

Abstract
Recently, gene set-based approaches have become very popular in gene expression profiling studies for assessing how genetic variants are related to disease outcomes. Since most genes are not differentially expressed, existing pathway tests considering all genes within a pathway suffer from considerable noise and power loss. Moreover, for a differentially expressed pathway, it is of interest to select important genes that drive the effect of the pathway. In this article, we propose an adaptive association test using double kernel machines (DKM), which can both select important genes within the pathway as well as test for the overall genetic pathway effect. This DKM procedure first uses the garrote kernel machines (GKM) test for the purposes of subset selection and then the least squares kernel machine (LSKM) test for testing the effect of the subset of genes. An appealing feature of the kernel machine framework is that it can provide a flexible and unified method for multi-dimensional modeling of the genetic pathway effect allowing for both parametric and nonparametric components. This DKM approach is illustrated with application to simulated data as well as to data from a neuroimaging genetics study.
Keywords: Double kernel machine, Garrote kernel machine, Least squares kernel machine, Subset testing, Thresholding
1 Introduction
Advances in high-throughout biotechnology over the last decades have culminated in large-scale genetic association studies, which have facilitated identification of many genetic variants associated with a range of complex traits in gene expression profiling experiments. An individual gene analysis approach is useful in identifying disease susceptibility genes. However, the major limitations of individual gene-based analysis are as follows. First, individual analysis is often too conservative due to multiple testing. Second, results of individual analysis are often hard to interpret and not reproducible across studies. Many genes found in the discovery phase are false positives and cannot be validated in other experiments. This is largely due to the restricted power to detect a small effect that an individual gene is truly associated with the outcome. Third, it is unlikely that individual genes work in isolation, and it is known that biological phenomena occur through the concerted expression of multiple genes. Such joint actions between genes cannot be captured in any individual gene analysis. Therefore analysis based on genetic pathways or gene sets has become popular recently (Cai et al. 2012; Wu et al. 2010).
The challenge in genetic pathway association studies is how to model and test for a complex pathway effect on a disease outcome. Given the fact that the outcome may rely on the genetic pathway in a complicated and unknown pattern, nonparametric methods are desirable here. Liu et al. (2007) proposed a kernel machine-based semiparametric approach for modeling the pathway effects. There are several appealing features of this kernel machine framework. First, it provides a nonparametric approach of modeling the pathway effect. It allows a flexible function for the joint effect of multiple genes within a pathway by specification of a kernel function that allows for nonlinear gene effects as well as complex interactions. Second, by taking the correlation between genes into account, kernel machine score test have improved power of detecting the pathway effect. Third, the kernel machine models can be easily connected with the linear mixed effects models which allows for a unified likelihood framework, in which parameter estimation and inference are feasible (Liu et al. 2007, 2008; Kwee et al. 2008).
Although the least squares kernel machine (LSKM) test proposed in Liu et al. (2007) enjoys those aforementioned benefits, but it has two major disadvantages. First, it is not robust against noisy variables. It has been shown that the association signal decreased with adding non-associated genetic variants (Wessel and Schork 2006; Wu et al. 2009). The existence of noisy variants in the genetic pathway will lower the power for detecting the pathway effect. As will be seen in the simulation studies, besides lower power, the LSKM test violates the nominal type I error rate if too many noisy variants are present in the pathway. To fix this problem, some pre-processing methods of filtering noisy genetic variants are needed. Second, the LSKM test can only give an overall p-value for whether the pathway has a significant effect on the disease outcome. In practice, for a genetic pathway with significant effects on a phenoAn Adaptive Genetic Association Test Using Double Kernel Machines 3 type, it is of interest to select important genes which drive the pathway effect.
The DKM approach we propose can address both these two issues. The DKM procedure utilizes two rounds of kernel machines. In the first round, we apply gene selection using the Garrote kernel machine (GKM) test of Maity and Lin (2011). For each gene in the pathway, we apply a GKM test and get a marginal p-value of that gene. A criterion is applied on those p-values to select a gene subset. In the second round, a LSKM test is performed on this selected subset, which gives an overall p-value of the pathway effect. Note that the information of the association between genes and the disease outcome is used twice, which will lead to an inflated type I error rate. There are in general two ways to fix this issue. The first is to perform the subset selection on an independent dataset as Kwee et al. (2008) did in order to determine the weight of each individual genetic variant. The other way is to perform some permutation-based methods to establish the significance of the DKM procedure (Pan and Shen 2011). In this paper, we use the first approach.
The rest of the paper is organized as follows. In Section 2, we first discuss the kernel machine framework and then review the LSKM test and the GKM test. In section 3, we describe our DKM test, which is a two-stage adaptive association testing procedure. The criterion of subset selection is also discussed. In Section 4, we evaluate the DKM test with simulation studies and compare our test to LSKM. The performance of the subset selection using GKM test is also evaluated in this simulation. Then our method is illustrated using data on a gene, GRIN2B, that was implicated in some initial neuroimaging genomics studies of Alzheimer’s disease in Section 5. The paper concludes with discussion in Section 6.
2 Kernel Machine Score Tests
2.1 A Semiparametric Kernel Machine Model
In this paper, a kernel machine is a symmetric and positive definite bivariate function k : χ ×χ → R; (x, y) ↦ → k(x, y). Here the positive definiteness implies the following, for any positive integer N and any set of distinct points {x1, · · ·, xN} ⊂ χ, the kernel matrix (Gram matrix) is positive definite. The kernel machine serves as a powerful dimension-reducing tool for modeling complex genetic pathway effect on a disease outcome. The dimension reduction is achieved by collapsing the comparison of the multidimensional genetic variants vector into a scalar using the kernel machine. The scalar serves as a measure of similarity between pairs of individuals in term of gene expressions within a pathway. This kernel machine-based similarity measure can be incorporated to test to what extent variation in the level of similarity exhibited by pairs of individual can explain other features (like disease status or a particular quantitative phenotype) those individuals possess. Most applications of kernel machines (Liu et al. 2007, 2008; Kwee et al. 2008; Wu et al. 2010; Maity and Lin 2011) rely on two results.
The first is the “kernel trick”. Using a kernel machine k corresponds to mapping the data from the input space χ into a possibly high-dimensional inner product space
 by a map Φ : χ →
and taking the inner product there. i.e.,
by a map Φ : χ →
and taking the inner product there. i.e.,
Φ is called the feature map associated with kernel machine k and
is called the reproducing kernel Hilbert space (RKHS) given that
is complete. The name RKHS comes form the following reproducing kernel property:
for any f ∈
. More details about RKHS and reproducing kernel can be found in Aronszajn (1950). The “kernel trick” means that these high-dimensional dot products can be computed within the original space by means of a kernel machine without having to compute the mapping explicitly. One implication of the“kernel trick” is that we are able to deal with nonlinear algorithm in χ by reducing them to linear ones in
. One of such example is the least square kernel machine (LSKM) regression in Liu et al. (2007).
Another important result is Mercer’s theorem (Cristianimi and Shawe-Taylor 2000). Under some regularity conditions, a symmetric positive definite kernel function k(·, ·) implicitly specifies a unique Hilbert functional space
spanned by a particular set of orthogonal basis functions
. Then any function f(z) ∈
can be represented as a linear combination of those basis functions by
. Moreover, according to Mercer’s theorem, we have a series expansion for the kernel k of the form
where
s are the eigenvalues of an integral operator induced by the kernel machine k. Mercer’s theorem characterizes the structure of the Hilbert functional space
spanned by kernel machine k. Every function in
can be expressed as a linear combination of the basis, which is called the primal representation. Equivalently, there is a dual representation which express each function f in
as a linear combination of the kernel as
for some x1, . . ., xL ∈ χ.
One of the most commonly used kernels is the Gaussian Kernel k(x, y) = exp{−ρ−1||x–y||2}, where ρ is a positive parameter and || · || is the L2 norm. The Gaussian kernel generates the function space spanned by radial basis functions (RBF) and ρ is called bandwidth or shape parameter in literature. See Bühmann (2003) for more details. Another widely used kernel is the dth polynomial kernel given by k(x, y) = (xT y+ρ)d, where ρ ≥ 0 and d is a positive integer. One example in this family is the linear kernel: k(x, y) = xT y + ρ. Other examples of kernel machine include the spline kernel, ANOVA kernel, tree kernel and graph kernel (Hofmann et al. 2008).
Suppose our data (yi, xi, zi) are a random sample from n individuals. Let yi be some quantitative phenotypic traits like body mass index (BMI) and blood pressure, which are continuous, xi be clinical covariates with dimension q, like age and gender. And zi be the genetic covariates with dimension p, like gene expressions within a pathway. Then we focus on the partial linear model in this paper:
| (1) |
where β is a q × 1 coefficients, f(·) is an unknown smooth function and ε are iid N(0, σ2) errors. Without loss of generality, we assume that the intercept β0 is included with the clinical covariates X instead of f(·). Moreover we assume that the nonparametric function f(·) lies in a RKHS generated by a kernel machine k(·, ·). Note that the RKHS is totally determined by the reproducing kernel, i.e., the choice of kernel function k determines the form of function f in the RKHS. So if one is interested in a linear form of function f, he can pick the kernel to be the linear kernel. The Gaussian kernel can be chosen if one is interested in a nonlinear relationship between y and z. As shown in model (1), the quantitative phenotype y depends on the genetic variants z through the function f and the pathway effect is tested via testing f(·) = 0.
2.2 LSKM Test of A Pathway Effect
Given observations , as shown in Liu et al. (2007), a LSKM estimator of β in (1) is obtained as
| (2) |
where λ is a tuning parameter which controls the tradeoff between goodness of fit and complexity of the model, K is the n × n Gram matrix with Kij = k(zi, zj ), and y = (y1, . . ., yn)T . The estimated function f(·) evaluated at the observed points z1, . . ., zn given by
| (3) |
For an arbitrary z, function f(·) is evaluated as
| (4) |
Note that (2), (3) and (4) involve the unknown tuning parameter λ and possible kernel parameters ρ. Moreover any inference or evaluation of those LSKM estimators also relies on the residual variance σ2. Estimation of those parameters is proceeded in the following way. Model (1) can be written as the following linear mixed model:
| (5) |
where Y is the response vector and X is the input matrix of clinical predictors. F ≡ (f(z1), . . ., f(zn))T is a n×1 vector of random effects from a multivariate normal distribution with mean 0 and covariance matrix σ2λ−1K and E is the error vector. It has been shown that the best least squares unbiased estimators of the fixed effects β and random effects F in (5) correspond with (2) and (3) (Liu et al. 2007). Note that the likelihood function of model (5) is a function of (λ, ρ, σ2). Therefore one way to estimate (λ, ρ, σ2) is via restricted maximum likelihood (REML, Harville 1977).
To test the effect of the genetic pathway in the LSKM framework, we test H0 : f(·) = 0 in our model (1). By the equivalence of the kernel machine model (1) and linear mixed effects model (5), the test of f(·) = 0 is equivalent to that of the variance component in the mixed effects model being 0. Thus is suffices to test τ ≡ σ2λ−1 = 0. The hypothesis H0 : τ = 0 is tested as a variance component score test in a unified (restricted) maximum likelihood framework. Readers can refer to Liu et al. (2007) for more details.
2.3 GKM Test of A Gene Effect Within A Pathway
Suppose the genetic pathway consists of p genes z1, . . ., zp. The LSKM test is able to test whether there is an effect of this whole pathway on the outcome y. To test the effect of a given gene in the pathway, we applied the GKM test in Maity and Lin (2011). A basic idea is that each time we remove one gene out of our model and then fit the reduced model. Comparing the fit of the full model and reduced model, a bigger difference indicates a more important gene has been removed from the full model. Let us take z1 as an example. The test that z1 has no effect is H0 : f(z1, · · ·, zp) = f(z2, · · ·, zp), where f is the function in equation (1). A functional z1 will tend to lead to a rejection of the null hypothesis. Maity and Lin (2011) conducted the test by introducing a nonnegative parameter δ which they called a garrote parameter. Then z1 is replaced by , and the corresponding new kernel is termed garrote kernel machine Kg with an extra parameter δ. In this setting, the original test is equivalent to the test . Like LSKM test, this new hypothesis is tested in a mixed effects model framework as a variance component score test. See Maity and Lin (2011) for more details.
The GKM test can evaluate a particular gene’s effect, so we can use the test to delete non-functional genes in the pathway. It has been observed in the genetic association test literature that the association signal decreased with adding non-associated genetic variants (Wessel and Schork 2006; Wu et al. 2009, 2010). It may thus be helpful in detecting the association by first using GKM to select a subset of the pathway and then performing the LSKM test on the reduced gene-set. Wu et al. (2009) proposed the sLDA which is based on a lasso algorithm to select the genes in the composite expression value, which is shown to have good performance in the linear case. However, the lasso-based sLDA method does not allow selection of features in a nonlinear genetic model with possible gene-gene interactions. GKM provides a perfect solution to this issue. Besides the advantage of not requiring a strong parametric assumption (like linearity in sLDA), the GKM test has addtional advantages. First, the test H0 : f(z1, · · ·, zp) = f(z2, · · ·, zp) may be a high or even infinite-dimensional problem while the test is only an one-dimensional testing problem. Second, there is a decrease in degrees of freedom due to the fact that GKM takes the correlations among genes into account, which boosts the power of the test as shown in simulation studies in Maity and Lin (2011).
3 DKM procedure
The DKM procedure we propose in this section can be taken as an adaptive version of the LSKM test to accomplish the dual goals of gene selection and testing genetic pathway effects. Suppose the pathway contains p genes. The DKM procedure works as follows. First, we apply the GKM test to each gene and obtain p p-values. Next, a certain criterion is applied to the p-values to select a subset of m genes. Finally, we apply the LSKM test on this new pathway consisting of the selected m genes. Since the GKM and LSKM tests have already been discussed, the most important aspect in DKM is the selection criterion. The essence of DKM is based on a technique called subset testing (Neyman 1937; Fan 1996; Kim and Arkitas 2011), which we now describe.
Neyman first used subset testing in a multivariate normal testing problem. Let X ~ N(θ, Ip) be a p-dimensional normal random vector. We want to test H0 : θ = 0 against H1 : θ ≠ 0. Let Xj and θj be the j-th component of X and θ respectively. When the dimensionality is high, testing all dimensions would accumulate stochastic errors which may deteriorate the performance of the testing procedure (Neyman, 1937). Instead, Neyman proposed testing the first m dimensions of subproblem. That is, H0 : θ1 = . . . = θm = 0, leading to the test statistics . This Neyman adaptive test is a kind of truncation test, for any dimensions larger than m are not considered. If there is some evidence showing that large s are located at small j′s, then the Neyman test may be an ideal choice to test such a problem. Inspired by the truncation test, related tests like the thresholding test (Fan 1996) and order-thresholding test (Kim and Arkitas 2011) have been proposed. The thresholding test selects those dimensions with values larger than some certain threshold value c and the order-thresholding selects those dimensions with k largest values. Both c and k can be estimated in a data-driven way.
These three techniques (truncation, thresholding and order-thresholding) are all possible candidates of the selection criterion in our DKM procedure. The truncation test may not be a good choice in our genetic association test framework since we have no idea of the location of functional genes in a pathway. Thresholding and order-thresholding seem to be more reasonable. Note that in both Fan (1996) and Kim and Akritas (2011), the assumption of independence across individual hypotheses is made. However, this independence assumption is violated in p GKM tests, since a GKM test for one gene uses all information contained in the rest (p–1) genes and there is a overlap of (p–2) common genes in two GKM tests. Some adjustment is needed before applying order-thresholding or thresholding. Examples include Monte Carlo-based method (Lin, 2005) and Bonferroni correction based on the effective number of independent tests (Nyholt, 2004). Here we make an assumption that such an adjustment is monotone in p-values, which may be reasonable. Let’s consider this in a multiple testing framework. Bonferroni method works in the case that individual tests are correlated. The adjustment is that we divide each individual p-value by the effective number of independent tests, which is of course monotone in p-values. Under this monotone adjustment assumption, it preserves the order of the p-values and there exists another threshold value which keep the same set of genes above (below) it.
One more issue is the estimation of cutoff point c in the thresholding method and the selected order k in the order-thresholding method. Note that the subset selection in DKM is directly performed on GKM p-values instead of GKM test statistic. This is because GKM test for different genes can yield test statistics that are dramatically different or even in completely different scale. The biggest difference of p-values and the normally distributed X’s as considered in Fan (1996) is that p-values are bounded in [0,1]. Hence the suggestion of estimating cutoff value c made in Fan (1996) is not valid in our case. However, there is no such issue in Kim and Akritas (2011) because they considered the order. An suggestion of estimating k data-adaptively is given by Kim and Akritas as follows. First, order the p GKM p-values from smallest to largest. Then pick the smallest p-values/genes, where is given by
where n is number of individual tests, Gn is the empirical cdf of the p-value vector P = (P1, . . ., Pn), the Pi’s are the p-values of the ith GKM test, and λ is the median of the Pi’s.
Finally, as observed in Kwee et al. (2008), if the GKM and LSKM tests are performed on the same dataset in our DKM procedure, it will lead to anticonservative tests. The first stage of GKM test is like training the model. If both GKM and LSKM are applied on the same dataset, then the supervised learning in GKM will lead to inflated type I error rate of the LSKM test on the second stage. Hence we strongly recommend that the first stage of GKM tests to be performed on some other independent datasets. Kwee et al. (2008) also discussed the availability of such independent datasets. Those datasets serve as some prior knowledge of the underlying genetic model. For some reason, if such information is not available, we recommend another permutation-based method to establish the significance of our DKM procedure. First, we apply both GKM and LSKM test on the original dataset. Denote the p-value of the LSKM test on the subset pobs. Second, we randomly shuffle y and x to yield a permuted version dataset {y(b), x(b), z}. DKM is performed on this permuted dataset and denote the p-value of LSKM test on this permuted dataset p(b). We repeat this process for b = 1, . . ., B times and the final p-value of DKM is . Such an approach was used in Pan and Shen (2011) and was shown to be able to protect the type I error rate. However, the drawback is the computational cost, since permutation requires re-estimation of the kernel matrix (both GKM and LSKM), parameters and so on.
4 Simulations
In this section, two sets of simulation studies were conducted. One was to compare the performance of testing-subset selection of thresholding and order-thresholding. The other was to compare the performance of LSKM test and DKM test in the presence of noisy variables. For a purpose of comparison, the way we generated our simulated data was similar to that in Liu et al. (2007). Kernel selection is an important issue in the kernel machine literature; however we do not pursue this goal in this paper. Throughout this simulation, the Gaussian kernel and the linear kernel were used.
4.1 Simulation Study I
In this part, we compared the selection performance of thresholding and order-thresholding. Our simulated data were generated from the following semiparametric model:
| (6) |
where ε ~ N(0, 1), zij ’s were generated from Uniform(0,1). To allow for clinical covariates x′s and genetic covariates z′s being correlated, xi was generated as xi = 3cos(zi1) + 2εi where εi were iid standard normal errors. The nonparametric function f(·) was allowed to have a complex form with nonlinear functions of z′s and interaction among the z′s. In this simulation study, the true model was given by:
| (7) |
The sample size was n = 60. In practice, the pathway will also contain nonfunctional genes. To mimic such a scenario, some noisy z′s were added in the simulation. Two different situations were considered. The first set of simulations contained p = 10 genes and the second set of simulations contained p = 20 genes. We assumed that the first 5 genes were the functional ones. Simulations were run on 100 datasets. For each dataset, certain genes were selected. We used Ni, i = 1, . . ., p to denote the number of times that gene i being selected. Figure 1 (Gaussian kernel) and Figure 2 (linear kernel) presented the histograms of those count variables.
Fig. 1.
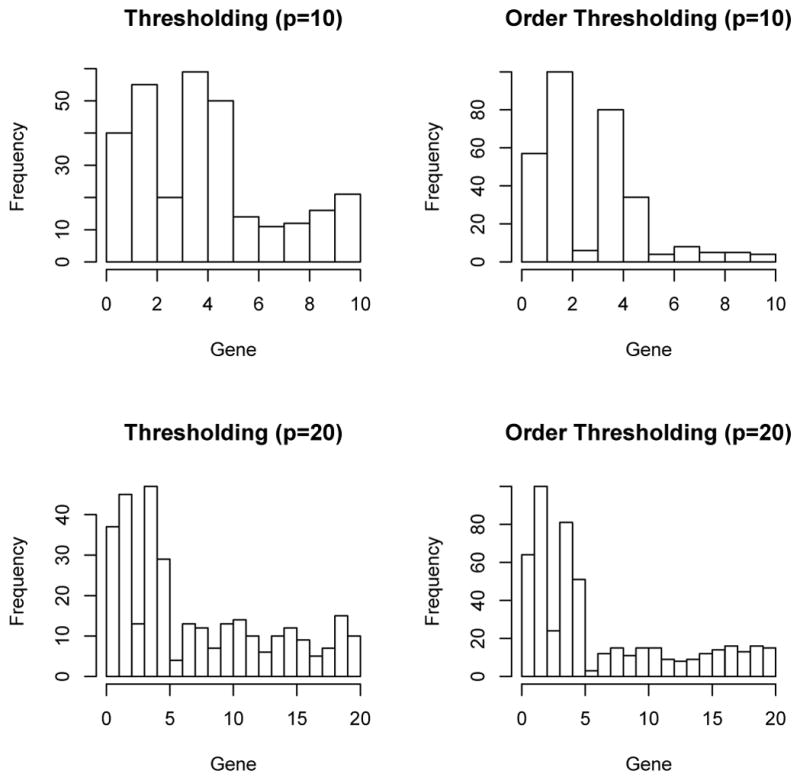
Histograms of number of times each gene being selected (N1, . . ., Np) when the Gaussian kernel is used.
Fig. 2.
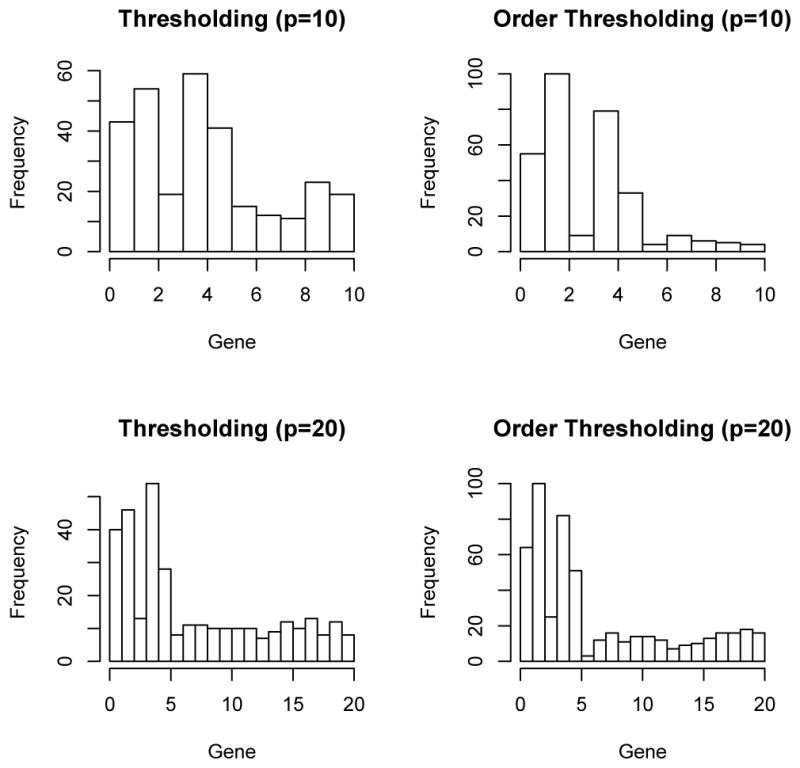
Histograms of number of times each gene being selected (N1, . . ., Np) when the linear kernel is used.
Based on both figures, order-thresholding outperforms thresholding. An interesting finding in both order-thresholding and thresholding is that, even though gene 3 is a functional gene in our simulation model (7), the strength of the association between the outcome and gene 3 is so weak that most times it is not selected. Regarding the issue of cut-off value of the thresholding procedure, the formula suggested in Fan (1996) does not work in our case because of the boundness of p-values, unlike the normal distributed test statistics in Fan (1996). We tried different values in our simulation and the results presented in the figures corresponds to a value of 0.05. That is, any genes with a GKM p-value smaller than 0.05 are selected in the subset used for the LSKM test on the second stage. Basically, if a higher cut-off (say, 0.1) is used, then the frequency of non-functional genes being selected would be larger and if a lower (say, 0.01) cut-off is used, then N1, . . . N5 would be smaller. No matter which value we picked, the comparison between thresholding and order-thresholding is similar. We can not find a cut-off value c such that thresholding outperforms order-thresholding in terms of subset selection. Hence, we choose order-thresholding as the subset selection criterion in our DKM approach in the simulations which follow.
4.2 Simulation Study II
In this section, we compared the type I error rate and power of our DKM test with those of the LSKM test. Both tests were performed at a bunch of ρ values as in Table 4 of Liu et al. (2007). In order to calculate the power of the test, we need to specify the alternatives. In this simulation we used the same testing structure as Liu et al. (2007) for a purpose of comparison. This testing setting was given by: f1(z) = af(z) where f was given in (7) and a = 0, 0.2, 0.4, 0.6, 0.8. Under this setting, we studied size of the test by generating data under a = 0 and power of the test by generating data under nonzero a’s. In order to preserve the type I error rate of DKM test, we performed the GKM-based gene selection and LSKM-based score test on two separate datasets. The results in the previous simulation section can be used as prior knowledge of the underlying genetic model. Based on the results shown in Figure 1 and Figure 2, we were confident to pick the testing-subset to be {1, 2, 4, 5}. For each a value, we generated 1000 datasets from the underlying model (7). The numerical empirical type I error rate and power were reported in Table 1 and Table 2. The a = 0 column is the empirical type I error rate and the non-zero a columns are the power.
Table 1.
Empirical size and power of DKM test and LSKM test when the Gaussian kernel k(x, y) = exp{ρ−1||x – y||2} is used. The above half is LSKM test and the below half is DKM test.
| ρ |
p = 10
|
p = 20
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| a = 0 | 0.2 | 0.4 | 0.6 | 0.8 | a = 0 | 0.2 | 0.4 | 0.6 | 0.8 | |
| 0.5 | 0.093 | 0.187 | 0.483 | 0.791 | 0.954 | 0.819 | 0.869 | 0.948 | 0.985 | 0.995 |
| 1 | 0.069 | 0.172 | 0.491 | 0.829 | 0.971 | 0.157 | 0.245 | 0.492 | 0.769 | 0.918 |
| 5 | 0.056 | 0.146 | 0.485 | 0.829 | 0.966 | 0.075 | 0.142 | 0.385 | 0.705 | 0.896 |
| 25 | 0.052 | 0.144 | 0.469 | 0.823 | 0.965 | 0.072 | 0.130 | 0.373 | 0.689 | 0.880 |
| 50 | 0.052 | 0.143 | 0.468 | 0.822 | 0.965 | 0.072 | 0.130 | 0.366 | 0.688 | 0.878 |
| 100 | 0.051 | 0.143 | 0.468 | 0.822 | 0.965 | 0.070 | 0.131 | 0.365 | 0.686 | 0.878 |
| 200 | 0.051 | 0.143 | 0.467 | 0.821 | 0.966 | 0.069 | 0.128 | 0.365 | 0.686 | 0.878 |
|
| ||||||||||
| 0.5 | 0.045 | 0.198 | 0.642 | 0.943 | 0.995 | 0.051 | 0.215 | 0.646 | 0.940 | 0.998 |
| 1 | 0.049 | 0.203 | 0.658 | 0.942 | 0.995 | 0.053 | 0.217 | 0.655 | 0.947 | 0.998 |
| 5 | 0.049 | 0.201 | 0.654 | 0.932 | 0.991 | 0.051 | 0.213 | 0.651 | 0.940 | 0.994 |
| 25 | 0.047 | 0.200 | 0.647 | 0.929 | 0.991 | 0.053 | 0.214 | 0.645 | 0.937 | 0.994 |
| 50 | 0.047 | 0.200 | 0.647 | 0.929 | 0.991 | 0.053 | 0.214 | 0.647 | 0.937 | 0.994 |
| 100 | 0.047 | 0.199 | 0.648 | 0.929 | 0.991 | 0.053 | 0.211 | 0.647 | 0.936 | 0.994 |
| 200 | 0.047 | 0.199 | 0.648 | 0.929 | 0.991 | 0.053 | 0.211 | 0.647 | 0.936 | 0.994 |
Table 2.
Empirical size and power of DKM test and LSKM test when the linear kernel k(x, y) = xT y + ρ is used.
| Method |
p = 10
|
p = 20
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| a = 0 | 0.2 | 0.4 | 0.6 | 0.8 | a = 0 | 0.2 | 0.4 | 0.6 | 0.8 | |
| LSKM | 0.051 | 0.144 | 0.467 | 0.821 | 0.966 | 0.069 | 0.128 | 0.363 | 0.686 | 0.878 |
|
| ||||||||||
| DKM | 0.048 | 0.199 | 0.648 | 0.929 | 0.991 | 0.053 | 0.211 | 0.646 | 0.936 | 0.994 |
For the Gaussian kernel, we can see that when ρ is small, the LSKM test is invalid since it has inflated type I error rate. Its type I error when ρ = 0.5 and p = 10 is 0.093 which is larger than the nominal level 0.05. The type I error of the LSKM test at ρ = 0.5 when p = 20 is even as high as 0.819. To explain the poor performance, we first note that association tests using kernel machines are effective when the kernel serves as a similarity measure between individuals. Note that the Gaussian kernel is given by:
If the dimensionality p is high and ρ is small, then k(x, y) → 0 for each individual pairs (x, y). That is each pair of individuals tend to be orthogonal to each other under such a similarity measure. For such a scenario (big p small ρ), the Gaussian kernel fails to provide a proper similarity measure between individual pairs. That explains why LSKM test fails. DKM, on the other hand, reduces the number of features from p to m and hence is able to alleviate the inflated type I error rate issue.
For the linear kernel , it can be seen in Table 2 that the test is not affected by the value of ρ (See the appendix for a proof of this result). Moreover, a similar pattern of inflated type I error rate at the presence of noisy variables is also observed. At p = 20, the empirical type I error rate is 0.069, higher than the nominal level 0.05. Applying our DKM procedure, this can be reduced to 0.053. The effect of increasing dimension on a linear kernel does exist, even though it is not as huge as that in a Gaussian kernel. The explanation why dimension makes a difference in a linear kernel is similar to that in a Gaussian kernel. Suppose x, y and z are three arbitrary p-dimensional measurements. When p is large, k(x, y), k(x, z) and k(y, z) will all be large. Hence k can not serve as an effective similarity measure to tell different measurements. By reducing the dimension, the differences among k(x, y), k(x, z) and k(y, z) will be relatively larger, hence, it is easier to distinguish different measurements.
To summarize, LSKM fails to protect the type I error rate and suffers from power loss in the presence of noisy variables. On the other hand, DKM can largely reduce the type I error rate of a corresponding LSKM test and have higher power in the same scenario. DKM performs much better than LSKM in the presence of noisy variables. The more noisy variables, the bigger the difference in performance between DKM and LSKM.
5 Application to GRIN2B Data
In the previous sections, we illustrated how our DKM procedure can be applied to pathway studies. The proposed method can be directly extended to other gene-based analysis like genome-wide association study (GWAS) (Wu et al. 2010). The set aggregation can be performed at other levels besides genetic pathways, like exons, SNPs-set and LD blocks. As a motivating example, we consider data from the Alzheimer’s Disease Neuroimaging Initiative (ADNI). A description of the study data and acknowledgment of investigators can be found in Appendix. In an earlier study, Stein et al. (2010) found that one SNP located in the GRIN2B gene is related to Alzheimer’s disease. Motivated by their study, we extracted SNPs based on the physical position of gene GRIN2B from National Center for Biotechnology Information (NCBI). We extracted the SNPs from 13714kb to 14133kb and found 119 SNPs. The raw dataset contained missing values in the SNP variables, so mean imputation was used to complete the dataset.
In this study, 4 clinical covariates were collected on 741 subjects. Those covariates were disease status based on baseline diagnosis, disease status based on 24 month later diagnosis, sex and age. We excluded the post-diagnosis disease status variable because of high missingness. The disease status based on baseline diagnosis had three levels: 1=normal control, 2=mild cognitive impairment (MCI) and 3=Alzheimer’s disease (AD). We created 2 indicator variables DS1 and DS2 to denote the effect of MCI and AD, respectively. The response variable was structural MRI expression from 119 regions of interest (ROI). To simplify the analysis, we computed the first standardized principal component of those 119 ROIs and used it as our response variable in the analysis. The goal was to evaluate whether the set of 119 SNPs for GRIN2B had an effect on ROI after adjusting for three covariates: disease status, sex and age. The model in this analysis was given by:
| (8) |
where h(·) was a nonparametric function in a functional space spanned by a certain kernel machine. In this GRIN2B data analysis, the Gaussian kernel and the linear kernel were used. We used both the LSKM and DKM method to fit model (8) and to test the SNP-set effect of h(·) = 0.
On the first stage, GKM tests were used to select a subset of the SNPs as the testing-subset. On the second stage, a LSKM score test was performed on this testing-subset. As discussed in the last paragraph in Section 3, in order to deal with the issue of inflated type I error rate and to avoid computationally expensive permutation methods, we split the data set into a training set and a test set. The training set was used in the GKM stage to determine which SNPs should be included in the testing-subset. The test set was used for the score test at the second stage. An issue was that different splits lead to different training sets, which further resulted in different testing-subsets. To fix this issue, we repeated the process of subset selection 100 times on different training sets to get a more stable result. The analysis was conducted in the following way. First, we split the 741 samples into a training set and a test set. We randomly chose 100 individuals as a training set pool. The remaining 641 individuals formed the test set. Second, each time we picked 60 of the 100 individuals as a training set to perform the GKM test, and repeated it 100 times. Each time we obtained 119 marginal p-values for each individual SNP and picked kn SNPs based on those p-values using the formula mentioned in Section 3. Third, we decided the final testing-subset based on these 100 runs. More details can be found in the next paragraph. Last, a LSKM test was conducted using the 641 samples in the test set.
Figure 3 presented the empirical distribution of s. The range of kn in a Gaussian kernel model was [10, 59] while that of a linear kernel was [10, 20]. The empirical distribution of kn in a linear model was more concentrated than that in a Gaussian kernel. This makes intuitive sense because the functional space spanned by a linear kernel is much simpler than that of a Gaussian kernel. Based on these empirical distributions, the final optimal is determined as the 90th percentile of the empirical distribution. We counted the number of times each SNP being selected in total out of those 100 runs and picked the top SNPs to form the testing-subset. For the Gaussian kernel , and for the linear kernel . The qq-plot of marginal GKM p-values of each SNP based on the whole training set was also presented in Figure 4. The deviations from the straight line were mostly minimal. Hence, the marginal p-values were basically distributed as Uniform(0,1).
Fig. 3.

Empirical distribution of the number of SNPs s being selected each time. The left panel is for the Gaussian kernel and the right panel is for the linear kernel.
Fig. 4.

QQ-plot of marginal p-values from GKM tests. The y-axis is sample quantiles and the x-axis is theoretical quantiles of the Uniform (0,1) distribution. The left panel is for the Gaussian kernel and the right panel is for the linear kernel.
SNP rs11055612 was found to be significant in Stein et al. (2010). We found that this SNP was included in the 39-SNP set in our Gaussian kernel model. However it was not in the 11-SNP set in our linear kernel model. A possible explanation is that data analyzed in Stein et al. (2010) included only one ROI while we used the first principal component from 119 ROIs. In a linear kernel model, the number of times SNP rs11055612 being selected ranked the 16th largest out of all 119 SNPs (the corresponding rank was 13th in a Gaussian kernel), which was close to the top 11 SNPs finally being selected in the testing-subset. Moreover, all 11 SNPs selected by a linear kernel were included in the 39-SNP set selected by the Gaussian kernel. This is reasonable because the functional space spanned by a Gaussian kernel is very general and can have a linear function as a special case. On the second stage, we performed kernel-based score tests on the selected SNPs subset. Results of those tests are reported in Table 3 and Table 4.
Table 3.
LSKM and DKM on the GRIN2B data using a Gaussian kernel.
| Method | Variable | Estimate | S.E. | P-value |
|---|---|---|---|---|
| LSKM | Intercept | -4.59 | 2.33 | 0.05 |
| DS1 | 0.17 | 0.37 | 0.64 | |
| DS2 | 1.30 | 0.36 | 0.0003 | |
| Sex | 0.13 | 0.30 | 0.68 | |
| Age | 0.05 | 0.02 | 0.02 | |
| h(·) | . | . | 0.041 | |
|
| ||||
| DKM | Intercept | -2.78 | 0.52 | 1.72e-7 |
| DS1 | 0.24 | 0.08 | 3.21e-03 | |
| DS2 | 0.63 | 0.09 | 1.55e-11 | |
| Sex | -0.32 | 0.07 | 5.09e-06 | |
| Age | 0.04 | 0.005 | 4.53e-14 | |
| h(·) | . | . | 0.067 | |
Table 4.
LSKM and DKM on the GRIN2B data using a Linear kernel.
| Method | Variable | Estimate | S.E. | P-value |
|---|---|---|---|---|
| LSKM | Intercept | -2.87 | 0.46 | 2.88e-10 |
| DS1 | 0.21 | 0.09 | 0.02 | |
| DS2 | 0.63 | 0.10 | 7.49e-10 | |
| Sex | -0.30 | 0.08 | 8.68e-5 | |
| Age | 0.04 | 0.005 | 1.76e-13 | |
| h(·) | . | . | 0.180 | |
|
| ||||
| DKM | Intercept | -2.83 | 0.44 | 1.45e-08 |
| DS1 | 0.20 | 0.09 | 0.02 | |
| DS2 | 0.62 | 0.10 | 1.22e-9 | |
| Sex | -0.30 | 0.08 | 7.39e-5 | |
| Age | 0.04 | 0.005 | 1.91e-13 | |
| h(·) | . | . | 0.695 | |
In Table 3 and Table 4, the estimates of those clinical covariates were based on the samples in the test set. The S.E. column was calculated based on the formulas in Liu et al. (2007). For the LSKM test of h(·) = 0, we used the estimate of ρ based on the training set. In Table 3, the DKM based standard errors of clinical covariates were much smaller than those of LSKM, which further resulted in much smaller p-values for the clinical covariates. However, such a phenomenon was not observed in Table 4 when a linear kernel was used. In a linear kernel model, h(SNP1, . . ., SNP119) = α1SNP1 + · · · + α119SNP119. Model (8) is quite simple and the reduction in the dimension of SNPs has little effect on the covariance of the clinical covariates part. In a Gaussian kernel model, h(SNP1, . . ., SNP119) can be much more complicated, including many interactions and nonlinear terms. By reducing the dimension of SNPs, we are fitting a much simpler model in DKM than in LSKM. Issues like the multi-collinearity and confounding are less likely to happen in DKM model. This explains why p-values of the clinical covariates in DKM are much smaller than that in LSKM. Another interesting founding is the p-values of the SNP-set. In Table 3, both p-values of LSKM and DKM are small and they are close. However, in Table 4, they are larger and the difference are also much larger. A possible reason is that a Gaussian kernel is able to capture a general relationship while a linear kernel can only works well when the linearity assumption holds. In this GRIN2B data, a linear kernel may fail to capture the effect of the SNPs on both stages of DKM. Some goodness of fit tests may be applied to test which kernel is better for this data; however this topic of kernel selection is out of the scope of this paper.
Our DKM method analyzed the GRIN2B data in a different way from Stein et al. (2010). In Stein et al. (2010), the authors performed an individual analysis and only one SNP survived the stringent multiple testing correction. However, in our DKM approach, we found a much bigger SNP-set, which largely improved the chance to detect the causal SNPs. After that, a kernel-based score test was performed to test whether this SNPs-set was associated with the phenotype. Further laboratory studies are required to explore a detailed relationship between the SNPs-set and the phenotype.
6 Discussion
In this paper, we have proposed an adaptive approach based on double kernel machines for assessing genetic pathway effect. This DKM method is particuAn Adaptive Genetic Association Test Using Double Kernel Machines 17 larly attractive in settings where the signal is moderate, that is, a few genes are functional or informative, contributing to a pathway’s significant effect, while others show little change relative to the noisiness of the data. The key advantage of DKM is that it can select informative genes within a pathway that drives the effect and then test for the significance of the pathway effect with improved power by eliminating non-informative genes. We illustrated the powerful results of the DKM test for detecting the pathway effect and gene selection within the pathway using both simulations and the GRIN2B data. All these numerical studies show that our DKM method has a good performance.
The motivation of introducing DKM is that we observed LSKM test being invalid in the presence of noisy variables especially in high-dimensional settings. The first stage of DKM serves as a dimension-reduction tool to solve such a potential issue in LSKM. Comparing with other similar tools like sLDA (Wu et al. 2009), the GKM has its advantages and perfectly fits within the kernel machine framework, as the order thresholding is able to select the true signals in our simulation studies in Section 4. Liu et al. (2007) also considered this problem. In their prostate cancer genetic pathway data analysis, they considered the performance of all possible 2p subsets, which is practically infeasible when p is large. In the ultra-high dimensional case, computational cost becomes an issue, because the DKM approach requires a garrote kernel machine test on each dimension. We recommend that one can first apply some fast dimension reduction or variable selection methods like screening (Fan and Lv, 2008), then use our DKM procedure for a testing purpose.
The DKM approach utilizes the same aspect of association information on its two stages. It is possible to get an inflated type I error rate. Pan and Shen (2011) proposed a permutation-based method to fix this issue. However, we strongly advocate that two stages of DKM should be performed on separate datasets to avoid the time-consuming permutation process. Another possible way to save the computing time is via parallel computing. Because the GKM test of each gene is independent in the sense that it does not rely on the result of other GKM tests. Hence we can break the whole genetic-set into several pieces and perform the first stage of GKM test in each piece simultaneously.
In this paper, we focus on a kernel machine model with a continuous outcome. Kernel model for binary and censored outcomes have also been developed in Liu et al. (2008) and Cai et al. (2011) respectively. It is possible to develop adaptive versions of such tests like what we did for the continuous outcome in this paper. Another issue involves kernel selection. There are also many papers using models based on other kernels. In this paper, we illustrate our DKM method using a Gaussian kernel and a linear kernel as two examples. It is of interest to know which kernel can have a best performance in this DKM approach. We leave these extensions for future work.
Acknowledgments
This research was supported by NIH grants CA129102. The authors thank the reviewers for helpful comments.
Appendix
A.1: Least squares kernel machine score test based on linear kernels
In this section, we prove that different linear kernels k(x, y; ρ) = xT y + ρ lead to the same LSKM score test. First let us recall the LSKM score test proposed in Liu et al. (2007). The test statistic of a LSKM score test is Q(β̂, σ̂2, ρ) where
β̂ and σ̂2 are the MLEs of β and σ2 under the null model y = xβ + ε. Liu et al. (2007) used a scaled chi-squared distribution to approximate the distribution of Q( β̂, σ̂2, ρ), where a and b are determined by matching the moments of Q and the scaled chi-squared distribution. It is easy to see that a = V ar(Q)/2E(Q) and b = 2E2(Q)/V ar(Q). Let X be the design matrix for the clinical covariates and Kρ be the kernel matrix, which depends on kernel parameter ρ. Denote P0 = I – X(XTX)−1XT . Then according to Liu et al. (2007):
| (9) |
Now consider two arbitrary linear kernels k(x, y, ρ1) and k(x, y, ρ2). Let Qi, Ki, ai and bi be some quantities corresponding to kernel i, i = 1, 2. Let A ≡ (1, . . ., 1)T . Then it is easy to see K2 = K1+(ρ2–ρ1)AAT . Moreover, P0 =  X⊥, where X⊥ denotes the projection matrix to the orthogonal complement of the space spanned by the columns of X. Note that A is the first column of X (we assume that the intercept is contained in the clinical part). Hence P0A = X⊥A = 0. Therefore,
X⊥, where X⊥ denotes the projection matrix to the orthogonal complement of the space spanned by the columns of X. Note that A is the first column of X (we assume that the intercept is contained in the clinical part). Hence P0A = X⊥A = 0. Therefore,
Plugging these results back to Eq. (9), one can show that E(Q2) = E(Q1) and V ar(Q2) = V ar(Q1). Hence a2 = a1 and b2 = b1. Because the residuals of the null model y = xβ + ε sum to 0, one can easily show that Q2 = Q1. That is, both the LSKM score test statistic and the null distribution of the test statistic are identical for two arbitrary linear kernels k(x, y, ρ1) and k(x, y, ρ2). Therefore, all linear kernels lead to the same LSKM score test.
A.2: Description of ADNI data
Data used in the preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.ucla.edu). The ADNI was launched in 2003 by the National Institute on Aging (NIA), the National Institute of Biomedical Imaging and Bioengineering (NIBIB), the Food and Drug Administration (FDA), private pharmaceutical companies and non-profit organizations, as a $ 60 million, 5- year public-private partnership. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early Alzheimer’s disease (AD). Determination of sensitive and specific markers of very early AD progression is intended to aid researchers and clinicians to develop new treatments and monitor their effectiveness, as well as lessen the time and cost of clinical trials.
The Principal Investigator of this initiative is Michael W. Weiner, MD, VA Medical Center and University of California San Francisco. ADNI is the result of efforts of many co-investigators from a broad range of academic institutions and private corporations, and subjects have been recruited from over 50 sites across the U.S. and Canada. The initial goal of ADNI was to recruit 800 subjects but ADNI has been followed by ADNI-GO and ADNI-2. To date these three protocols have recruited over 1500 adults, ages 55 to 90, to participate in the research, consisting of cognitively normal older individuals, people with early or late MCI, and people with early AD. The follow up duration of each group is specified in the protocols for ADNI-1, ADNI-2 and ADNI-GO. Subjects originally recruited for ADNI-1 and ADNI-GO had the option to be followed in ADNI-2. For up-to-date information, see www.adni-info.org.
A.3: Acknowledgments to ADNI
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; BioClinica, Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; GE Healthcare; Innogenetics, N.V.; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles. This research was also supported by NIH grants P30 AG010129 and K01 AG030514.
Footnotes
Conflict of Interest: none declared.
Contributor Information
Xiang Zhan, Email: xyz5074@psu.edu, Department of Statistics, Pennsylvania State University, University Park, PA 16802, U.S.A. Tel.: +1-8143213493, Fax: +1-8148636699.
Michael P. Epstein, Email: mpepste@emory.edu, Department of Human Genetics, Emory University, Atlanta, GA 30322, U.S.A
Debashis Ghosh, Email: ghoshd@psu.edu, Department of Statistics, Department of Public Health Sciences, Pennsylvania State University, University Park, PA 16802, U.S.A.
References
- 1.Aronszajn N. Theory of reproducing kernels. Trans Am Math Soc. 1950;68:337–404. [Google Scholar]
- 2.Bühmann MD. Radial Basis Functions. Cambridge, U.K: 2003. [Google Scholar]
- 3.Cristianini N, Shawe-Tayor J. An Introduction to Support Vector Machines. Cambridge, U.K: 2000. [Google Scholar]
- 4.Cai T, Lin X, Carroll RJ. Identifying genetic marker sets associated with phenotypes via an efficient adaptive score test. Biostatistics. 2012;13:776–790. doi: 10.1093/biostatistics/kxs015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai T, Tonini G, Lin X. Kernel machine approach to testing the significance of multiple genetic markers for risk prediction. Biometrics. 2011;67:975–986. doi: 10.1111/j.1541-0420.2010.01544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fan J. Test of significance based on wavelet thresholding and Neyman’s truncation. J Am Stat Assoc. 1996;91:674–688. [Google Scholar]
- 7.Fan J, Lv J. Sure independence screening for ultrahigh dimensional feature space. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 2008;70:849–911. doi: 10.1111/j.1467-9868.2008.00674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harville DA. Maximum likelihood approaches to variance component estimation and to related problems. J Am Stat Assoc. 1977;72:320–338. [Google Scholar]
- 9.Hofmann T, Schölkopf B, Smola AJ. Kernel method in machine learning. The Annals of Statistics. 2008;36:1171–1220. [Google Scholar]
- 10.Kim MH, Akritas MG. Order thresholding. The Annals of Statistics. 2010;38:2314–2350. [Google Scholar]
- 11.Kwee LC, Liu D, Lin X, Ghosh D, Epstein MP. A powerful and flexible multilocus association test for quantitative traits. Am J Hum Genet. 2008;82:386–397. doi: 10.1016/j.ajhg.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin D. An efficient Monte Carlo approach to assessing statistical significance in genomic studies. Bioinformatics. 2005;21:781–787. doi: 10.1093/bioinformatics/bti053. [DOI] [PubMed] [Google Scholar]
- 13.Liu D, Lin X, Ghosh D. Semiparametric regression of multi-dimensional genetic pathway data: Least squares kernel machine and linear mixed models. Biometrics. 2007;63:1079– 1088. doi: 10.1111/j.1541-0420.2007.00799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu D, Ghosh D, Lin X. Estimation and testing for the effect of a genetic pathway on a disease outcome using logistic kernel machine regression via logistic mixed models. BMC Bioinformatics. 2008;9:292. doi: 10.1186/1471-2105-9-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maity A, Lin X. Powerful tests for detecing a gene effect in the presence of possible gene-gene interactions using garrote kernel machines. Biometrics. 2011;67:1271–1284. doi: 10.1111/j.1541-0420.2011.01598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neyman J. Smooth test for goodness of fit. Scandinavian Actuarial Journal. 1937;3–4:149–199. [Google Scholar]
- 17.Nyholt D. A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. The American Journal of Human Genetics. 2004;74:765–769. doi: 10.1086/383251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan W, Shen X. Adaptive tests for association analysis of rare variants. Genetic Epidemiology. 2011;35:381–388. doi: 10.1002/gepi.20586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stein JL, Hua X, Morra JH, et al. Genome-wide analysis reveals novel genes influencing temporal lobe structure with relevance to neurodegeneration in Alzheimer’s disease. Neurolmage. 2010;51:542–554. doi: 10.1016/j.neuroimage.2010.02.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wessel J, Schork NJ. Generalized gonomic distance-based regression methodology for multilocus association analysis. Am J Hum Genet. 2006;79:792–806. doi: 10.1086/508346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu MC, Kraft P, Epstein MP, Taylor DM, Chanock SJ, Hunter DJ, Lin X. Powerful SNP-set analysis for case-control genome-wide association studies. Am J Hum Genet. 2010;86:929–942. doi: 10.1016/j.ajhg.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu MC, Zhang L, Wang Z, Christiani DC, Lin X. Sparse linear discriminant analysis for simultaneous testing for the significance of a gene set/pathway and gene selection. Bioinformatics. 2009;25:1145–1151. doi: 10.1093/bioinformatics/btp019. [DOI] [PMC free article] [PubMed] [Google Scholar]