

SUMMARY
In nuclear pre-messenger RNA splicing, introns are excised by the spliceosome, a multi-megadalton machine composed of both proteins and small nuclear RNAs (snRNAs). Over thirty years ago, following the discovery of self-splicing group II intron RNAs, the snRNAs were hypothesized to catalyze splicing. However, no definitive evidence for a role of either RNA or protein in catalysis by the spliceosome has been reported to date. By using metal rescue strategies, here we show that the U6 snRNA catalyzes both splicing reactions by positioning divalent metals that stabilize the leaving groups during each reaction. Strikingly, all of the U6 catalytic metal ligands we identified correspond to the ligands observed to position catalytic, divalent metals in crystal structures of a group II intron RNA. These findings indicate that group II introns and the spliceosome share common catalytic mechanisms, and likely common evolutionary origins. Our results demonstrate that RNA mediates catalysis within the spliceosome.
Nuclear pre-mRNA splicing (Fig. 1a) is a crucial determinant of the export, translation, stability, and diversity of eukaryotic messages1, but the spliceosome is the only major cellular machinery2 required for gene expression for which the catalytic components remain undefined. Nevertheless, for three decades, there has been widespread speculation that nuclear pre-mRNA splicing is catalyzed by RNA.
Figure 1. Chemistry of pre-mRNA splicing and U2/U6 model showing sites sensitive to sulfur substitutions and rescued by thiophilic metal.
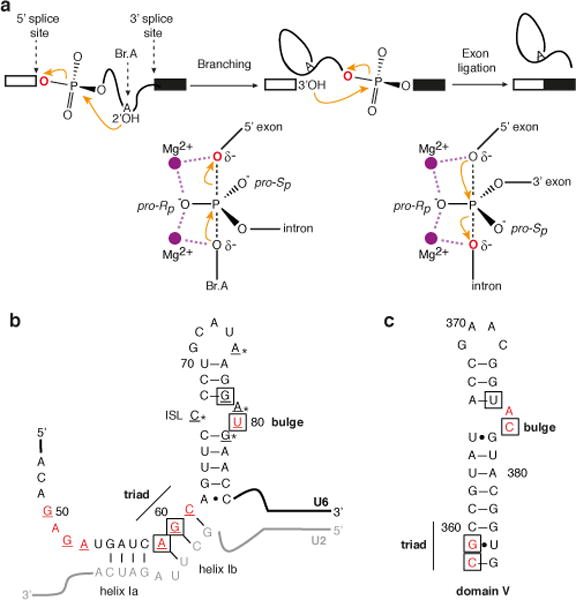
a, Reaction scheme (top) and transition state diagrams (bottom) for the two steps of nuclear pre-mRNA splicing. b–c, Models of the U2/U6 structure from the spliceosome (b) and domain V of a group II intron (c). Nucleotides where sulfur substitutions were shown previously to interfere with splicing12–16 are coloured red; underlined U6 nucleotides were tested in this study; * indicates those positions where only the pro-Sp oxygen was substituted with sulfur; and boxed nucleotides provide ligands for metals at the catalytic stage (Fig. 2, ref. 26).
This speculation arose from the discovery of self-splicing RNAs, the identification of snRNA components of the spliceosome, and the finding that pre-mRNA introns and group II introns both splice through an intermediate having a lariat structure3,4 (Fig. 1a). Since then, genetic, biochemical, and NMR data have shown that the snRNAs share functional and structural similarity with the catalytic core of group II introns5–10. Similarly to the catalytic domain V of group II introns, U2/U6 helix Ib and the intramolecular stem-loop (ISL) of U6 adopt a secondary structure having a conserved bulge and AGC triad sensitive to phosphorothioate substitutions and important for both steps of splicing5–8,10–16 (Fig. 1b,c). Extending the parallel, a recent crystal structure of a central splicing factor, Prp8, revealed domains similar to those found in cofactors of group II introns17.
Consistent with a catalytic role for RNA in the spliceosome, in the absence of spliceosomal proteins, U2 and U6 can base-pair and fold in vitro into a structure that catalyzes reactions similar to the two steps of pre-mRNA splicing18,19, although the relevance of such protein-free, minimal model systems for understanding spliceosomal catalysis has been questioned (ref. 20; cf. 21).
Whether or not through RNA, the catalytic centre of the spliceosome, like that of group II introns, functions by positioning catalytic metals. Steitz and Steitz first proposed that the two phosphotransesterifications of splicing are catalyzed by a two-metal mechanism22, in which one metal stabilizes the nucleophile and the second metal stabilizes the leaving group (Fig. 1a). Indeed, in human spliceosomes, as well as group II introns23, divalent metals stabilize the leaving group during each step of splicing24,25.
Intriguingly, recent crystal structures of a group II intron have revealed that domain V utilizes five non-bridging phosphate oxygens to coordinate two metals 3.9 Å apart26,27 – positioned to effect catalysis by the two-metal mechanism22. By analogy, the snRNAs have been suggested to similarly position metals, consistent with early phosphorothioate substitution studies in U6 (refs. 12,13). However, only residue U80, situated in the U6 ISL, has been shown to interact with a metal14,28 and it has remained unclear whether U80 positions a structural or a catalytic metal. Thus, despite work highlighting similarities between self-splicing RNA and the snRNAs, there is still no direct evidence that the snRNAs mediate splicing catalysis.
Definitive evidence for a direct role for metals coordinated by the RNA in the catalysis of self-splicing group I introns has come from metal rescue strategies29–31. These approaches, validated by subsequent structural studies31,32, enabled the direct linkage of metal ligands in the ribozyme to the splice sites. Application of such strategies in an investigation of pre-mRNA splicing has been hindered by proofreading and discard mechanisms that compete with catalysis during both steps of splicing28,33–35. Here, by disabling such proofreading, we implemented metal rescue strategies in the fully assembled spliceosome and identified the direct effectors of splicing catalysis. Our results provide definitive evidence that snRNAs interact with the splice sites directly through catalytic metals during both chemical steps of splicing, establishing that the spliceosome utilizes RNA to catalyze splicing.
U6 binds metals during both splicing steps
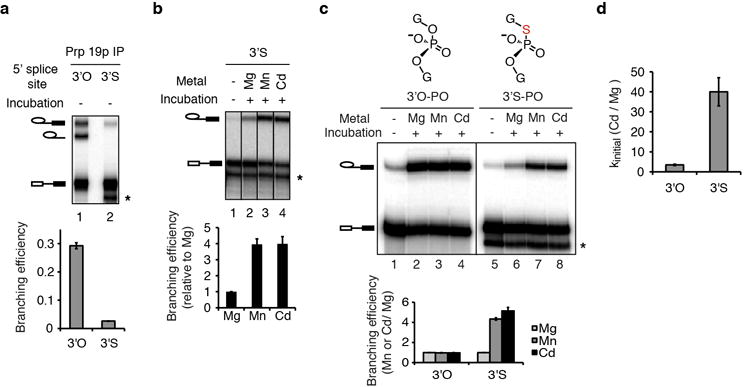
To identify components that mediate metal catalysis in the spliceosome, we employed metal rescue approaches31 (Fig. 2a) to find metal ligands that function at the catalytic stage and subsequently test these ligands for a direct role in positioning catalytic metals (Fig. 1a). The similarities between U6 and the catalytic domain V of group II introns suggest U6 as the best candidate for providing metal ligands that function at the catalytic stage (Fig. 1b,c). Indeed, early sulfur substitution studies implicated eight oxygens in the phosphodiester backbone of U6 as important for splicing12,13 (Fig. 1b). While informative, these studies assayed sulfur substitutions only at the pro-Rp oxygen (referred to as PS(Rp), Fig. 2a) and subsequent studies have only revealed rescue by thiophilic metals for U80-PS(Sp) (refs. 14,28).
Figure 2. U6 snRNA positions metals important for both steps of splicing.
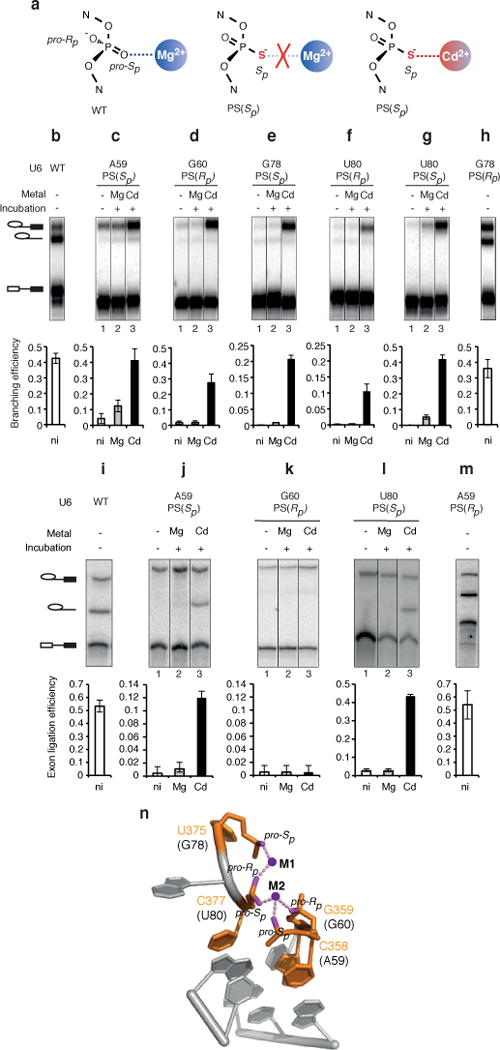
a, Metal rescue strategy, indicating the non-bridging oxygens that were substituted with sulfur and the consequence of a substitution for binding of Mg2+ or the thiophilic metal Cd2+. b–m, Cd2+ rescues the branching defects (b–h) and exon ligation defects (i–m) induced by specific sulfur substitutions. Values represent averages; error bars, s.d. (n=3 for b–h; n=2 for i–m); ni, no incubation. n, Structure of the domain V metal binding core (PDB 3EOH, ref. 26). Residue numbers are shown for the group II intron, with the corresponding U6 residues in parentheses.
To identify metal ligands, we assayed splicing in S. cerevisiae extracts reconstituted with U6 bearing individual sulfur substitutions covering both the Rp and Sp diastereomers at twenty positions, based on previous studies on pre-mRNA and group II splicing12–16,26 (Fig. 1b,c). To focus on ligand-metal interactions important during catalysis, we restricted our analysis to spliceosomes that had already undergone catalytic activation36 by affinity-purifying spliceosomes using tagged Prp19p (Extended Data Fig. 1a–c). We then assayed for rescue in the absence ATP and soluble factors, thus ensuring that spliceosomes had progressed beyond the final ATP-dependent activation step, while at the same time eliminating ATP-dependent proofreading mechanisms28,34 to enhance the potential for rescue (Extended Data Fig. 1d, Supplementary Note 1).
Five of the twenty tested substitutions conferred strong branching defects (Fig. 1b). In addition to the sulfur substitutions G60-PS(Rp), U80-PS(Sp), and U80-PS(Rp) (Fig. 2b,h vs. d, f, g – first lanes) (refs. 12–14), the substitutions G78-PS(Sp) and A59-PS(Sp) also caused branching defects in extract in Mg2+ (Fig. 2c, e – first lanes, Extended Data Fig. 2a).
Importantly, in addition to spliceosomes containing U80-PS(Sp) (Fig. 2g; refs. 14,28), affinity-purified spliceosomes containing any of the other four sulfur substitutions that conferred a branching defect could catalyze branching in Cd2+ much more efficiently than in Mg2+ (Fig. 2c,d,e,f). Thus, all five oxygens sensitive to sulfur substitution bind metals important for branching at the catalytic stage.
To test whether these non-bridging oxygens implicated in branching also function in exon ligation, we assembled each of the twenty sulfur-substituted spliceosomes on a model substrate, chased spliceosomes through branching, and assayed for exon ligation after affinity purification to remove ATP. Only spliceosomes containing the five sulfur substitutions implicated in branching failed to catalyze exon ligation in Mg2+ (Fig. 2i vs. j, k, l – first lanes; data not shown). In addition to U80-PS(Sp) spliceosomes (Fig. 2i, m vs. l; ref. 28), A59-PS(Sp) spliceosomes catalyzed exon ligation in Cd2+ (Fig. 2j). While G60-PS(Rp), G78-PS(Sp), and U80-PS(Rp) failed to catalyze exon ligation even in Cd2+ (Fig. 2k; data not shown), these sulfur substitutions exhibited more stringent requirements for metal rescue in branching, failing to tolerate the less thiophilic Mn2+, whereas U80-PS(Sp) and A59-PS(Sp) spliceosomes did (Extended Data Fig. 2b). Therefore, the nonrescuable defects of these sulfur substitutions in exon ligation remain consistent with a role for the substituted oxygens as metal ligands during exon ligation (Supplementary Note 2). Thus, at least two of the oxygens that bind a metal important for branching also bind a metal important for exon ligation.
Overall our analysis shows that five non-bridging oxygens in U6 are bona fide metal ligands in the spliceosome (Fig. 2). These ligands function after catalytic activation, implicating a structural or direct catalytic role for these ligands during the catalytic stage. Strikingly, these ligands correspond directly and stereospecifically to the oxygens that coordinate two divalent metals in domain V of a group II intron, as revealed by X-ray crystallography26,27 (Fig. 2n). This parallel suggested that the U6 ligands that function at the catalytic stage may function analogously to domain V metal ligands by directly positioning the catalytic metals required for splicing.
U6 positions catalytic metals during branching
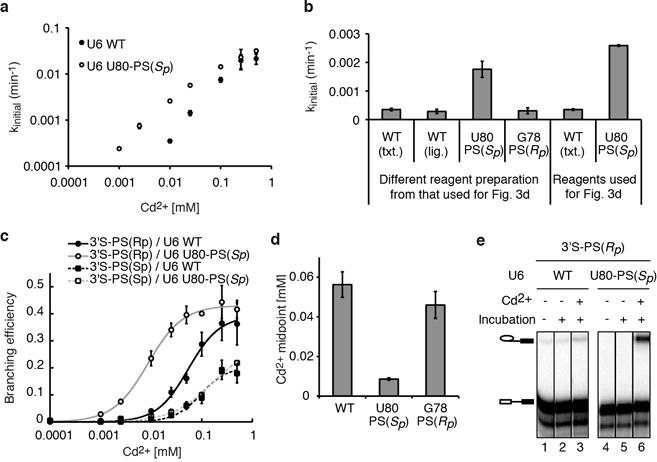
Experiments with the Tetrahymena group I intron have established biochemical signatures, for identifying ligands that position catalytic metals: sulfur substitution of ligands to a catalytic metal can be rescued more strongly by thiophilic metal or with increased specificity for Cd2+ when the substrate ligands to that metal are also substituted with sulfur31. Consequently, to determine whether the U6 snRNA metal ligands function catalytically during branching, we first identified a substrate sensitive to the identity of catalytic metals required for branching. A pre-mRNA substrate bearing a double sulfur substitution at both the leaving group and the non-bridging pro-Rp oxygen at the 5′ splice site (3′S-PS(Rp)) was branched efficiently only in the presence of Cd2+ (Fig. 3a), indicating that in yeast, as in mammals24 (cf. 37), divalent metals interact with the scissile phosphate (see also Supplementary Notes 3, 4 and Extended Data Figs. 3–4). We used the 3′S-PS(Rp) substrate as a reporter for catalytic metal interactions between the spliceosome and the 5′ splice site during branching.
Figure 3. U6 snRNA positions catalytic metals during branching.
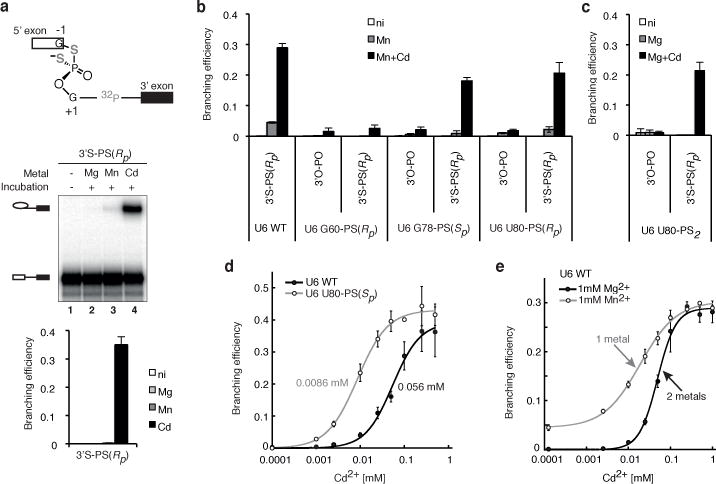
a, The 3′S-PS(Rp) substrate is sensitive to catalytic metal interactions. b,c, The 3′S-PS(Rp) substrate improves rescue of G78-PS(Sp), U80-PS(Rp), and U80-PS2 spliceosomes. d, U80-PS(Sp) reduces the Cd2+ concentration required for branching of the 3′S-PS(Rp) substrate. e, The 3′S-PS(Rp) substrate is rescued by Cd2+ bound at two distinct sites. In d and e, Hill fits are shown (solid curves); in e, titration midpoints are indicated. In b and e, a lower pH (7.0) was used compared to Fig. 2 (pH 8.5) to sensitize the system for improved rescue. Values represent averages; error bars, s.d. (n=3); ni, no incubation.
These experiments revealed several catalytic interactions between U6 and the 5′ splice site. Under sensitized conditions (Fig. 3 legend), the 3′S-PS(Rp) substrate strongly improved Cd2+-mediated rescue for G78-PS(Sp) and U80-PS(Rp) spliceosomes (Fig. 3b). In contrast to the 3′S-PS(Rp) substrate, the 3′S-PS(Sp) substrate, having the catalytically insignificant pro-Sp oxygen substituted with sulfur (Extended Data Fig. 3), did not improve Cd2+-mediated rescue for G78-PS(Sp) and U80-PS(Rp) (Extended Data Fig. 5a,b). These data indicate that the G78 pro-Sp and U80 pro-Rp oxygens interact with the 5′ splice site through catalytic metals during branching.
The 3′S-PS(Rp) substrate also rescued sulfur substitutions that could not be rescued on their own. Substitution of both the pro-Sp and pro-Rp oxygens at U80 with sulfur (U80-PS2) impaired branching of the 3′O-PO substrate, but unlike the individual substitutions, Cd2+ could not rescue branching (Fig. 3c). Strikingly, however, the 3′S-PS(Rp) substrate allowed robust Cd2+ rescue of branching for U80-PS2 (Fig. 3c), providing evidence that both of the U80 non-bridging oxygens bind catalytic metals during branching.
The 3′S-PS(Rp) substrate improved rescue of these sulfur substitutions in U6 in a specific manner. The modified substrate did not improve branching for U6 variants that compromised branching due to base mutations (Extended Data Fig. 5c,d, Supplementary Note 5) and did not improve Cd2+ rescue for G60-PS(Rp) (Fig. 3b). Thus the enhanced rescue of G78-PS(Sp), U80-PS(Rp), and U80-PS2 spliceosomes conferred specifically by the 3′S-PS(Rp) substrate bears the hallmark of ligands linked by common metals29–31 and indicates that the corresponding oxygens in U6 position catalytic metals during branching.
An implication of this conclusion is that a sulfur substitution in U6 might also reduce the Cd2+ concentration required for rescue of a substrate with sulfur substitutions at the 5′ splice site, by enhancing Cd2+ occupancy of a common metal binding site. Indeed, with the 3′S-PS(Rp) substrate, substitution of the pro-Sp oxygen of U80 with sulfur decreased the Cd2+ titration midpoint for rescue by 6-fold compared to U6 WT (Fig. 3d, Supplementary Note 6), and, at a limiting Cd2+ concentration (10 μM), increased the rate of branching 7-fold compared to U6 WT (Extended Data Fig. 6a,b), demonstrating that U80-PS(Sp) increased binding of Cd2+ at the catalytic core.
Our analysis has also revealed that branching requires two, distinct catalytic metals (cf. ref. 22). With the 3′S-PS(Rp) substrate, Cd2+ titrations indicated that rescue required binding of Cd2+ to two sites, when Cd2+ was the only thiophilic metal present (Fig. 3e). In contrast, when Mn2+ was also present, rescue still required binding of Cd2+ but to only one site (Fig. 3e; Extended Data Fig. 7a–c), demonstrating that one metal site could bind Mn2+ while the other site could not. Given that Cd2+ is expected to bind a site containing multiple sulfur ligands while Mn2+ is not38, these and additional data indicate that one metal interacts with both sulfur atoms of the 3′S-PS(Rp) substrate (referred to as M1) (Extended Data Fig. 7e,f), while the second metal interacts with the non-bridging Rp sulfur (M2). Further evidence suggests that the U80 pro-Rp and G78 pro-Sp oxygens also interact with the M1 site, while the U80 pro-Sp oxygen interacts with the second metal site (M2) (Extended Data Figs. 7,8, Supplementary Note 7). Thus, at least three of the five identified U6 ligands coordinate two distinct catalytic metals that interact with the scissile phosphate of the 5′ splice site during branching (Supplementary Note 8, Extended Data Figs. 5e–g, 6e).
U6 positions a catalytic metal during exon ligation
To determine whether the U6 metal ligands function during exon ligation by directly binding catalytic metals, we tested for biochemical signatures31 as above, that would link the U6 ligands to the catalytic metal that interacts with the 3′ splice site during exon ligation.
First, we introduced a sulfur substitution at the 3′ splice site leaving group to sensitize splicing of a model substrate to binding of the catalytic metal that interacts with this group. Additionally, we introduced a mutation of the 3′ splice site consensus sequence (UAG to UAc) to stall spliceosomes prior to exon ligation34 regardless of whether the 3′-oxygen leaving group at the 3′ splice site was substituted with sulfur or oxygen (3′S or 3′O). Exon ligation of this substrate can proceed only in the absence of ATP due to proofreading34. While exon ligation of affinity-purified UAc-3′O spliceosomes proceeded in the absence of ATP, the 3′S substitution compromised exon ligation when only Mg2+ was present (Fig. 4a). When Mn2+ or Cd2+ was present, spliceosomes assembled on the UAc-3′S substrate catalyzed exon ligation 4- to 6-fold more efficiently (Fig. 4a), indicating that a divalent metal stabilizes the leaving group in yeast, as in mammals25 (see also Supplementary Note 9 and Extended Data Fig. 9a–f). Because the UAc-3′S substrate specifically required thiophilic, catalytic metals, we used it as a reporter for catalytic metal interactions between the spliceosome and the 3′ splice site during exon ligation (Supplementary Note 10).
Figure 4. U6 snRNA positions a catalytic metal during exon ligation.
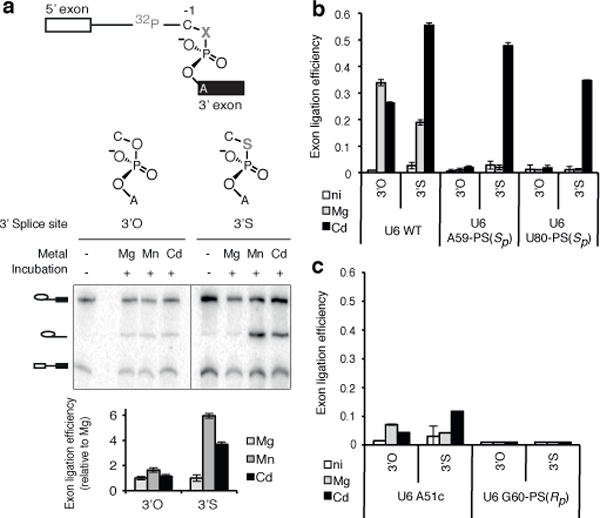
a, The UAc-3′S substrate is sensitive to catalytic metal interactions. b–c, The UAc-3′S substrate specifically improves rescue of U6 A59-PS(Sp) and U80-PS(Sp) spliceosomes. Values represent averages; error bars, s.d. (n=3); ni, no incubation.
After assembling spliceosomes containing U6 sulfur substitutions on this sulfur-substituted substrate, we probed for exon ligation at the catalytic stage (i.e., in the absence of ATP and soluble factors). As expected from splicing assays of the wild-type UAG-3′O substrate (Fig. 2j,l), exon ligation of the mutated UAc-3′O substrate in Mg2+ occurred with exceedingly low efficiency with spliceosomes containing U6 A59-PS(Sp) or U80-PS(Sp) (Fig. 4b). However, in contrast to exon ligation assays of the wild-type UAG-3′O substrate (Fig. 2j,l), exon ligation of the mutated UAc-3′O substrate by these sulfur-substituted spliceosomes was not rescued by Cd2+ (Fig. 4b), indicating that Cd2+ could no longer rescue the U6 sulfur substitutions in the context of the substrate base mutation. Nevertheless, when the substrate also contained sulfur in the leaving group position (UAc-3′S), Cd2+, but not Mg2+, strongly stimulated exon ligation by the sulfur-substituted spliceosomes (Fig. 4b, Extended Data Fig. 9g). This stronger rescue by Cd2+ of a sulfur-substituted ligand in the presence of a second sulfur-substituted ligand constitutes a signature for a functional and physical link between two ligands that bind the same metal29–31. These data therefore establish that the U6 A59 pro-Sp and U80 pro-Sp oxygens interact with the catalytic metal that stabilizes the leaving group during exon ligation.
The signature was specific to the U6 A59-PS(Sp) or U80-PS(Sp) spliceosomes and was not observed for other spliceosomes that were compromised for exon ligation. Unlike the U6 sulfur-substituted spliceosomes assembled on the UAc-3′S substrate, the exon ligation defect39 of U6 A51c spliceosomes was not substantially suppressed in Cd2+ by the UAc-3′S substrate (Fig. 4c). Furthermore, the exon ligation defect of G60-PS(Rp) was not suppressed by the UAc-3′S substrate in Cd2+ (Fig. 4c; Extended Data Fig. 9h; Supplementary Note 11), indicating that the substrate sulfur substitution alone does not enable exon ligation of compromised spliceosomes. These results and a metal specificity switch induced by U80-PS(Sp) (Extended Data Fig. 8 and Supplementary Note 12) validate the evidence that the U6 A59 pro-Sp and U80 pro-Sp oxygens interact with a catalytic metal during exon.
Through a comprehensive analysis, encompassing every substrate allowed by sulfur chemistry (Extended Data Figs. 5g, 9h), we have found that four of the five metal ligands in U6 position catalytic metals during the catalytic stages of splicing. Even though metal rescue strategies were insufficient to establish direct evidence linking the fifth ligand (G60 pro-Sp) to a splice site (Supplementary Note 2), the configuration of the RNA catalytic core implied by our data is consistent with the fifth ligand also binding a catalytic metal (see below). Importantly, at least two of the U6 metal ligands function in both steps of splicing and at least one of these, U80 pro-Sp, positions a catalytic metal during both branching and exon ligation (M2, Fig. 5a,b). These and other findings (Supplementary Note 13) support a model22 in which both splicing reactions are catalyzed by a common, two-metal catalytic core, rather than two independent active sites, and thereby implicate a rearrangement of the substrate to sequentially accommodate mutually exclusive interactions between U6 and the 5′ and 3′ splice sites. Further, underscoring the importance of disabling fidelity mechanisms in our approach, these results establish that the spliceosome proofreads catalytic interactions (cf. 28).
Figure 5. Model for catalytic metal interactions during pre-mRNA splicing and comparison to the domain V catalytic core of group II introns.
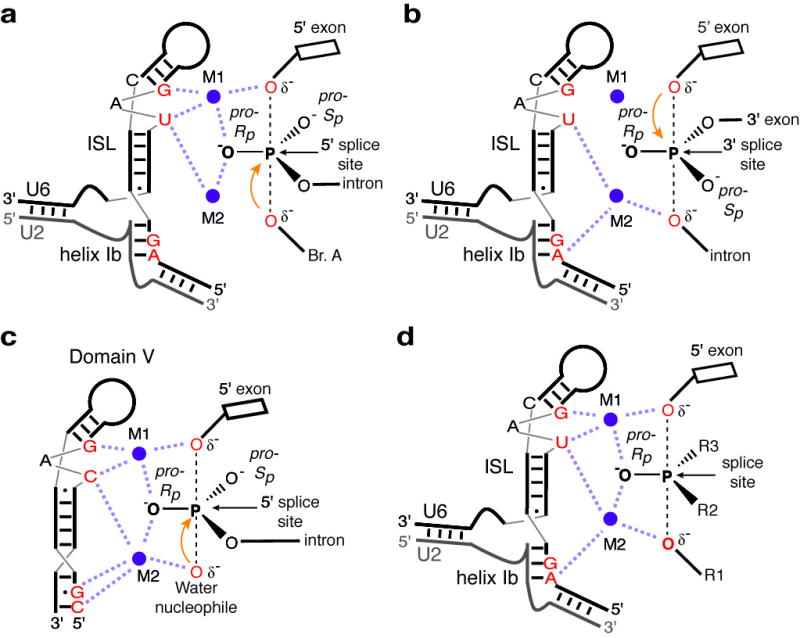
a–b, Catalytic metal interactions during branching (a) and exon ligation (b). c, Model of domain V before hydrolytic branching (PDB 4FAQ, ref. 27). d, Two-metal model for the RNA catalytic core of the spliceosome. For branching, R1 represents the 2′ hydroxyl of the branch adenosine; R2, the intron; and R3, the pro-Sp oxygen. For exon ligation R1 represents the 3′ oxygen leaving group; R2, the pro-Sp oxygen; and R3, the 3′ exon. Throughout, the reactive oxygens are coloured red, the pre-mRNA scissile phosphate is depicted in a transition state, and interactions between specific ligands and the reactive oxygens mediated by M1 and M2 are shown as light magenta dashed lines.
Discussion
Our results indicate that the metals that mediate splicing catalysis through interactions with the scissile phosphates are bound by the spliceosome through ligands in the U6 snRNA, thus demonstrating definitively that RNA directly mediates catalysis in the spliceosome (Fig. 5d). Notably, all of the five U6 metal ligands that function in pre-mRNA splicing (Fig. 2) correspond directly to catalytic metal ligands observed in domain V in structures of a group II intron (Figs. 1b,c; 2n; 5c, refs. 26,27). Moreover, our findings imply an equivalent orientation of the substrate relative to the metal binding core in Fig. 5. Taken together, our data support a model for a single, two-metal active site (Fig. 5d; Supplementary Note 13; ref. 22), which is consistent with recent chemical probing data40, and validate (cf. 25) the long-standing hypothesis that the spliceosome shares a similar, RNA-based catalytic core and mechanism with group II introns5,6,13,18,22,27,41 (Supplementary Note 14). This RNA-based mechanism is sufficient to effect metal catalysis of pre-mRNA splicing, without the need for direct protein involvement.
Given this evidence, it is noteworthy that the RNaseH-like domain of Prp8, which interacts with all reactive sites of the substrate, can bind a metal in crystallo42. Further, mutations that compromise metal binding in crystallo impair exon ligation, leading to the possibility that Prp8 may play a catalytic role during exon ligation42. Nevertheless, we have not found any evidence for a direct metal-mediated catalytic interaction between the Prp8 metal binding site and the 3′ splice site (Supplementary Note 15; Extended Data Fig. 10). Thus, Prp8 may primarily promote formation of the RNA catalytic core without being a part of it; indeed, a recent structure comprising most of Prp8 has led to the suggestion that Prp8 forms a scaffold for the spliceosome’s catalytic core, analogously to protein co-factors of group II introns17. A recent model of the catalytic core of the human spliceosome based on RNA structure probing provides further support for this interpretation40,43.
Overall our data indicate that the spliceosome, like the ribosome44,45, uses RNA to effect catalysis in the context of a complex RNP assembly. Moreover, the common catalytic mechanism used by the spliceosome and group II introns is consistent with a common evolutionary origin between the spliceosome and these ancient RNA retroelements46,47. Our findings thus support the idea that modern RNP enzymes evolved from a primordial “RNA world” (ref. 48), in which catalysis was performed exclusively by RNA.
Methods Summary
U6 was depleted from Saccharomyces cerevisiae splicing extracts and splicing activity reconstituted with synthetic U6 snRNA, essentially as described28. Spliceosomes were assembled on modified model pre-mRNA substrates: UBC4, for experiments probing branching, or ACT1, for experiments probing exon ligation, both synthesized by splint-mediated ligation49. Oligonucleotides containing specific 5′ or 3′ splice site modifications were synthesized in house, as described previously50. Assembled spliceosomes were isolated by affinity purification with Prp19p (ref. 36), washed to remove ATP, and chased as described28, at room temperature, in the absence of ATP, at pH 7.0, 8.0, or 8.5. All experiments were repeated with at least two independent extract preparations. Data were quantified using ImageQuant TL (Amersham Biosciences).
METHODS
Strains
Experiments probing branching were performed using S. cerevisiae strain yJPS1405, which was derived from BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0, Open Biosystems), by integrating a biotinylation signal C-terminal to the PRP19 locus using KanMX6 as a selective marker; the integration fragment, including the biotinylation signal, an ADH1 terminator, and the KanMX sequences were amplified from pFA6-HTB-KanMX (ref. 51).
Experiments probing exon ligation were performed using the previously described S. cerevisiae strain yJPS860, containing a C-terminal TAP tag on Prp19 (ref. 34).
Experiments involving Prp8 were performed using S. cerevisiae strains yJPS1471 (PRP8 wild-type) and yJPS1472 (PRP8 D1853C), both derived from yJPS86034. Briefly, the entire coding sequence of PRP8 was replaced in yJPS860 with LEU2, after transformation with PRP8 on a URA3 plasmid (bJPS1874), yielding yJPS1470. Then yJPS1471 and 1472 were derived from yJPS1470 by plasmid shuffle on 5-FOA after transformation with bJPS2263 (PRP8 wild-type on a HIS3 plasmid, same as pJU186, ref. 52) or bJPS2589 (PRP8 D1853C). bJPS2589 was derived from pJU186 (ref. 52) by quick change mutagenesis and confirmed by sequencing.
Pre-mRNA splicing substrates
Branching experiments were performed using UBC4 pre-mRNA53 truncated at the 5′ and 3′ ends down to 20nt exons. Exon ligation experiments were performed using ACT1 pre-mRNA. Pre-mRNA substrates were prepared by splint-mediated ligation using T4 RNA ligase 2 (NEB) or T4 DNA ligase (in house).
Oligonucleotides UBC4-5′E, 5′Ev2, UBC4-M, and UBC4-M1 were purchased from Dharmacon. Oligonucleotide UBC4 M2, bearing various modifications, was synthesized in house and purified by HPLC on a DNAPac P-100 column using the Waters 2795 system54. Oligonucleotides ACT1-3′O-pc, ACT1-3′S-pc, ACT1-UAc-3′O, and ACT1-UAc-3′S were synthesized in house. The correct identity (position and chirality) of specific sulfur modifications was verified by mass spectrometry, silver or iodine cleavage, and analytical T1 digestion55 (data not shown). Oligos M, M1, and M2 were phosphorylated with unlabeled ATP prior to ligation. The UBC4-TX transcript was synthesized by in vitro transcription using a PCR-derived template that started transcription at position +37 of the intron and ended at the last nucleotide of the exon; the template was generated using primers T7 UBC4 37–135 F and HindIII UBC4 R. The transcript was gel purified, treated with calf inorganic phosphatase (NEB), and 5′ phosphorylated with γ-32P-ATP (Perkin Elmer, 6000 Ci/ mmol) prior to ligation.
The ACT1-5′-end plasmid was constructed by stepwise PCR from plasmid pJPS149 (ref. 34) to generate a DNA template containing an EcoRI site, the T7 promoter, ACT1 nucleotides 1–373, an HDV ribozyme sequence56, and a HindIII site. The template was then cloned into vector pUC19. The ACT1-1-373 transcript was synthesized by in vitro transcription from the ACT1-5′-piece plasmid linearized with HindIII. In cases where HDV cleavage was inefficient during transcription, the RNA was resuspended in 10mM Tris (pH 7.5) and 20 mM MgCl2. Ribozyme cleavage was induced via 2–4 cycles of 90°C for 1 minute, room temperature for 15 minutes, and 37°C for 15 minutes. The buffer conditions were then adjusted for T4 PNK (NEB) treatment of the transcript to remove the 2′-3′-cyclic phosphate left by the ribozyme. The ACT1-392-590 transcript was synthesized by in vitro transcription using a PCR-derived template generated using plasmid bJPS149 (ref. 34). As the subsequent ligation requires a 5′-monophosphate group, a 4-fold excess of GMP over GTP was included in the transcription reaction.
For a typical UBC4 ligation reaction, 20 pmol of UBC4 5′E, 10 pmol of UBC4 M, M1, or M2 and 20 pmol of UBC4 TX were hybridized to 10 pmol of UBC4 splint in buffer TEN (10 mM Tris-HCl, pH 7.5; 1 mM EDTA; 66 mM NaCl) on a thermal cycler by heating to 90°C for 2 minutes followed by reduction of the temperature by 1°C for 1 minute in 72 sequential steps, to a final temperature of 18°C. T4 RNA ligase 2 (10–20 U) was then added and reactions were incubated at 37°C for 6 hours. Ligated, full length UBC4 pre-mRNA was purified on an 8% denaturing polyacrylamide gel and recovered by passive elution in TE buffer at 4°C overnight (10 mM Tris-HCl pH 7.5; 1 mM EDTA).
For a typical ACT1 ligation reaction, 500 pmol of ACT1-1-373, 50 pmol of oligonucleotide ACT1-3′O, and 500 pmol of ACT1-392-590 were hybridized to 50 pmol of ACT1 splint in buffer TEN50 (10 mM Tris-HCl, pH 7.5; 1 mM EDTA; 50 mM NaCl) on a thermal cycler by heating to 90°C for 2 minutes followed by reduction of the temperature by 1°C for 1 minute to 24°C, then cooling to 4°C for five minutes. T4 DNA ligase (~100 pmol, synthesized in house) was then added and reactions were incubated at 37°C for four hours. The ligation reactions were DNase treated (RNase-free DNase, Promega) for 15 minutes to remove splint, phenol-chloroform extracted and ethanol precipitated before purification on 6% denaturing polyacrylamide gel. Bands containing full-length ACT1 pre-mRNA were excised and recovered by passive elution in TEN250 buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA, 250 mM NaCl) overnight at 4°C.
Splicing extracts and U6 depletion and reconstitution
Splicing extracts were prepared using the liquid nitrogen method, as described52. U6 depletion and reconstitution was performed essentially as described28, with the following modifications. The U6 d1 oligonucleotide was titrated for each extract to optimize depletion and reconstitution of U6; typically, 0.8 μM U6 d1 was optimal. After depletion of U6, to promote degradation of the U6 d1 oligonucleotide before addition of the synthetic U6, DNase I (Ambion, 0.05 U/ μL final) was added together with the glucose used to deplete ATP. Additionally, to ensure complete inactivation of the U6 d1 oligonucleotide, an oligonucleotide antisense to U6 d1 (U6 αd1, 0.3 μM final)57 was added immediately prior to reconstitution and incubated on ice for 5 minutes. For reconstitution U6 was added back to a final concentration of 0.2 – 0.3 μM. Modified U6 was constructed by splint-mediated ligation essentially as described28. Wild type U6 was synthesized by in vitro transcription according to standard procedures using pJPS488 linearized with DraI as template58. There was no difference in splicing efficiency between spliceosomes reconstituted with wild type U6 made by ligation or by in vitro transcription (data not shown and Extended Data Fig. 6b).
In vitro splicing
In vitro splicing was performed essentially as described28 using 32P-labeled substrates (0.2 – 0.4 nM). In Fig. 2c,g, extracts were pre-incubated with 2mM EDTA at 4°C for 30 minutes before assembling the reactions; MgCl2 was adjusted to 3.3 mM during the splicing reaction (compared to standard 2.5 mM) to compensate for the final EDTA concentration (0.8 mM). Reactions were usually incubated in extract for 25 minutes prior to affinity purification of assembled complexes, except for experiments in Fig. 3a and Extended Data Fig. 3, where initial incubation was performed for 10 minutes. Assembled and stalled complexes were affinity-purified by incubating the splicing reactions for 1–3 hours at 4°C with a 10–20% reaction volume of streptavidin-agarose (Thermo Scientific) or IgG-Sepharose (GE) slurry pre-washed twice with 25–50 volumes of IPP150 (10 mM Tris-HCl pH 8.0; 150 mM NaCl; 0.01% NP-40 substitute (Fluka)). Following immunoprecipitation, beads were washed at 4°C twice with 50 volumes of buffer PK (3% PEG8000, 60mM potassium phosphate, pH 7.0), and spliceosomes were assayed for splicing in the absence of ATP (except where noted), in buffer PK (pH 7.0, 8.0, or 8.5, as indicated) with various amounts of the indicated metals and EDTA (where noted) at room temperature (23–24°C) with constant rotation, for 60–90 minutes. Metal solutions were prepared from corresponding powdered salts of at least 99.99% purity (Sigma).
Affinity-purified spliceosomes were incubated as follows. In Figs. 2c, 3a – buffer PK (pH 8.5, with 0.5 mM EDTA) with 1 mM MgCl2 and 1 mM of the indicated metals; in Fig. 2d–g – buffer PK (pH 8.5) with 0.5 mM MgCl2 and 0.5 mM of the indicated metals; in Fig. 2j–l, 4b–c – buffer PK (pH 7.0) with 2 mM MgCl2 and 0.1 mM CdCl2; in Fig. 2k,l spliceosomes containing A59-PS(Sp) and U80-PS(Sp) were chased through branching in the presence of 0.1 mM CdCl2 before affinity purification. In Fig. 3b,e – buffer PK (pH 7.0) with 1 mM MnCl2 alone or in combination with 1 mM CdCl2; in Fig. 3c – buffer PK (pH 8.5) with 0.5 mM MgCl2 and 0.5 mM CdCl2; in Fig. 3d – buffer PK (pH 8.5) with 0.5 mM MgCl2 and various amounts of CdCl2, as indicated. In Fig. 4a – buffer PK (pH 7.0) with 1 mM MgCl2 and 0.1 mM CdCl2 or 0.1 mM MnCl2.
Following in vitro splicing, products were separated on 15% denaturing polyacrylamide gels for UBC4 substrates or 6% denaturing polyacrylamide gels for ACT1 substrates.
Cwc25p depletion and reconstitution
For depletion of Cwc25p, splicing extracts were prepared fresh, dialyzed against buffer D (ref. 52), and then incubated at 4°C for 1.5 hours with 50% volume equivalent of protein A-sepharose (Sigma) (1:1 slurry in IPP150) conjugated to anti-Cwc25 serum (gift from S.-C. Cheng, ref. 59) and pre-equilibrated in buffer D by rotation at 4°C for 15 minutes. Beads were removed by centrifugation at 800× g for 4 minutes and the supernatant was used as depleted extract.
rCwc25p extended with a C-terminal six-histidine tag was expressed in E. coli BL21(DE3)pLysS transformed with pET15b (ref. 59; a gift from S.-C. Cheng). Induction in E. coli and purification by Ni2+-NTA affinity chromatography were performed essentially as described60 but binding and washing was performed manually. Following elution from the Ni2+-NTA resin, the protein was further purified by glycerol gradient centrifugation to more than 90% purity (as estimated by Coomassie Blue staining).
To prepare the heat-soluble extract fraction (HP), yeast splicing extracts were incubated at 90°C for 5 minutes and insoluble material was removed by centrifugation at 16000 × g for 5 minutes. The supernatant was used as HP.
For complementation of affinity purified spliceosomes from extracts immunodepleted of Cwc25p, spliceosomes were washed twice with 50 volumes of buffer DK (20mM HEPES-KOH, pH 7.9; 60mM potassium phosphate, pH 7.0; 50mM NaCl; 0.2mM EDTA) and splicing was assayed in buffer DK, in the presence of rCwc25-6His (0.7–1.4 μM) and a heat soluble extract fraction (HP; typically 1–2 μL for a 50 μL splicing reaction).
To determine the rates of branching in Extended Data Fig. 4d, reactions were pre-incubated with rCwc25p and HP in the absence of metal at room temperature for 5 minutes to allow binding and temperature equilibration. Splicing was initiated by addition of metals; aliquots were removed at various times, immediately quenched in STOP buffer (50mM NaOAc, pH 5.2; 1mM EDTA; 0.1% SDS; 0.1 mg/ mL glycogen), and placed on ice prior to phenol extraction.
Oligonucleotides
The following oligonucleotides were used for synthesis of UBC4 substrates:
UBC4 5′E: 5′-GAACUAAGUGAUCUAGAAAGG-3′
UBC4 5′Ev2: 5′-GAACUAAGUGAUCUA-3′
UBC4 M: 5′-UAUGUCUAAAGUUAU-3′
UBC4 M1: 5′-GAAAG(3′S)GUAUGUCUAAAGUUAU-3′
UBC4 M2: 5′- GAAAG(3′S-PS)GUAUGUCUAAAGUUAU-3′
UBC4 Splint: 5′-CACGCATTTGAAACGTGGCCATAACTTTAGACATACCTTTCTAGATCACTTATTC-3′ (ligation splint)
T7 UBC4 37-135 F: 5′-TAATACGACTCACTATAGGCCACGTTTCAAATGC-3′ (forward primer to generate the template UBC4 TX)
Hind III UBC4 R: 5′-ATAAGCTTAACATGAAGTAGGTGGATCTC-3′ (reverse primer to generate the template for UBC4 TX).
The following oligonucleotides were used for synthesis of ACT1 substrates:
ACT1-3′O-pc: 5′-UUUA(2′-o-nitrobenzyl-G)AGGUUGCUGCUUU-3′
ACT1-3′S-pc: 5′-UUUA(3′S, 2′-o-nitrobenzyl-G)AGGUUGCUGCUUU-3′
ACT1-UAc-3′O: 5′-UUUACACGUUGCUGCUUU-3′
ACT1-UAc-3′S: 5′-UUUA(3′S-C)AGGUUGCUGCUUU-3′
ACT1-UAc-splint: 5′-GAACCGTTATCAATAACCAAAGCAGCAACGTGTAAACATATAATATAGCAACAAAAA-3′
ACT1-splint: 5′-GAACCGTTATCAATAACCAAAGCAGCAACCTCTAAACATATAATATAGCAACAAAAA-3′
act1-3′-end-for-1: 5′-TAATACGACTCACTATAGGTTATTGAT AACGGTTATTG-3′ (forward DNA primer to generate ACT1 392–590 transcription template)
act1-3′-end-for-2: 5′-GAAATTAATACGACTCACTATAGGTTATTG-3′ (forward DNA primer to generate ACT1 392–590 transcription template)
act1-3′-end-rev: 5′-mUmUGGGCTGCAGGTCGAGCTC-3′ (reverse DNA primer to generate ACT1 392-590 transcription template)
EcoRI+T7:5′-AGTGAATTCCCTTAATACGACTCACTATAGG-3′ (forward DNA primer to generate ACT1 1-373-HDV template)
Forward long: 5′-CGTAATACGACTCACTATAGGGCGAATTGG-3′ (forward DNA primer to generate ACT1 1-373-HDV template)
Reverse-A-25: 5′-CGAGGAGGCTGGGAGCATGCCGGCCCATATAATATAGCAACAAAAAGAAT-3′ (reverse DNA primer to generate ACT1 1-373-HDV template)
Reverse 0-50: 5′-CCGGAATGTTGCCCAGCCGGCGCCGCGAGGAGGCTGGGAGCATGCCGGCC-3′ (reverse DNA primer to generate ACT1 1-373-HDV template)
Reverse 25-75: 5′-GTGCGTCCCATTCGCCATTACCGGACGGTCCGGAATGTTGCCCAGCCGGCGCCG-3′ (reverse DNA primer to generate ACT1 1-373-HDV template)
Reverse final: 5′-GTGCGTCCCATTCGCCATTACCCG-3′ (reverse DNA primer to generate ACT1 1-373-HDV template).
Data Analysis
Gels were dried, exposed to storage phosphor screens (Amersham Biosciences) for 24–48h hours, and scanned using a Typhoon Trio phosphorimager (Amersham Biosciences). Bands were quantified using ImageQuant TL with an automated rolling ball algorithm for background subtraction. The efficiency of branching was calculated as LI/(LI+P), if EI represented less than 1% of all species or (LI+EI)/(LI+EI+P) in all other cases; LI, lariat intermediate; EI, excised intron; P, pre-mRNA. The efficiency of exon ligation was calculated as EI/(EI+LI).
The rescue midpoint for the metal titration curves in Fig. 3d and Extended Data Fig. 6c,d was obtained by fitting the rescue profiles to the general Hill equation A=Amax*xn/(xn+Kn), where Amax is the splicing efficiency at saturation, K is the rescue midpoint, x is the CdCl2 concentration, and n is the Hill coefficient. Curves in Fig. 3e and Extended Data Fig. 7a were fit to the following equations: y = E0 + E*x / (x+KCd) (1 metal model) or y = E0 + E*x2 / (x2+KCd2) (2 metal model), where y is the branching efficiency, E0 is the extent of branching in the absence of Cd2+, E is the extent of branching at saturating Cd2+, and KCd is the apparent transition midpoint for Cd2+ binding. Fits using the general Hill equation: y = E0 + E*xn / (xn+KCdn), where n is the number of metal sites titrated, gave the same number of sites (within fit error) as those assuming a fixed n (data not shown).
Initial rates in Extended Data Figs. 4d and 6a–b were obtained by fitting the linear portion of the splicing time courses to the equation A=A0+kinitial*t, where A is the splicing efficiency at time t and A0 is the extent of splicing at time 0. Rates in Extended Data Fig. 9 were obtained by fitting the splicing time courses to the equation E = E0 + A(1-e−kt) where E is the extent of reaction, E0 is the extent at time = 0, A is the amplitude, k is the rate of reaction, and t is the time in minutes.
Extended Data
Extended Data Figure 1. Affinity purification of spliceosomes with Prp19 requires reconstitution with U6 snRNA and enhances the potential to detect rescue by thiophilic metals.
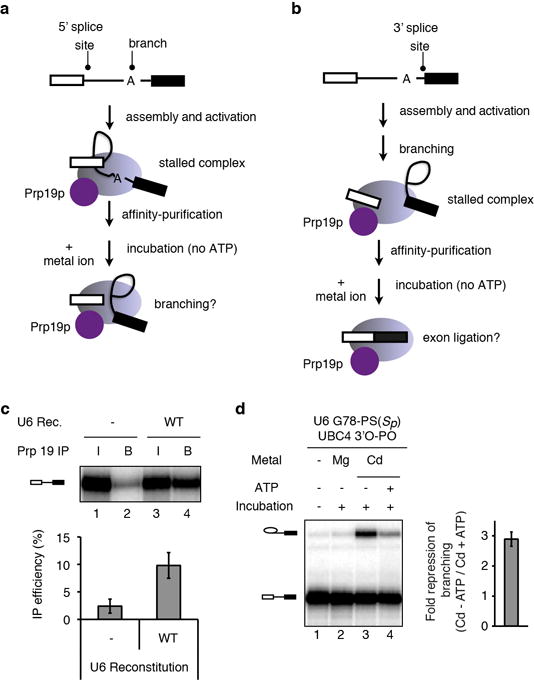
a–b, Schemes depicting experimental strategy for staging spliceosomes to monitor branching (a) or exon ligation (b) in the absence of ATP. Spliceosomes are depicted as light magenta ovals and Prp19p as a magenta circle. Following affinity purification of spliceosomes via Prp19p (ref. 36), beads were washed to remove ATP and soluble factors, and metals ions were added to assay for splicing. c, Prp19p-mediated affinity purification of activated spliceosomes, reflected by immunoprecipitation of pre-mRNA, is specific for properly reconstituted complexes. Note that the affinity purification allows quantification of the branching efficiency for activated complexes, independently of any effects on assembly. RNA from 10% of the reaction (input, I) or from beads after affinity purification (B) was extracted and analyzed by denaturing PAGE. (top) raw data; (bottom) quantification of IP efficiency; Rec., reconstitution. d, ATP represses the Cd2+ rescue for G78-PS(Sp) spliceosomes. Spliceosomes were assayed as in Fig. 2e; for lane 4, 2mM ATP-Mg2+ was also present during the incubation. (left) representative gel; (right) quantification of the extent of ATP repression. Values are averages; error bars, s.d. (n=3).
Extended Data Figure 2. Broad rescue specificity of A59-PS(Sp) and U80-PS(Sp) spliceosomes.
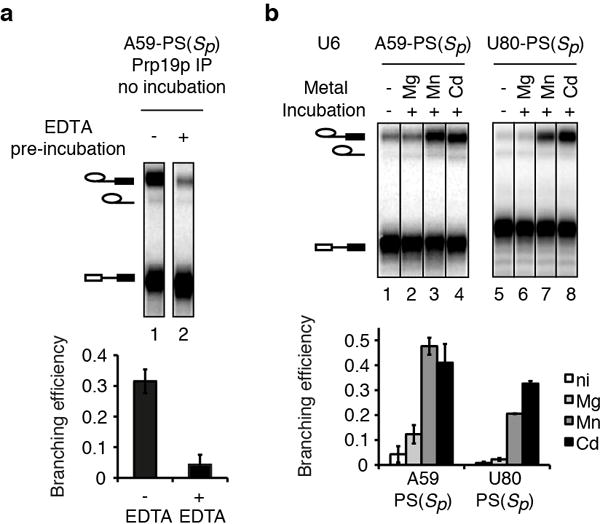
a, Pre-incubation with EDTA reveals a branching defect for A59-PS(Sp). Extracts were pre-incubated at 4°C in the presence or absence of 2 mM EDTA and then incubated with the 3′O substrate. After affinity-purification, branching efficiency was quantified without further incubation (bottom). A representative gel is shown (top). EDTA pre-incubation caused an 8-fold reduction in branching efficiency, suggesting that splicing extracts may contain a thiophilic metal that supports branching by A59-PS(Sp) spliceosomes; note that pre-mRNA still immunoprecipitated efficiently, indicating catalytic activation of the spliceosome. In contrast EDTA pre-incubation has no effect on U6 WT spliceosomes (data not shown). b, The branching defects for A59-PS(Sp) spliceosomes are rescued by either Mn2+ or Cd2+. Assays were as in Fig. 2c. A representative gel (top) and quantification (bottom) are shown; ni, no incubation. Both Mn2+ and Cd2+ strongly rescue A59-PS(Sp) and U80-PS(Sp) spliceosomes (lanes 3,4,7,8), suggesting that even the weaker Mn2+-S interaction38 at these positions can support branching. This broad specificity for branching may also explain why A59-PS(Sp) and U80-PS(Sp) spliceosomes also catalyzed exon ligation in the presence of thiophilic metals, whereas G60-PS(Rp), G78-PS(Sp), and U80-PS(Rp) spliceosomes, for which branching was only rescued in the presence of Cd2+, stalled after branching. Values are averages; error bars, s.d. (n=3).
Extended Data Figure 3. A divalent metal binds the 5′ splice site pro-Rp oxygen during branching.
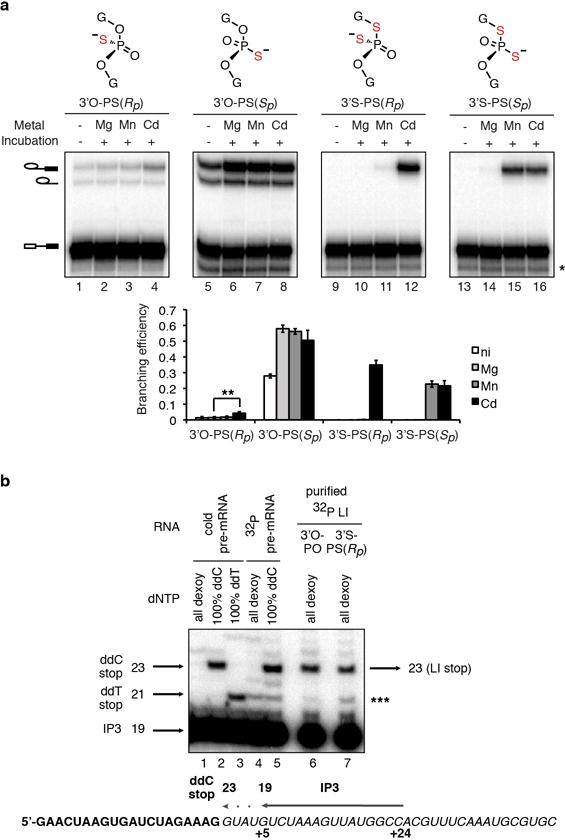
a, UBC4 pre-mRNAs bearing the indicated modifications at the 5′ splice site (top panel) were assayed as in Fig. 3a. The band marked * results from 5′ to 3′ exonucleolytic degradation that is blocked by the sulfur; ** denotes statistical significance of Cd2+ rescue compared to Mg2+ splicing (p = 0.004, paired, 1-tailed t-test, n=3). The data from Fig. 3a are reproduced here to aid comparison. Values are averages; error bars, s.d. (n=3). b, Mapping of the site of branching. Purified, 32P-labeled, intact lariat intermediates (LI ~ 0.2 fmol) resulting from branching of the UBC4 3′O-PO or 3′S-PS(Rp) substrates were used as templates for reverse transcriptase using primer IP3 (~10 fmol), which binds at nucleotides +5 to +24 of the intron (see lower diagram). Lanes 6 and 7 show that the major RT stop occurs at the same position when either the 3′O-PO or 3′S-PS(Rp) lariats are used as template. This stop migrates at the expected position, which is the position of the ddC stop resulting from extension of primer IP3 with pre-mRNA as template and at the expected position and therefore corresponds to position +1 of the intron, the expected branch site. Lower diagram shows mapping of the primer and expected RT stop onto the UBC4 pre-mRNA sequence; bold exon; italics intron. Note that the band marked *** is present in the 32P pre-mRNA lane and thus is likely a nonspecific band resulting from contamination with pre-mRNA degradation products that can anneal to the primer and serve as template.
Extended Data Figure 4. The UBC4 3′S substrate can be branched efficiently only in the presence of thiophilic metals.
a, A sulfur at the 5′ splice site leaving group alone blocked splicing in extract. Following affinity-purification branching efficiency was quantified (bottom) without further incubation. Note that immunoprecipitation of pre-mRNA indicated that the sulfur did not block the catalytic activation of the spliceosome. b, Thiophilic metals rescued the branching defect of the 3′S substrate. Spliceosomes were assayed in buffer PK (pH 7.0) in the presence of 2 mM total metal (1 mM MgCl2 plus 1 mM MnCl2 or 1 mM CdCl2) for 1.5 hours. The bar graph quantifies the relative stimulation by specific metals (normalized to Mg2+). c–d, Thiophilic metals specifically stimulate branching of the 3′S, but not the 3′O substrate. Affinity-purified spliceosomes from extracts depleted of Cwc25p, to stall spliceosomes independent of the sulfur substitution, were incubated as in Fig. 3a except that washes and incubation were done in buffer DK without EDTA and with 1 mM MgCl2; rCwc25 as well as an “HP” extract fraction were also added to complement (Supplementary Note 4; Supplementary Methods). Quantification of the thiophilic metal stimulation relative to Mg2+ is shown both for reaction endpoints (c) and for the rate of branching (d). Values are averages; error bars, s.d. (n=3). The band marked * results from 5′ to 3′ exonucleolytic degradation that is blocked by the sulfur.
Extended Data Figure 5. The 3′S-PS(Rp) substrate specifically improves rescue for spliceosomes containing U6 sulfur substitutions that compromise catalytic metal binding.

a–b, Spliceosomes were assayed as in Fig. 3b. Note that the 3′S-PS(Sp) substrate does not significantly improve Cd2+ rescue when compared to the 3′O-PO substrate (a), despite having similar reactivity to the 3′S-PS(Rp) substrate with U6 WT (b). c, The U6 double mutation U80g/C61g permitted both spliceosome assembly and activation, as reflected by the stable association of Prp19p with the splicing substrate. 10% of the RNA in the input (I) for the IP or 100% of the RNA associated with affinity-purified spliceosomes (B) were analyzed by dPAGE (top). Total IP efficiency was quantitated for all splicing species combined (bottom). d, The 3′S-PS(Rp) substrate did not significantly improve splicing for U80g and U80g/C61g spliceosomes. Assays were as in Fig. 3b. e–f, Representative raw data for Fig. 3. Assays were as in Fig. 3. In e, for U6 WT lanes 1–3 and 4–6 were taken from two different gels, for G78-PS(Sp) lanes 7–12 and 13–15 were taken from two different gels. In all other cases the lanes for different substrates assembled with spliceosomes bearing the same U6 modification were taken from the same gel. Values are averages; error bars, s.d. (n=3 for a,b,d; n=2 for c). g, Summary of combinations of sulfur substitutions in U6 and the substrate tested for rescue of branching. The “+” sign indicates that branching was observed in the presence of thiophilic metal.
Extended Data Figure 6. U6 snRNA positions catalytic metals during branching: controls for the U80-PS(Sp) induced shift in the Cd2+ transition midpoint for rescue of the 3′S-PS(Rp) substrate.
a, The shift induced by U80 in the Cd2+ midpoint for rescue was also observed when reaction rates, instead of amplitudes, were compared. Initial rates are plotted versus CdCl2 concentration. Assays were as in Fig. 3d. Values are averages; error bars represent s.d. (n=2). Initial rates, rather than apparent overall rates, were used here because the branching efficiency did not level off by 120 minutes at Cd2+ concentrations below 0.025mM (assuming an endpoint of ~ 0.4). In support of this approach, at saturating Cd2+ both wild-type and U80-PS(Sp) spliceosomes branched similar fractions of the 3′S-PS(Rp) substrate (see c), indicating that addition of a sulfur in U6 within the catalytic core did not necessarily alter the population of complexes that are competent for catalysis. b, Transcribed (txt.) and ligated (lig.) wild-type U6 behaved similarly relative to U80-PS(Sp) for branching of the 3′S-PS(Rp) substrate at a limiting Cd2+ concentration. Initial rates are shown for branching of the 3′S-PS(Rp) substrate in the presence of 0.01 mM Cd2+. c, A sulfur at U80 pro-Sp shifts the Cd2+ titration midpoint for rescue of the 3′S-PS(Rp) substrate relative to the 3′S-PS(Sp) substrate. Assays were as in Fig. 3d; the 3′S-PS(Rp) data from Fig. 3d are shown again here for comparison. Curves represent Hill fits to the data. Values are averages; error bars, s.d. (n=3). Although the Cd2+ titration for rescue of U80-PS(Sp) spliceosomes assembled on the 3′S-PS(Sp) substrate did not plateau under our experimental conditions (panel c), the data nevertheless set a lower limit for the apparent transition midpoint; further, this midpoint is equal to or greater than that observed for U6 wild-type spliceosomes, indicating that the shift by U80-PS(Sp) of the Cd2+ transition midpoint for rescue was specific for the 3′S-PS(Rp) substrate. d, With the 3′S-PS(Rp) substrate, U80-PS(Sp) decreased the Cd2+ titration midpoint for rescue of branching by 6-fold. The apparent midpoint for G78-PS(Rp) spliceosomes is shown as an additional specificity control for the shift observed for U80-PS(Sp) (actual titration data not shown). The apparent Cd2+ rescue midpoints were obtained by fitting titration curves to the general Hill equation (see Supplementary Methods). Error bars represent error of the Hill fit. e, Representative raw data for Fig. 3d. Assays were in the presence of 0.01 mM Cd2+, where indicated.
Extended Data Figure 7. Further evidence for two distinct catalytic metal sites during branching.
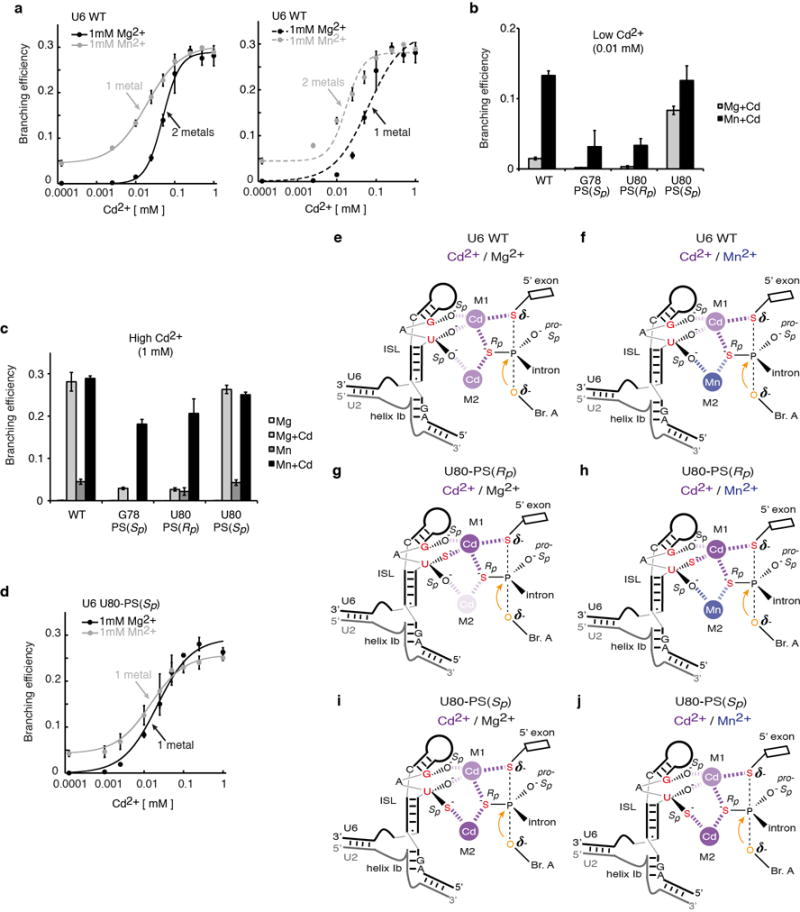
a–d, Branching requires two catalytic divalent metals (a). The 5′ splice site pro-Rp oxygen and the U80 pro-Sp oxygen interact with a metal distinct from the metal that interacts with the 5′ splice site pro-Rp oxygen and the U80 pro-Rp and G78 pro-Sp oxygens (b–d). See Supplementary Note 7 for discussion. Spliceosomes were assayed for as in Fig. 3e; where indicated, MgCl2 or MnCl2 were present at 1mM. Curves represent Hill fits to the data. The data in a are reproduced here from Fig. 3e to aid comparison. In panels a–d, values are averages; error bars, s.d. (n=3). Cd2+ was limiting in b to sensitize the assay to binding of a second metal, and Cd2+ was saturating in c to show that G78-PS(Sp) and U80-PS(Sp) have the potential to be rescued at levels comparable to U6 WT. Panel d shows that U80-PS(Sp) eliminates the affect of Mn2+ on the titration curves. e–j, Metal binding during branching for different combinations of sulfur substitutions in the substrate and U6 in the presence of the indicated metals, as reflected by the data in panels a–d. Panels e and f reflect data in a,b,c; g and h, data in b,c; i and j, data in b,c,d. Relevant U6 ligands are colored red in each panel; the nucleophile is colored orange. Metals are colored magenta (Cd2+) and blue (Mn2+), and their interactions with specific U6 ligands are depicted as dashed lines, with differential shading intensity meant to illustrate differences in the expected strength of interaction with oxygen vs. sulfur, as inferred from studies with model compounds38,61. Shading of metals bound at M1 and M2 is further adjusted to reflect experimental observations. Panels for G78-PS(Sp) would look the same as those for U80-PS(Rp) (g and h).
Extended Data Figure 8. A sulfur at the 3′ splice site, but not 5′ splice site leaving group alters the metal specificity for rescue of U80-PS(Sp) spliceosomes: further evidence that U80 interacts with a catalytic metal during exon ligation.
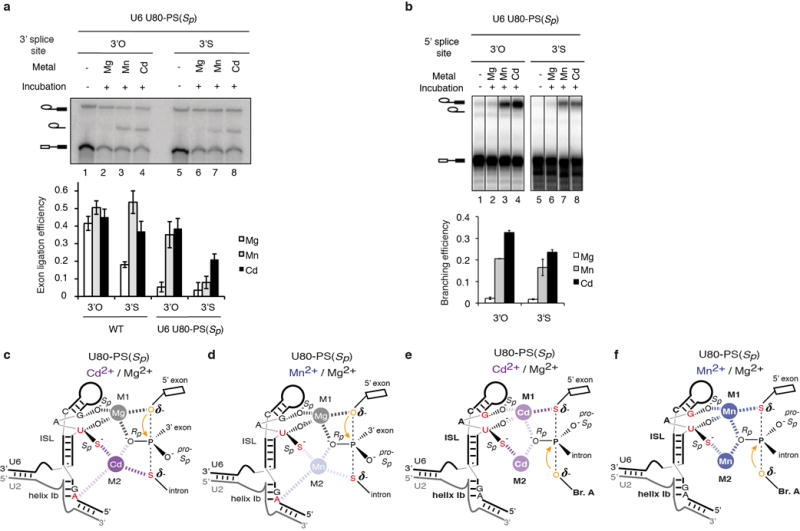
a, A sulfur at the 3′ splice site leaving group during exon ligation alters the metal specificity for rescue of U80-PS(Sp) spliceosomes. Assays were as in Fig. 2j. The bar graph depicts quantification of exon ligation efficiency. b, A sulfur at the 5′ splice site leaving group during branching does not alter the metal specificity for rescue of U80-PS(Sp) spliceosomes. Assays were as in Fig. 3a, except with the 3′S-PO substrate. Values are averages; error bars, s.d (n=3). c–f, Inferred metal binding during exon ligation (c–d) and branching (e–f) for the indicated combinations of sulfur substitutions in the substrate and U6 in the presence of the indicated metals. Relevant U6 ligands are colored red in each panel; the nucleophile is colored orange. Metals and ligand interactions are coloured as in Extended Data Fig. 7.
Extended Data Figure 9. Thiophilic metals rescue exon ligation for substrates containing a sulfur at the 3′ splice site leaving group in both a mutated and wild-type 3′ splice site context.
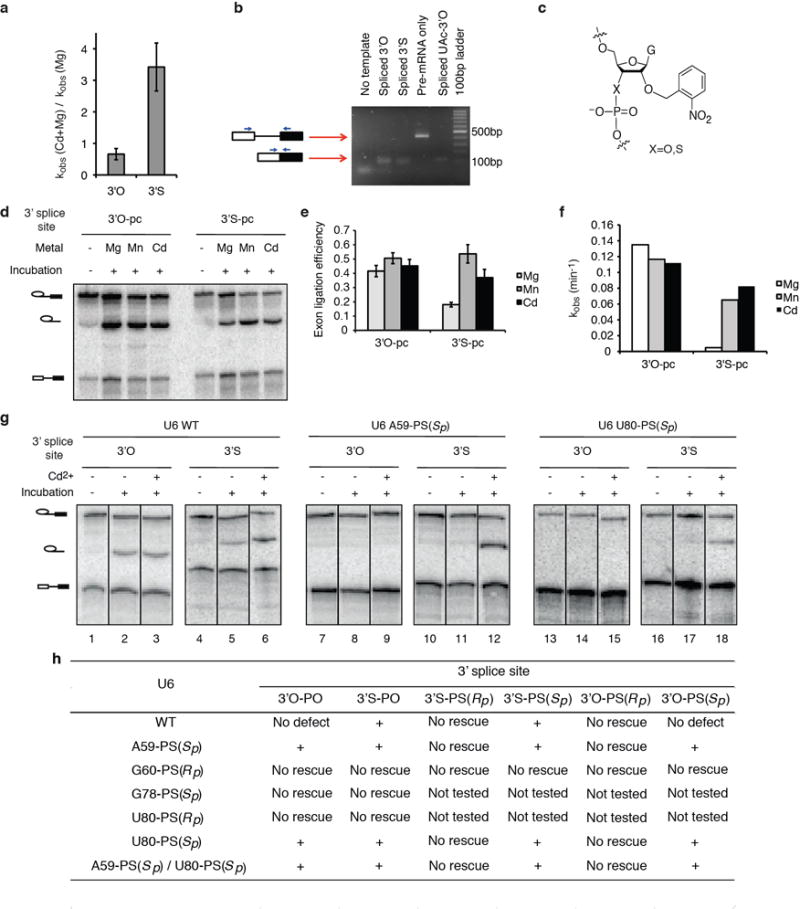
a, Cd2+ specifically stimulates the rate of exon ligation for the mutated, UAc-3′S substrate. Assays were as in Fig. 4a. b, Exon ligation occurs at the correct site for the 3′S substrate. RNA from affinity-purified spliceosomes assembled on ACT1-3′O, ACT1-3′S, and ACT1-UAc-3′O and chased as in Fig. 4a was subjected to RT-PCR using the primers depicted in blue arrows. c, Diagram of the photocaged linkage at the 3′ splice site. d–f, Cd2+ specifically stimulates the rate of exon ligation for the wild-type, UAG-3′S substrate. Assays were as in Fig. 4a, except that before addition of divalent metals, samples were irradiated with 308nm light on ice for five minutes to remove the photocage. Shown are a representative gel (d), quantification of the reaction end points (e) and quantification of reaction rates (f). g, Representative raw data for Fig. 4b; bands within each set came from non-adjacent lanes on the same gel. Values are averages; error bars, s.d. (n=3). h, Summary of combinations of sulfur substitutions in U6 and the substrate tested for rescue of exon ligation. The “+” sign indicates that exon ligation was observed in the presence of thiophilic metal.
Extended Data Figure 10. Residue D1853 of the RNaseH-like domain of Prp8 does not play a direct role in metal-mediated catalysis of exon ligation.
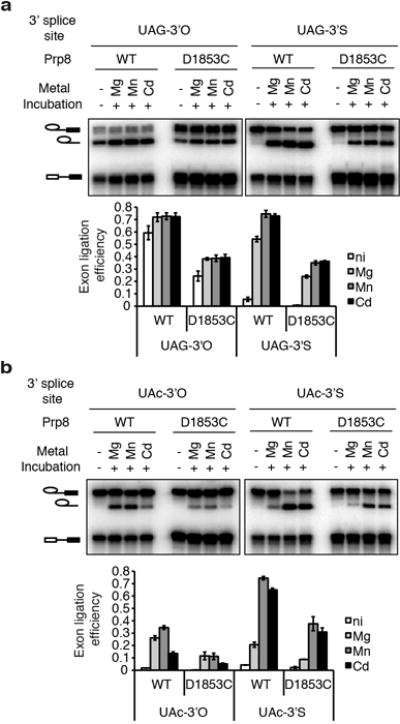
a–b, Spliceosomes assembled on the indicated ACT1 UAG (a) or UAc (b) 3′O or 3′S substrates were assayed as in Fig. 2j, in the absence of ATP. Splicing extracts were prepared from either a wild-type PRP8 strain or a mutant strain having the prp8-D1853C mutation. See Supplementary Note 15 for details and a discussion. Values are averages; error bars, s.d. (n=2); ni, no incubation.
Supplementary Material
Acknowledgments
We thank C. Guthrie for plasmids; S.-C. Cheng for anti-Cwc25p serum; D. Semlow for strains; J. Olvera for reagents and experimental assistance; R.-J. Lin for sharing unpublished data; members of the Staley and Piccirilli labs for critical discussions; and D. Herschlag, A. Macmillan, and T. Nilsen for comments on the manuscript. N.T. was supported by an NSF Graduate Research Fellowship and by a CBI Training Grant (5T32GM008720). This work was funded by a grant from the Chicago Biomedical Consortium, with support from The Searle Funds at the Chicago Community Trust, to J.P.S, A.S. Mankin, and E.J. Sontheimer, and by a grant from the National Institutes of Health (R01GM088656) to J.P.S. and J.A.P.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions S.M.F., N.T., T.N., J.P.S., and J.A.P. designed the study; T.N. and P.K. performed initial screening of U6 sulfur substitutions; S.M.F. performed all experiments related to branching; N.T. performed all experiments related to exon ligation; S.M.F. and N.T. together performed Prp8p experiments; J.L., N.S.L., and Q.D. synthesized RNA oligonucleotides; and S.M.F., N.T., J.P.S., and J.A.P. analyzed the data and wrote the manuscript.
Author Information The authors declare no competing financial interests.
References
- 1.Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Sharp PA. On the origin of RNA splicing and introns. Cell. 1985;42:397–400. doi: 10.1016/0092-8674(85)90092-3. [DOI] [PubMed] [Google Scholar]
- 4.Cech TR. The generality of self-splicing RNA: relationship to nuclear mRNA splicing. Cell. 1986;44:207–210. doi: 10.1016/0092-8674(86)90751-8. [DOI] [PubMed] [Google Scholar]
- 5.Madhani HD, Guthrie C. A novel base-pairing interaction between U2 and U6 snRNAs suggests a mechanism for the catalytic activation of the spliceosome. Cell. 1992;71:803–817. doi: 10.1016/0092-8674(92)90556-r. [DOI] [PubMed] [Google Scholar]
- 6.Shukla GC, Padgett RA. A catalytically active group II intron domain 5 can function in the U12-dependent spliceosome. Mol Cell. 2002;9:1145–1150. doi: 10.1016/s1097-2765(02)00505-1. [DOI] [PubMed] [Google Scholar]
- 7.Hilliker AK, Staley JP. Multiple functions for the invariant AGC triad of U6 snRNA. RNA. 2004;10:921–928. doi: 10.1261/rna.7310704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mefford MA, Staley JP. Evidence that U2/U6 helix I promotes both catalytic steps of pre-mRNA splicing and rearranges in between these steps. RNA. 2009;15:1386–1397. doi: 10.1261/rna.1582609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burke JE, Sashital DG, Zuo X, Wang YX, Butcher SE. Structure of the yeast U2/U6 snRNA complex. RNA. 2012;18:673–683. doi: 10.1261/rna.031138.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun JS, Manley JL. A novel U2-U6 snRNA structure is necessary for mammalian mRNA splicing. Genes Dev. 1995;9:843–854. doi: 10.1101/gad.9.7.843. [DOI] [PubMed] [Google Scholar]
- 11.Chanfreau G, Jacquier A. Catalytic site components common to both splicing steps of a group II intron. Science. 1994;266:1383–1387. doi: 10.1126/science.7973729. [DOI] [PubMed] [Google Scholar]
- 12.Fabrizio P, Abelson J. Thiophosphates in yeast U6 snRNA specifically affect pre-mRNA splicing in vitro. Nucleic Acids Res. 1992;20:3659–3664. doi: 10.1093/nar/20.14.3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu YT, Maroney PA, Darzynkiwicz E, Nilsen TW. U6 snRNA function in nuclear pre-mRNA splicing: a phosphorothioate interference analysis of the U6 phosphate backbone. RNA. 1995;1:46–54. [PMC free article] [PubMed] [Google Scholar]
- 14.Yean SL, Wuenschell G, Termini J, Lin RJ. Metal-ion coordination by U6 small nuclear RNA contributes to catalysis in the spliceosome. Nature. 2000;408:881–884. doi: 10.1038/35048617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gordon PM, Piccirilli JA. Metal ion coordination by the AGC triad in domain 5 contributes to group II intron catalysis. Nat Struct Biol. 2001;8:893–898. doi: 10.1038/nsb1001-893. [DOI] [PubMed] [Google Scholar]
- 16.Boudvillain M, Pyle AM. Defining functional groups, core structural features and inter-domain tertiary contacts essential for group II intron self-splicing: a NAIM analysis. EMBO J. 1998;17:7091–7104. doi: 10.1093/emboj/17.23.7091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galej WP, Oubridge C, Newman AJ, Nagai K. Crystal structure of Prp8 reveals active site cavity of the spliceosome. Nature. 2013;493:638–643. doi: 10.1038/nature11843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valadkhan S, Manley JL. Splicing-related catalysis by protein-free snRNAs. Nature. 2001;413:701–707. doi: 10.1038/35099500. [DOI] [PubMed] [Google Scholar]
- 19.Jaladat Y, Zhang B, Mohammadi A, Valadkhan S. Splicing of an intervening sequence by protein-free human snRNAs. RNA biology. 2011;8:372–377. doi: 10.4161/rna.8.3.15386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith DJ, Konarska MM. A critical assessment of the utility of protein-free splicing systems. RNA. 2009;15:1–3. doi: 10.1261/rna.1322709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valadkhan S, Manley JL. The use of simple model systems to study spliceosomal catalysis. RNA. 2009;15:4–7. doi: 10.1261/rna.1425809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steitz TA, Steitz JA. A general two-metal-ion mechanism for catalytic RNA. Proc Natl Acad Sci USA. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sontheimer EJ, Gordon P, Piccirilli J. Metal ion catalysis during group II intron self-splicing: parallels with the spliceosome. Genes Dev. 1999;13:1729–1741. doi: 10.1101/gad.13.13.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sontheimer EJ, Sun S, Piccirilli JA. Metal ion catalysis during splicing of premessenger RNA. Nature. 1997;388:801–805. doi: 10.1038/42068. [DOI] [PubMed] [Google Scholar]
- 25.Gordon PM, Sontheimer EJ, Piccirilli JA. Metal ion catalysis during the exon-ligation step of nuclear pre-mRNA splicing: extending the parallels between the spliceosome and group II introns. RNA. 2000;6:199–205. doi: 10.1017/s1355838200992069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toor N, Keating KS, Taylor SD, Pyle AM. Crystal structure of a self-spliced group II intron. Science. 2008;320:77–82. doi: 10.1126/science.1153803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marcia M, Pyle AM. Visualizing group II intron catalysis through the stages of splicing. Cell. 2012;151:497–507. doi: 10.1016/j.cell.2012.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koodathingal P, Novak T, Piccirilli JA, Staley JP. The DEAH box ATPases Prp16 and Prp43 cooperate to proofread 5′ splice site cleavage during pre-mRNA splicing. Mol Cell. 2010;39:385–395. doi: 10.1016/j.molcel.2010.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shan SO, Yoshida A, Sun S, Piccirilli JA, Herschlag D. Three metal ions at the active site of the Tetrahymena group I ribozyme. Proc Natl Acad Sci USA. 1999;96:12299–12304. doi: 10.1073/pnas.96.22.12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Forconi M, Lee J, Lee JK, Piccirilli JA, Herschlag D. Functional identification of ligands for a catalytic metal ion in group I introns. Biochemistry. 2008;47:6883–6894. doi: 10.1021/bi800519a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frederiksen JK, Piccirilli JA. Identification of catalytic metal ion ligands in ribozymes. Methods. 2009;49:148–166. doi: 10.1016/j.ymeth.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo F, Gooding AR, Cech TR. Structure of the Tetrahymena ribozyme: base triple sandwich and metal ion at the active site. Mol Cell. 2004;16:351–362. doi: 10.1016/j.molcel.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 33.Burgess SM, Guthrie C. A mechanism to enhance mRNA splicing fidelity: the RNA-dependent ATPase Prp16 governs usage of a discard pathway for aberrant lariat intermediates. Cell. 1993;73:1377–1391. doi: 10.1016/0092-8674(93)90363-u. [DOI] [PubMed] [Google Scholar]
- 34.Mayas RM, Maita H, Staley JP. Exon ligation is proofread by the DExD/H-box ATPase Prp22p. Nat Struct Mol Biol. 2006;13:482–490. doi: 10.1038/nsmb1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Semlow DR, Staley JP. Staying on message: ensuring fidelity in pre-mRNA splicing. Trends in Biochemical Sciences. 2012;37:263–273. doi: 10.1016/j.tibs.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan SP, Kao DI, Tsai WY, Cheng SC. The Prp19p-associated complex in spliceosome activation. Science. 2003;302:279–282. doi: 10.1126/science.1086602. [DOI] [PubMed] [Google Scholar]
- 37.Moore MJ, Sharp PA. Evidence for two active sites in the spliceosome provided by stereochemistry of pre-mRNA splicing. Nature. 1993;365:364–368. doi: 10.1038/365364a0. [DOI] [PubMed] [Google Scholar]
- 38.Pecoraro VL, Hermes JD, Cleland WW. Stability constants of Mg2+ and Cd2+ complexes of adenine nucleotides and thionucleotides and rate constants for formation and dissociation of MgATP and MgADP. Biochemistry. 1984;23:5262–5271. doi: 10.1021/bi00317a026. [DOI] [PubMed] [Google Scholar]
- 39.Fabrizio PP, Abelson JJ. Two domains of yeast U6 small nuclear RNA required for both steps of nuclear precursor messenger RNA splicing. Science. 1990;250:404–409. doi: 10.1126/science.2145630. [DOI] [PubMed] [Google Scholar]
- 40.Anokhina M, et al. RNA structure analysis of human spliceosomes reveals a compact 3D arrangement of snRNAs at the catalytic core. EMBO J. 2013 doi: 10.1038/emboj.2013.198. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cech TR. The chemistry of self-splicing RNA and RNA enzymes. Science. 1987;236:1532–1539. doi: 10.1126/science.2438771. [DOI] [PubMed] [Google Scholar]
- 42.Schellenberg MJ, et al. A conformational switch in PRP8 mediates metal ion coordination that promotes pre-mRNA exon ligation. Nat Struct Mol Biol. 2013;20:728–734. doi: 10.1038/nsmb.2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bonnal S, Valcárcel J. RNAtomy of the spliceosome’s heart. EMBO J. doi: 10.1038/emboj.2013.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nissen P, Hansen J, Ban N, Moore PB, Steitz TA. The structural basis of ribosome activity in peptide bond synthesis. Science. 2000;289:920–930. doi: 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- 45.Beringer M, Rodnina MV. The ribosomal peptidyl transferase. Mol Cell. 2007;26:311–321. doi: 10.1016/j.molcel.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 46.Sharp PA. Five easy pieces. Science. 1991;254:663. doi: 10.1126/science.1948046. [DOI] [PubMed] [Google Scholar]
- 47.Martin W, Koonin EV. Introns and the origin of nucleus-cytosol compartmentalization. Nature. 2006;440:41–45. doi: 10.1038/nature04531. [DOI] [PubMed] [Google Scholar]
- 48.Joyce GF. The antiquity of RNA-based evolution. Nature. 2002;418:214–221. doi: 10.1038/418214a. [DOI] [PubMed] [Google Scholar]
- 49.Moore M, Sharp P. Site-specific modification of pre-mRNA: the 2′-hydroxyl groups at the splice sites. Science. 1992;256:992–997. doi: 10.1126/science.1589782. [DOI] [PubMed] [Google Scholar]
- 50.Yoshida A, Sun S, Piccirilli JA. A new metal ion interaction in the Tetrahymena ribozyme reaction revealed by double sulfur substitution. Nat Struct Biol. 1999;6:318–321. doi: 10.1038/7551. [DOI] [PubMed] [Google Scholar]
- 51.Tagwerker C, et al. HB tag modules for PCR-based gene tagging and tandem affinity purification in Saccharomyces cerevisiae. Yeast. 2006;23:623–632. doi: 10.1002/yea.1380. [DOI] [PubMed] [Google Scholar]
- 52.Umen JG, Guthrie C. A novel role for a U5 snRNP protein in 3′ splice site selection. Genes Dev. 1995;9:855–868. doi: 10.1101/gad.9.7.855. [DOI] [PubMed] [Google Scholar]
- 53.Abelson J, Hadjivassiliou H, Guthrie C. Preparation of fluorescent pre-mRNA substrates for an smFRET study of pre-mRNA splicing in yeast. Meth Enzymol. 2010;472:31–40. doi: 10.1016/S0076-6879(10)72017-6. [DOI] [PubMed] [Google Scholar]
- 54.Frederiksen JK, Piccirilli JA. Separation of RNA phosphorothioate oligonucleotides by HPLC. Meth Enzymol. 2009;468:289–309. doi: 10.1016/S0076-6879(09)68014-9. [DOI] [PubMed] [Google Scholar]
- 55.Loverix S, Winqvist A, Strömberg R, Steyaert J. Mechanism of RNase T1: concerted triester-like phosphoryl transfer via a catalytic three-centered hydrogen bond. Chem Biol. 2000;7:651–658. doi: 10.1016/s1074-5521(00)00005-3. [DOI] [PubMed] [Google Scholar]
- 56.Schürer H, Lang K, Schuster J, Mörl M. A universal method to produce in vitro transcripts with homogeneous 3′ ends. Nucleic Acids Res. 2002;30:56e–56. doi: 10.1093/nar/gnf055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dery KJ, Yean SL, Lin RJ. Assembly and glycerol gradient isolation of yeast spliceosomes containing transcribed or synthetic U6 snRNA. Methods Mol Biol. 2008;488:41–63. doi: 10.1007/978-1-60327-475-3_4. [DOI] [PubMed] [Google Scholar]
- 58.Fabrizio P, McPheeters DS, Abelson J. In vitro assembly of yeast U6 snRNP: a functional assay. Genes Dev. 1989;3:2137–2150. doi: 10.1101/gad.3.12b.2137. [DOI] [PubMed] [Google Scholar]
- 59.Chiu YF, et al. Cwc25 is a novel splicing factor required after Prp2 and Yju2 to facilitate the first catalytic reaction. Mol Cell Biol. 2009;29:5671–5678. doi: 10.1128/MCB.00773-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Warkocki Z, et al. Reconstitution of both steps of Saccharomyces cerevisiae splicing with purified spliceosomal components. Nat Struct Mol Biol. 2009;16:1237–1243. doi: 10.1038/nsmb.1729. [DOI] [PubMed] [Google Scholar]
- 61.Sigel RKO, Song B, Sigel H. Stabilities and structures of metal ion complexes of adenosine 5′- O-thiomonophosphate (AMPS2−) in comparison with those of its parent nucleotide (AMP2−) in aqueous solution. J Am Chem Soc. 1997;119:744–755. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.