

Abstract
We performed fine-mapping of 39 established type 2 diabetes (T2D) loci in 27,206 cases and 57,574 controls of European ancestry. We identified 49 distinct association signals at these loci, including five mapping in/near KCNQ1. “Credible sets” of variants most likely to drive each distinct signal mapped predominantly to non-coding sequence, implying that T2D association is mediated through gene regulation. Credible set variants were enriched for overlap with FOXA2 chromatin immunoprecipitation binding sites in human islet and liver cells, including at MTNR1B, where fine-mapping implicated rs10830963 as driving T2D association. We confirmed that this T2D-risk allele increases FOXA2-bound enhancer activity in islet- and liver-derived cells. We observed allele-specific differences in NEUROD1 binding in islet-derived cells, consistent with evidence that the T2D-risk allele increases islet MTNR1B expression. Our study demonstrates how integration of genetic and genomic information can define molecular mechanisms through which variants underlying association signals exert their effects on disease.
INTRODUCTION
Genome-wide association studies (GWAS) of common variants, defined by minor allele frequency (MAF) ≥5%, have been successful in identifying loci contributing to type 2 diabetes (T2D) susceptibility1–5. GWAS loci are typically represented by a “lead” SNP with the strongest signal of association in the region. However, lead SNPs may not directly impact disease susceptibility, but instead be proxies for causal variants because of linkage disequilibrium (LD). Interpretation may be further complicated by the presence of more than one causal variant at a locus, possibly acting through the joint effects of alleles on the same haplotype. This complex genetic architecture would result in multiple “distinct” association signals at the same locus, which can only be delineated, statistically, through conditional analyses.
With the exception loci where the lead SNPs are protein altering variants, including PPARG6, KCNJ11-ABCC87, SLC30A88, and GCKR9, the mechanisms by which associated alleles influence T2D susceptibility are largely unknown. At other loci, direct biological interpretation of the effect of genetic variation on T2D is more challenging because the association signals mostly map to non-coding sequence. While recent reports have demonstrated a relationship between T2D-associated variants and transcriptional enhancer activity, particularly in human pancreatic islets, liver cells, adipose tissue, and muscle10–14, the DNA-binding proteins through which these effects are mediated remain obscure. Localisation of non-coding causal variants may highlight the specific regulatory elements they perturb, and potentially the genes through which they operate, providing valuable insights into the pathophysiological basis of T2D susceptibility at GWAS loci.
To improve the localisation of potential causal variants for T2D, and characterise the mechanisms through which they alter disease risk, we performed comprehensive fine-mapping of 39 established loci through high-density imputation into 27,206 cases and 57,574 controls from 23 studies of European ancestry, genotyped with the Metabochip15 (Supplementary Tables 1 and 2). Within each locus, we aimed to: (i) evaluate the evidence for multiple distinct association signals through conditional analyses; (ii) undertake fine-mapping by defining credible sets of variants that account for ≥99% of the probability of driving each distinct association signal; and (iii) interrogate credible sets for functional and regulatory annotation to provide insight into the mechanisms through which variants driving association signals influence disease risk.
RESULTS
Imputation into Metabochip fine-mapping regions
The Metabochip includes high-density coverage of 257 “fine-mapping regions” that have been previously associated with 23 metabolic, cardiovascular, and anthropometric traits15. SNPs in these regions were selected using reference data from the HapMap16 and the 1000 Genomes (1000G) Project17. At design, 27 T2D susceptibility loci were selected for fine-mapping. However, subsequent T2D GWAS efforts have identified additional loci that overlap 12 further fine-mapping regions that were initially selected for other traits (Supplementary Table 3). To enhance coverage of variation in the fine-mapping regions, we undertook imputation into the Metabochip scaffold up to the 1000G phase 1 integrated reference panel (March 2012 release)18, including multi-ethnic haplotypes to reduce error rates19 (Online Methods).
The quality of imputation was variable across studies, particularly for MAF<5% variants, and dependent on the scaffold sample size (Supplementary Table 4). We defined variants to be “well-imputed” at widely-used thresholds20 of IMPUTEv221 info≥0.4 or minimac22 r2≥0.3 in at least 80% of the total effective sample size (Neff≥59,122) across studies. With this definition, 99.4% and 89.0%, respectively, of common and low-frequency (0.5%≤MAF<5%) variants in 1000G European ancestry haplotypes were well imputed, and therefore retained for downstream association analyses. Within studies, imputation quality was consistent across loci, despite the differential priority of fine-mapping regions and their coverage of variation at design (Supplementary Table 5). 1000G imputation into the Metabochip scaffold thus provides near complete coverage of common and low-frequency variation across the 39 T2D susceptibility loci, and supports direct interrogation of the majority of variants with MAF≥0.5% in European ancestry populations.
Distinct association signals at T2D susceptibility loci
The first step in fine-mapping GWAS loci is to delineate distinct association signals arising from multiple causal variants in the same region, which can efficiently be achieved through approximate conditioning with GCTA23. Within each T2D fine-mapping region, we identified distinct signals attaining “locus-wide” significance (represented by an index variant with pJ<10−5 in the joint association model) by applying GCTA in two stages (Online Methods). First, we selected index variants on the basis of fixed-effects meta-analysis across Metabochip studies. Second, we performed in silico replication of the index variants in a validation meta-analysis of an additional 19,662 T2D cases and 115,140 controls from 10 GWAS of European ancestry (Supplementary Tables 1, 2, and 6). Finally, because GCTA is only an approximation, we confirmed the association of each index variant through exact conditional analysis across Metabochip studies (Online Methods, Supplementary Table 7).
The most dramatic delineation of distinct association signals was observed for the region flanking KCNQ1, where five non-coding index variants attained locus-wide significance (Table 1, Supplementary Figure 1). Distinct association signals represented by three of the index variants have been reported in previous GWAS of European4 and East Asian24 ancestry: rs74046911 (pJ=3.6×10−26, r2=0.98 with East Asian lead SNP, rs2237897) and rs2237895 (pJ=2.1×10−9, r2=0.75 with one European lead SNP, rs163184), both of which map to a <50kb intronic recombination interval of KCNQ1; and chr11:2692322:D (pJ=7.2×10−16, r2=0.59 with second European lead SNP, rs231361), which resides in the KCNQ1OT1 transcript that controls regional imprinting25. The remaining two distinct association signals at this locus are novel. The first, indexed by rs458069 (pJ=3.2×10−6), maps to the same <50kb recombination interval as rs74046911 and rs2237895, but is in only weak LD with both (r2=0.02 and r2=0.25, respectively). The second, indexed by rs2283220 (pJ=2.2×10−7), resides in a neighbouring intron of KCNQ1, outside of the <50kb recombination interval (Supplementary Figure 1).
Table 1.
Established T2D susceptibility loci with multiple distinct signals of association at locus-wide significance in the GCTA joint regression model (pJ<10−5).
| Locus | Index variant | Chr | Position (b37) | Risk allele | Other allele | Metabochip GCTA joint model 27,206 cases and 57,574 controls |
Validation GCTA joint model 19,662 cases and 115,140 controls |
Combined GCTA joint model 46,868 cases and 172,714 controls |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RAF | OR (95% CI) | pJ | RAF | OR (95% CI) | pJ | OR (95% CI) | pJ | ||||||
| DGKB | rs10276674 | 7 | 14,922,007 | C | T | 0.183 | 1.08 (1.04–1.11) | 4.5×10−6 | 0.216 | 1.09 (1.05–1.12) | 1.3×10−6 | 1.08 (1.06–1.11) | 2.8×10−11 |
| rs1974620 | 7 | 15,065,467 | T | C | 0.519 | 1.06 (1.04–1.09) | 1.6×10−6 | 0.515 | 1.05 (1.03–1.08) | 0.00014 | 1.06 (1.04–1.08) | 1.0×10−9 | |
| CDKN2B | rs10811660 | 9 | 22,134,068 | G | A | 0.830 | 1.32 (1.27–1.38) | 2.4×10−44 | 0.817 | 1.21 (1.17–1.26) | 2.6×10−21 | 1.27 (1.23–1.30) | 1.1×10−61 |
| rs10757283 | 9 | 22,134,172 | T | C | 0.437 | 1.14 (1.10–1.17) | 7.4×10−18 | 0.436 | 1.11 (1.07–1.14) | 1.3×10−10 | 1.12 (1.10–1.14) | 3.6×10−26 | |
| KCNQ1 | chr11:2692322:D | 11 | 2,692,322 | D | R | 0.374 | 1.08 (1.05–1.10) | 3.5×10−8 | 0.413 | 1.09 (1.06–1.12) | 1.2×10−8 | 1.08 (1.06–1.10) | 2.3×10−15 |
| rs2283220 | 11 | 2,755,548 | A | G | 0.661 | 1.06 (1.03–1.09) | 0.000016 | 0.710 | 1.05 (1.02–1.08) | 0.0031 | 1.06 (1.03–1.08) | 2.4×10−7 | |
| rs2237895 | 11 | 2,857,194 | C | A | 0.428 | 1.08 (1.05–1.11) | 6.6×10−7 | 0.433 | 1.07 (1.03–1.10) | 2.8×10−4 | 1.07 (1.05–1.10) | 5.3×10−10 | |
| rs74046911 | 11 | 2,858,636 | C | T | 0.951 | 1.32 (1.24–1.40) | 1.7×10−17 | 0.943 | 1.25 (1.17–1.34) | 4.8×10−10 | 1.29 (1.23–1.35) | 9.6×10−26 | |
| rs458069 | 11 | 2,858,800 | G | C | 0.707 | 1.06 (1.03–1.10) | 0.00026 | 0.707 | 1.07 (1.03–1.11) | 0.00085 | 1.06 (1.04–1.09) | 1.0×10−6 | |
| HNF1A | rs1169288 | 12 | 121,416,650 | C | A | 0.334 | 1.10 (1.07–1.13) | 5.4×10−10 | 0.316 | 1.08 (1.05–1.12) | 2.8×10−6 | 1.09 (1.07–1.12) | 8.1×10−15 |
| rs1800574 | 12 | 121,416,864 | T | C | 0.027 | 1.21 (1.11–1.31) | 5.2×10−6 | 0.020 | 1.23 (1.12–1.35) | 0.000026 | 1.22 (1.14–1.29) | 5.1×10−10 | |
| chr12:121440833:D | 12 | 121,440,833 | R | D | 0.416 | 1.06 (1.03–1.09) | 0.000028 | 0.382 | 1.08 (1.04–1.11) | 2.5×10−6 | 1.07 (1.05–1.09) | 2.9×10−10 | |
| MC4R | chr18:57739289:D | 18 | 57,739,289 | D | R | 0.234 | 1.05 (1.02–1.09) | 0.00079 | 0.254 | 1.07 (1.03–1.10) | 0.000059 | 1.06 (1.04–1.08) | 1.9×10−7 |
| rs17066842 | 18 | 58,040,624 | G | A | 0.961 | 1.13 (1.06–1.21) | 0.00033 | 0.948 | 1.11 (1.04–1.19) | 0.0012 | 1.12 (1.07–1.17) | 1.4×10−6 | |
| GIPR | rs4399645 | 19 | 46,166,073 | T | C | 0.395 | 1.07 (1.04–1.10) | 4.4×10−7 | 0.441 | 1.05 (1.01–1.08) | 0.0046 | 1.06 (1.04–1.08) | 1.4×10−8 |
| rs2238689 | 19 | 46,178,661 | C | T | 0.425 | 1.09 (1.07–1.12) | 9.7×10−12 | 0.424 | 1.07 (1.04–1.10) | 9.0×10−6 | 1.08 (1.06–1.11) | 8.3×10−16 | |
| HNF4Aa | rs1800961 | 20 | 43,042,364 | T | C | 0.034 | 1.16 (1.09–1.24) | 0.000011 | 0.041 | 1.16 (1.08–1.25) | 0.000051 | 1.16 (1.10–1.22) | 2.3×10−9 |
Each distinct association signal was represented by an index variant in the GCTA joint regression model on the basis of: (i) summary statistics from a combined meta-analysis of 46,868 cases and 172,714 controls of European ancestry; and (ii) reference genotype data from GoDARTS (3,298 cases and 3,708 controls of European ancestry from the UK) to approximate LD across fine-mapping regions.
Chr: chromosome. RAF: risk allele frequency. OR: odds-ratio for risk allele. CI: confidence interval.
The previously reported T2D GWAS SNP at the HNF4A locus (rs4812829) is not included in the fine-mapping region. However, the reported index variant, rs1800961, is independent of the GWAS SNP, and thus represents a novel distinct association signal at this locus.
At the HNF1A locus, we observed three distinct association signals (Table 1, Supplementary Figure 2), represented by index variants that are in only weak LD with the previously reported lead GWAS SNP, rs12427353. They include two non-synonymous variants, rs1169288 (pJ=4.4×10−14, r2=0.09, HNF1A p.I27L) and rs1800574 (pJ=4.2×10−10, r2=0.01, HNF1A p.A98V), and one inter-genic SNP, chr12:121440833:D (pJ=2.9×10−10, r2=0.19).
We also observed four loci with two distinct association signals (CDKN2A-B, DGKB, MC4R and GIPR), each represented by non-coding index variants (Table 1, Supplementary Figure 3). The index variants at the CDKN2A-B locus represent the known T2D haplotype association signal mapping to a 12kb inter-genic recombination interval26–28. Previous European ancestry GWAS meta-analyses4 have highlighted a potential distinct association signal, located upstream of the recombination interval in the non-coding CDKN2B-AS1 (ANRIL) transcript. However, our conditional analyses indicate that the association in this region can be fully explained by the two index SNPs in the recombination interval, which when considered together, fully extinguish the CDKN2B-AS1 signal (Supplementary Figure 4). The index variants at DGKB and MC4R also confirm previously reported distinct association signals at these loci in European ancestry GWAS meta-analyses4. At the GIPR locus, the two index variants (rs2238689, pJ=8.3×10−16; rs4399645, pJ=1.4×10−8) are not in strong LD with the previously reported4 lead SNP (rs8108269; r2=0.43 with rs2238689, r2=0.00 with rs4399645), but together can better explain the T2D association signal in this region.
Finally, we observed a novel distinct association signal at the HNF4A locus, represented by the coding index variant rs1800961 (pJ=1.4×10−9, HNF4A p.T139I, referred to as p.T130I in some previous studies29). Unfortunately, this fine-mapping region was included on Metabochip for high-density lipoprotein cholesterol15,30 (Supplementary Table 3), and does not include the previously reported4 lead T2D SNP at this locus, rs4812829, precluding conditional analyses in these data. However, rs4812829 is not in LD with our index variant (r2=0.02), suggesting that there are at least two distinct T2D association signals at the HNF4A locus.
Of the 49 distinct association signals achieving locus-wide significance across T2D loci represented on Metabochip (five at KCNQ1, three at HNF1A, two each at CDKN2A-B, DGKB, MC4R and GIPR, and one each at the remainder), only three index variants are not common (Supplementary Table 6, Supplementary Figure 5): rs1800574 (MAF=2.2%, OR=1.21) for one signal at the HNF1A locus; rs1800961 (MAF=3.9%, OR=1.16) at the HNF4A locus; and rs17066842 (MAF=4.8%, OR=1.12) for one signal at the MC4R locus.
Localising variants driving T2D association signals
We used statistical evidence of association from the meta-analysis of Metabochip studies to construct 99% “credible sets” of variants28 that are most likely to drive the 49 distinct signals (Online Methods, Supplementary Table 8, Supplementary Figure 6). For ten distinct association signals, mapping to nine loci, the 99% credible set included no more than ten variants (Table 2, Supplementary Table 9). The greatest refinement was observed at the MTNR1B locus, where the credible set included only the index variant, rs10830963, accounting for more than 99.8% of the posterior probability of driving the association signal (πC). Small credible sets were also observed for the association at TCF7L2 (three variants, indexed by rs7903146, mapping to 4.3kb), and one signal at KCNQ1 (three variants, indexed by rs74046911, mapping to just 200bp). The 99% credible sets for both distinct association signals at CDKN2A-B together included just 11 variants in total, and map to less than 2kb.
Table 2.
Distinct association signals at established T2D susceptibility loci for which the 99% credible set contains no more than ten variants.
| Locus | Index variant | Chr | Position (b37) | Risk allele | Other allele | RAF | p-value | OR (95% CI) | 99% credible set | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNPs | Interval (bp) | Interval start (bp) | Interval stop (bp) | |||||||||
| MTNR1B | rs10830963 | 11 | 92,708,710 | G | C | 0.283 | 2.9×10−12 | 1.10 (1.07–1.13) | 1 | 1 | 92,708,710 | 92,708,710 |
| TCF7L2 | rs7903146 | 10 | 114,758,349 | T | C | 0.260 | 5.8×10−120 | 1.39 (1.35–1.43) | 3 | 4,279 | 114,754,071 | 114,758,349 |
| KCNQ1 | rs74046911 | 11 | 2,858,636 | C | T | 0.951 | 5.9×10−18 | 1.33 (1.25–1.42) | 3 | 197 | 2,858,440 | 2,858,636 |
| ZBED3 | rs7732130 | 5 | 76,435,004 | G | A | 0.278 | 6.4×10−10 | 1.09 (1.06–1.12) | 5 | 10,056 | 76,424,949 | 76,435,004 |
| CDKN2A-B | rs10757283 | 9 | 22,134,172 | T | C | 0.437 | 2.8×10−19 | 1.14 (1.11–1.18) | 5 | 1,007 | 22,133,645 | 22,134,651 |
| SLC30A8 | rs13266634 | 8 | 118,184,783 | C | T | 0.676 | 1.3×10−18 | 1.13 (1.10–1.16) | 6 | 33,133 | 118,184,783 | 118,217,915 |
| CDKN2A-B | rs10811660 | 9 | 22,134,068 | G | A | 0.830 | 7.0×10−43 | 1.32 (1.27–1.37) | 6 | 1,397 | 22,132,698 | 22,134,094 |
| HNF1B | rs4430796 | 17 | 36,098,040 | G | A | 0.455 | 6.3×10−12 | 1.09 (1.07–1.12) | 7 | 5,791 | 36,097,775 | 36,103,565 |
| CDKAL1 | rs35261542 | 6 | 20,675,792 | A | C | 0.280 | 9.6×10−23 | 1.15 (1.12–1.18) | 8 | 30,073 | 20,673,880 | 20,703,952 |
| GLIS3 | chr9:4294707:I | 9 | 4,294,707 | I | R | 0.360 | 6.5×10−8 | 1.07 (1.05–1.10) | 10 | 15,453 | 4,283,137 | 4,298,589 |
Association summary statistics and credible set construction are based on the meta-analysis of Metabochip studies in 27,206 cases and 57,574 controls of European ancestry. In loci with multiple distinct signals of association, results are presented from exact conditional analysis after adjusting for all other index variants in the fine-mapping region. In loci with a single signal of association, results are presented from unconditional analysis.
Chr: chromosome. RAF: risk allele frequency. OR: odds-ratio for risk allele. CI: confidence interval.
We performed functional annotation of credible variants to search for evidence that association signals are driven by coding alleles. Across the 49 signals, only nine coding variants attained πC>1% (Supplementary Table 10), including six previously reported non-synonymous T2D-risk alleles at PPARG6, KCNJ11-ABCC87,31,32, SLC30A88,33, and GCKR9,34. The remaining three coding alleles were the index variants for association signals mapping to HNF4A (p.T139I, rs1800961, πC=97.4%) and HNF1A (p.I27L, rs1169288, πC=75.5%; p.A98V, rs1800574, πC=34.0%). Our findings are supported by earlier studies, which reported nominal evidence for association of these three coding variants with T2D and defects in insulin secretion in vivo, and demonstrated reduced transcriptional activity of HNF1A target genes using in vitro assays29,35. These data provide strong evidence that HNF4A and HNF1A are T2D effector transcripts at these loci, a view further supported by the known impact of rare, loss of function mutations in these genes on maturity onset diabetes of the young36,37. Given the near complete coverage of common and low-frequency variants in fine-mapping regions after 1000G imputation, it is unlikely that additional distinct signals in established T2D susceptibility loci represented on the Metabochip are driven by coding variation with MAF≥0.5%, confirming reports that these associations are most likely to be mediated by effects on gene regulation10,13,14,38.
Regulatory mechanisms underlying T2D association signals
We sought to understand the regulatory mechanisms through which variants at the 39 established T2D susceptibility loci influence disease by intersecting the 99% credible sets for each distinct association signal with chromatin immunoprecipitation sequence (ChIP-seq) data for 165 transcription factors, chromatin state maps from 12 cell types, and long non-coding RNA transcripts from 25 cell types (Online Methods, Supplementary Table 11). We applied an enrichment procedure that compared the mean posterior probability of driving the association signal for credible set variants directly overlapping sites for each regulatory annotation with a null distribution obtained from randomly shifted site locations within 100kb in either direction.
We first applied this procedure to chromatin state and non-coding RNA elements using the 19,266 credible set variants for all 49 distinct association signals (Supplementary Figure 7). Using a Bonferroni correction for the 37 tested cell type annotations (p<0.0014), variants in pancreatic islet enhancer elements14 had significantly higher posterior probability of driving association signals than that expected from the null distribution (1.97-fold, p=0.00022). We also observed nominal evidence for enrichment of the posterior probability of driving association signals among variants in human islet and hepatocellular carcinoma (HepG2) promoters10,14 (p=0.0052 and p=0.0064, respectively). However, there was no corresponding enrichment of variants in regulatory elements for other cell types or in non-coding transcripts. These results are consistent with previous studies supporting a contribution of regulatory enhancer and promoter variants to T2D susceptibility in specific cell types11–14.
We next sought to gain insight into the transcription factors these regulatory variants perturb, and applied the same procedure to ChIP-seq binding data for 165 proteins (Figure 1, Supplementary Figure 8). Using a Bonferroni correction for the 165 tested proteins (p<0.00030), the 89 credible set variants overlapping 57 FOXA2 ChIP-seq binding sites, assayed in human HepG210 and islet14 cells, had significantly higher posterior probability of driving association signals than expected from the null distribution (8.24-fold, p=0.00028). The enrichment of FOXA2 ChIP-seq sites was exclusive to those shared with at least one other factor (9.18-fold, p=0.00028) compared to those that were not (1.12-fold, p=0.11). FOXA2 enrichment was also more pronounced among sites identified in pancreatic islets (15.43-fold, p=0.00045) than in HepG2 cells (4.55-fold, p=0.011). To exclude the possibility that this enrichment in HepG2 cells was driven by artefacts caused by a cultured cell line, we compared FOXA2 HepG2 sites to those previously assayed in primary liver39. We observed significant intersection of the HepG2 and liver FOXA2 sites that overlapped credible set variants (p=1.5×10−9). Consequently, we detected similar FOXA2 enrichment among sites detected in liver (3.63-fold, p=0.061) to that observed in HepG2 cells. We also compared FOXA2 ChIP-seq sites, genome-wide, from liver, HepG2 and islet cells (Supplementary Figure 9). The number of sites varied across cell types (8,023 for liver, 40,866 for HepG2, and 27,291 for islets), which is likely due, in part, to technical differences including sequencing platform, depth and read length. However, the intersection of FOXA2 sites between each pair of cell types was highly significant (p<2.2×10−16).
Figure 1. FOX2A bound sites are a genomic marker of T2D risk variants.
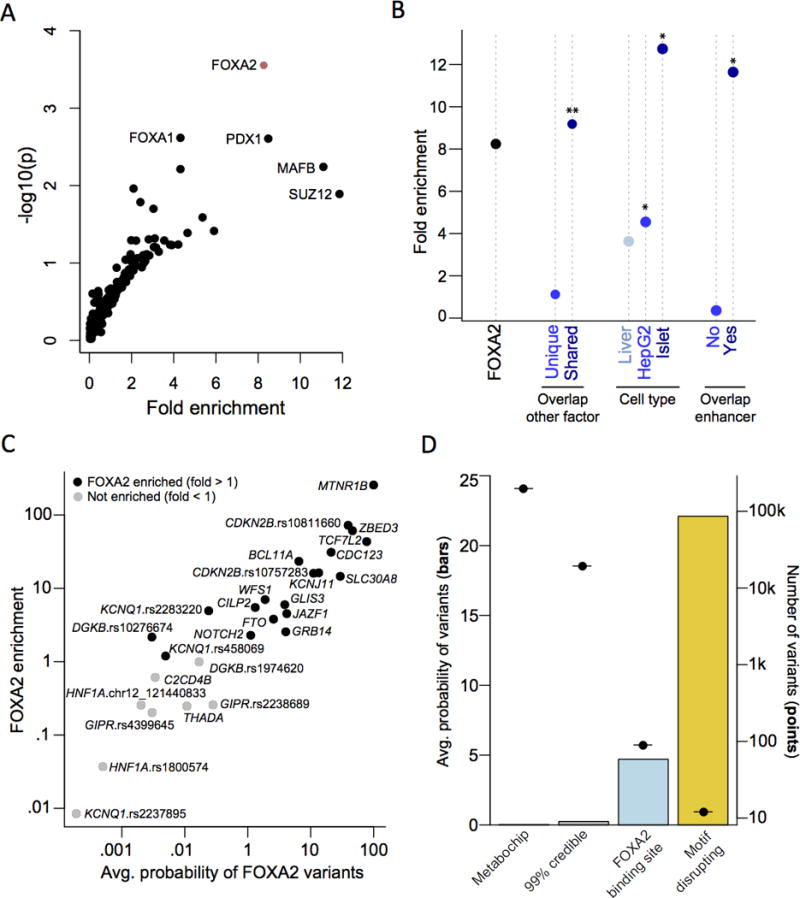
(A) Variants in ChIP-seq binding sites for 165 proteins were tested for enrichment of posterior probabilities compared to variants in shifted sites. Variants in FOXA2 ChIP-seq sites were significantly enriched (p<0.00030). (B) FOXA2 ChIP-seq sites were partitioned based on overlap with other genomic features. There was stronger enrichment in: (i) FOXA2 sites overlapping a ChIP-seq site for another protein compared to unique sites; (ii) sites identified in primary islets compared to HepG2 or primary liver cells; and (iii) sites overlapping islet enhancers compared to those that did not (**p<0.00030; *p<0.05). (C) Variants at each signal were tested for FOXA2 enrichment. Nineteen signals had greater enrichment than expected compared to shifted sites; at 15 signals this enrichment was nominally-significant (p<0.05). (D) FOXA2-bound variants disrupting recognition motifs have an increased probability of being causal.
Given the preponderance of T2D-associated variants for islet enhancers, we next tested to what extent FOXA2 enrichment is driven by co-localisation with these genomic features14. Variants in FOXA2-bound sites were not enriched for posterior probability of driving association signals after removing enhancer sites (0.36-fold, p=0.69). Conversely, variants in islet enhancers remained nominally enriched when removing FOXA2 sites (1.65-fold, p=0.014). These results suggest that FOXA2 binding assayed by ChIP-seq, at a subset of enhancer element locations that are often shared by other proteins, is a genomic marker of variants with an increased posterior probability of driving T2D association signals.
Having demonstrated global over-representation for FOXA2 ChIP-seq binding by considering all loci simultaneously, we applied the same procedure to the 99% credible sets of each distinct association signal, separately, to identify those with the strongest evidence for local enrichment (Figure 1). We observed over-representation of credible set variants in islet or HepG2 FOXA2 sites for 19 association signals, 15 of which attained nominal significance (p<0.05). A total of 41 credible set variants at these 19 distinct association signals overlap a FOXA2 ChIP-seq site in at least one of the two cell types (Supplementary Table 12). Of these, 12 variants were predicted to disrupt de novo recognition motifs (for FOXA2 and other factors) that were enriched in FOXA2-bound sequence (Table 3, Supplementary Table 13). The mean posterior probability of driving the association (πC) for these 12 variants was 22.0% on the basis of genetic fine-mapping (Figure 1), more than four times greater than for those in FOXA2 ChIP-seq sites that were not motif-disrupting at the same signals (mean πC of 5.2%, p=0.024). Furthermore, 11 of these 12 variants also overlapped an enhancer element in islets (9 variants) or HepG2 cells (6 variants), indicating that they are in transcriptionally active regions (Table 3). They include two variants with experimentally validated differences in regulatory activity: rs7903146 (πC=77.6%) at TCF7L240 and rs11257655 (πC=21.1%) at CDC12341. They also include rs10830963, the index variant at the MTNR1B locus, which accounts for 99.8% of the posterior probability of driving the association signal on the basis of genetic fine-mapping. These results suggest that FOXA2 binding patterns can be used to highlight specific variants that are potentially causal for T2D susceptibility through altered regulatory binding.
Table 3.
Motif-altering credible set variants in FOXA2 sites.
| Locus | Index variant | Motif-altering variant | Chr | Position (b37) | Posterior probability (πC) | Motif-altering allele | Chromatin state |
|---|---|---|---|---|---|---|---|
| MTNR1B | rs10830963 | rs10830963 | 11 | 92708710 | 0.998 | G | Islet-enhancer, HepG2-enhancer |
| TCF7L2 | rs7903146 | rs7903146 | 10 | 114,758,349 | 0.78 | T | Islet-enhancer |
| SLC30A8 | rs13266634 | rs13266634 | 8 | 118,184,783 | 0.29 | T | Islet-enhancer |
| CDKN2B | rs10811660 | rs10811660 | 9 | 22,134,068 | 0.24 | A | Islet-enhancer |
| CDC123 | rs11257658 | rs11257655 | 10 | 12,307,894 | 0.21 | T | Islet-enhancer, HepG2-enhancer |
| JAZF1 | rs1513272 | rs849133 | 7 | 28,192,280 | 0.042 | T | Islet-enhancer |
| KCNQ1 | rs2283220 | rs231907 | 11 | 2,752,130 | 0.031 | T | HepG2-enhancer |
| FTO | rs9927317 | rs9940128 | 16 | 53,800,754 | 0.027 | G | Islet-enhancer, HepG2-enhancer |
| FTO | rs9927317 | rs9939973 | 16 | 53,800,568 | 0.025 | G | Islet-enhancer, HepG2-enhancer |
| KCNQ1 | rs458069 | rs78688069 | 11 | 2,752,183 | 0.0006 | A | HepG2-enhancer |
| KCNQ1 | rs458069 | rs190728714 | 11 | 2,813,084 | 0.00042 | G | Islet-enhancer |
| DGKB | rs10276674 | rs7798360 | 7 | 15,055,972 | 0.00005 | G | - |
Chr: chromosome.
Altered regulatory activity of the MTNR1B credible variant
To demonstrate how local enrichment of FOXA2 binding can be used to highlight regulatory mechanisms through which credible variants might impact T2D susceptibility, we focussed on the MTNR1B locus. Variants mapping to this region have amongst the strongest known effects on both T2D risk6 and fasting plasma glucose concentration42, and physiological data indicate an impact of MTNR1B on both insulin secretion and insulin action43. The lone credible variant at MTNR1B, rs10830963, overlaps a FOXA2 ChIP-seq binding site, and the risk allele, G, is predicted to create a recognition motif that matches the consensus sequence of NEUROD1 and several other factors (Figure 2, Supplementary Table 13). We tested in silico predictions of protein binding at rs10830963 via electrophoretic mobility shift assay (EMSA) with 25bp probe fragments surrounding each allele in human pancreatic islet beta-cell (EndoC-βH1)44 or human liver HepG2 cell extracts. We observed allele-specific binding with extracts from both cell lines (Figure 2, Supplementary Figure 10).
Figure 2. The lone variant in the 99% credible set at the MTNR1B locus affects FOXA2-bound enhancer activity.
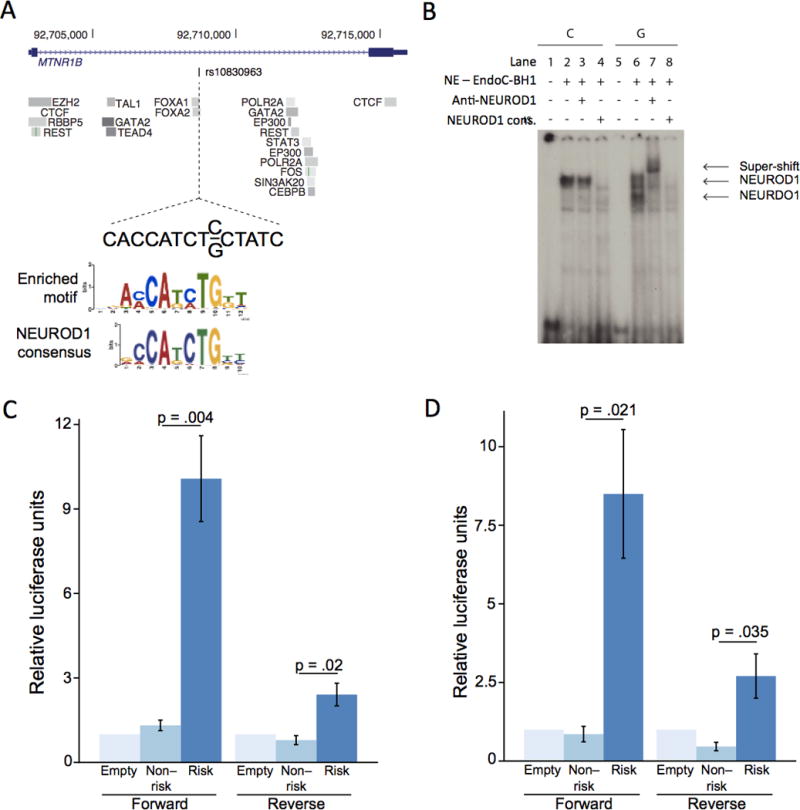
(A) The intronic variant, rs10830963, has 99.8% probability of driving the association signal at the MTRN1B locus. This variant overlaps a FOXA2 binding site, and the risk allele G is predicted to create a de novo recognition motif, which closely matches the NEUROD1 consensus. (B) Electrophoretic mobility shift assay of a 25bp fragment surrounding both alleles in EndoC-βH1 cell extracts. Proteins were bound to both alleles. In the presence of a NEUROD1 antibody, only the risk allele band was super-shifted, and in the presence of an unlabelled NEUROD1 consensus probe, the signal was competed away. NE: nuclear extract. (C, D) The 224bp sequence surrounding each allele was cloned into a luciferase reporter construct containing a minimal promoter and tested for luciferase activity in (C) EndoC-βH1 and (D) HepG2 cells (n=3 for each cell type). Results are presented as mean ± standard error. The risk allele had significantly increased enhancer activity over the protective allele in both forward and reverse orientations in both cell types.
To determine the specific protein(s) bound at each allele, we then performed supershift experiments using antibodies directed against NEUROD1, FOXA2, and three other factors (TAL1, PTF1A, and YY1), whose consensus binding sequences resemble the recognition motif (Online Methods). We observed a shift in the presence of the NEUROD1 antibody on the risk allele in EndoC-βH1 extracts, which could be competed away by an excess of unlabelled NEUROD1 consensus sequence probe (Figure 2). None of the tested antibodies (including NEUROD1) shifted the risk allele band in HepG2 cell extracts (Supplementary Figure 10). These results demonstrate that, in vitro, the risk allele of rs10830963 preferentially binds NEUROD1 in islet-derived cells, and binds a protein not identified from known recognition motifs in liver-derived cells.
To relate allelic differences in protein binding to genomic activity at this site, we cloned a 224bp region surrounding rs10830963 into a luciferase reporter vector containing a minimal promoter, and tested its enhancer activity in EndoC-βH1 and HepG2 cell lines. Consistent with in silico predictions, we observed a significant (p<0.05) increase in luciferase expression on the risk allele compared to the protective allele in both cell lines (Figure 2). Furthermore, RNA-seq data reported from human islets have linked the T2D risk allele of rs10830963 to increased expression of MTNR1B45,46. Taken together, these results suggest that the G allele of rs10830963 increases T2D risk through increased FOXA2-bound enhancer activity, potentially mediated through NEUROD1 binding in islets, and consequently higher expression of MTNR1B.
Candidate effector genes at FOXA2 enriched T2D signals
We hypothesised that the locus-specific effects of murine transcription factor knockout models would mimic patterns of binding enrichment at human disease loci. We thus attempted to relate FOXA2 binding at the 19 FOXA2-enriched association signals (Figure 1) to target effector genes using previously reported pancreatic islet expression profiles from wild-type and Foxa1/2-null mice47 (Online Methods). Syntenic genes mapping within 500kb of the credible set at the 19 FOXA2-enriched signals were significantly down-regulated (45.2% decrease) in Foxa1/2 knockout mice (Supplementary Figure 11) compared to those genome-wide (0.021% increase, p=0.012), whilst those mapping within 500kb of the other 30 T2D association signals were not (2.25% decrease, p=0.20). We observed a consistent down-regulation (39.6% decrease) when considering only those genes mapping closest to each FOXA2-enriched signal, compared to those genome-wide (0.021% increase, p=0.0021). Thus, data related to altered gene expression in Foxa1/2 knockout mice support patterns of FOXA2 binding site enrichment in humans.
We next identified specific genes at the 19 FOXA2-enriched association signals that were down-regulated in Foxa1/2 knockout mice, which might represent effector transcripts for these loci (Supplementary Table 14). Several of these genes have been previously implicated as likely effector transcripts in humans, including TCF7L248,49 (57% decrease), KCNJ117,50 (38% decrease), and SLC30A851 (135% decrease). These data also implicate novel candidate effector genes at FOXA2-enriched association signals (Supplementary Table 14). For example, in Foxa1/2 knockout mice, there is a marked down-regulation of Reg4 (1,415% decrease), which maps to a syntenic region at the FOXA2-enriched NOTCH2 GWAS locus, highlighting REG4 as a likely effector transcript in humans. Additional examples of candidate effector genes include IGF2 at the KCNQ1 locus (135% decrease), and CAMK1D at the CDC123 locus (81% decrease). Together, these results provide additional support for the importance of FOXA2 binding at a subset of T2D susceptibility loci, and further highlight specific genes through which regulatory variants in these regions may operate.
DISCUSSION
We have undertaken comprehensive fine-mapping of 39 established T2D susceptibility loci in 27,206 cases and 57,574 controls of European ancestry, and have demonstrated that multiple distinct association signals in these regions is a common phenomenon. Index variants for just three of the 49 distinct association signals are not common, despite near complete coverage of variation with MAF≥0.5% in fine-mapping regions after 1000G imputation. Although we cannot evaluate the impact of rare variation (MAF<0.5%) in established T2D susceptibility loci without large-scale re-sequencing, our data strongly argue against a role for low-frequency variants of large effect via synthetic association52. We have demonstrated that seven distinct association signals, mapping to six T2D susceptibility loci represented on the Metabochip, are likely to be driven by coding alleles, including novel index variants mapping to HNF1A and HNF4A. Outside of these regions, our fine-mapping confirms previous reports that T2D association signals are primarily driven by non-coding alleles, with effects that are mediated through gene regulation10,13,14,38.
We have demonstrated, by genomic annotation and functional assays, that FOXA2 binding assayed by ChIP-seq can be used to pinpoint candidate causal regulatory elements, providing routes to understanding the biology of specific T2D susceptibility loci. These elements highlight variants and effector transcripts through which association signals are mediated, via altered binding of either FOXA2, directly, or another transcription factor. For example, at the MTNR1B locus, the risk allele of the lone credible variant, rs10830963, which drives the T2D association signal, preferentially binds NEUROD1 in islet-derived cells in vitro, and increases FOXA2-bound enhancer activity in human islet and liver-derived cells. These data are consistent with previous reports correlating the risk allele with higher MTNR1B expression45,46, and not loss of function53, and suggest altered NEUROD1 binding in islets contributes to T2D susceptibility at this locus. Further experiments will be required to establish that our in vitro findings regarding NEUROD1 binding can be confirmed in vivo. However, our attempts to perform ChIP-Seq in primary islet samples of the defined MTNR1B genotype were repeatedly unsuccessful, owing to a lack of a suitable NEUROD1 antibody. These studies are further complicated by the limited availability of primary human islets, and the slow division rate of human islet derived cell-lines is an impediment to the implementation of genome-editing technologies.
FOXA2 is a pioneer factor that binds native chromatin and bookmarks genomic regions for transcriptional activity54, and is involved in pancreatic and hepatic development55,56. FOXA2 is also expressed in other T2D-relevant cell types, such as adipocytes. Future studies will be required to elucidate the extent to which FOXA2 binding events across cell types influence disease risk. Foxa2 null mice have impaired insulin secretion47, and common variants at the FOXA2 locus are associated with fasting plasma glucose concentrations42,57. Our findings are thus consistent with the involvement of FOXA2 in maintaining normal glucose homeostasis. Common T2D-associated variants at FOXA2 have also been reported in South Asians58, although they do not attain genome-wide significance in the largest GWAS for the disease from multiple ancestry groups1–5, and therefore require further replication. Enrichment of FOXA2 binding has also been reported within genomic intervals containing GWAS signals for endocrine, neuropsychiatric, cardiovascular and cancer traits59. Our study has the advantage that we consider only those FOXA2 sites that directly overlap variants that drive association signals by first fine-mapping GWAS loci, thereby providing more targeted credible sets for functional enrichment. Nevertheless, the results of these studies, taken together, suggest a possible role for FOXA2 across a broad spectrum of complex human phenotypes.
In conclusion, we have highlighted that FOXA2 binding patterns can be used to inform future hypothesis-driven investigation of the variants, genes, and molecular mechanisms underlying T2D association signals mapping to non-coding sequence. Continued identification of the effector transcripts at these non-coding association signals will require the use of expression QTL and knockout models, in combination with high-throughput experimental data derived from chromatin conformation capture techniques, such as Capture-C. Our findings support the use of transcription factor binding events as a means to partition susceptibility loci, potentially residing in distinct pathways, within disease-relevant cell types. Finally, our study demonstrates the utility of fine-mapping through integration of genetic and genomic information from relevant tissues and cellular models to elucidate the pathophysiology of complex human diseases, thus offering a promising avenue for translation of GWAS findings for clinical utility.
ONLINE METHODS
Ethics statement
All human research was approved by the relevant institutional review boards, and conducted according to the Declaration of Helsinki. All participants provided written informed consent.
Metabochip imputation and association analysis
We considered a total of 27,206 T2D cases and 57,574 controls from 23 studies from populations of European ancestry (Supplementary Table 1), all genotyped with the Metabochip. Sample and variant quality control was performed within each study (Supplementary Table 2). To improve the quality of the genotype scaffold in each study, variants were subsequently removed if: (i) allele frequencies differed from those for European ancestry haplotypes from the 1000 Genomes Project Consortium phase 1 integrated reference panel (March 2012 release)18 by more than 20%; AT/GC variants had MAF>40% because of potential undetected errors in strand alignment; or (iii) MAF<1% because of difficulties in calling rare variants. Each scaffold was then imputed up to up to the phase 1 integrated reference panel (all ancestries, March 2012 release) from the 1000 Genomes Project Consortium18, using IMPUTEv221 or minimac22. Within each study, well-imputed variants (IMPUTEv221 info>0.4 or minimac22 r2>0.3) were tested for T2D association under an additive model after adjustment for study-specific covariates (Supplementary Table 2), including principal components to adjust for population structure. Association summary statistics for each variant for each study were corrected for residual population structure using the genomic control inflation factor60 obtained from 3,598 independent (r2<0.05) QT-interval variants, which were not expected to be associated with T2D4 (Supplementary Table 2). We then combined association summary statistics for each variant across studies via fixed-effects inverse-variance weighted meta-analysis. The results of the meta-analysis were subsequently corrected by a second round of QT-interval genomic control (λQT=1.18) to account for structure between studies. Variants were excluded from downstream analyses if they were reported in less than 80% of the total effective sample size, defined as Neff = 4×Ncases×Ncontrols/(Ncases+Ncontrols), thus removing those that were not well imputed in the majority of studies.
Identification of distinct association signals in established GWAS loci
We used GCTA23 to select index variants in each of the 39 established loci represented on Metabochip with nominal evidence of association (pJ<0.001) with T2D in an approximate joint regression model. The GCTA model made use of: (i) summary statistics from the fixed-effects meta-analysis Metabochip studies; and (ii) genotype data for 3,298 T2D cases and 3,708 controls of UK ancestry from GoDARTS as a reference for LD across each fine-mapping region. For comparison, we also obtained association summary statistics for the selected index variants from the GCTA joint regression model on the basis of genotype data from an alternative reference consisting of 4,435 T2D cases and 5,757 controls of Finnish ancestry from FUSION (Supplementary Table 15, Supplementary Figure 12). Selected index variants were then carried forward for in silico follow-up in validation meta-analysis.
The validation meta-analysis consisted of 19,662 T2D cases and 115,140 controls from 10 GWAS from populations of European ancestry, genotyped with a range of genome-wide arrays (Supplementary Table 1). Sample and variant quality control was performed within each study (Supplementary Table 2). Each scaffold was then imputed up to the phase 1 integrated reference panel (all ancestries, March 2012 release) from the 1000 Genomes Project Consortium18, using IMPUTEv221 or minimac22. Within each study, well-imputed variants (IMPUTEv221 info≥0.4 or minimac22 r2≥0.3) were tested for T2D association under an additive model after adjustment for study-specific covariates (Supplementary Table 2), including principal components to adjust for population structure. Association summary statistics for each variant for each study were corrected for residual population structure using the genomic control inflation factor60 (Supplementary Table 2). We then combined association summary statistics for each variant across studies via fixed-effects inverse-variance weighted meta-analysis.
Association summary statistics for the selected index variants from the Metabochip and validation meta-analyses were next combined via fixed-effects inverse-variance weighted meta-analysis. In each of the 39 established loci represented on Metabochip, GCTA23 was used to select index variants with locus-wide evidence of association (pJ<10−5) in the approximate joint regression model on the basis of: (i) summary statistics from the combined meta-analysis; and (ii) genotype data for 3,298 T2D cases and 3,708 controls from GoDARTS as a reference for LD across each fine-mapping region.
For established loci with multiple index variants selected at locus-wide significance from the GCTA approximate joint regression model in combined meta-analysis, we performed exact conditioning within each Metabochip study (Supplementary Table 7). To obtain the association signal attributed to a specific index variant, high-quality variants (IMPUTEv221 info>0.4 or minimac22 r2>0.3) were tested for T2D association under an additive model after adjustment for study-specific covariates (Supplementary Table 2) and genotypes at other selected index variants in the fine-mapping region. Association summary statistics for each study were corrected for residual population structure using the QT interval genomic control inflation factor obtained in the Metabochip meta-analysis. For each association signal, summary statistics for each variant were then combined across discovery studies via fixed-effects inverse-variance meta-analysis, and subsequently corrected by a second round of QT-interval genomic control (λQT=1.18).
Credible set construction
In an ideal fine-mapping experiment, we would calculate the posterior probability of driving each distinct association signal for all variants mapping to a locus. However, the posterior probability is determined by the association signal effect size of the variant and the corresponding standard error, which is also impacted by the quality of imputation across studies, amongst other factors. To minimise the impact of imputation quality on fine-mapping, we therefore retained only those variants that were directly typed and/or well imputed in at least 80% of the total effective sample size. Assuming that the variant driving an association signal meets these quality criteria, the probability that it would be contained within the 99% credible set would be ~0.99.
For each distinct signal, we first calculated the posterior probability, πCj, that the jth variant is driving the association, given by
where the summation is over all retained variants in the fine-mapping region. In this expression, Λj is the approximate Bayes’ factor61 for the jth variant, given by
where βj and Vj denote the estimated allelic effect (log-OR) and corresponding variance from the meta-analysis across Metabochip studies. In loci with multiple distinct signals of association, results are presented from exact conditional meta-analysis after adjusting for all other index variants in the fine-mapping region. In loci with a single association signal, results are presented from unconditional meta-analysis. The parameter ω denotes the prior variance in allelic effects, taken here to be 0.0461. The 99% credible set29 for each signal was then constructed by: (i) ranking all variants according to their Bayes’ factor, Λj; and (ii) including ranked variants until their cumulative posterior probability of driving the association attained or exceeded 0.99.
Genomic annotation data and enrichment analyses
We obtained genomic annotation data for transcription factor binding sites (TFBS) assayed through ChIP experiments from multiple sources. We used sites from the ENCODE Project Consortium10 for 161 proteins available from the UCSC human genome browser. We also obtained raw ChIP and input sequence data for additional factors assayed in primary pancreatic islets14. We then processed these additional factors using protocols employed by the ENCODE Project Consortium10. First, sequence reads were aligned to the human genome (hg19) using BWA62 with sex-specific references, and were then converted to BAM files using SAMtools63 after removing duplicate and non-uniquely mapped reads. Binding sites were called from reads of each replicate, as well as reads pooled across all replicates, using SPP64. Raw sites from each replicate of a protein were compared using an irreproducible discovery rate65 (IDR) threshold of 0.02. The resulting number of sites passing this IDR threshold was then used to filter the pooled sites of a protein. The set of sites were further filtered for artefacts using a blacklist of genomic regions from the ENCODE Project Consortium. Sites from all sources for each protein, including ENCODE, were then combined. The complete set of 165 proteins employed in these analyses is presented in Supplementary Table 11. In addition, we obtained FOXA2 ChIP-seq sites that were previously identified in human liver39 and lifted their positions to hg19.
We obtained annotation data for five histone modifications (H3K4me1, H3K4me3, H3K27ac, H3K36me3, and H3K27me3) and CTCF binding assayed from ChIP experiments. We used data from 9 cell types from ENCODE10 (Gm12878, K562, Hepg2, Hsmm, Huvec, Nhek, Nhlf, h1Hesc, and Hmec); we also obtained raw ChIP data assayed in primary pancreatic islets14 and pre-mature and mature human adipose stromal cells66. We mapped reads to hg19 using BWA62, and used the resulting mapped reads from these 12 cell types as input to ChromHMM67. We assigned states based on the following chromatin signatures: active promoter (H3K4me3 and H3K27ac); strong enhancer 1 (H3K4me3, H3K27ac, and H3K4me1); strong enhancer 2 (H3K27ac and H3K4me1); weak enhancer (H3K4me1); poised promoter (H3K27me3, H3K4me3, and H3K4me1); repressed (H3K27me3); insulator (CTCF); and transcription (H3K36me3). For each cell type, we pooled the three enhancer states into one enhancer category, and the two promoter states into one promoter category. We also identified long non-coding RNA data from the Human Body Map (UCSC genome browser) and from pancreatic islets68.
For each genomic annotation, we tested for overall enrichment of the posterior probability that overlapping variants in the 99% credible sets are driving distinct association signals (πC). We first calculated the mean posterior probability (mean πC) over the set of variants overlapping a given annotation. We then generated a null distribution of the mean posterior probability (mean πC) by: (i) shifting the genomic locations of binding sites a random distance within 100kb in either direction; (ii) recalculating the mean posterior probability for 99% credible set variants overlapping shifted sites; and (iii) repeating this procedure 100,000 times. We estimated the fold-enrichment of each overlap by calculating the expected null posterior probability, and dividing the observed probability by the expected probability. We calculated a p-value for the enrichment by the proportion of permutations for which the expected posterior probability of driving the association signal was greater than or equal to that observed. We considered cell type annotations to be significantly enriched if the p-value was less than 0.05/37 = 0.0014 (Bonferroni correction for 37 annotations). We considered TFBS annotations to be significantly enriched if the p-value was less than 0.05/165 = 0.00030 (Bonferroni correction for 165 factors). We next partitioned binding sites into those that are “shared” with another factor (i.e. genomic interval intersects a site for at least one other factor), and those that are “unique”. We also partitioned binding sites based on overlap with islet enhancer elements. For each factor with significant enrichment across all credible sets (FOXA2), we applied the same enrichment analysis, but restricted to credible set variants for each distinct association signal, separately.
We assessed the evidence for intersection in FOXA2 ChIP-seq sites from islets14, HepG210, and liver39, genome-wide and overlapping credible set variants, using BEDtools69.
Motif analysis
We conducted recognition motif enhancement analyses for the set of FOXA2 ChIP-seq binding sites. First, we obtained repeat-masked genomic sequence underlying each site using the UCSC human genome browser. We scanned sequences for enrichment in these motifs using MEME-ChIP70, which uses up to 100bp surrounding the mid-point of each site. This resulted in 198 enriched motifs with E-value (expected number of hits) less than 0.05 (Supplementary Table 16). We compared each motif to those known from JASPAR71, ENCODE10, and Homer72 using Tomtom73.
Second, we identified variants in FOXA2 ChIP-seq sites predicted to disrupt an enriched recognition motif by: (i) scanning a 25bp of sequence flanking each variant allele using FIMO74 (p<0.0001); and (ii) retaining variants in highly conserved positions (entropy less than 0.5). For the 12 variants at FOXA2-enriched signals disrupting at least one recognition motif (Table 3, Supplementary Table 14), we compared their posterior probabilities of driving the association (πC) with those for non-disrupting variants in FOXA2 ChIP-seq sites at the same signals using a two-sided Wilcoxon rank-sum test.
Electrophoretic mobility shift assays
EMSA was performed using nuclear extracts from human HepG2 and EndoC-βH1 cells. HepG2 cells were the generous gift of the Ratcliffe laboratory75 and authenticated by genotyping in the MHC region. Endo-βH1 cells were obtained from Endocells and have been previously authenticated44. Both cell lines were tested and found negative for mycoplasma contamination. Nuclear extracts were incubated with32 P gamma-ATP end-labeled double-stranded DNA probes (PerkinElmer, MA). The forward strand probe sequences used are presented in Supplementary Table 17.
For each lane of the EMSA, 5μg of nuclear extract was incubated with 100 fmol labeled probes in a 10ul binding reaction containing 10mM Tris-HCl pH7.5, 4% glycerol, 1mM MgCl2, 0.5mM EDTA, 0.5mM DTT, 50mM NaCl and 1μg poly(dI-dC). For competition assays unlabeled probe at 100-fold excess was added to the binding reaction before addition of labeled probes. For super-shift assays the nuclear extract was pre-incubated with 1μg antibody for 30 minutes on ice before the probe was added. The following antibodies were used: anti-NEUROD1 (sc-1084X, Santa Cruz Biotechnology, Texas), anti-PTF1A (sc-98612X, Santa Cruz Biotechnology, Texas), anti-HNF3B (FOXA2) (sc-6554X, Santa Cruz Biotechnology, Texas), anti-YY1 (sc281X, Santa Cruz Biotechnology, Texas), anti-TAL1 (sc12984X, Santa Cruz Biotechnology, Texas), normal rabbit Ig (sc-2027, Santa Cruz Biotechnology, Texas), normal goat Ig (sc-2028, Santa Cruz Biotechnology, Texas).
Luciferase activity
We synthesised 224bp nucleotide sequences containing either the risk or protective allele of the MTNR1B enhancer sequence rs10830963 in either the forward or reverse orientation by GeneArt (Life Technologies). Complementary single-stranded oligos were then annealed and sub-cloned into the minimal promoter-driven luciferase vector pGL4.23 (Promega) using Nhel and Xhol. Isolated clones were verified by sequencing.
For luciferase assays, human liver HepG2 and human beta-cell EndoC-βH145 cells were counted and seeded into 24 well trays (Corning) at 1.5×105 (HepG2) or 1.4×105 (EndoC-βH1) cells/well. Transfections were performed in triplicate with either Lipofectamine 2000 (HepG2) or Fugene 6 (EndoC-βH1) as per manufacturer’s instructions. Cells were transfected with 700ng pGL4.23 DNA containing the protective or risk MTNR1B enhancer sequence in either the forward or reverse orientation, or an equivalent amount of empty vector DNA, plus 10ng pRL-SV40 DNA (Promega) as a transfection control, per well. Cells were lysed 48 hours post-transfection and analysed for Firefly and Renilla luciferase activities using the Dual Luciferase Assay System (Promega) as per manufacturer’s instructions, in half-volume 96 well tray format on an Enspire Multimode Plate Reader (Perkin Elmer). Firefly luciferase activity was normalised to Renilla luciferase activity for each well, and the results expressed as a mean normalised activity relative to empty vector-transfected cells. All experiments were performed three times in triplicate. A two-sided unpaired t-test was used to compare luciferase activity between alleles.
Mouse gene expression analysis
We obtained fold-changes in pancreatic islet gene expression in wild type compared to Foxa1/Foxa2-null mice47. We used ENSEMBL to map mouse genes to human orthologs. We filtered for human genes annotated as protein coding in GENCODE. This filtering resulted in 4,629 human protein coding genes for analysis.
First, we calculated the genomic interval spanned by the variants in each credible set. We expanded this interval for 500kb on either side, and identified the set of genes overlapping this region using BEDtools69. To account for syntenic differences in gene order between species, we retained only those genes that were: (a) on the same chromosome; and (b) in exactly the same relative order in both mouse and human genomes. At the GIPR locus, one of the genes was ordered differently and thus removed from the analysis. At two loci, KCNJ11 and HNF1A, at least one of the genes was located on a different part of the same chromosome, and at another locus, GCK, genes were located on different chromosomes. For these three loci, we retained only those genes that were at the same chromosomal location to the interval covered by the credible set for the association signal for that locus (by lifting over from hg19 to mouse build mm10). Second, for each distinct association signal, we identified the closest gene to the index variant using BEDtools69. We then partitioned distinct association signals into those with evidence for enriched FOXA2 binding (fold-enrichment >1) and those without, counting each gene only once in a given group. For each analysis, we converted the fold-changes to percentages, and compared the percent change in expression using a one-sided Wilcoxon rank-sum test between genes in each partition and all 4,629 protein coding genes.
Supplementary Material
Acknowledgments
Funding for the research undertaken in this study has been received from: Academy of Finland (including grant numbers 77299, 102318, 10493, 118065, 123885, 124243, 129293, 129680, 136895, 139635, 211119, 213506, 251217, and 263836); Agence National de la Recherche; Association de Langue Francaise pour l’Etude du Diabete et des Maladies Metaboliques; Association Diabete Risque Vasculaire; Association Francaise des Diabetiques; Association of Danish Pharmacies; Augustinus Foundation; Becket Foundation; British Diabetes Association (BDA) Research; British Heart Foundation; Central Norway Health Authority; Central Finland Hospital District; Center for Inherited Disease Research (CIDR); City of Kuopio; City of Leutkirch; Copenhagen County; the Danish Centre for Evaluation and Health Technology Assessment; Danish Council for Independent Research; Danish Heart Foundation; Danish Research Councils; Deutsche Forschungsgemeinschaft (including project ER 155/6-2); the Diabetes Research Foundation; Diabetes UK; Doris Duke Charitable Foundation; Erasmus Medical Center; Erasmus University; Estonian Government (SF0180142s08); European Commission (including ENGAGE HEALTH-F4-2007-201413, FP7-201413, FP7-245536, EXGENESIS LSHM-CT-2004-005272, FP6 LSHM_CT_2006_037197, LSHM-CT-2007-037273, Directorate C-Public Health 2004310, DG XII); European Regional Development Fund; Federal Ministry of Education and Research, Germany (including FKZ 01GI1128 and FKZ 01EO1001); Federal Ministry of Health, Germany; Finnish Diabetes Association; Finnish Diabetes Research Foundation; Finnish Foundation for Cardiovascular Research; Finnish Medical Society; Folkhalsan Research Foundation; Foundation for Life and Health in Finland; Foundation for Old Servants; Fredrick och Ingrid Thuring Foundation; French Region of Nord Pas de Calais (Contrat de Projets Etat-Region); German Center for Diabetes Research; German Research Council (including grant number GRK1041); German National Genome Research Network; Groupe d’Etude des Maladies Metaboliques et Systemiques; Health Care Centers in Vasa, Narpes and Korsholm; the Health Foundation; Heinz Nixdorf Foundation; Helmholtz Zentrum Munchen; Helsinki University Central Hospital Research Foundation; Hospital District of Southwest Finland; Ib Henriksens Foundation; IngaBritt and Arne Lundberg’s Research Foundation (including grant number 359); Karolinska Institutet; Knut and Alice Wallenberg Foundation (including grant number KAW 2009.0243); Kuopio University Hospital; Lundbeck Foundation; Magnus Bergvall Foundation; Medical Faculty of the University Duisburg-Essen; Medical Research Council, UK (including grant numbers G0000649 and G0601261); Ministry for Health, Welfare and Sports, Netherlands; Ministry of Education and Culture, Finland (including grant number 722 and 627;2004–2011); Ministry of Education, Culture and Science, Netherlands; Ministry of Health and Prevention, Denmark; Ministry of Social Affairs and Health, Finland; Ministry of Innovation, Science, Research and Technology of North Rhine-Westphalia, Germany; Munich Center of Health Sciences; Municipal Health Care Center and Hospital in Jakobstad; Municipality of Rotterdam; Narpes Health Care Foundation; National Health Screening Service of Norway; National Heart, Lung, and Blood Institute, USA (including grant numbers/contracts HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, HHSN268201100012C, N01HC25195, N02HL64278, R01HL087641, R01HL59367, and R01HL086694); National Human Genome Research Initiative, USA (including grant numbers/contracts U01HG004402 and N01HG65403); National Institute for Diabetes and Digestive and Kidney Diseases, USA (including grant numbers R01-DK078616, U01-DK085526, K24-DK080140, and R01-DK073490); National Institute for Health and Welfare, Finland; National Institutes of Health, USA (including grant numbers/contracts HHSN268200625226C, UL1RR025005, R01DK062370, R01DK072193, 1Z01HG000024, AG028555, AG08724, AG04563, AG10175, AG08861, U01HG004399, DK58845, CA055075, DK085545, and DK098032); Netherlands Genomics Initiative; Netherlands Organisation for the Health Research and Development; Netherlands Organisation of Scientific Research NOW Investments (including numbers 175.010.2005.011, 911-03-012, 050-060-810); Nord-Trondelag County Council; Nordic Center of Excellence in Disease Genetics; Norwegian Institute of Public Health; Norwegian Research Council; Novo Nordisk Foundation; Ollquist Foundation; Oxford NIHR Biomedical Research Centre; Paavo Nurmi Foundation; Paivikki and Sakari Sohlberg Foundation; Perklen Foundation; Pirkanmaa Hospital District, Finland; Programme Hospitalier de Recherche Clinique; Programme National de Recherche sur la Diabete; Research Institute for Diseases in the Elderly (including grant number 014-93-015); Robert Dawson Evans Endowment, Department of Medicine, Boston University School of Medicine and Boston Medical Center; Royal Swedish Academy of Sciences; Sarstedt AG & Co., Germany; Signe and Ane Gyllenberg Foundation; Sigrid Juselius Foundation; Slottery Machine Association, Finland; Social Insurance Institution of Finland; South OstroBothnia Hospital District; State of Baden-Wurttemberg, Germany; Stockholm County Council (including grant number 560183); Swedish Cultural Foundation, Finland; Swedish Diabetes Foundation; Swedish e-science Research Center ; Swedish Foundation for Strategic Research; Swedish Heart-Lung Foundation; Swedish Research Council (including grant numbers SFO EXODIAB 2009-1039, 521-2010-3490, 521-2007-4037, 521-2008-2974, ANDIS 825-2010-5983, LUDC 349-2008-6589, and 8691); Swedish Society of Medicine; Tore Nilsson Foundation; Torsten and Ragnar Soderbergs Stiftelser (including grant number MT33/09); University Hospital Essen; University of Tromso; UCL NIHR Biomedical Research Centre; UK NIHR Cambridge Biomedical Research Centre; Uppsala University; Uppsala University Hospital; Vaasa Hospital District; Velux Foundation; Wellcome Trust (including the Biomedical Collections Grant GR072960, and grant numbers 076113, 083948, 090367, 090532, 083270, 086596, 098017, 095101, 098051, 098381). The authors are grateful to Raphael Scharfmann (INSERM U1016, Cochin Institute Paris) for the gift of EndoC BH-1 cells and for providing technical support with their maintenance. The authors thank Paul Johnson and the Oxford National Institute for Health Research Biomedical Research Centre funded Islet Isolation facility for providing human islets for this study. Detailed acknowledgements are provided in the Supplementary Note.
Footnotes
URLs
Endocells (www.endocells.com)
AUTHOR CONTRIBUTIONS
Writing group. K.J.G., T.F., Y.Lee, A.R., R.M., M.Reschen, A.L.G., D.A., M.Boehnke, T.M.T., M.I.M., A.P.M.
Central meta-analysis group. K.J.G., T.F., Y.Lee, R.M., A.Mahajan, A.Locke, N.W.R., N.R., T.M.T., M.I.M., A.P.M.
Annotation and functional analysis group. K.J.G., A.R., M.Reschen, S.K.T., J.K.R., N.L.B., M.v.d.B., A.C., I.D., E.Birney, L.Pasquali, J.Ferrer, C.A.O’C., A.L.G., M.I.M.
Validation meta-analysis group. R.M., R.A.S., I.P., L.J.S., A.P.M.
Metabochip cohort-level primary analysis. Y.Lee, T.G., T.S., D.T., L.Y., H.G., S.Wahl, M.F., R.J.S., H.Kestler, H.Chheda, L.E., S.G., T.M.T., A.P.M.
Validation cohort-level primary analysis. V.Steinthorsdottir, G.T., L.Q., L.C.K., E.v.L., S.M.W., M.Li, H.Chen, C.Fuchsberger, P.Kwan, C.M., M.Linderman, Y.Lu
Metabochip design. H.M.K., B.F.V.
Cohort sample collection, genotyping, phenotyping, or additional analysis. B.F.V., G.R.A., P.A., D.B., B.B., R.B., M.Blüher, H.B., L.L.B., E.P.B., N.B., J.C., G.C., P.S.C., M.C.C., D.J.C., A.T.C., R.M.v.D., A.S.F.D., M.D., S.E., J.G.E., T.E., E.E., J.Fadista, J.Flannick, P.Fontanillas, C.Fox, P.W.F., K.G., C.G., B.G., O.G., G.B.G., N.G., C.J.G., M.H., C.T.H., C.H., O.L.H., A.B.H., S.E.H., D.J.H., A.U.J., A.J., M.E.J., T.J., W.H.L.K, N.D.K., L.K., N.K., A.K., P.Kovacs, P.Kraft, J.Kravic, C.Langford, K.L., L.Liang, P.L., C.M.L., E.L., A.Linneberg, C.-T.L., S.L., J.L., V.L., S. Mӓnnistö, O.McLeod, J.M., E.M., G.M., T.W.M., M.M.-N., C.N., M.M.N., N.N.O., K.R.O., D.P., S.P., L.Peltonen, J.R.B.P., C.G.P.P., M.Roden, D.Ruderfer, D.Rybin., Y.T.v.d.S., B.S., G.Sigurđsson, A.S., G.Steinbach, P.S., K.Strauch, H.M.S., Q.S., B.T., E.Tikkanen, A.T., J.Trakalo, E.Tremoli, T.T., R.W., S.Wiltshire, A.R.W., E.Z.
Validation cohort principal investigators. R.L., J.D., J.C.F., E.Boerwinkle, J.S.P., C.v.D., E.S., J.B.M., F.B.H., U.T., K.Stefansson, P.D., P.J.D., T.M.F., A.T.H., I.B., C.Langenberg, N.J.W., M.Boehnke, M.I.M.
Metabochip cohort principal investigators. T.A.L., R.R., M.S., N.L.P., L.Lind, S.K.-K., E.K.-H., T.E.S., J.S., J.Kuusisto, M.Laakso, A.Metspalu, R.E., K.-H.J., S.Moebus, S.R., V.Salomaa, E.I., B.O.B., R.N.B., F.S.C., K.L.M., H.Koistinen, J.Tuomilehto, K.H., I.N., P.D., P.J.D., T.M.F., A.T.H., U.d.F., A.H., T.I., A.P., S.C., R.S., P.Froguel, O.P., T.H., A.D.M., C.N.A.P., S.K., O.Melander, P.M.N., L.C.G., I.B., C.Langenberg, N.J.W., D.A., M.Boehnke, M.I.M.
Project management. K.J.G., A.L.G., D.A., M.Boehnke, T.M.T., M.I.M., A.P.M.
DIAGRAM Consortium management. D.A., M.Boehnke, M.I.M.
COMPETING FINANCIAL INTERESTS
V.Steinthorsdottir, G.T., A.K., U.T., and K.Stefansson are employed by deCODE Genetics/Amgen inc. I.B. and spouse own stock in GlaxoSmithKline and Incyte.
References
- 1.Kooner JS, et al. Genome-wide association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nat Genet. 2011;43:984–989. doi: 10.1038/ng.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cho YS, et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in East Asians. Nat Genet. 2012;44:67–72. doi: 10.1038/ng.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Voight BF, et al. Twelve type 2 diabetes susceptibility loci identified through large scale association analysis. Nat Genet. 2010;42:579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morris AP, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44:981–990. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahajan A, et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet. 2014;46:234–244. doi: 10.1038/ng.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Altshuler D, et al. The common PPARγ Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet. 2000;26:76–80. doi: 10.1038/79216. [DOI] [PubMed] [Google Scholar]
- 7.Gloyn AL, et al. Large-scale association studies of variants in genes encoding the pancreatic-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) conӿrm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes. 2003;52:568–572. doi: 10.2337/diabetes.52.2.568. [DOI] [PubMed] [Google Scholar]
- 8.Sladek R, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–885. doi: 10.1038/nature05616. [DOI] [PubMed] [Google Scholar]
- 9.Dupuis J, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes. Nat Genet. 2010;42:105–116. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elbein SC, Gamazon ER, Das SK, Rasouli N, Kern PA, Cox NJ. Genetic risk factors for type 2 diabetes: a trans-regulatory genetic architecture? Am J Hum Genet. 2012;91:466–477. doi: 10.1016/j.ajhg.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trynka G, et al. Chromatin marks identify critical cell types for fine-mapping complex trait variants. Nat Genet. 2013;45:124–130. doi: 10.1038/ng.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parker SCJ, et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbour human disease risk variants. Proc Natl Acad Sci USA. 2013;110:17921–17926. doi: 10.1073/pnas.1317023110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pasquali L, et al. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat Genet. 2014;46:136–143. doi: 10.1038/ng.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Voight BF, et al. The Metabochip, a custom genotyping array for genetic studies of metabolic, cardiovascular, and anthropometric traits. PLoS Genet. 2012;8:e1002793. doi: 10.1371/journal.pgen.1002793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.International HapMap Project Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet. 2010;11:499–511. doi: 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 20.Winkler TW, et al. Quality control and conduct of genome-wide association meta-analyses. Nat Protoc. 2014;9:1192–1212. doi: 10.1038/nprot.2014.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44:955–959. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang J, et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet. 2012;44:369–375. doi: 10.1038/ng.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Unoki H, et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet. 2008;40:1098–1102. doi: 10.1038/ng.208. [DOI] [PubMed] [Google Scholar]
- 25.Fitzpatrick GV, Soloway PD, Higgins MJ. Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat Genet. 2002;32:426–431. doi: 10.1038/ng988. [DOI] [PubMed] [Google Scholar]
- 26.Zeggini E, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shea J, et al. Comparing strategies to fine-map the association of common SNPs at chromosome 9p21 with type 2 diabetes and myocardial infarction. Nat Genet. 2011;43:801–805. doi: 10.1038/ng.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maller JB, et al. Bayesian refinement of association signals for 14 loci in 3 common diseases. Nat Genet. 2012;44:1294–1301. doi: 10.1038/ng.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jafar-Mohammadi B, et al. A role for coding functional variants in HNF4A in type 2 diabetes susceptibility. Diabetologia. 2011;54:111–119. doi: 10.1007/s00125-010-1916-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teslovich TM, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Florez JC, et al. Haplotype structure and genotype-phenotype correlations of the sulfonylurea receptor and the islet ATP-sensitive potassium channel gene. Diabetes. 2004;53:1360–1368. doi: 10.2337/diabetes.53.5.1360. [DOI] [PubMed] [Google Scholar]
- 32.Hamming KS, et al. Co-expression of the type 2 diabetes susceptibility gene variants KCNJ11 E23K and ABCC8 S1369A alter the ATP and sulfonylurea sensitivities of the ATP-sensitive K+ channel. Diabetes. 2009;58:2419–2424. doi: 10.2337/db09-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nicolson TJ, et al. Insulin storage and glucose homeostasis in mice null for the granule zinc transporter ZnT8 and studies of the type 2 diabetes-associated variants. Diabetes. 2009;58:2070–2083. doi: 10.2337/db09-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beer NL, et al. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum Mol Genet. 2009;18:4081–4088. doi: 10.1093/hmg/ddp357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holmkvist J, et al. Common variants in HNF-1 alpha and risk of type 2 diabetes. Diabetologia. 2006;49:2882–2891. doi: 10.1007/s00125-006-0450-x. [DOI] [PubMed] [Google Scholar]
- 36.Yamagata K, et al. Mutations in the hepatocyte nuclear factor-1 alpha gene in maturity-onset diabetes of the young (MODY3) Nature. 1996;384:455–458. doi: 10.1038/384455a0. [DOI] [PubMed] [Google Scholar]
- 37.Yamagata K, et al. Mutations in the hepatocyte nuclear factor-4 alpha gene in maturity-onset diabetes of the young (MODY1) Nature. 1996;384:458–460. doi: 10.1038/384458a0. [DOI] [PubMed] [Google Scholar]
- 38.Gusev A, et al. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am J Hum Genet. 2014;95:535–552. doi: 10.1016/j.ajhg.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soccio RE, Tuteja G, Everett LJ, Li Z, Lazar MA, Kaestner KH. Species-specific strategies underlying conserved functions of metabolic transcription factors. Mol Endocrinol. 2011;25:694–706. doi: 10.1210/me.2010-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gaulton KJ, et al. A map of open chromatin in human pancreatic islets. Nat Genet. 2010;42:255–259. doi: 10.1038/ng.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fogarty MP, Cannon ME, Vadlamudi S, Gaulton KJ, Mohlke KL. Identification of a regulatory variant that binds FOXA1 and FOXA2 at the CDC123/CAMK1D type 2 diabetes GWAS locus. PLoS Genet. 2014;10:e1004633. doi: 10.1371/journal.pgen.1004633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manning AK, et al. A genome-wide approach accounting for body-mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet. 2012;44:659–669. doi: 10.1038/ng.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dimas AS, et al. Impact of type 2 diabetes susceptibility variants on quantitative glycemic traits reveals mechanistic heterogeneity. Diabetes. 2014;63:2158–2171. doi: 10.2337/db13-0949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ravassard P, et al. A genetically engineered human pancreatic β cell line exhibiting glucose-inducible insulin secretion. J Clin Invest. 2011;121:3589–3597. doi: 10.1172/JCI58447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fadista J, et al. Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc Natl Acad Sci USA. 2014;111:13924–13929. doi: 10.1073/pnas.1402665111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lyssenko V, et al. Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nat Genet. 2009;41:82–88. doi: 10.1038/ng.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao N, et al. Foxa1 and Foxa2 maintain the metabolic and secretory features of the mature beta-cell. Mol Endocrinol. 2010;24:1594–1604. doi: 10.1210/me.2009-0513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou Y, et al. TCF7L2 is a master regulator of insulin production and processing. Hum Mol Genet. 2014;23:6419–6431. doi: 10.1093/hmg/ddu359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lyssenko V, et al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J Clin Invest. 2007;117:2155–2163. doi: 10.1172/JCI30706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gloyn AL, et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med. 2004;350:1838–1849. doi: 10.1056/NEJMoa032922. [DOI] [PubMed] [Google Scholar]
- 51.Flannick J, et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat Genet. 2014;46:357–363. doi: 10.1038/ng.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8:e1000294. doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bonnefond A, et al. Rare MTNR1B variants impairing melatonin receptor 1B function contribute to type 2 diabetes. Nat Genet. 2012;44:297–301. doi: 10.1038/ng.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zaret KS, Carroll JS. Pioneer transcription factors: establishing a competence for gene expression. Genes Dev. 2011;25:2227–41. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gao N, LeLay J, Vatamaniuk MZ, Rieck S, Friedman JR, Kaestner KH. Dynamic regulation of Pdx1 enhancers by Foxa1 and Foxa2 is essential for pancreas development. Genes Dev. 2008;22:3435–3448. doi: 10.1101/gad.1752608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee CS, Friedman JR, Fulmer JT, Kaestner KH. The initiation of liver development is dependent on Foxa transcription factors. Nature. 2005;435:944–947. doi: 10.1038/nature03649. [DOI] [PubMed] [Google Scholar]
- 57.Scott RA, et al. Large-scale association analyses identify new loci influencing glycaemic traits and provide insight into the underlying biological pathways. Nat Genet. 2012;44:991–1005. doi: 10.1038/ng.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tabassum R, Chavali S, Dwivedi OP, Tandon N, Bharadwaj D. Genetic variants of FOXA2: risk of type 2 diabetes and effect on metabolic traits in North Indians. J Hum Genet. 2008;53:957–965. doi: 10.1007/s10038-008-0335-6. [DOI] [PubMed] [Google Scholar]
- 59.Johnson ME, Schug J, Wells AD, Kaestner KH, Grant SF. Genome-wide analyses of ChIP-Seq derived FOXA2 DNA occupancy in liver points to genetic networks underpinning multiple complex traits. J Clin Endocrinol Metab. 2014;99:E1580–E1585. doi: 10.1210/jc.2013-4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 61.Wakefield JA. Bayesian measure of the probability of false discovery in genetic epidemiology studies. Am J Hum Genet. 2007;81:208–227. doi: 10.1086/519024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinf. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li H, et al. The sequence alignment/map format and SAMtools. Bioinf. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kharchenko PV, Tolstorukov MY, Park PJ. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol. 2008;26:1351–1359. doi: 10.1038/nbt.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li Q, Brown JB, Huang H, Bickel PJ. Measuring reproducibility of high-throughput experiments. Ann App Stat. 2011;5:1752–1779. [Google Scholar]
- 66.Mikkelsen TS, et al. Comparative epigenomic analysis of murine and human adipogenesis. Cell. 2010;143:156–169. doi: 10.1016/j.cell.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol. 2010;28:817–825. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Morán I, et al. Human β cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metab. 2012;16:435–448. doi: 10.1016/j.cmet.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinf. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Machanick P, Bailey TL. MEME-ChiP. Motif analysis of large DNA datasets. Bioinf. 2011;27:1696–1697. doi: 10.1093/bioinformatics/btr189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mathelier A, et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 2014;42:D142–D147. doi: 10.1093/nar/gkt997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and b-cell identifies. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bailey TL, et al. MEME Suite: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinf. 2011;27:1017–1018. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pugh CW, Tan CC, Jones RW, Ratcliffe PJ. Functional analysis of an oxygen-regulated transcriptional enhancer lying 3′ to the mouse erythropoietin gene. Proc Natl Acad Sci USA. 1991;88:10553–10557. doi: 10.1073/pnas.88.23.10553. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.