

Abstract
We report herein the use of Medicago sativa alfalfa shoot biomass for the removal of gold from aqueous solutions. The accumulation process involves the reduction of Au(III) to colloidal Au(0) and is shown to increase at elevated temperatures and at lower pH. X-ray absorption spectroscopy (XAS) was used to determine that gold(III) was reduced to form gold(0) colloids, which varied in size depending on the pH of the initial solution. The gold cluster radius was 6.2 ± 1 Å at pH 5 and 9.0 ± 1 Å at pH 2. Our findings indicate that essentially another layer of gold atoms was deposited onto the colloid surface at pH 2. Possible mechanisms of bioreduction and accumulation are discussed.
Introduction
Current technology for the recovery of gold from ores involves the use of chemical methods, such as cyanidation and thiourea leaching, which present environmental and health risks (1–4). Increasing demands for gold, because of its vast industrial applications, has inspired the development of novel methods for gold recovery from solution that are both environmentally friendly and less threatening to the population (5).
Sequestering metal ions using living or nonliving plants is a cost-effective means of removing waste metals via intracellular accumulation and/or surface adsorption (6–13). However, the use of living plants, phytoremediation, is often a slow and time-consuming process. The use of nonliving plant materials for surface adsorption of metal contaminants, phytofiltration, offers several advantages over living plants (14). The plants cell walls have a variety of functional groups that can bind and transform metals during uptake from waste streams; they are cost-effective, easily regenerated, and depending on the plant used, have different metal specificities (14). The mechanism of adsorption of metalions to nonliving biomass materials is believed to occur at the cell wall via ion exchange, complexation, electrostatic binding, and/or precipitation (15).
Gold and colloidal gold have been extensively studied as therapeutic agents, nanoparticles that possess novel catalytic, optical, magnetic, and electronic properties, and as labels for X-ray crystallography and electron microscopy (16, 17). The accumulation of precious metals by plants has been studied since the 1900s for prospecting purposes (18, 19). We have shown that alfalfa biomass is able to adsorb a variety of heavy metals from aqueous solutions (9, 20). Biosorption of precious metals from aqueous solutions using algae and higher plant biomass materials has applications in the recovery of trace metals from mining effluents and are starting to appear in the literature (6, 21–28). The accumulation of gold using microbacteria such as bacteria, algae, yeast, and fungi was recently reviewed (29). Some general observations on gold biosorption are as follows: (i) Au(III) is initially reduced to Au(I) followed by a sometimes slower reduction to Au(0); (ii) the effect of pH on Au(III) accumulation depends on the microorganism, species, and mechanism of uptake; and (iii) multiple binding sites and adsorption mechanisms have been proposed for Au(III) accumulation. Both algae and alfalfa biomass samples turn purple (similar to the purple of Cassius) upon exposure to Au(III), characteristic of the formation of colloidal Au(0) (22, 23). In previous experiments, we noted that alfalfa biomass accumulated appreciable amounts of gold(III) ions (40 mg of gold/g of alfalfa) from aqueous solutions and that the biomass turned purple, indicating the formation of colloidal Au(0) (23). Transmission electron microscopy (TEM) experiments confirmed the formation of colloidal Au(0) (24). This process is rapid, nearly pH independent (pH 2–6), and 87% of the gold was recovered upon regeneration (23). Algae biomass was also shown to have a similar high affinity for gold(I) and gold(III) in aqueous solutions (22, 25). XAS studies determined that gold(III) was reduced to gold(I) by the algae and that binding occurred via a ligand exchange mechanism involving sulfur or nitrogen ligands on the algae (25). A large temperature dependence on Au(III) binding by Spirulina was noted in which gold(0) colloids were produced at 55 °C as evidenced by the purple color of the algae (6). Niu et al. have shown that the main mechanism of gold biosorption by certain algal biomass involved electrostatic interactions between cationic functional groups of the biomass and anionic Au(I) (30).
The objectives of this work are to further characterize the mechanism(s) of gold(III) biosorption and reduction by nonliving Medicago sativa alfalfa shoot biomass. The effects of temperature and pH on Au(III) accumulation were studied via batch experiments. X-ray absorption spectroscopy (XAS) was used to characterize the gold colloids and determine the effect of pH on the size of the colloids. Possible mechanism(s) of gold(III) bioreduction and biosorption are discussed herein.
Experimental Methods
Sample Collection
The Malone population of Medicago sativa (alfalfa shoots) samples were acquired and prepared using previously published procedures (23). The shoots were oven dried at 90 °C for 1 week and ground with a Wiley mill to pass a 100-mesh screen.
Temperature-Dependent Studies
The alfalfa biomass (300 mg) was washed twice with 0.01 M HCl and once with deionized water to remove any soluble biomolecules. Aqueous biomass suspensions (5 g/mL) were prepared, and the solution pH was adjusted to 5.0 using 0.01 M NaOH or to 2.0 using 0.01 M HCl. The samples were centrifuged at 3000 rpm for 5 min, and the supernatants were removed. A 0.3 mM gold(III) solution was prepared using K(Au(III)Cl4), and the pH was adjusted to either 5.0 or 2.0. The biomass and gold(III) solutions were separately equilibrated for 1 h prior to mixing. The samples were equilibrated on a rocker at their respective temperatures for an additional hour. The samples were kept in a refrigerator (experiment at 4 °C) and in a temperature controlled-oven (experiment at 55 °C). The samples were then centrifuged, and the amount of gold in the supernatants was determined using flame atomic absorption spectroscopy as previously described (23). The final solution pH values were unchanged during the experiments (data not shown). Three samples per temperature (4, 25, and 55 °C) were measured to obtain better statistics.
X-ray Absorption Spectroscopy
The X-ray absorption spectra were measured at room temperature at beam line X-18B at the National Synchrotron Light Source. Data were collected with Si 111 monochromator crystals with slits adjusted to give ~1–2 eV resolution. All of the samples were measured in transmission mode using standard ion detectors at the Au LIII edge. The K(Au(III)Cl4) standard was measured as a solid on tape. Silica-immobilized biomass samples were prepared using previously published procedures (23). A 100-mg sample of immobilized Malone shoots was reacted with 30 mL of a 1000 ppm gold(III) solution [as K(Au(III)Cl4)] at pH 2 and pH 5. The Au biomass samples were washed with distilled water prior to use and were measured as solid powders in 5 × 15 × 1 mm cells with tape windows. The absolute energy positions were calibrated against the inflection point energy for Au foil, 11921 eV. The data were analyzed with the extended X-ray absorption fine structure (EXAFS) analysis package using standard methods (31–33). The E0 values were determined from the absorption edge inflection point. Quantitative comparison between unknown and standards was accomplished with nonlinear fits based on the general EXAFS equation and verified with theoretical simulations carried out with FEFF 3.11 an ab initio curved-wave single scattering EXAFS simulation code (34, 35).
A general model developed by Borowski to determine the size of small Cu clusters using a spherical cluster model is shown in eq 1 (36). This method was useful in determining the size of small clusters [radius (R) < 25 Å] and was used in these studies to determine the size of the Au colloids formed:
| (1) |
where the radius of the cluster is R, nearest neighbor distance is di, the reduced cluster size is ρ ≡ di/R, and the average number of neighbors is .
Equation 1 can be rewritten as
This is rearranged to
This model should also apply to gold(0) clusters since they are also face-centered-cubic (fcc) structures (24). The values for and di are obtained from the nonlinear least-squares fitting results for the isolated first shell EXAFS oscillations, and the cluster size was determined graphically by plotting F(R) versus R. An estimate of the uncertainty in the cluster size determination resulting from the uncertainties in N and d was generated via a Monte Carlo procedure. A large number of trial values of N and d were generated with a Gaussian noise model with mean and standard deviation as given by the experimental results and the effect on R analyzed.
Results and Discussion
Gold(III) removal from aqueous solutions by alfalfa biomass was nearly independent of pH, although there was a slight increase in uptake at pH 2 (23). The effect of temperature on Au(III) removal is shown in Figure 1 at pH 2 and pH 5. There is an increase in Au removal with increased temperature and at lower pH. The current experiments were performed at higher initial Au(III) concentrations (0.3 vs 0.1 mM) than in prior studies (23). At low Au(III) concentrations the biomass is effective at bioreduction and recovery of gold ions, whereas at higher concentrations other factors must be present. The pH dependence on Au(III) removal is sensitive to the initial Au(III) concentration, possibly due to saturation of the biomass metal ion binding or reduction sites. The pH dependence could mean that a combination of ligands with different pKa values are involved in the uptake of gold ions by alfalfa as they would have varied affinities and reduction capacities depending on pH. Possible mechanisms of gold(III) accumulation that take into account the pH dependence are discussed below.
FIGURE 1.
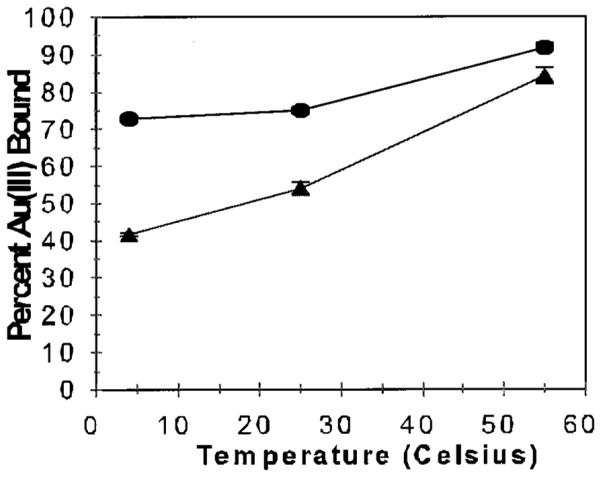
Effect of temperature on Au(III) accumulation by alfalfa biomass at pH 2.0 (•) and pH 5.0 (▲). Confidence interval is 95%, as represented by the error bars.
XAS was used to investigate the nature of the gold(III) accumulation by alfalfa biomass shoots as a function of pH. The samples used in the XAS experiments were silica immobilized biomass, which does not alter the biomass adsorption chemistry (23). The X-ray absorption near edge structure (XANES) provides information about the metals’ oxidation state, coordination number, and geometry; the extended X-ray absorption fine structure (EXAFS), which is produced by the backscattering of photoelectrons from nearest neighbor atoms, provides structural information for the metal coordination environment (31, 32). XAS techniques have proven instrumental in determining the metal adsorption environment and oxidation state along with possible mechanisms of heavy metal remediation by biological tissues (12, 20, 25, 37–40). The XANES spectra of silica-immobilized alfalfa reacted with Au(III) solution at pH 2.0 and pH 5.0, gold foil, and KAu(III)Cl4 are shown in Figure 2.
FIGURE 2.
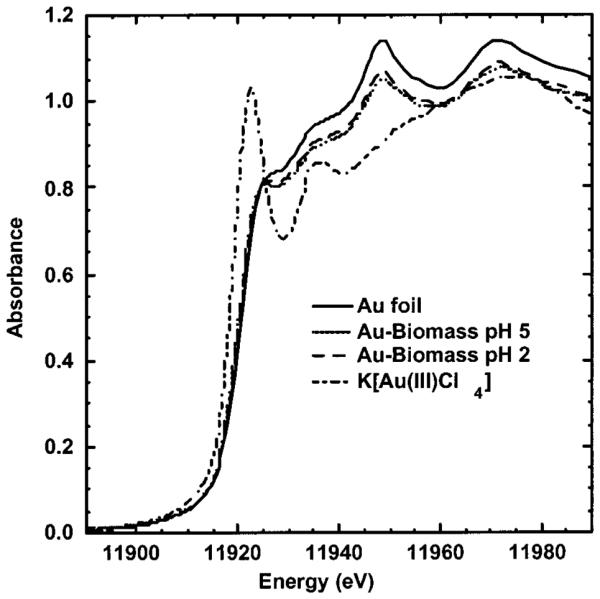
XANES spectra for Au foil, Au biomass at pH 2.0 and pH 5.0, and K[Au(III)Cl4].
The gold(III) model compound [K(Au(III)Cl4)] absorption edge (~11917 eV) shifts to lower energy as compared to the Au(0) foil, in contrast with the usual pattern of positively charged ions having higher edge energies than their neutral parent species. This unusual edge shift for Au(III) results from a low-energy dipole allowed 2p to 5d transition, which is allowed for the d8 species and forbidden for d10 Au(0) and Au(I) (25). The inflection points (11921 eV) for both of the Au biomass samples are identical, within experimental error, to the Au foil, thus indicating that the Au(III) is reduced to Au(0). The Au biomass samples also have XANES maxima near 11945 and 11967 eV, which are also diagnostic of Au(0) (41). This confirms that the Au(III) reacted with the alfalfa biomass at both pH values was reduced to Au(0) and that there is < 10% Au(III) and/or Au(I) remaining on the biomass.
Since the Au(III) is reduced to Au(0), analysis of the isolated first shell EXAFS oscillations can be used to determine the size of the Au(0) particles. A general method developed by Borowski (see Experimental Methods) has been used to determine the size of small Cu clusters using a spherical cluster model (36). This method, based on the coordinative unsaturation of atoms on the surface of nanoclusters, is useful in determining the size of small clusters [radii (R) < 25 Å] and was applied to determine the size of the gold colloids. The average number of neighbors, , and the nearest neighbor distance, di, were determined from nonlinear least-squares fitting results for the isolated first shell EXAFS oscillations, see Table 1. The k3 weighted EXAFS oscillations and FT-EXAFS data for Au foil and the Au(III) exposed to the alfalfa biomass are shown Figures 3 and 4, respectively. The radius of the Au(0) cluster, R, was determined graphically by plotting F(R) versus R (see Figure 5). The Au(0) cluster size was dependent on the pH of the solution, 9.0 ± 1 Å at pH 2 and 6.2 ± 1 Å at pH 5. The difference in the colloidal gold radius at pH 2 vs pH 5 is approximately equal to another layer of gold atoms deposited on the surface.
TABLE 1.
EXAFS Fitting Results for Au Foil and Au Biomass Samplesa
| compound |
d ± 0.02 (Å) |
N ± 0.25 | Δσ2 ± 0.002 (Å2) |
χ2
(gof) |
r ± 1 (Å) |
|---|---|---|---|---|---|
| Au foil | 2.85 | 11.5 | 0.008 | 0.015 | |
| Au biomass, pH 2 | 2.85 | 9.19 | 0.007 | 0.053 | 9.0 |
| Au biomass, pH 5 | 2.86 | 7.91 | 0.007 | 0.093 | 6.2 |
N is the coordination number per metal. The metal-coordination atom distance (d ± 0.02 Å) is determined from fits of the EXAFS data (using FEFF 3.11-generated standards). Δσ2 is the relative mean square deviation in d(the square of the Debye–Waller factor), Δσ2 = Δσ2unknown – Δσ2standard. Δχ2gof is a relative goodness-of-fit statistic, defined as σ(exp - fit)2/σexp2. The value of r is the estimated radius of the cluster size. Experimental details: k range ~3.3–12, Hanning window 0.5 Å−1; weighting k3, d window ~0.8–2.2Å for first shell fits, Hanning windows ~0.1 Å.
FIGURE 3.
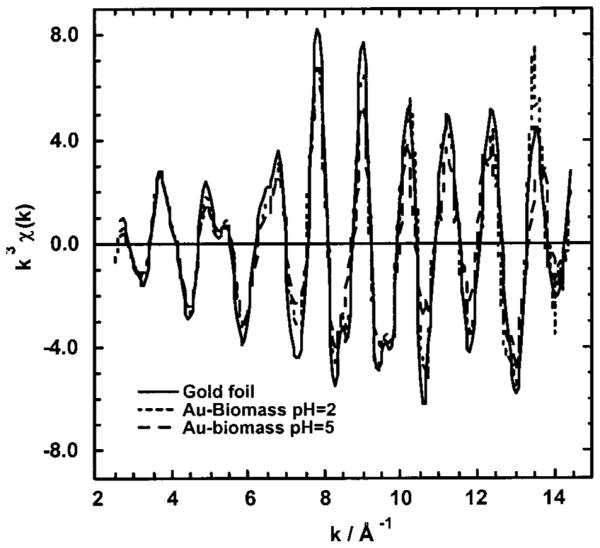
Isolated k3 EXAFS oscillations for Au foil and Au biomass at pH 2.0 and pH 5.0.
FIGURE 4.
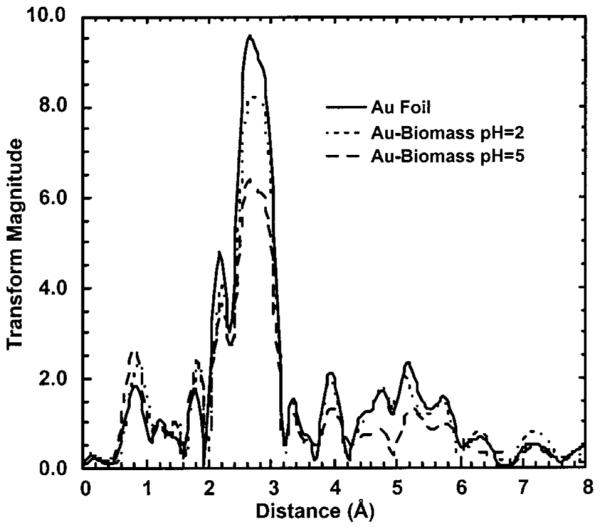
Fourier transform EXAFS for Au foil and Au biomass at pH 2.0 and pH 5.0.
FIGURE 5.
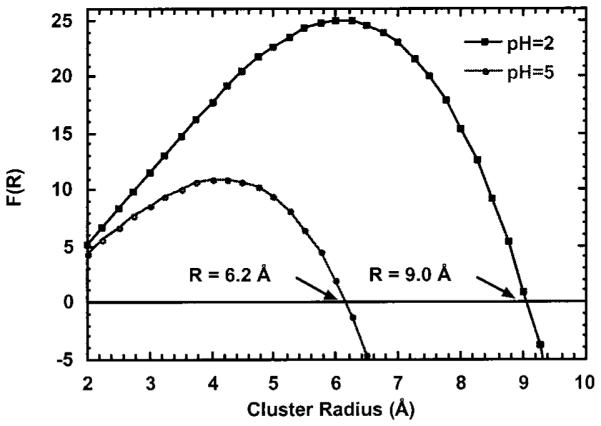
Plot of F(R) versus R, cluster radius (Å), for the Au colloids formed by reacting Malone alfalfa shoot biomass with Au(III) at pH 2.0 and pH 5.0.
The exact mechanism of Au(III) reduction and accumulation by the biomass is unknown but must incorporate the aqueous chemistry of gold ions and colloids into the mechanism. Gold(III) (AuCl4−) is a powerful oxidizing agent and is reduced to Au(0) (42). Since Au(I) and Au(III) are both considered soft metal ions, interactions with soft sulfur or nitrogen ligands are more likely to occur. The protein residues cysteine and methionine are capable of reducing Au(III) to Au(I), and even disulfide reduces Au(III) in vivo (43–46). AuIIICl4− will oxidize methionine to sulfoxide along with the formation of Au(0) (43). For aqueous AuIIICl4− solutions, the rate of hydrolysis of the chlorides depends on both the pH and the chloride concentrations. At pH 2 and high chloride concentrations, the chloride ions do not dissociate, whereas at higher pH and chloride concentrations, the chloride ions are replaced by water (47, 48). At pH 5, hydrolysis of one of the chlorides on AuCl4− affords a neutral AuCl3(H2O) species. Depending on which species is present in solution, different mechanisms can be envisioned. If AuIIICl4− is reduced to AuICl2−, this compound is unstable in water and disproportionates to form Au(0) and Au(III) (49). The stability of both Au(III) and Au(I) are sensitive to the identity of the ligands. Cyanide and sulfur ligands tend to stabilize Au(I) in water, whereas nitrogen ligands stabilize Au(III) (49). If the initial step of the reaction involved covalent Au(III) biomass binding, the nature of the ligands could alter the stability of the Au(III) complex toward reduction and further reaction. Also, the reaction mechanism depends on whether the Au(I) is bound to the biomass or free in solution. In short, the solution pH, which can alter both the biomass surface charge and the gold’s charge, will be instrumental in determining the mode of reaction.
The chemistry of gold colloids has been extensively studied, and in order to better understand the mechanism(s) of colloidal biomass uptake, the chemistry of colloids must be considered (17). There are numerous papers dealing with theoretical models for adsorption of colloids on surfaces and the experimental factors, such as pH, ionic strength, concentration, and immersion time, that influence surface coverage (17, 50, 51). Thiols are commonly used to reduce Au(III) to gold particles, and the gold colloid size can be controlled by the strength of the reducing agent (17). The isoelectric point of gold colloids is about 2, meaning they are negatively charged due to adsorption of anions in the pH range studied due to a layer of AuICl2−, Cl−, or Au(OH)2− around the gold(0) (52). At pH 2, the amine residues are protonated (positively charged) and can lead to electrostatic interaction with the negatively charged gold colloid, which would explain the slight pH dependence for gold accumulation by the biomass. Photochemical reduction of Au(III) to Au colloids should also be considered since we have previously shown that the biomass did not turn as intensively purple upon exclusion of light from the reaction mixture (23). This reduction process is generally a slower process and results in smaller colloids (53).
Therefore, the mechanism of Au(III) reduction most likely involves oxidation of biomass functional groups, such as cysteine or methionine. Time-dependent XAS experiments, at shorten time periods, might allow detection of intermediates in this reduction process. The pH and temperature dependence of Au(III) accumulation tends to indicate that more than one mechanism is involved and that the chemistry of colloidal gold must be considered. At pH 2, gold colloids are negatively charged, as are AuCl2− and AuCl3− ions that are present in solution, while the biomass is positively charged due to the protonation of the amine residues. The interaction is thus electrostatic, involving ion pairing (RN–H+⋯A−). At pH 5, the Au(III) can exist as either AuCl4− or AuCl3(H2O), which will interact differently with the biomass leading to different mechanisms of reduction. Since Au(III) is a strong oxidizing agent, the available biomass residues will depend on the pH of the solution. The X-ray absorption spectroscopic studies revealed that Au(III) is reduced to Au(0), and at long time periods, there was no evidence for either Au(III) or Au(I) bound to the biomass. XAS spectral analysis was used to determine the size of the gold colloids, which were smaller at pH 5.0 than at pH 2.0, corroborating the changes in gold(III) adsorption seen in the temperature studies. At pH 2, the colloidal gold radius indicates that another layer of gold atoms is deposited onto the surface. Experiments involving chemically blocking the biomass functional groups and time-dependent XAS are planned to determine how these variables may affect the Au(III) reduction and accumulation. Alfalfa biomass has potential as a phytofilter for the reduction and recuperation of gold ions from the mineral refining industry and mining effluents through “green” technologies.
Acknowledgments
The authors acknowledge the financial support from the Center for Environmental Resource Management (CERM) at the University of Texas at El Paso through funding from the HBCU/OMI Environmental Technology Consortium, which is funded by the Department of Energy and the Office of Exploratory Research of the U.S. Environmental Protection Agency (Cooperative Agreement CR-819849-01-4). J.L.G.-T. acknowledges the financial support from the National Institutes of Health (Grant GM08012-25) and the BEST Program, Department of Defense, Army Corps of Engineers. The work performed at Brookhaven National Laboratory, National Synchrotron Light Source, was supported by the Department of Energy (Contract DE- ACO2-76CH00016).
Literature Cited
- (1).Deschenes G. Hydrometallurgy. 1989;20:180. [Google Scholar]
- (2).Addison R. Am. Min. Congr. J. 1980:47. [Google Scholar]
- (3).Cho EH, Pitt CH. The Adsorption of Gold and Silver Cyanide from Solution by Activated Charcoal, Gold, Silver, Uranium and Coal Geology, Mining, Extraction and the Environment. The American Institute of Mining, Metallurgical and Petroleum Engineers, Inc.; New York: 1983. pp. 114–133. [Google Scholar]
- (4).Carson BL, Ellis HV, McCann JL. Toxicology and Biological Monitoring of Metals in Humans. Lewis Publishers; Chelsea, MI: 1986. pp. 107–111. [Google Scholar]
- (5).Lucas JM. Gold Mineral Facts and Problems; Bureau of Mines Preprint from Bulletin 675. U.S. Department of the Interior; Washington, DC: 1985. pp. 1–16. [Google Scholar]
- (6).Lujan JR, Darnall DW, Stark PC, Rayson GD, Gardea-Torresdey JL. Solvent Extr. Ion Exch. 1994;12:803. [Google Scholar]
- (7).Gardea-Torresdey JL, Tiemann KJ, Gonzales JH, Henning JA, Towsend MS. J. Hazard. Mater. 1996;48:181. [Google Scholar]
- (8).Tiemann KJ, Gardea-Torresdey JL, Gamez G, Dokken K, Canno-Agluilera, Renner MW, Furenlid LR. Environ. Sci. Technol. 2000;34:693. doi: 10.1021/es991325m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gardea-Torresdey JL, Tiemann KJ, Gonzales JH, Henning JA, Towsend MS. Solvent Extr. Ion Exch. 1996;14:119. [Google Scholar]
- (10).Gardea-Torresdey JL, Tiemann KJ, Gonzales JH, Henning JA, Towsend MS, Canno-Aguilera I. J. Hazard. Mater. 1996;49:205. [Google Scholar]
- (11).Schnoor JL, Licht LA, McCutcheon SC, Wolfe NF, Carreira LH. Environ. Sci. Technol. 1995;29:318A. doi: 10.1021/es00007a747. [DOI] [PubMed] [Google Scholar]
- (12).Salt DE, Prince RE, Baker AJ, Raskin I, Pickering I. Environ. Sci. Technol. 1999;33:713. [Google Scholar]
- (13).Salt DE, Smith RD, Raskin I. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1998;49:643. doi: 10.1146/annurev.arplant.49.1.643. [DOI] [PubMed] [Google Scholar]
- (14).Drake LR, Rayson GD. Anal. Chem. 1996;68:22A. doi: 10.1021/ac960569b. [DOI] [PubMed] [Google Scholar]
- (15).Volesky B, Holan ZR. Biotechnol. Prog. 1995;11:235. doi: 10.1021/bp00033a001. [DOI] [PubMed] [Google Scholar]
- (16).Shaw CF. Chem. Rev. 1999;99:2589. [Google Scholar]
- (17).Colloidal Gold . In: Principles, Methods and Applications. Hayat MA, editor. Academic; San Diego: 1989. [Google Scholar]
- (18).Girling CA, Peterson PJ. Gold Bull. 1980;13:151. [Google Scholar]
- (19).Rapson WS. Gold Bull. 1982;15:19. [Google Scholar]
- (20).Tiemann KJ, Gardea-Torresdey JL, Gamez G, Dokken K, Sias S, Renner MW, Furenlid LR. Environ. Sci. Technol. 1999;33:150. doi: 10.1021/es9804722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Nakajima A, Sakaguchi T. J. Chem. Technol. Biotechnol. 1993;57:321. doi: 10.1002/jctb.280570405. [DOI] [PubMed] [Google Scholar]
- (22).Greene B, Hosea M, McPherson R, Henzl M, Alexander MD, Darnall DW. Environ. Sci. Technol. 1986;20:627. doi: 10.1021/es00148a014. [DOI] [PubMed] [Google Scholar]
- (23).Gardea-Torresdey JL, Tiemann KJ, Gamez G, Dokken K, Pingitore NE. Adv. Environ. Res. 1999;3:83. [Google Scholar]
- (24).Gardea-Torresdey JL, Tiemann KJ, Gamez G, Dokken K, Tehuacanero S, Jose-Yacaman M. J. Nanopart. Res. 1999;1:397. [Google Scholar]
- (25).Watkins JW, Elder RC, Greene B, Darnall DW. Inorg. Chem. 1987;26:1147. [Google Scholar]
- (26).Kuyucak N, Volesky B. Biorecovery. 1989;1:189. [Google Scholar]
- (27).Kuyucak N, Volesky B. Biorecovery. 1989;1:205. [Google Scholar]
- (28).Kuyucak N, Volesky B. Biorecovery. 1989;1:218. [Google Scholar]
- (29).Savaidis I, Karamushka VI, Lee H, Trevors JT. Biometals. 1998;11:69. [Google Scholar]
- (30).Niu H, Volesky B. J. Chem. Technol. Biotechnol. 1999;74:778. [Google Scholar]
- (31).Stern EA, Heald SM. In: Handbook on Synchrotron Radiation. Koch EE, editor. Springer-Verlag; Amsterdam: 1983. [Google Scholar]
- (32).Teo BK. EXAFS: Basic Principles and Data Analysis. Springer-Verlag; Berlin: 1986. [Google Scholar]
- (33).Bouldin CE, Elam WT, Furenlid LR. Physica B. 1995:739. 208&209. [Google Scholar]
- (34).Rehr JJ, Balci M, Pramod K, Koch P, Lex J, Ermer O. J. Am. Chem. Soc. 1991;113:5135. [Google Scholar]
- (35).Mustre de Leon J, Rehr JJ, Zabinsky SI. Phys. Rev. B. 1991;44:4146. doi: 10.1103/physrevb.44.4146. [DOI] [PubMed] [Google Scholar]
- (36).Borowski M. J. Phys. IV. 1997;7:C259. [Google Scholar]
- (37).Lytle MC, Lytle FW, Yang N, Qian J, Hansen D, Zayed A, Terry N. Environ. Sci. Technol. 1998;32:3087. [Google Scholar]
- (38).Gadd GM. In: Microbial control of heavy metal pollution in Microbial Mineral Recovery. Gadd GM, Jones CW, Watson Craik I, editors. Cambridge University Press; Cambridge: 1992. pp. 59–88. [Google Scholar]
- (39).Kelley C, Mielke R, Dimaquibo D, Curtis AJ, DeWitt JG. Environ. Sci. Technol. 1999;33:1439. [Google Scholar]
- (40).Mukerji I, Andrews JC, DeRose VJ, Latimer MJ, Yachandra VK, Sauer K, Klein MP. Biochemistry. 1994;33:9712. doi: 10.1021/bi00198a042. [DOI] [PubMed] [Google Scholar]
- (41).Shaw CF, Schaeffer NA, Elder RC, Eidsness MK, Trooster JM, Calis GHM. J. Am. Chem. Soc. 1984;106:3511. [Google Scholar]
- (42).Moodley KG, Nicol MJ. J. Chem. Soc., Dalton Trans. 1977:993. [Google Scholar]
- (43).Isab AA, Sadler PJ. Biochim. Biophys. Acta. 1997;492:322. doi: 10.1016/0005-2795(77)90083-6. [DOI] [PubMed] [Google Scholar]
- (44).Shaw CF, Cancro MP, Witkiewicz PL, Eldridge MK. Inorg. Chem. 1980;19:3198. [Google Scholar]
- (45).Saito KM. Res. Commun. Mol. Pathol. Pharmacol. 1996;1:101. [PubMed] [Google Scholar]
- (46).Witkiewicz PL, Chaw CF. J. Chem. Soc. Chem. Commun. 1981:1111. [Google Scholar]
- (47).Canovese L, Cattolini L, Maranoni G. J. Chem. Soc., Dalton Trans. 1985:731. [Google Scholar]
- (48).Calamai P, Carotti S, Guerri A, Messori L, Mini E, Orioli P, Speroni GP. J. Inorg. Biochem. 1997;66:103. doi: 10.1016/s0162-0134(96)00190-0. [DOI] [PubMed] [Google Scholar]
- (49).Sadler PJ. Struct. Bonding. 1976;29:171. [Google Scholar]
- (50).Johnson PR, Elimelech M. Langmuir. 1995;11:801. [Google Scholar]
- (51).Zhu T, Fu X, Mu T, Wang J, Liu Z. Langmuir. 1999;15:5197. [Google Scholar]
- (52).Thompson DW, Collins IR. J. Colloid Interface Sci. 1991;152:197. [Google Scholar]
- (53).Mayer ABR, Mark JE. Eur. Polym. J. 1998;34:103. [Google Scholar]