degradable forms of silica can potentially reduce these inflammatory events, opening routes for utility in biomedical applications. Molecular silicates are not detrimental to the human environment at current levels of exposure, suggesting that degradation products could be well-tolerated [256]. A prerequisite, however, for successful development of degradable nanosilica constructs is to understand the biological fate of their degradation process and products.
8. Designing nanomaterials that reduce phagocytic recognition
Innovating the “bioinvisibility” of nanomaterials in the host to reduce macrophage recognition in vivo might yield new insights. Grafted and coated PEG and other hydrophilic polymer brush layers are commonly used in drug delivery to facilitate enhanced particle circulation times and reduced protein adsorption but this strategy has only produced incremental improvements after several decades of refinement. These systems are still recognized eventually by the innate immune system, taken up by phagocytes and rapidly removed from circulation. The most reported half-life for clinically used PEGylated liposomes is ~24 hours. Some suggestions have been made that this is due to anti-PEG antibodies against these polymers and other heavily used pharmaceutical excipients [257] but this is recently questioned[258].
The concept of host “bioinvisibility” requires a different and distinct approach to make a clinically significant performance improvement. One approach seeks to mask the foreign particle surface with self-identifying proteins. By utilizing pathogenic or eukaryotic mimicry, reduction of complement activation and local recognition and uptake may be possible. For example, Factor H is a cofactor for Factor I that mediates C3b cleavage and dissociation of the Bb complex, inactivating the complement pathway [239]. Pathogens that utilize Factor H binding in their immune defense mechanisms include HIV-1 and streptococcal M6. Researchers have utilized sialic acid, the main component on the surfaces of these pathogens, to bind Factor H in an effort to evade complement activation and thus immune recognition. This component is also over-expressed in erythrocytes; as erythrocytes age they undergo programmed desialyation and initiate complement activation and phagocytic destruction [259].
CD47 is a potential marker of “self” and potential surface ligand to promote long circulation of nanomaterials. It is over-expressed on erythrocytes, facilitating their evasion of the immune system during their long circulation period by binding to the SIRP-alpha receptor [260]. Pathogens have also exploited this system, over-expressing CD47 on some viruses such as smallpox to evade recognition [261]. The active binding sequence of this protein has been recently identified and attached to surfaces of nanoparticles, reducing MPS uptake and drastically increasing circulation times [262]. In general, these results reflect a newer approach for utilization of host “self” recognition markers to reduce immune recognition, complement activation and phagocytic recognition.
9. Drug Delivery Systems Targeting Macrophages or Utilizing Macrophages as a Delivery Platform
Nanoparticles residing in macrophages in clearance organs by virtue of abundant uptake mechanisms could be useful in certain disease states as a “targeted” therapeutic approach. Several labs have harnessed this power to target tumor-associated macrophages in cancer[263, 264], human immunodeficiency virus (HIV)[265–267], bacterial/fungal infections[220, 268, 269], among others. Additionally, more recently, utilization of macrophages themselves as mobile, disease-homing drug delivery vehicles has been discussed [270, 271]. This illustrates the power of targeting macrophages for therapeutic purposes, exploiting a formidable host particle collection mechanism for therapeutic gain.
10. Current clinically relevant nanoparticle-macrophage interactions
Therapeutic capacity and clearance mechanisms have been linked with macrophage activity in clinically relevant nanomaterial therapies. For example, liposomal chemotherapeutic intravenous formulations have been prescribed clinically on- and off-label for several years (e.g. Doxil™). These formulations exhibit increased plasma half-life as patients’ age increases or monocytic activity decreases (Figure 6)[3, 4]. Decreased particle clearance appears to be a direct result of reduced patient phagocytic competence with age, while increased phagocytic activity appears to increase clearance rates [3, 4]. This propensity for clearance provides clinical evidence for nanoparticle-MPS interactions that might be better studied as a basis for patient-specific factors, and thus hold clues for better particle designs.
Figure 6.
Patients (n=22) injected with CKD-602 (PEGylated liposomal camptothecin analog) were monitored for particle clearance and drug release versus decrease in monocytes over the course of treatment. Increased monocytic activities in cancer patients reduced the area under the curve (AUC, bioavailability), or the amount of circulating nanoparticles in plasma. This MPS effect clearly implicates involvement of host macrophages in the clearance of clinical nanoparticle formulations. Reprinted with permission from [3].
Environmentally sourced nanoparticle-MPS interactions in patients induce well-known inflammatory morbidities [49–51]. These mechanistically well-understood and clinically relevant disease states, including silicosis, mesothelioma and pneumoconiosis, present after years of initial particle exposure. While the characteristics of environmental particles responsible for these diseases differ from particles engineered for human exposures today, the initial phases of these diseases follow similar patterns to that observed in acute toxicity studies using engineered nanoparticles. These include local fibrotic lesions with increased recruitment of polymorphonuclear- and antigen-presenting cells, macrophages, and dendritic cells [51]. It has been argued that engineered nanomaterials are “inert” suggesting that large aggregates or microparticulates are responsible for these disease states, and that smaller nanoscale materials bear little physical resemblance and are therefore completely cleared without lasting toxicities. Unfortunately chronic toxicity studies following engineered nanoparticle exposure are not well studied within the literature leaving gaps in knowledge and unanswered questions regarding lasting toxicities.
Generally, fibrotic mechanisms in tissues such as the lung proceed over decades; little evidence supports that under certain circumstances smaller non-clearable agents would not elicit similar responses [272]. For example, tuberculosis (TB) is a chronic inflammatory fibrotic disease state, with hallmark chronic macrophage involvement, frustration, and foreign body reactions producing life-long lung lesions. TB pathogens lie within the typically clearable particle size range; however, the pathogen is not cleared.[138] To compensate, the body initiates fibrotic encapsulating lesions as the control mechanism [273]. The body may utilize similar mechanisms for non-clearable, enduring, engineered nanomaterials.
Similar mechanisms could be initiated in other particulate exposures in different organ systems. For example, silica nanoparticles induce release of inflammatory mediators such as interleukins, TNF alpha, transforming growth factor (TGF), monocyte chemoattractant proteins, and other inflammatory mediators [15, 20, 250]. After silica nanoparticle treatment, direct correlations are observed between the release of these factors and migration, proliferation, and differentiation of inflammatory and immunological cells [20, 224]. These acute inflammatory events reflect potential for local recruitment and involvement of phagocytes in particle clearance, and eventual development of chronic inflammation, should clearance and excretion not succeed as physiologically required.
11. Conclusions
Nanoparticle use in imaging, vaccines, drug delivery and theranostic systems has the potential to produce new breakthroughs for medical capabilities. However, long-standing limitations for particle-host interactions remain as formidable barriers for concept translation. Few nanoparticle systems have distinguished themselves in medical performance in full physiological test beds from decades of studies. Additionally, the enormous complexity of nanoparticle chemistries, surface modifications, ligand and bioconjugate attachments and relatively incomplete analytical and physicochemical characterization together complicate descriptions and understandings for how these nanoparticles interact with the biological environments and specifically with macrophages. Observations of in vitro and in vivo behaviors produce correlative explanations for mechanisms and hypotheses for improving performance. Molecular bases for understanding and therefore for rational design principles for future nanoparticles are lacking. How to produce specific biological engagement while minimizing non-specific interactions that plague these systems demands more attention. The ability to engineer nanoparticle systems to overcome and avoid deleterious toxic effects or potentially utilize those effects therapeutically requires more knowledge of how current nanoparticles and macrophages interact. This should allow extraction of essential elements of control for this interaction, especially as it regards nanoparticle phagocyte clearance and accumulation in vivo. Moving forward, consideration of biological pathogen interactions, persistence and clearance studies, known clinical effects from environmental nanoparticle exposure, and increasing definition of the molecular requirements for eluding elimination by establishing “self” should produce important new clues for the engineered nanoparticle community. Ultimately, nanomaterials selective in their interactions with proteins and specific in engagement with cell receptors might be anticipated to control and guide in vivo targeting to cells with high affinity and specificity while reducing the primarily non-specific default pathways to the MPS and phagocytic cells that dictates the in vivo fates of current nanoparticles.
Highlights.
Diverse populations of different macrophages very effectively clear nanomaterials using specific and non-specific uptake mechanisms.
Nanomaterials are very efficiently scavenged from circulating blood and tissues by macrophages resident in tissue and filtration organs (MPS system), severely limiting particle targeting.
To avoid rapid uptake, clearance and to improve targeting, nanomaterials that avoid both specific and non-specific macrophage recognition must be developed to improve intra- and extra-cellular targeting.
Long-term residence of non-degradable systems within macrophages in clearance organs poses a unique challenge and could initiate inflammatory mechanisms.
Acknowledgments
Support was provided by National Institutes of Health grants R01ES024681 (to HG) and R01EB000894 (to DWG) and a predoctoral fellowship (W81XWH-11-1-0057 to HH) from the Department of Defense Breast Cancer Research Program. DWG acknowledges support for the George S. and Dolores Eccles Foundation (USA).
Biographies

Heather Herd Gustafson received her B.S.E. in Biomedical Engineering from Case Western Reserve University and Ph.D. in Bioengineering from The University of Utah. Her dissertation work focused on understanding macrophage nanoparticle interactions. During the course of her PhD she was awarded both a Whitaker Foundation Fellowship and Department of Defense Predoctoral Fellowship to elucidate the role that physicochemical characteristics played on nanoparticle intracellular uptake and fate within macrophages. She is currently a postdoctoral fellow at The Fred Hutchinson Cancer Research Institute working on the role that macrophages play in tumor development.

Dolly Holt-Casper received her B.S. from Utah State University in Biological Engineering and M.S. and Ph.D. in Bioengineering at the University of Utah. She served as a fellow for both the American Heart Association and NSF IMSURE nanotechnology program. Her PhD focused on analyzing the foreign body response to various biomaterials including nanoparticles. She also completed a postdoctoral fellowship in Tissue Engineering from the University of Utah, working on creating 3D scaffolds for cardiac, cartilage, and adipose tissue regeneration. She received a Women Tech Award for academic excellence in the State of Utah, has filed 2 patents, and has won several idea and business plan competitions.

David W. Grainger is a University Distinguished Professor and George S. and Dolores Doré Eccles Presidential Endowed Chair in Pharmaceutics and Pharmaceutical Chemistry, and Professor of Bioengineering at the University of Utah. Grainger’s research focuses on improving drug delivery methods, implanted medical device and diagnostics performance, and nanomaterials toxicity. Grainger has published 176 research papers and 23 book chapters on biomaterials innovation in medicine and biotechnology, and novel surface chemistry. He has won research awards, including the 2013 Excellence in Surface Science Award (Surfaces in Biomaterials Foundation), the 2007 Clemson Award for Basic Research (Society for Biomaterials), and the 2005 American Pharmaceutical Research and Manufacturer’s Association’s award for “Excellence in Pharmaceutics”, and several teaching awards.

Hamid Ghandehari is a Professor at the Departments of Bioengineering and Pharmaceutics and Pharmaceutical Chemistry, Director of Utah Center for Nanomedicine and Co-Founder and Co-Director of the Nano Institute of Utah at the University of Utah. His research focuses on the design of new polymers for gene therapy of head and neck cancer, targeted delivery of polymer therapeutics to solid tumors, oral delivery of chemotherapeutics, and assessing the biocompatibility of silica and dendritic nanoconstructs. Hamid is Editor in Chief of Advanced Drug Delivery Reviews, Fellow of the American Institute for Medical and Biological Engineering, the American Association of Pharmaceutical Scientists, and Controlled Release Society, Member of Center for Scientific Review College of Reviewers at the NIH, and serves on boards of several drug delivery journals and organizations. He has published nearly 150 peer reviewed articles. He received his BS in Pharmacy and PhD in Pharmaceutics and Pharmaceutical Chemistry from the University of Utah.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Allen TM, Cullis PR. Science. 2004;303:1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 2.Etheridge ML, Campbell SA, Erdman AG, Haynes CL, Wolf SM, McCullough J. Nanomedicine. 2013;9:1–14. doi: 10.1016/j.nano.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caron WP, Song G, Kumar P, Rawal S, Zamboni WC. Clin Pharmacol Ther. 2012;91:802–812. doi: 10.1038/clpt.2012.12. [DOI] [PubMed] [Google Scholar]
- 4.Caron WP, Lay JC, Fong AM, La-Beck NM, Kumar P, Newman SE, Zhou H, Monaco JH, Clarke-Pearson DL, Brewster WR, Van Le L, Bae-Jump VL, Gehrig PA, Zamboni WC. J Pharmacol Exp Ther. 2013;347:599–606. doi: 10.1124/jpet.113.208801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He X, Nie H, Wang K, Tan W, Wu X, Zhang P. Anal Chem. 2008;80:9597–9603. doi: 10.1021/ac801882g. [DOI] [PubMed] [Google Scholar]
- 6.Huang X, Li L, Liu T, Hao N, Liu H, Chen D, Tang F. ACS Nano. 2011;5:5390–5399. doi: 10.1021/nn200365a. [DOI] [PubMed] [Google Scholar]
- 7.Kumar R, Roy I, Ohulchanskky TY, Vathy LA, Bergey EJ, Sajjad M, Prasad PN. ACS Nano. 2010;4:699–708. doi: 10.1021/nn901146y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu T, Li L, Teng X, Huang X, Liu H, Chen D, Ren J, He J, Tang F. Biomaterials. 2011;32:1657–1668. doi: 10.1016/j.biomaterials.2010.10.035. [DOI] [PubMed] [Google Scholar]
- 9.Liu Y, Hu Y, Huang L. Biomaterials. 2014;35:3027–3034. doi: 10.1016/j.biomaterials.2013.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohno K, Akashi T, Tsujii Y, Yamamoto M, Tabata Y. Biomacromolecules. 2012;13:927–936. doi: 10.1021/bm201855m. [DOI] [PubMed] [Google Scholar]
- 11.Owens DE, 3rd, Peppas NA. Int J Pharm. 2006;307:93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 12.Albanese A, Sykes EA, Chan WC. ACS Nano. 2010;4:2490–2493. doi: 10.1021/nn100776z. [DOI] [PubMed] [Google Scholar]
- 13.Carter JM, Driscoll KE. J Environ Pathol Toxicol Oncol. 2001; 20(Suppl 1):33–43. [PubMed] [Google Scholar]
- 14.Cho WS, Choi M, Han BS, Cho M, Oh J, Park K, Kim SJ, Kim SH, Jeong J. Toxicol Lett. 2007;175:24–33. doi: 10.1016/j.toxlet.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 15.Driscoll KE. Toxicol Lett. 2000;112–113:177–183. doi: 10.1016/s0378-4274(99)00282-9. [DOI] [PubMed] [Google Scholar]
- 16.Gazi U, Martinez-Pomares L. Immunobiology. 2009;214:554–561. doi: 10.1016/j.imbio.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 17.Nishanth RP, Jyotsna RG, Schlager JJ, Hussain SM, Reddanna P. Nanotoxicology. 2011;5:502–516. doi: 10.3109/17435390.2010.541604. [DOI] [PubMed] [Google Scholar]
- 18.Orr GA, Chrisler WB, Cassens KJ, Tan R, Tarasevich BJ, Markillie LM, Zangar RC, Thrall BD. Nanotoxicology. 2011;5:296–311. doi: 10.3109/17435390.2010.513836. [DOI] [PubMed] [Google Scholar]
- 19.Pajarinen J, Kouri VP, Jamsen E, Li TF, Mandelin J, Konttinen YT. Acta Biomater. 2013;9:9229–9240. doi: 10.1016/j.actbio.2013.06.027. [DOI] [PubMed] [Google Scholar]
- 20.Park EJ, Park K. Toxicol Lett. 2009;184:18–25. doi: 10.1016/j.toxlet.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 21.Sharma G, Valenta DT, Altman Y, Harvey S, Xie H, Mitragotri S, Smith JW. J Control Release. 2010;147:408–412. doi: 10.1016/j.jconrel.2010.07.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Velard F, Braux J, Amedee J, Laquerriere P. Acta Biomater. 2013;9:4956–4963. doi: 10.1016/j.actbio.2012.09.035. [DOI] [PubMed] [Google Scholar]
- 23.Bae YH, Park K. J Control Release. 2011;153:198–205. doi: 10.1016/j.jconrel.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Florence AT. J Control Release. 2012;164:115–124. doi: 10.1016/j.jconrel.2012.03.022. [DOI] [PubMed] [Google Scholar]
- 25.Crommelin DJ, Florence AT. Int J Pharm. 2013;454:496–511. doi: 10.1016/j.ijpharm.2013.02.020. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Grainger DW. FCHEM. 2014;8:265–275. [Google Scholar]
- 27.Dobrovolskaia MA, McNeil SE. Nat Nanotechnol. 2007;2:469–478. doi: 10.1038/nnano.2007.223. [DOI] [PubMed] [Google Scholar]
- 28.Dobrovolskaia MA, Aggarwal P, Hall JB, McNeil SE. Mol Pharm. 2008;5:487–495. doi: 10.1021/mp800032f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dobrovolskaia MA, McNeil SE. J Control Release. 2013;172:456–466. doi: 10.1016/j.jconrel.2013.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dobrovolskaia MA, Germolec DR, Weaver JL. Nat Nanotechnol. 2009;4:411–414. doi: 10.1038/nnano.2009.175. [DOI] [PubMed] [Google Scholar]
- 31.Mortimer GM, Butcher NJ, Musumeci AW, Deng ZJ, Martin DJ, Minchin RF. ACS Nano. 2014;8:3357–3366. doi: 10.1021/nn405830g. [DOI] [PubMed] [Google Scholar]
- 32.Jenkin CR, Rowley D. J Exp Med. 1961;114:363–374. doi: 10.1084/jem.114.3.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pouliquen D, Le Jeune J, Perdrisot R, Ermias A, Jallet P. J Magn Reson Im. 1991;9:275–283. doi: 10.1016/0730-725x(91)90412-f. [DOI] [PubMed] [Google Scholar]
- 34.Briley-Saebo K, Bjornerud A, Grant D, Ahlstrom H, Berg T, Kindberg GM. Cell Tissue Res. 2004;316:315–323. doi: 10.1007/s00441-004-0884-8. [DOI] [PubMed] [Google Scholar]
- 35.Aggarwal P, Hall JB, McLeland CB, Dobrovolskaia MA, McNeil SE. Adv Drug Deliv Rev. 2009;61:428–437. doi: 10.1016/j.addr.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Albanese A, Tang PS, Chan WC. Annu Rev Biomed Eng. 2012;14:1–16. doi: 10.1146/annurev-bioeng-071811-150124. [DOI] [PubMed] [Google Scholar]
- 37.Greish K, Thiagarajan G, Ghandehari H. Methods Mol Biol. 2012;926:235–253. doi: 10.1007/978-1-62703-002-1_17. [DOI] [PubMed] [Google Scholar]
- 38.Karmali PP, Simberg D. Expert Opin Drug Deliv. 2011;8:343–357. doi: 10.1517/17425247.2011.554818. [DOI] [PubMed] [Google Scholar]
- 39.Walkey CD, Olsen JB, Guo H, Emili A, Chan WC. J Am Chem Soc. 2012;134:2139–2147. doi: 10.1021/ja2084338. [DOI] [PubMed] [Google Scholar]
- 40.Fubini B, Hubbard A. Free Radic Biol Med. 2003;34:1507–1516. doi: 10.1016/s0891-5849(03)00149-7. [DOI] [PubMed] [Google Scholar]
- 41.Zhu M, Nie G, Meng H, Xia T, Nel A, Zhao Y. Acc Chem Res. 2013;46:622–631. doi: 10.1021/ar300031y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khan MI, Mohammad A, Patil G, Naqvi SA, Chauhan LK, Ahmad I. Biomaterials. 2012;33:1477–1488. doi: 10.1016/j.biomaterials.2011.10.080. [DOI] [PubMed] [Google Scholar]
- 43.Lunov O, Syrovets T, Loos C, Nienhaus GU, Mailander V, Landfester K, Rouis M, Simmet T. ACS Nano. 2011;5:9648–9657. doi: 10.1021/nn203596e. [DOI] [PubMed] [Google Scholar]
- 44.Wang F, Gao F, Lan M, Yuan H, Huang Y, Liu J. Toxicol In Vitro. 2009;23:808–815. doi: 10.1016/j.tiv.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 45.Xia T, Kovochich M, Brant J, Hotze M, Sempf J, Oberley T, Sioutas C, Yeh JI, Wiesner MR, Nel AE. Nano Lett. 2006;6:1794–1807. doi: 10.1021/nl061025k. [DOI] [PubMed] [Google Scholar]
- 46.Allison AC. Arch Intern Med. 1971;128:131–139. [PubMed] [Google Scholar]
- 47.Mossman B, Light W, Wei E. Annu Rev Pharmacol Toxicol. 1983;23:595–615. doi: 10.1146/annurev.pa.23.040183.003115. [DOI] [PubMed] [Google Scholar]
- 48.Allison AC, Harington JS, Birbeck M. J Exp Med. 1966;124:141–154. doi: 10.1084/jem.124.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fujimura N. Curr Opin Pulm Med. 2000;6:140–144. doi: 10.1097/00063198-200003000-00010. [DOI] [PubMed] [Google Scholar]
- 50.Mossman BT, Churg A. Am J Respir Crit Care Med. 1998;157:1666–1680. doi: 10.1164/ajrccm.157.5.9707141. [DOI] [PubMed] [Google Scholar]
- 51.Byrne JD, Baugh JA. Mcgill J Med. 2008;11:43–50. [PMC free article] [PubMed] [Google Scholar]
- 52.Nishimori H, Kondoh M, Isoda K, Tsunoda S, Tsutsumi Y, Yagi K. Eur J Pharm Biopharm. 2009;72:496–501. doi: 10.1016/j.ejpb.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 53.Rimal B, Greenberg AK, Rom WN. Curr Opin Pulm Med. 2005;11:169–173. doi: 10.1097/01.mcp.0000152998.11335.24. [DOI] [PubMed] [Google Scholar]
- 54.Choi HS, Ashitate Y, Lee JH, Kim SH, Matsui A, Insin N, Bawendi MG, Semmler-Behnke M, Frangioni JV, Tsuda A. Nat Biotechnol. 2010;28:1300–1303. doi: 10.1038/nbt.1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Terzano C, Di Stefano F, Conti V, Graziani E, Petroianni A. Eur Rev Med Pharmacol Sci. 2010;14:809–821. [PubMed] [Google Scholar]
- 56.Burke B, Lewis CE. The macrophage. Oxford University Press; New York: 2002. [Google Scholar]
- 57.Shi C, Pamer EG. Nat Rev Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Murray PJ, Wynn TA. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matsui H, Ito T, Ohnishi S. J Cell Sci. 1983;59:133–143. doi: 10.1242/jcs.59.1.133. [DOI] [PubMed] [Google Scholar]
- 60.Michell RH, Pancake SJ, Noseworthy J, Karnovsky ML. J Cell Biol. 1969;40:216–224. doi: 10.1083/jcb.40.1.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Epelman S, Lavine KJ, Randolph GJ. Immunity. 2014;41:21–35. doi: 10.1016/j.immuni.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mosser DM, Edwards JP. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. J Pathol. 2013;229:176–185. doi: 10.1002/path.4133. [DOI] [PubMed] [Google Scholar]
- 64.Locati M, Mantovani A, Sica A. Adv Immunol. 2013;120:163–184. doi: 10.1016/B978-0-12-417028-5.00006-5. [DOI] [PubMed] [Google Scholar]
- 65.Nau GJ, Richmond JF, Schlesinger A, Jennings EG, Lander ES, Young RA. Proc Natl Acad Sci U S A. 2002;99:1503–1508. doi: 10.1073/pnas.022649799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Akira S, Uematsu S, Takeuchi O. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 67.Anderson JM, Rodriguez A, Chang DT. Semin Immunol. 2008;20:86–100. doi: 10.1016/j.smim.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cassetta L, Cassol E, Poli G. Sci World. 2011;11:2391–2402. doi: 10.1100/2011/213962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martinez FO, Sica A, Mantovani A, Locati M. Front Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 70.Herd HL, Bartlett KT, Gustafson JA, McGill LD, Ghandehari H. Biomaterials. 2015;53:574–582. doi: 10.1016/j.biomaterials.2015.02.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shepard JL, Zon LI. Curr Opin Hematol. 2000;7:3–8. doi: 10.1097/00062752-200001000-00002. [DOI] [PubMed] [Google Scholar]
- 72.Wynn TA, Chawla A, Pollard JW. Nature. 2013;496:445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Haldar M, Murphy KM. Immunol Rev. 2014;262:25–35. doi: 10.1111/imr.12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Davies LC, Jenkins SJ, Allen JE, Taylor PR. Nat Immunol. 2013;14:986–995. [Google Scholar]
- 75.Juliano R. Adv Drug Deliv Rev. 1988;2:31–54. [Google Scholar]
- 76.Borchardt G, Brandriss S, Kreuter J, Margel S. J Drug Target. 1994;2:61–77. doi: 10.3109/10611869409015894. [DOI] [PubMed] [Google Scholar]
- 77.Moghimi SM, Muir IS, Illum L, Davis SS, Kolb-Bachofen V. Biochim Biophys Acta. 1993;1179:157–165. doi: 10.1016/0167-4889(93)90137-e. [DOI] [PubMed] [Google Scholar]
- 78.Shibata Y, Foster LA, Metzger WJ, Myrvik QN. Infect Immun. 1997;65:1734–1741. doi: 10.1128/iai.65.5.1734-1741.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Holt DJ, Grainger DW. Biomaterials. 2012;33:7497–7507. doi: 10.1016/j.biomaterials.2012.06.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ruffell B, Affara NI, Coussens LM. Trends Immunol. 2012;33:119–126. doi: 10.1016/j.it.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege JL, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, Suttles J, Udalova I, van Ginderachter JA, Vogel SN, Wynn TA. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hoppstadter J, Seif M, Dembek A, Cavelius C, Huwer H, Kraegeloh A, Kiemer AK. Front Pharmacol. 2015;6:55. doi: 10.3389/fphar.2015.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Andrade J, Hlady V. Biopolymers/Non-Exclusion HPLC. Springer; 1986. Protein adsorption and materials biocompatibility: a tutorial review and suggested hypotheses; pp. 1–63. [Google Scholar]
- 84.Andrade JD, Hlady VL, Van Wagenen RA. Pure Appl Chem. 1984;56:1345–1350. [Google Scholar]
- 85.Senior J, Delgado C, Fisher D, Tilcock C, Gregoriadis G. Biochim Biophys Acta. 1991;1062:77–82. doi: 10.1016/0005-2736(91)90337-8. [DOI] [PubMed] [Google Scholar]
- 86.Senior J, Crawley JC, Gregoriadis G. Biochim Biophys Acta. 1985;839:1–8. doi: 10.1016/0304-4165(85)90174-6. [DOI] [PubMed] [Google Scholar]
- 87.Chao Y, Karmali PP, Mukthavaram R, Kesari S, Kouznetsova VL, Tsigelny IF, Simberg D. ACS Nano. 2013;7:4289–4298. doi: 10.1021/nn400769e. [DOI] [PubMed] [Google Scholar]
- 88.Nel AE, Madler L, Velegol D, Xia T, Hoek EM, Somasundaran P, Klaessig F, Castranova V, Thompson M. Nat Mater. 2009;8:543–557. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 89.Lynch I, Dawson KA. Nano Today. 2008;3:40–47. [Google Scholar]
- 90.Grainger DW, Castner DG. Adv Mater. 2008;20:867–877. [Google Scholar]
- 91.Absolom DR. Methods Enzymol. 1986;132:281–318. doi: 10.1016/s0076-6879(86)32015-9. [DOI] [PubMed] [Google Scholar]
- 92.Vertegel AA, Siegel RW, Dordick JS. Langmuir. 2004;20:6800–6807. doi: 10.1021/la0497200. [DOI] [PubMed] [Google Scholar]
- 93.Davidson WS, Jonas A, Clayton DF, George JM. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 94.Roach P, Farrar D, Perry CC. J Am Chem. 2006;128:3939–3945. doi: 10.1021/ja056278e. [DOI] [PubMed] [Google Scholar]
- 95.Lord MS, Foss M, Besenbacher F. Nano Today. 2010;5:66–78. [Google Scholar]
- 96.Dolatshahi-Pirouz A, Rechendorff K, Hovgaard MB, Foss M, Chevallier J, Besenbacher F. Colloids Surf B. 2008;66:53–59. doi: 10.1016/j.colsurfb.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 97.Walkey CD, Chan WC. Chem Soc Rev. 2012;41:2780–2799. doi: 10.1039/c1cs15233e. [DOI] [PubMed] [Google Scholar]
- 98.Banda NK, Mehta G, Chao Y, Wang G, Inturi S, Fossati-Jimack L, Botto M, Wu L, Moghimi SM, Simberg D. Part Fibre Toxicol. 2014;11:64. doi: 10.1186/s12989-014-0064-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Monopoli MP, Aberg C, Salvati A, Dawson KA. Nat Nanotechnol. 2012;7:779–786. doi: 10.1038/nnano.2012.207. [DOI] [PubMed] [Google Scholar]
- 100.Gordon S. Cell. 2002;111:927–930. doi: 10.1016/s0092-8674(02)01201-1. [DOI] [PubMed] [Google Scholar]
- 101.Janeway CA, Jr, Medzhitov R. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 102.Boller T, Felix G. Annu Rev Plant Biol. 2009;60:379–406. doi: 10.1146/annurev.arplant.57.032905.105346. [DOI] [PubMed] [Google Scholar]
- 103.Kumagai Y, Akira S. J Allergy Clin Immunol. 2010;125:985–992. doi: 10.1016/j.jaci.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 104.Singh H. Encyclopedia of Systems Biology. 2013:517–517. [Google Scholar]
- 105.Collier MA, Gallovic MD, Peine KJ, Duong AD, Bachelder EM, Gunn JS, Schlesinger LS, Ainslie KM. Expert Rev Anti Infect Ther. 2013;11:1225–1235. doi: 10.1586/14787210.2013.845524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. EMBO J. 2008;27:1110–1121. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Anand PK, Tait SW, Lamkanfi M, Amer AO, Nunez G, Pages G, Pouyssegur J, McGargill MA, Green DR, Kanneganti TD. J Biol Chem. 2011;286:42981–42991. doi: 10.1074/jbc.M111.310599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lu J, Li Z, Zink JI, Tamanoi F. Nanomedicine. 2012;8:212–220. doi: 10.1016/j.nano.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schroder K, Tschopp J. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 110.Moon JJ, Suh H, Polhemus ME, Ockenhouse CF, Yadava A, Irvine DJ. PLoS One. 2012;7:e31472. doi: 10.1371/journal.pone.0031472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Demento SL, Eisenbarth SC, Foellmer HG, Platt C, Caplan MJ, Mark Saltzman W, Mellman I, Ledizet M, Fikrig E, Flavell RA, Fahmy TM. Vaccine. 2009;27:3013–3021. doi: 10.1016/j.vaccine.2009.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Turner MW. Immunol Today. 1996;17:532–540. doi: 10.1016/0167-5699(96)10062-1. [DOI] [PubMed] [Google Scholar]
- 113.Chavez-Santoscoy AV, Roychoudhury R, Pohl NL, Wannemuehler MJ, Narasimhan B, Ramer-Tait AE. Biomaterials. 2012;33:4762–4772. doi: 10.1016/j.biomaterials.2012.03.027. [DOI] [PubMed] [Google Scholar]
- 114.Weis WI, Drickamer K. Structure. 1994;2:1227–1240. doi: 10.1016/S0969-2126(94)00124-3. [DOI] [PubMed] [Google Scholar]
- 115.Connaris H, Crocker PR, Taylor GL. J Biol Chem. 2009;284:7339–7351. doi: 10.1074/jbc.M807398200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jain SK, Gupta Y, Jain A, Saxena AR, Khare P, Jain A. Nanomedicine. 2008;4:41–48. doi: 10.1016/j.nano.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 117.Kaur A, Jain S, Tiwary AK. Acta Pharm. 2008;58:61–74. doi: 10.2478/v10007-007-0045-1. [DOI] [PubMed] [Google Scholar]
- 118.Luo Y, Zhou H, Krueger J, Kaplan C, Lee SH, Dolman C, Markowitz D, Wu W, Liu C, Reisfeld RA. J Clin Invest. 2006;116:2132–2141. doi: 10.1172/JCI27648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Keler T, Ramakrishna V, Fanger MW. Expert Opin Biol Ther. 2004;4:1953–1962. doi: 10.1517/14712598.4.12.1953. [DOI] [PubMed] [Google Scholar]
- 120.Gary-Bobo M, Mir Y, Rouxel C, Brevet D, Basile I, Maynadier M, Vaillant O, Mongin O, Blanchard-Desce M, Morere A, Garcia M, Durand JO, Raehm L. Angew Chem Int Ed Engl. 2011;50:11425–11429. doi: 10.1002/anie.201104765. [DOI] [PubMed] [Google Scholar]
- 121.Kanno S, Furuyama A, Hirano S. Toxicol Sci. 2007;97:398–406. doi: 10.1093/toxsci/kfm050. [DOI] [PubMed] [Google Scholar]
- 122.Peiser L, Mukhopadhyay S, Gordon S. Curr Opin Immunol. 2002;14:123–128. doi: 10.1016/s0952-7915(01)00307-7. [DOI] [PubMed] [Google Scholar]
- 123.Canton J, Neculai D, Grinstein S. Nat Rev Immunol. 2013;13:621–634. doi: 10.1038/nri3515. [DOI] [PubMed] [Google Scholar]
- 124.Pluddemann A, Neyen C, Gordon S. Methods. 2007;43:207–217. doi: 10.1016/j.ymeth.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 125.Lunov O, Zablotskii V, Syrovets T, Rocker C, Tron K, Nienhaus GU, Simmet T. Biomaterials. 2011;32:547–555. doi: 10.1016/j.biomaterials.2010.08.111. [DOI] [PubMed] [Google Scholar]
- 126.Le Duc G, Vander Elst L, Colet JM, Roch A, Gillis P, Le Bas JF, Muller RN. J Magn Reson Imaging. 2001;13:619–626. doi: 10.1002/jmri.1087. [DOI] [PubMed] [Google Scholar]
- 127.Aderem A, Underhill DM. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- 128.Bell NC, Minelli C, Shard AG. Anal Methods. 2013;5:4591–4601. [Google Scholar]
- 129.Khlebtsov N, Bogatyrev V, Khlebtsov B, Dykman L, Englebienne P. Colloid Journal. 2003;65:622–635. [Google Scholar]
- 130.Bee JS, Chiu D, Sawicki S, Stevenson JL, Chatterjee K, Freund E, Carpenter JF, Randolph TW. J Pharm Sci. 2009;98:3218–3238. doi: 10.1002/jps.21768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Taylor PR, Martinez-Pomares L, Stacey M, Lin HH, Brown GD, Gordon S. Annu Rev Immunol. 2005;23:901–944. doi: 10.1146/annurev.immunol.23.021704.115816. [DOI] [PubMed] [Google Scholar]
- 132.Lesniak A, Fenaroli F, Monopoli MP, Aberg C, Dawson KA, Salvati A. ACS Nano. 2012;6:5845–5857. doi: 10.1021/nn300223w. [DOI] [PubMed] [Google Scholar]
- 133.Dutta D, Sundaram SK, Teeguarden JG, Riley BJ, Fifield LS, Jacobs JM, Addleman SR, Kaysen GA, Moudgil BM, Weber TJ. Toxicol Sci. 2007;100:303–315. doi: 10.1093/toxsci/kfm217. [DOI] [PubMed] [Google Scholar]
- 134.Herd H, Daum N, Jones AT, Huwer H, Ghandehari H, Lehr CM. ACS Nano. 2013;7:1961–1973. doi: 10.1021/nn304439f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Herd H, Ghandehari H. Synthetic and toxicological characteristics of silica nanomaterials for imaging and drug delivery applications. CRC Press; Boca Raton, FL: 2011. [Google Scholar]
- 136.Walters MI, Papadimitriou JM, Spector W. CRC critical reviews in toxicology. 1978;5:377–421. doi: 10.3109/10408447809081012. [DOI] [PubMed] [Google Scholar]
- 137.Monks J, Rosner D, Geske FJ, Lehman L, Hanson L, Neville M, Fadok V. Cell Death & Differentiation. 2005;12:107–114. doi: 10.1038/sj.cdd.4401517. [DOI] [PubMed] [Google Scholar]
- 138.de Jonge MR, Koymans LH, Guillemont JE, Koul A, Andries K. Proteins. 2007;67:971–980. doi: 10.1002/prot.21376. [DOI] [PubMed] [Google Scholar]
- 139.Doherty GJ, McMahon HT. Annu Rev Biochem. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- 140.Mayor S, Parton RG, Donaldson JG. Cold Spring Harb Perspect Biol. 2014;6:a016758. doi: 10.1101/cshperspect.a016758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kumari S, Mg S, Mayor S. Cell Res. 2010;20:256–275. doi: 10.1038/cr.2010.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mooren OL, Galletta BJ, Cooper JA. Annu Rev Biochem. 2012;81:661–686. doi: 10.1146/annurev-biochem-060910-094416. [DOI] [PubMed] [Google Scholar]
- 143.Sandvig K, Pust S, Skotland T, van Deurs B. Curr Opin Cell Biol. 2011;23:413–420. doi: 10.1016/j.ceb.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 144.McMahon HT, Boucrot E. Nat Rev Mol Cell Biol. 2011;12:517–533. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- 145.Canton I, Battaglia G. Chem Soc Rev. 2012;41:2718–2739. doi: 10.1039/c2cs15309b. [DOI] [PubMed] [Google Scholar]
- 146.Parton RG, Richards AA. Traffic. 2003;4:724–738. doi: 10.1034/j.1600-0854.2003.00128.x. [DOI] [PubMed] [Google Scholar]
- 147.Pelkmans L, Burli T, Zerial M, Helenius A. Cell. 2004;118:767–780. doi: 10.1016/j.cell.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 148.Gratton SE, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, DeSimone JM. Proc Natl Acad Sci U S A. 2008;105:11613–11618. doi: 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Biochem J. 2004;377:159–169. doi: 10.1042/BJ20031253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chithrani BD, Ghazani AA, Chan WCW. Nano Letters. 2006;6:662–668. doi: 10.1021/nl052396o. [DOI] [PubMed] [Google Scholar]
- 151.Chithrani BD, Chan WC. Nano Lett. 2007;7:1542–1550. doi: 10.1021/nl070363y. [DOI] [PubMed] [Google Scholar]
- 152.Chung TH, Wu SH, Yao M, Lu CW, Lin YS, Hung Y, Mou CY, Chen YC, Huang DM. Biomaterials. 2007;28:2959–2966. doi: 10.1016/j.biomaterials.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 153.Champion JA, Mitragotri S. Proc Natl Acad Sci U S A. 2006;103:4930–4934. doi: 10.1073/pnas.0600997103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Champion JA, Katare YK, Mitragotri S. Proc Natl Acad Sci U S A. 2007;104:11901–11904. doi: 10.1073/pnas.0705326104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Decuzzi P, Ferrari M. Biophys J. 2008;94:3790–3797. doi: 10.1529/biophysj.107.120238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jiang W, Kim BYS, Rutka JT, Chan WCW. Nat Nanotechnol. 2008;3:145–150. doi: 10.1038/nnano.2008.30. [DOI] [PubMed] [Google Scholar]
- 157.Haensler J, Szoka FC., Jr Bioconjug Chem. 1993;4:372–379. doi: 10.1021/bc00023a012. [DOI] [PubMed] [Google Scholar]
- 158.Fischer D, Bieber T, Li Y, Elsasser HP, Kissel T. Pharm Res. 1999;16:1273–1279. doi: 10.1023/a:1014861900478. [DOI] [PubMed] [Google Scholar]
- 159.Roser M, Fischer D, Kissel T. Eur J Pharm Biopharm. 1998;46:255–263. doi: 10.1016/s0939-6411(98)00038-1. [DOI] [PubMed] [Google Scholar]
- 160.Chellat F, Merhi Y, Moreau A, Yahia L. Biomaterials. 2005;26:7260–7275. doi: 10.1016/j.biomaterials.2005.05.044. [DOI] [PubMed] [Google Scholar]
- 161.Yuan H, Li J, Bao G, Zhang S. Phys Rev Lett. 2010;105:138101. doi: 10.1103/PhysRevLett.105.138101. [DOI] [PubMed] [Google Scholar]
- 162.Deretic V, Saitoh T, Akira S. Nat Rev Immunol. 2013;13:722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hamilton RF, Jr, Thakur SA, Mayfair JK, Holian A. J Biol Chem. 2006;281:34218–34226. doi: 10.1074/jbc.M605229200. [DOI] [PubMed] [Google Scholar]
- 164.Petrovski G, Zahuczky G, Majai G, Fesus L. Autophagy. 2007;3:509–511. doi: 10.4161/auto.4731. [DOI] [PubMed] [Google Scholar]
- 165.Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, Suffredini AF. Blood. 2003;101:2652–2660. doi: 10.1182/blood-2002-05-1300. [DOI] [PubMed] [Google Scholar]
- 166.Zhang XD, Wu D, Shen X, Liu PX, Fan FY, Fan SJ. Biomaterials. 2012;33:4628–4638. doi: 10.1016/j.biomaterials.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 167.Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA, Yen Y, Heidel JD, Ribas A. Nature. 2010;464:1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Vercauteren D, Deschout H, Remaut K, Engbersen JF, Jones AT, Demeester J, De Smedt SC, Braeckmans K. ACS Nano. 2011;5:7874–7884. doi: 10.1021/nn2020858. [DOI] [PubMed] [Google Scholar]
- 169.Herd HL, Malugin A, Ghandehari H. J Control Release. 2011;153:40–48. doi: 10.1016/j.jconrel.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Levine B, Mizushima N, Virgin HW. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Stern ST, Adiseshaiah PP, Crist RM. Part Fibre Toxicol. 2012;9:20. doi: 10.1186/1743-8977-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kanduc D, Mittelman A, Serpico R, Sinigaglia E, Sinha AA, Natale C, Santacroce R, Di Corcia MG, Lucchese A, Dini L, Pani P, Santacroce S, Simone S, Bucci R, Farber E. Int J Oncol. 2002;21:165–170. [PubMed] [Google Scholar]
- 173.Perez-Carrion MD, Perez-Martinez FC, Merino S, Sanchez-Verdu P, Martinez-Hernandez J, Lujan R, Cena V. J Neurochem. 2012;120:259–268. doi: 10.1111/j.1471-4159.2011.07556.x. [DOI] [PubMed] [Google Scholar]
- 174.Vernon PJ, Tang D. Antioxid Redox Signal. 2013;18:677–691. doi: 10.1089/ars.2012.4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Wu L, Zhang Y, Zhang C, Cui X, Zhai S, Liu Y, Li C, Zhu H, Qu G, Jiang G. ACS Nano. 2014;8:2087–2099. doi: 10.1021/nn500376w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Gupta A, Pant G, Mitra K, Madan J, Chourasia MK, Misra A. Mol Pharm. 2014;11:1201–1207. doi: 10.1021/mp4006563. [DOI] [PubMed] [Google Scholar]
- 178.Kruth HS, Chang J, Ifrim I, Zhang WY. Eur J Cell Biol. 1999;78:91–99. doi: 10.1016/S0171-9335(99)80010-7. [DOI] [PubMed] [Google Scholar]
- 179.Kruth HS, Skarlatos SI, Lilly K, Chang J, Ifrim I. J Cell Biol. 1995;129:133–145. doi: 10.1083/jcb.129.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Motskin M, Müller KH, Genoud C, Monteith AG, Skepper JN. Biomaterials. 2011;32:9470–9482. doi: 10.1016/j.biomaterials.2011.08.060. [DOI] [PubMed] [Google Scholar]
- 181.Kirchhausen T. Annu Rev Cell Dev Biol. 1999;15:705–732. doi: 10.1146/annurev.cellbio.15.1.705. [DOI] [PubMed] [Google Scholar]
- 182.Benjaminsen RV, Mattebjerg MA, Henriksen JR, Moghimi SM, Andresen TL. Mol Ther. 2013;21:149–157. doi: 10.1038/mt.2012.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Szewczyk A, Wojtczak L. Pharmacol Rev. 2002;54:101–127. doi: 10.1124/pr.54.1.101. [DOI] [PubMed] [Google Scholar]
- 184.Boddapati SV, D’Souza GG, Erdogan S, Torchilin VP, Weissig V. Nano Lett. 2008;8:2559–2563. doi: 10.1021/nl801908y. [DOI] [PubMed] [Google Scholar]
- 185.Mukhopadhyay A, Weiner H. Adv Drug Deliv Rev. 2007;59:729–738. doi: 10.1016/j.addr.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hoshino A, Fujioka K, Oku T, Nakamura S, Suga M, Yamaguchi Y, Suzuki K, Yasuhara M, Yamamoto K. Microbiol Immunol. 2004;48:985–994. doi: 10.1111/j.1348-0421.2004.tb03621.x. [DOI] [PubMed] [Google Scholar]
- 187.Tkachenko AG, Xie H, Liu Y, Coleman D, Ryan J, Glomm WR, Shipton MK, Franzen S, Feldheim DL. Bioconjug Chem. 2004;15:482–490. doi: 10.1021/bc034189q. [DOI] [PubMed] [Google Scholar]
- 188.Misra R, Sahoo SK. Eur J Pharm Sci. 2010;39:152–163. doi: 10.1016/j.ejps.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 189.Pouton CW, Wagstaff KM, Roth DM, Moseley GW, Jans DA. Adv Drug Deliv Rev. 2007;59:698–717. doi: 10.1016/j.addr.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 190.Murphy MP, Smith RA. Adv Drug Deliv Rev. 2000;41:235–250. doi: 10.1016/s0169-409x(99)00069-1. [DOI] [PubMed] [Google Scholar]
- 191.Jones CF, Grainger DW. Adv Drug Deliv Rev. 2009;61:438–456. doi: 10.1016/j.addr.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Pulskamp K, Diabaté S, Krug HF. Toxicol Lett. 2007;168:58. doi: 10.1016/j.toxlet.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 193.Petri-Fink A, Steitz B, Finka A, Salaklang J, Hofmann H. Eur J Pharm Biopharm. 2008;68:129–137. doi: 10.1016/j.ejpb.2007.02.024. [DOI] [PubMed] [Google Scholar]
- 194.Malugin A, Ghandehari H. Nanosci Nanotechnol Lett. 2011;3:309–313. [Google Scholar]
- 195.Joshi GN, Knecht DA. Apoptosis. 2013;18:271–285. doi: 10.1007/s10495-012-0798-y. [DOI] [PubMed] [Google Scholar]
- 196.Winterbourn CC. Toxicol Lett. 1995;82–83:969–974. doi: 10.1016/0378-4274(95)03532-x. [DOI] [PubMed] [Google Scholar]
- 197.Dalal NS, Shi XL, Vallyathan V. J Toxicol Environ Health. 1990;29:307–316. doi: 10.1080/15287399009531393. [DOI] [PubMed] [Google Scholar]
- 198.Lin W, Huang YW, Zhou XD, Ma Y. Toxicol Appl Pharmacol. 2006;217:252–259. doi: 10.1016/j.taap.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 199.Waters KM, Masiello LM, Zangar RC, Tarasevich BJ, Karin NJ, Quesenberry RD, Bandyopadhyay S, Teeguarden JG, Pounds JG, Thrall BD. Toxicol Sci. 2009;107:553–569. doi: 10.1093/toxsci/kfn250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Shahbazi MA, Hamidi M, Makila EM, Zhang H, Almeida PV, Kaasalainen M, Salonen JJ, Hirvonen JT, Santos HA. Biomaterials. 2013;34:7776–7789. doi: 10.1016/j.biomaterials.2013.06.052. [DOI] [PubMed] [Google Scholar]
- 201.Lee S, Yun HS, Kim SH. Biomaterials. 2011;32:9434–9443. doi: 10.1016/j.biomaterials.2011.08.042. [DOI] [PubMed] [Google Scholar]
- 202.Fischer HC, Hauck TS, Gomez-Aristizabal A, Chan WC. Adv Mater. 2010;22:2520–2524. doi: 10.1002/adma.200904231. [DOI] [PubMed] [Google Scholar]
- 203.Bregoli L, Chiarini F, Gambarelli A, Sighinolfi G, Gatti AM, Santi P, Martelli AM, Cocco L. Toxicology. 2009;262:121–129. doi: 10.1016/j.tox.2009.05.017. [DOI] [PubMed] [Google Scholar]
- 204.Oberdorster G, Maynard A, Donaldson K, Castranova V, Fitzpatrick J, Ausman K, Carter J, Karn B, Kreyling W, Lai D, Olin S, Monteiro-Riviere N, Warheit D, Yang H I.R.F.R.S.I.N.T.S.W. Group. Part Fibre Toxicol. 2005;2:8. doi: 10.1186/1743-8977-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hauck TS, Anderson RE, Fischer HC, Newbigging S, Chan WC. Small. 2010;6:138–144. doi: 10.1002/smll.200900626. [DOI] [PubMed] [Google Scholar]
- 206.Naito M, Hasegawa G, Ebe Y, Yamamoto T. Med Electron Microsc. 2004;37:16–28. doi: 10.1007/s00795-003-0228-x. [DOI] [PubMed] [Google Scholar]
- 207.Wake K, Decker K, Kirn A, Knook D, McCuskey R, Bouwens L, Wisse E. Int Rev Cytol. 1988;118:173–229. doi: 10.1016/s0074-7696(08)60875-x. [DOI] [PubMed] [Google Scholar]
- 208.Baffy G. J Hepatol. 2009;51:212–223. doi: 10.1016/j.jhep.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Moghimi SM, Hunter AC, Murray JC. Pharmacol Rev. 2001;53:283–318. [PubMed] [Google Scholar]
- 210.Longmire M, Choyke PL, Kobayashi H. Nanomedicine. 2008;3:703–717. doi: 10.2217/17435889.3.5.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Hardonk MJ, Harms G, Koudstaal J. Histochemistry. 1985;83:473–477. doi: 10.1007/BF00509211. [DOI] [PubMed] [Google Scholar]
- 212.Sadauskas E, Danscher G, Stoltenberg M, Vogel U, Larsen A, Wallin H. Nanomedicine. 2009;5:162–169. doi: 10.1016/j.nano.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 213.Gad SC, Sharp KL, Montgomery C, Payne JD, Goodrich GP. Int J Toxicol. 2012;31:584–594. doi: 10.1177/1091581812465969. [DOI] [PubMed] [Google Scholar]
- 214.Arvizo RR, Miranda OR, Moyano DF, Walden CA, Giri K, Bhattacharya R, Robertson JD, Rotello VM, Reid JM, Mukherjee P. PLoS One. 2011;6:e24374. doi: 10.1371/journal.pone.0024374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Souris JS, Lee CH, Cheng SH, Chen CT, Yang CS, Ho JA, Mou CY, Lo LW. Biomaterials. 2010;31:5564–5574. doi: 10.1016/j.biomaterials.2010.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.He Q, Zhang Z, Gao F, Li Y, Shi J. Small. 2011;7:271–280. doi: 10.1002/smll.201001459. [DOI] [PubMed] [Google Scholar]
- 217.Kim ST, Saha K, Kim C, Rotello VM. Acc Chem Res. 2013;46:681–691. doi: 10.1021/ar3000647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Sarparanta M, Bimbo LM, Rytkonen J, Makila E, Laaksonen TJ, Laaksonen P, Nyman M, Salonen J, Linder MB, Hirvonen J, Santos HA, Airaksinen AJ. Mol Pharm. 2012;9:654–663. doi: 10.1021/mp200611d. [DOI] [PubMed] [Google Scholar]
- 219.Gaucher G, Asahina K, Wang J, Leroux JC. Biomacromolecules. 2009;10:408–416. doi: 10.1021/bm801178f. [DOI] [PubMed] [Google Scholar]
- 220.Wingard JR, White MH, Anaissie E, Raffalli J, Goodman J, Arrieta A L.A.A.C.S. Group. Clin Infect Dis. 2000;31:1155–1163. doi: 10.1086/317451. [DOI] [PubMed] [Google Scholar]
- 221.Sankhala K, Mita A, Adinin R, Wood L, Beeram M, Bullock S, Yamagata N, Matsuno K, Fujisawa T, Phan A. J Clin Oncol. 2009;27:2535. [Google Scholar]
- 222.Leamon CP, Reddy JA. Adv Drug Deliv Rev. 2004;56:1127–1141. doi: 10.1016/j.addr.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 223.Puvanakrishnan P, Park J, Chatterjee D, Krishnan S, Tunnell JW. Int J Nanomedicine. 2012;7:1251–1258. doi: 10.2147/IJN.S29147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Cho M, Cho WS, Choi M, Kim SJ, Han BS, Kim SH, Kim HO, Sheen YY, Jeong J. Toxicol Lett. 2009;189:177–183. doi: 10.1016/j.toxlet.2009.04.017. [DOI] [PubMed] [Google Scholar]
- 225.Hirst SM, Karakoti A, Singh S, Self W, Tyler R, Seal S, Reilly CM. Environ Toxicol. 2013;28:107–118. doi: 10.1002/tox.20704. [DOI] [PubMed] [Google Scholar]
- 226.Zreiqat H, Kumar RK, Markovic B, Zicat B, Howlett CR. J Biomed Mater Res A. 2003;65:109–117. doi: 10.1002/jbm.a.10441. [DOI] [PubMed] [Google Scholar]
- 227.Garrigues GE, Cho DR, Rubash HE, Goldring SR, Herndon JH, Shanbhag AS. Biomaterials. 2005;26:2933–2945. doi: 10.1016/j.biomaterials.2004.06.034. [DOI] [PubMed] [Google Scholar]
- 228.Neun BW, Dobrovolskaia MA. Methods Mol Biol. 2011;697:237–245. doi: 10.1007/978-1-60327-198-1_25. [DOI] [PubMed] [Google Scholar]
- 229.Thomas SN, van der Vlies AJ, O’Neil CP, Reddy ST, Yu SS, Giorgio TD, Swartz MA, Hubbell JA. Biomaterials. 2011;32:2194–2203. doi: 10.1016/j.biomaterials.2010.11.037. [DOI] [PubMed] [Google Scholar]
- 230.Szebeni J, Baranyi L, Savay S, Milosevits J, Bunger R, Laverman P, Metselaar JM, Storm G, Chanan-Khan A, Liebes L, Muggia FM, Cohen R, Barenholz Y, Alving CR. J Liposome Res. 2002;12:165–172. doi: 10.1081/lpr-120004790. [DOI] [PubMed] [Google Scholar]
- 231.Pedersen MB, Zhou X, Larsen EKU, Sørensen US, Kjems J, Nygaard JV, Nyengaard JR, Meyer RL, Boesen T, Vorup-Jensen T. J Immun. 2010;184:1931–1945. doi: 10.4049/jimmunol.0902214. [DOI] [PubMed] [Google Scholar]
- 232.Ross GD, Reed W, Dalzell JG, Becker SE, Hogg N. J Leukoc Biol. 1992;51:109–117. doi: 10.1002/jlb.51.2.109. [DOI] [PubMed] [Google Scholar]
- 233.Moghimi SM, Szebeni J. Prog Lipid Res. 2003;42:463–478. doi: 10.1016/s0163-7827(03)00033-x. [DOI] [PubMed] [Google Scholar]
- 234.van Lookeren Campagne M, Wiesmann C, Brown EJ. Cell Microbiol. 2007;9:2095–2102. doi: 10.1111/j.1462-5822.2007.00981.x. [DOI] [PubMed] [Google Scholar]
- 235.Corot C, Robert P, Idee JM, Port M. Adv Drug Deliv Rev. 2006;58:1471–1504. doi: 10.1016/j.addr.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 236.Hamidi M, Azadi A, Rafiei P. Drug Deliv. 2006;13:399–409. doi: 10.1080/10717540600814402. [DOI] [PubMed] [Google Scholar]
- 237.Yoo JW, Chambers E, Mitragotri S. Curr pharm design. 2010;16:2298–2307. doi: 10.2174/138161210791920496. [DOI] [PubMed] [Google Scholar]
- 238.Moghimi SM, Hedeman H, Christy NM, Illum L, Davis SS. J Leukoc Biol. 1993;54:513–517. doi: 10.1002/jlb.54.6.513. [DOI] [PubMed] [Google Scholar]
- 239.Moghimi SM, Andersen AJ, Ahmadvand D, Wibroe PP, Andresen TL, Hunter AC. Adv Drug Deliv Rev. 2011;63:1000–1007. doi: 10.1016/j.addr.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 240.Greish K, Thiagarajan G, Herd H, Price R, Bauer H, Hubbard D, Burckle A, Sadekar S, Yu T, Anwar A. Nanotoxicology. 2012;6:713–723. doi: 10.3109/17435390.2011.604442. [DOI] [PubMed] [Google Scholar]
- 241.Radomski A, Jurasz P, Alonso-Escolano D, Drews M, Morandi M, Malinski T, Radomski MW. Br J Pharmacol. 2005;146:882–893. doi: 10.1038/sj.bjp.0706386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Movat HZ, Weiser WJ, Glynn MF, Mustard JF. J Cell Biol. 1965;27:531–543. doi: 10.1083/jcb.27.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Jones CF, Campbell RA, Brooks AE, Assemi S, Tadjiki S, Thiagarajan G, Mulcock C, Weyrich AS, Brooks BD, Ghandehari H, Grainger DW. ACS Nano. 2012;6:9900–9910. doi: 10.1021/nn303472r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Jones CF, Campbell RA, Franks Z, Gibson CC, Thiagarajan G, Vieira-de-Abreu A, Sukavaneshvar S, Mohammad SF, Li DY, Ghandehari H, Weyrich AS, Brooks BD, Grainger DW. Mol Pharm. 2012;9:1599–1611. doi: 10.1021/mp2006054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Greish K, Thiagarajan G, Herd H, Price R, Bauer H, Hubbard D, Burckle A, Sadekar S, Yu T, Anwar A, Ray A, Ghandehari H. Nanotoxicology. 2012;6:713–723. doi: 10.3109/17435390.2011.604442. [DOI] [PubMed] [Google Scholar]
- 246.Ilinskaya AN, Dobrovolskaia MA. Nanomedicine (Lond) 2013;8:969–981. doi: 10.2217/nnm.13.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Park J, Lim DH, Lim HJ, Kwon T, Choi JS, Jeong S, Choi IH, Cheon J. Chem Commun (Camb) 2011;47:4382–4384. doi: 10.1039/c1cc10357a. [DOI] [PubMed] [Google Scholar]
- 248.Lim DH, Jang J, Kim S, Kang T, Lee K, Choi IH. Biomaterials. 2012;33:4690–4699. doi: 10.1016/j.biomaterials.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 249.Wilbourn JD, McGregor DB, Partensky C, Rice JM. Environ Health Perspect. 1997;105:756–759. doi: 10.1289/ehp.97105756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Ovrevik J, Refsnes M, Namork E, Becher R, Sandnes D, Schwarze PE, Lag M. Toxicology. 2006;227:105–116. doi: 10.1016/j.tox.2006.07.029. [DOI] [PubMed] [Google Scholar]
- 251.Lenz AG, Krombach F, Maier KL. Free Radic Biol Med. 1992;12:1–10. doi: 10.1016/0891-5849(92)90052-i. [DOI] [PubMed] [Google Scholar]
- 252.Kleinman MT, Araujo JA, Nel A, Sioutas C, Campbell A, Cong PQ, Li H, Bondy SC. Toxicol Lett. 2008;178:127–130. doi: 10.1016/j.toxlet.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Lacasse Y, Martin S, Gagne D, Lakhal L. Cancer Causes Control. 2009;20:925–933. doi: 10.1007/s10552-009-9296-0. [DOI] [PubMed] [Google Scholar]
- 254.Hnizdo E, Vallyathan V. Occup Environ Med. 2003;60:237–243. doi: 10.1136/oem.60.4.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Cocco P, Dosemeci M, Rice C. Med Lav. 2007;98:3–17. [PubMed] [Google Scholar]
- 256.Iler RK. The chemistry of silica. John Wiley and Sons, Inc; 1979. [Google Scholar]
- 257.Garay RP, El-Gewely R, Armstrong JK, Garratty G, Richette P. Expert Opin Drug Deliv. 2012;9:1319–1323. doi: 10.1517/17425247.2012.720969. [DOI] [PubMed] [Google Scholar]
- 258.Schellekens H, Hennink WE, Brinks V. Pharm Res. 2013;30:1729–1734. doi: 10.1007/s11095-013-1067-7. [DOI] [PubMed] [Google Scholar]
- 259.Burger P, Hilarius-Stokman P, de Korte D, van den Berg TK, van Bruggen R. Blood. 2012;119:5512–5521. doi: 10.1182/blood-2011-10-386805. [DOI] [PubMed] [Google Scholar]
- 260.Weiskopf K, Ring AM, Ho CCM, Volkmer JP, Levin AM, Volkmer AK, Özkan E, Fernhoff NB, van de Rijn M, Weissman IL. Science. 2013;341:88–91. doi: 10.1126/science.1238856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Smith CA, Wiley S, Kaykas A, Probst P. Google Patents. United States: 2007. [Google Scholar]
- 262.Rodriguez PL, Harada T, Christian DA, Pantano DA, Tsai RK, Discher DE. Science. 2013;339:971–975. doi: 10.1126/science.1229568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Niu M, Naguib YW, Aldayel AM, Shi YC, Hursting SD, Hersh MA, Cui Z. Mol Pharm. 2014;11:4425–4436. doi: 10.1021/mp500565q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Zhu S, Niu M, O’Mary H, Cui Z. Mol Pharm. 2013;10:3525–3530. doi: 10.1021/mp400216r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Duzgunes N, Pretzer E, Simoes S, Slepushkin V, Konopka K, Flasher D, de Lima MC. Mol Membr Biol. 1999;16:111–118. doi: 10.1080/096876899294832. [DOI] [PubMed] [Google Scholar]
- 266.Dutta T, Garg M, Jain NK. Eur J Pharm Sci. 2008;34:181–189. doi: 10.1016/j.ejps.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 267.Schafer V, von Briesen H, Andreesen R, Steffan AM, Royer C, Troster S, Kreuter J, Rubsamen-Waigmann H. Pharm Res. 1992;9:541–546. doi: 10.1023/a:1015852732512. [DOI] [PubMed] [Google Scholar]
- 268.Couvreur P, Fattal E, Andremont A. Pharm Res. 1991;8:1079–1086. doi: 10.1023/a:1015885814417. [DOI] [PubMed] [Google Scholar]
- 269.Clemens DL, Lee BY, Xue M, Thomas CR, Meng H, Ferris D, Nel AE, Zink JI, Horwitz MA. Antimicrob Agents Chemother. 2012;56:2535–2545. doi: 10.1128/AAC.06049-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Escobar G, Gentner B, Naldini L, Mazzieri R. Oncoimmunology. 2014;3:e28696. doi: 10.4161/onci.28696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Batrakova EV, Gendelman HE, Kabanov AV. Expert Opin Drug Deliv. 2011;8:415–433. doi: 10.1517/17425247.2011.559457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Shive MS, Anderson JM. Adv Drug Deliv Rev. 1997;28:5–24. doi: 10.1016/s0169-409x(97)00048-3. [DOI] [PubMed] [Google Scholar]
- 273.Basaraba RJ. Tuberculosis (Edinb) 2008;88(Suppl 1):S35–47. doi: 10.1016/S1472-9792(08)70035-0. [DOI] [PubMed] [Google Scholar]