

Abstract
We introduce the modification of bacteriophage particles with aptamers for the use as bioanalytical reporters, and demonstrate the use of these particles in ultrasensitive lateral flow assays. M13 phage displaying an in vivo biotinylatable peptide (AviTag) genetically fused to the phage tail protein pIII were used as reporter particle scaffolds, with biotinylated aptamers attached via avidin-biotin linkages, and horseradish peroxidase (HRP) reporter enzymes covalently attached to the pVIII coat protein. These modified viral nanoparticles were used in immunochromatographic sandwich assays for the direct detection of IgE and of the penicillin-binding protein from Staphylococcus aureus (PBP2a). We also developed an additional lateral flow assay for IgE, in which the analyte is sandwiched between immobilized anti-IgE antibodies and aptamer-bearing reporter phage modified with HRP. The limit of detection of this LFA was 0.13 ng/mL IgE, ~100 times lower than those of previously reported IgE assays.
INTRODUCTION
Lateral flow assays (LFAs), in which reporter particles are transported by capillary wicking in a porous material and accumulate to form a visible line in the presence of analyte, are widely used because of their convenience, low cost, and adaptability to use at the point-of-care 1-3. LFAs most often employ antibodies as recognition elements, but nucleic acid recognition elements have increasingly been employed, not just in lateral flow nucleic acid hybridization assays 4, but also in protein detection assays where target-specific RNA or DNA aptamers serve as the molecular recognition element 1,5-9. Signal readout is most commonly achieved by using visible nanoparticles (gold, blue latex, or carbon). But even if the classic LFA is a brilliant approach to delivering assays in a rapid and easy-to-use format, the LFA limits of detection (LOD) lag behind more complex laboratory methods, e.g. ELISA. Thus the development of alternative LFA readout reporters to increase assay sensitivity is a topic of great commercial and academic interest.
Bacteriophages have been explored in recent years for use as reporters for immunoassays that enable both improved recognition and enhanced signal readout. The surface of phage, consisting of multiple copies of identical coat proteins, can be readily modified using well-established chemistries 10-13. M13 phage, in particular, have been modified with recognition elements (e.g. antibodies) and/or reporter elements (e.g. Horseradish peroxidase (HRP), fluorophores), and have been successfully used in various protein or small molecule detection assays, e.g. enzyme-linked immunosorbent assays (ELISA) 14-16, microarrays 17 or other protein sensors 18,19. PCR amplification of the phage genetic material has led to the development of ultra-sensitive immuno-phage PCR assays by ourselves and others 20-22.
We recently introduced the use of engineered bacteriophage particles as reporters in lateral flow assays 23-25. M13 phage particles functionalized with both target-specific antibodies and enzyme reporters were integrated into an immunochromatographic lateral flow assay (LFA) for the detection of a model virus (MS2), leading to greatly enhanced detection sensitivity (one thousand-fold better than a conventional gold nanoparticle LFA using the same antibodies). Our strategy often relies upon a phage-displayed “AviTag” peptide 26-28 which can be biotinylated by biotin ligase, either in vivo during phage assembly in Escherichia coli or in vitro through treatment of purified phage particles with the enzyme 24. This strategy also allows the convenient modification of phage reporter particles with any biotinylated molecular recognition agent, including antibodies and (as introduced here) aptamers.
Here we report the use of engineered phage particles modified with aptamers on their tail proteins and bearing reporter enzymes, in lateral flow assays with improved limits of detection, compared to a conventional gold nanoparticle assay. The conjugation of aptamers to phage constitutes a novel and promising approach to the point-of-care detection of proteins. This approach may also readily be extended to the development of higher-sensitivity lateral flow nucleic acid hybridization assays.
EXPERIMENTAL SECTION
Materials and Reagents
Polyclonal anti-IgE antibody was purchased from Fitzgerald Industries International (Acton, MA, USA). Human immunoglobulin E (IgE) was obtained from Abcam (Cambridge, MA, USA). An oligonucleotide bearing a 5’ biotin and TEG (triethylene glycol) spacer with sequence matching that of a previously-reported DNA aptamer binding human IgE 29 was purchased from Integrated DNA Technologies (Coralville, IA, USA) (Figure 1). In addition, an aptamer specific for the Staphylococcus aureus penicillin-binding protein 2a (PBP2a) (5’-biotinylated; proprietary sequence), was purchased from Base Pair Technologies (Pearland, TX, USA). It is a DNA aptamer that was selected from a randomized 32-mer library against MRSA (Methicillin-resistant Staphylococcus aureus) protein. Since IgE aptamer is a DNA aptamer, a DNA aptamer (PBP2a) was used as an appropriate control for the aptamer-phage LFA. PBP2a protein was purchased from Ray Biotech (Norcross, GA, USA).
Figure 1.
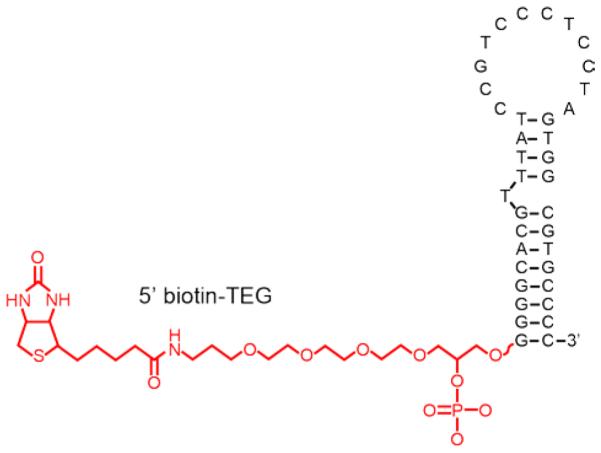
Biotin-TEG IgE aptamer (secondary structure adapted from Reference 29).
Propagation and modification of SAM-AviTag Phage
SAM (for serine-amber)-AviTag M13 phage, a gift from Dr. Brian Kay, UIC (Chicago, IL, USA) 26,27,30, are derivatives of M13 phage displaying an AviTag peptide (a 15-amino-acid peptide that is a substrate for biotin ligase (BirA)) on the phage coat protein, pIII. The phage were propagated, titered, biotinylated and conjugated with horseradish peroxidase largely as previously described 24. Briefly, phage were initially grown on E. coli TG1 in LB medium at 37 °C, and this infected culture was then transferred to 500 mL 2xTY 31 medium in a 2 L flask and incubated overnight at 37 °C on a shaker. After centrifugation, the phage-containing supernatant was passed through a 0.45 μm filter and phage were purified by precipitation with 20% polyethylene glycol (PEG) in 2.5 M NaCl. Phage titers were determined by plaque assay on X-Gal/IPTG plates 32. The pIII protein of SAM-AviTag phage was enzymatically biotinylated using E. coli biotin ligase (Avidity, Aurora, CO) according to the manufacturer’s instructions; biotin ligase also can be prepared in-house as previously described 20. The efficiency of biotinylation was determined through an ELISA on streptavidin-coated microtiter plates (StreptaWell High Bind, Roche Applied Science, Indianapolis, IN). Subsequently, phage were activated using Traut’s reagent (2- iminothiolane-HCl, Thermo Fisher Scientific, Waltham, MA). 100 μL of a 1×1011 phage per mL solution were suspended in 800 μL phosphate-buffered saline (PBS), pH 7.4, 3 mM EDTA with a 20-fold molar excess of Traut's reagent. This reaction was continued for 90 min at 25 °C on a rotator. Excess Traut's reagent was removed using a 10 kDa filter (Millipore, Billerica, MA). HRP (Sigma-Aldrich, St. Louis, MO) was modified with maleimide groups using sulfo-SMCC (succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate, Sigma-Aldrich) by mixing HRP and sulfo-SMCC at a final concentration of 22 μM and 1.14 mM, respectively and incubating for 30 min. Excess sulfo-SMCC was removed using a 10 kDa filter. Traut’s reagent-activated phage and maleimide-HRP were then incubated together for 90 min at 25 °C (270,000 molecules of HRP were offered per phage). Uncoupled HRP was then removed using 100 kDa filters (Millipore). Subsequently, 1 μL of 1 mg/mL of Neutravidin (Thermo Fisher Scientific) was then offered to the HRP-labeled phage and bound through the biotinylated AviTag displayed on phage protein pIII (100 molecules of Neutravidin were offered per phage). Uncoupled Neutravidin was removed using 100 kDa filters (Millipore).
Aptamer modification of avidin phage
Biotinylated IgE aptamer (100 μl, 100 nM in PBS, pH 7.0, 1 mM MgCI2) was first heat-denatured at 95 °C for 10 min (to disrupt any pre-existing higher-order structures that might interfere with phage functionalization and to allow the 5’biotin to interact freely with its avidin binding partner 33) and then slowly renatured at room temperature. Subsequently, the biotinylated IgE aptamer was added to 1012 neutravidin-functionalized phage in 100 μL PBS. The complex was incubated for 2 hrs at 25 °C before unconjugated IgE aptamer was removed using PEG precipitation (Figure 2). As a control, phage were functionalized with the anti-PBP2a aptamer in the same fashion. A series of 10-fold dilutions (105 to 1010 phage/mL) of unmodified phage (titer obtained by plaque counting) was used as PCR calibration standards and the concentration of the various phage constructs was determined against that standard curve. The AviTag-targeted PCR primers were: Forward: 5’-GTTGTTTCTTTCTATTCTCACTCC-3’ and Reverse: 5’- CAGACGTTAGTAAATGAATTTTCTG -3’. For the phage amplification 0.1 μL of a 10 μM stock of each primer were combined with 10 μL 2xPCR mix (Brilliant III Ultra-Fast SYBR mix, Agilent, Santa Clara, CA), 4.8 μL ultra-pure (RNase and DNase free) water and 5 μL of each phage sample to achieve a total volume of 20 μL. The PCR conditions were: 10 min at 95 °C, 40 cycles of 30 sec at 95 °C, 30 sec at 62 °C and 30 sec at 72 °C, followed by a dissociation step to detect possible contaminations (1 min at 95 °C, 30 sec at 55 °C, and 30 sec at 95 °C).
Figure 2.
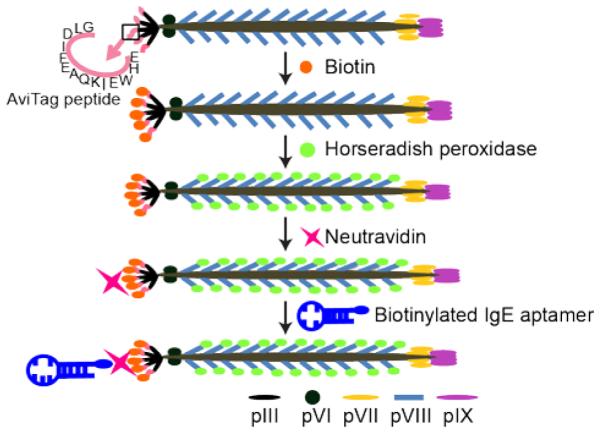
Functionalization of M13 phage with IgE aptamers and Horseradish peroxidase (HRP). Phage displaying AviTag peptides are biotinylated using Biotin ligase. Biotinylated phage are covalently modified with HRP on the major coat protein pVIII before neutravidin is bound to biotinylated AviTag on HRP-labeled phage. Neutravidin-functionalized phage are then conjugated with the biotinylated IgE aptamer.
Preparation of LFA strips
Figure 3 depicts the schematic of the aptamer-phage LFA. Polyclonal anti-human IgE rabbit serum (Fitzgerald Industries International, Acton, MA; 5 μl, diluted 100-fold in 50 mM sodium acetate buffer (pH 3.6) based on preliminary screening experiments) and 5 μL of rabbit polyclonal anti-M13 IgG antibodies (Novus Biologicals, Littleton, CO; 0.1 μg/μL in 50 mM sodium acetate buffer (pH 3.6)) were used to make test and control lines respectively on Fusion 5 membranes (GE Healthcare, Piscataway, NJ) using a Lateral Flow Reagent Dispenser (Claremont BioSolutions, Upland, CA) equipped with an external syringe pump (Chemyx, Stafford, TX). The Fusion 5 membrane was cut into 7 mm × 50 mm strips using a guillotine paper cutter. CF5 membrane (GE Healthcare) was used as the absorbent pad (7 mm × 25 mm) and Fusion 5 membrane (7 mm × 10 mm) was used as a sample pad. The strips were allowed to dry for 1 hr at 25 °C before use.
Figure 3.

Aptamer-phage lateral flow assay. The M13 phage SAM-AviTag protein, pIII, was specifically biotinylated and functionalized with analyte-specific biotinylated aptamers, and the M13 coat proteins were functionalized with Horseradish peroxidase. The detection line contains IgE-specific antibodies, and the control line has anti-M13 antibodies. Captured HRP-labeled phage were detected on the test and control lines using the chromogenic substrate, 3’,3,5,5’-tetramethylbenzidine (TMB).
Lateral flow assay with aptamer-phage
A 10-fold dilution series of human IgE was prepared in PBS, pH 7.0 with 0.5% bovine serum albumin (BSA). Each IgE sample (100 μL) was dispensed onto the sample application area of a prepared LFA strip. The strips were then washed with 200 μL LFA buffer (50-μl aliquots; 4x) PBS, pH 7.0; 1% Tween-20; 1% Triton; 0.1% PEG-3350) followed by 10 μL of the aptamer-phage reporter construct (~109 phage), and finally with 500 μL LFA buffer (50-μl aliquots; 10x). Signals were visualized by spotting 25 μL TMB Liquid Substrate System for Membranes (Sigma-Aldrich, St. Louis, MO) on each line. The signals were allowed to develop for 10 min and then the strips were scanned on a Perfection V600 flatbed color scanner (Epson, Long Beach, CA). The scanned images were analyzed using ImageJ’s Gel Analysis Tool and by plotting the line intensity profile. The intensity of each line was given by the area under each peak that was numerically integrated using ImageJ’s Gel Analysis Toolbox. The ratio of the intensity of the test line to that of the control line for each strip (T/C) was used as an indicator of signal strength over the background.
RESULTS AND DISCUSSION
Evaluation of aptamer-phage for direct detection of spotted IgE and PBP2a in LFA
Initial LFA experiments tested the binding of the anti-IgE and anti-PBP2a aptamer-phage directly to IgE and PBP2a proteins, respectively, spotted on a Fusion 5 membrane. For the IgE experiments, 5 μL IgE (0.2 mg/mL) and 5 μL anti-M13 antibodies (0.1 mg/mL) in 50 mM sodium acetate buffer (pH 3.6) were spotted on the test and control lines, respectively, of a Fusion 5 membrane. For the anti-PBP2 aptamer-phage, 5 μL PBP2a protein (0.2 mg/mL) was spotted on the test line and anti-M13 antibodies on the control line. The strips were dried for 1 hr at room temperature. Aptamer-phage dilutions (10 μl) ranging from 107 to 109 phage were dispensed onto the sample application area of the LFA strip. The strips were washed with 500 μL LFA buffer and signals were obtained by spotting TMB Liquid Substrate on each line. As shown in Figure 4, a clear signal was obtained at the anti-M13 control line for all samples containing >107 aptamer-phage. This result confirms that the aptamer-phage moved successfully through the membrane. The signals obtained at the test lines using 108 and 107 aptamer-phage were weak, but with 109 phage the signals for both IgE and PBP2a were clear and distinguishable from other aptamer-phage concentrations (Figure 4). Tests with phage modified with HRP but no aptamer gave no visible test line signal with either protein, thus confirming that M13 phage without aptamers are not retained non-specifically by IgE or PBP2a protein at the test line but still yield a specific signal at the anti-M13 control line.
Figure 4.

Direct detection of spotted target analytes (PBP2a (A) or IgE (B)) with aptamer-phage. (A) PBP2a protein and anti-M13 antibodies were spotted on the test and control lines, respectively. (B) IgE protein and anti-M13 antibodies were spotted on the test and control lines, respectively. Different concentrations of aptamer-phage constructs were then passed through the membranes. Phage bearing HRP reporters but no aptamers (HRP-M13) served as a specificity control.
Lateral flow sandwich immuno-aptamer assay for IgE detection
We next developed a sandwich IgE LFA based on the IgE-dependent capture of phage modified with anti-IgE aptamers and HRP on anti-IgE polyclonal antibodies at a test line on Fusion 5 strips. Serial dilutions of the IgE test analyte ranging from 0.013-130 ng/mL were prepared in PBS at pH 7.0 with 0.5% BSA. Each IgE sample (100 μL) was dispensed onto the sample application area of the prepared LFA strips. A clear signal on the anti-M13 antibody control line for all samples confirmed that the phage reporters had successfully moved through the membrane. A signal clearly distinguishable from the no-IgE control was obtained at the test line of the strip with 0.13 ng/mL IgE (0.68 pM) (Figure 5, Table S1). The relative intensities of the test lines were divided by the control line intensities (T/C) to adjust for any background and this ratio was used as the reported LFA signal. The limit of detection (LOD) was determined using Student’s t-test from five independent replicate LFAs. We report the LOD for IgE detection using aptamer-phage to be at least 0.13 ng/mL, defined as the lowest tested concentration that was significantly different from the negative control with no IgE at a significance level of α=0.05. Table S2 in the supplementary material shows that the average T/C differs between the negative and the positive control at a significant level of α=0.05, and we thus reject the null hypothesis for all the IgE concentrations except for 0.013 ng/mL. Therefore the LOD of our assay is concluded to be at or lower than 0.13 ng/mL, in that the positive control signal from 0.13 ng/mL of the analyte concentration is statistically distinguishable from the negative control.
Figure 5.
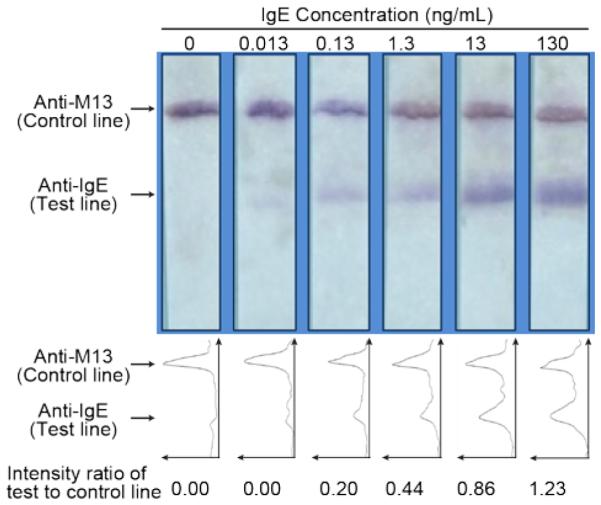
Lateral flow assay with aptamer-phage for IgE detection. Various concentrations of IgE (volume: 10 μL) were detected using anti-IgE antibodies at the test line and IgE aptamer-HRP-phage reporters. Control line consisted of anti-M13 antibodies. Line intensity profiles as evaluated by ImageJ density analysis of the LFA strips are shown below each strip. The area under each peak was numerically integrated using the ImageJ Gel Analysis Toolbox to give the intensity for that line. The intensity of the test line divided by the intensity of the control line for each strip is shown below each line intensity profile.
Compared to previously reported assays using the same anti-IgE aptamer, our phage-based LFA achieves a greater sensitivity in detecting IgE with a reduction in the LOD by ~230 times compared to the assay reported by Tran et al. 34 (30 ng/mL), who used an aptamer-based impedimetric biosensor using a nanocrystalline diamond (NCD) film as a working electrode to detect IgE and by ~8 times compared to the assay reported by Wei et al. 35 (10 ng/mL) who used silver nanoparticles modified with the IgE aptamers to detect captured IgE on microarrays with metal-enhanced fluorescence. It is noteworthy that Tran et al. 34 reported similar LOD for the detection of IgE in buffer and in human serum using this aptamer. Initial testing with human IgE spiked into human serum confirms that these samples are compatible with phage transport through the LFA strip, preservation of LOD, and specific capture of the phage constructs on the test line (preliminary data not shown).
Currently the most popular FDA-cleared assay for IgE detection in serum is the ELISA-based ImmunoCAP system, which requires considerable investment in instrumentation and a moderately high cost per assay with a detection range of total IgE of 0-100 kU/I (0-240 ng/mL; 1 kU/I = 2.4 ng/mL). ImmunoCAP Rapid is a related commercially available qualitative lateral flow test that measures specific IgE in whole blood and its sensitivity ranges between 0.72-1.68 ng/mL. The ALFA (Allergy Lateral Flow Assay) Total IgE test from Dr. Fooke Laboratories (GmbH) reports a detection sensitivity of 12 ng/mL in serum or whole blood.
Aptamer-phage specificity
The specificity of the aptamer-phage LFA was tested by spotting the unrelated murine IgG1 anti-hen egg lysozyme antibody, HyHEL-5 36, onto the test line. Neither the no-IgE samples, nor 13 ng/mL IgE gave any signals at the HyHEL-5 test line, implying that neither IgE nor aptamer-phage bind non-specifically to this unrelated antibody (Figure 6, Strips 1 and 2). Furthermore, no signal was observed when HyHEL-5 was passed as a mock-analyte on an LFA membrane with an anti-IgE test line followed by anti-IgE aptamer-phage (Figure 6, Strip 7). The strips where IgE was added confirmed that IgE binds specifically to the anti-IgE test line (Figure 6, Strips 3, 4 and 5) and functionalized aptamer-phage are subsequently retained, hence demonstrating the specific performance of the aptamer-phage LFA for IgE detection.
Figure 6.

Control for non-specific binding of IgE and aptamer-phage to an unrelated protein (the murine anti-lysozyme IgG antibody HyHEL-5). Strips 1 & 2; HyHEL-5 was spotted on the test line and anti-M13 antibodies on the control line. IgE protein (100 μL in LFA buffer) was passed through the membrane and aptamer-phage were added as reporters. No signal was observed on the HyHEL-5 test line. Strips 3, 4 & 5; To confirm specificity and sensitivity of the assay, varied concentrations of IgE were passed through the membrane. Strip 6; Competition assay where free IgE aptamer was passed on the strip prior to offering the aptamer-phage construct; 130 ng/mL IgE in 100 μL of LFA buffer were passed, followed by soluble IgE aptamer (10 μM aptamer in 100 μL of LFA buffer) and then the aptamer-phage construct was offered. No signal was observed on the test line confirming that soluble anti-IgE aptamer competes with aptamer-phage for the available binding sites on the IgE protein. Strip 7; 100 μL of HyHEL-5 in LFA buffer was passed through the membrane with anti-IgE and anti-M13 lines and detected using the IgE aptamer-phage reporter. No signal was observed on the HyHEL-5 test line, confirming specificity of the assay. Line intensity profiles as evaluated by ImageJ density analysis of the LFA strips. The area under each peak was numerically integrated using the ImageJ Gel Analysis Toolbox to give the intensity for that line. The intensity of the test line divided by the intensity of the control line for each strip is shown below each line intensity profile.
The specificity of the IgE LFA was also demonstrated by a competition assay (Figure 6, Strip 6). Here IgE (130 ng/mL) was passed through the membrane (with anti-IgE test line and anti-M13 control line) followed by an excess amount of soluble anti-IgE aptamer (10 μM aptamer in 100 μL of LFA buffer) to saturate the binding sites on the captured IgE protein and subsequently adding the anti-IgE aptamer-phage reporters. We previously confirmed that IgE was captured in the anti-IgE test line, as shown in Figure 6, Strips 3, 4, and 6. However, the excess of competing soluble IgE aptamer occupies the sites on the captured IgE, so anti-IgE phage cannot bind and hence no signal was observed, indicating that phage do not bind non-specifically by interactions other than those mediated by aptamer-IgE recognition.
PBP2a aptamer-phage as specificity control for IgE-aptamer-phage reporters
As a control for the specificity of the IgE aptamer-phage construct, we used an aptamer-phage functionalized with a proprietary aptamer recognizing the Staphylococcus aureus penicillin-binding protein, PBP2a. This PBP2a-M13 phage construct was prepared in a fashion analogous to that used for the anti-IgE aptamer-phage construct.
IgE (13 ng in 100 μL of LFA buffer) was passed through the strip, followed by the PBP2a-M13 phage construct. As expected, the anti-PBP2a aptamer on the phage did not recognize the IgE protein captured on the test line and hence there was no signal (Figure 7, Strip 6). To confirm the performance of the PBP2a-M13 phage construct, strips were made with PBP2a protein and anti-M13 antibodies on the test and control lines, respectively (Figure 7, Strip 2). A clear signal was obtained on the strip where the PBP2a-M13 phage construct were applied, indicating detection of the target PBP2a protein. As a further control, the strip with HRP-phage reporters bearing no PBP2a aptamers did not show any signal, indicating that phage do not bind non-specifically to the PBP2a protein (Figure 7, Strip 1). The strips where IgE was added reveal that IgE binds specifically to the anti-IgE test line (Figure 7, Strips 4 & 5) and functionalized aptamer-phage are subsequently retained there, hence demonstrating the efficient performance of the aptamer-phage LFA for IgE detection. The test and control lines were quantified by ImageJ density’s analysis function.
Figure 7.
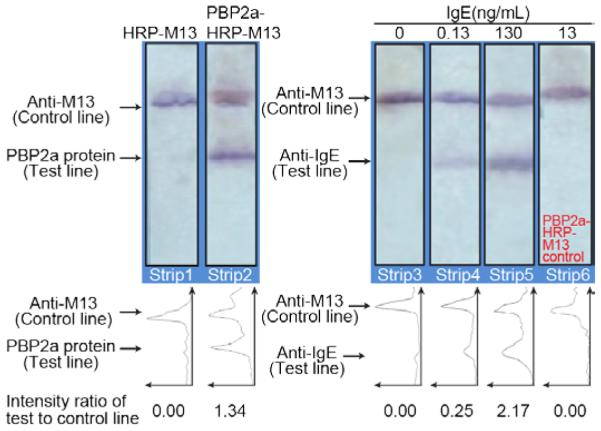
PBP2a aptamer-phage as a specificity control for IgE-aptamer-phage reporters. Strips 1 & 2; Strips with PBP2a protein and anti-M13 antibodies on the test and control lines, respectively, were made to evaluate the binding of PBP2a-M13 to PBP2a protein. A clear signal on the test line of strip 2 indicates binding of the PBP2a-phage to the PBP2a protein. Strip 1 with HRP-phage devoid of any signal, confirms that phage do not bind non-specifically to the PBP2a protein. Strips 3, 4 & 5 show the various concentrations of IgE protein (100 μL in LFA buffer) that were used. IgE binds on the anti-IgE test line. Signals on the control lines indicate the proper functioning of the assay. Strip 6; A PBP2a-M13 phage construct was passed through a strip with anti-IgE and anti-M13 lines. The anti-PBP2a aptamer on the phage did not bind to the IgE protein on the test line as indicated by the absence of any signal on the test line. Line intensity profiles as plotted by ImageJ density analyses of the LFA strips are shown below each figure. The area under each peak was numerically integrated using the ImageJ Gel Analysis Toolbox, and the average intensity of the test line divided by the intensity of the control line for each strip is also shown below each line intensity profile.
CONCLUSIONS
We have demonstrated a novel, specific and highly sensitive lateral flow assay for the detection of proteins using viral nanoparticles as scaffolds bearing aptamers as bio-recognition elements. With further refinement, the use of aptamer-phage as reporters in lateral flow assays promises a convenient, cost-effective and specific framework for the detection of analytes. Compared to antibodies, aptamers have lower production cost, and can be easily and rapidly be selected and synthesized. They are chemically more stable, and can be selected to have high affinity towards their targets. The 2,700 copies of the M13 coat protein provide multiple sites for enzyme conjugation, and thus an enhanced signal per individual affinity agent is generated, resulting in increased sensitivities. Phage nanoparticles represent versatile scaffolds with high colloidal stability and very low non-specific binding that can accommodate a variety of recognition and reporter elements, thus making them broadly applicable and ultrasensitive bio-detection reporters.
While the aim of the present study was primarily to demonstrate the feasibility and performance of aptamer-phage LFAs, the success of this approach suggests that phage bearing nucleic acids also could serve as the basis of high-sensitivity lateral-flow nucleic acid assays. It is also worth noting that the detection of IgE, used as a model here, has clinical value in itself. IgE is known to play an important role in the allergic response and is widely reported as a marker of atopic diseases such as asthma, dermatitis and pollenosis 37 . Testing of specific IgE, particularly at low levels of IgE, can be helpful in evaluating a patient's sensitivity profile and risk factors for other severe conditions such as anaphylaxis.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by in part by the Welch Foundation [E-1264], NIH [U54 AI057156 and 1R21AI111120-01A1] and National Science Foundation [CBET-1511789]. The contents of the paper are solely the responsibility of the authors and do not necessarily represent the official views of the funding agencies. Postdoctoral scholarships for AEV Hagström from the Olle Engkvist Byggmästare Foundation and Carl Trygger Foundation are gratefully acknowledged. We thank Bill Jackson for helpful discussions.
Footnotes
Supporting Information.
Details on test line intensity profiles and Student t-tests. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Gubala V, Harris LF, Ricco AJ, Tan MX, Williams DE. Anal Chem. 2012;84:487–515. doi: 10.1021/ac2030199. [DOI] [PubMed] [Google Scholar]
- (2).Ngom B, Guo Y, Wang X, Bi D. Anal Bioanal Chem. 2010;397:1113–1135. doi: 10.1007/s00216-010-3661-4. [DOI] [PubMed] [Google Scholar]
- (3).Anfossi L, Baggiani C, Giovannoli C, D'Arco G, Giraudi G. Anal Bioanal Chem. 2013;405:467–480. doi: 10.1007/s00216-012-6033-4. [DOI] [PubMed] [Google Scholar]
- (4).Carter DJ, Cary RB. Nucleic Acids Res. 2007;35:e74. doi: 10.1093/nar/gkm269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Liu G, Mao X, Phillips JA, Xu H, Tan W, Zeng L. Anal Chem. 2009;81:10013–10018. doi: 10.1021/ac901889s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Fang Z, Wu W, Lu X, Zeng L. Biosens Bioelectron. 2014;56:192–197. doi: 10.1016/j.bios.2014.01.015. [DOI] [PubMed] [Google Scholar]
- (7).Wong RC, Tse HY. Lateral flow immunoassay. Springer; New York: 2009. [Google Scholar]
- (8).Bruno JG. Pathogens. 2014;3:341–355. doi: 10.3390/pathogens3020341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Chen A, Yang S. Biosens Bioelectron. 2015;71:230–242. doi: 10.1016/j.bios.2015.04.041. [DOI] [PubMed] [Google Scholar]
- (10).Li K, Chen Y, Li S, Nguyen HG, Niu Z, You S, Mello CM, Lu X, Wang Q. Bioconjug Chem. 2010;21:1369–1377. doi: 10.1021/bc900405q. [DOI] [PubMed] [Google Scholar]
- (11).Sapsford KE, Soto CM, Blum AS, Chatterji A, Lin T, Johnson JE, Ligler FS, Ratna BR. Biosens Bioelectron. 2006;21:1668–1673. doi: 10.1016/j.bios.2005.09.003. [DOI] [PubMed] [Google Scholar]
- (12).Strable E, Finn MG. Curr Top Microbiol Immunol. 2009;327:1–21. doi: 10.1007/978-3-540-69379-6_1. [DOI] [PubMed] [Google Scholar]
- (13).Soto CM, Ratna BR. Curr Opin Biotech. 2010;21:426–438. doi: 10.1016/j.copbio.2010.07.004. [DOI] [PubMed] [Google Scholar]
- (14).Kim HJ, Ahn KC, Gonzalez-Techera A, Gonzalez-Sapienza GG, Gee SJ, Hammock BD. Anal Biochem. 2009;386:45–52. doi: 10.1016/j.ab.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kim HJ, Rossotti MA, Ahn KC, Gonzalez-Sapienza GG, Gee SJ, Musker R, Hammock BD. Anal Biochem. 2010;401:38–46. doi: 10.1016/j.ab.2010.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Brasino M, Lee JH, Cha JN. Anal Biochem. 2015;470:7–13. doi: 10.1016/j.ab.2014.10.006. [DOI] [PubMed] [Google Scholar]
- (17).Domaille DW, Lee JH, Cha JN. Chem Commun (Camb) 2013;49:1759–1761. doi: 10.1039/c3cc38871a. [DOI] [PubMed] [Google Scholar]
- (18).Park JS, Cho MK, Lee EJ, Ahn KY, Lee KE, Jung JH, Cho Y, Han SS, Kim YK, Lee J. Nat Nanotechnol. 2009;4:259–264. doi: 10.1038/nnano.2009.38. [DOI] [PubMed] [Google Scholar]
- (19).Lee J-W, Song J, Hwang MP, Lee KH. Int J Nanomedicine. 2013;8:3917–3925. doi: 10.2147/IJN.S51894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Litvinov J, Hagstrom AE, Lopez Y, Adhikari M, Kourentzi K, Strych U, Monzon FA, Foster W, Cagle PT, Willson RC. Biotechnol Lett. 2014;36:1863–1868. doi: 10.1007/s10529-014-1555-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kim HJ, McCoy M, Gee SJ, Gonzalez-Sapienza GG, Hammock BD. Anal Chem. 2011;83:246–253. doi: 10.1021/ac102353z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhang H, Xu Y, Huang Q, Yi C, Xiao T, Li Q. Chem Commun (Camb) 2013;49:3778–3780. doi: 10.1039/c3cc40688a. [DOI] [PubMed] [Google Scholar]
- (23).Kim J, Adhikari M, Dhamane S, Hagstrom AE, Kourentzi K, Strych U, Willson RC, Conrad JC. ACS Appl Mater Interfaces. 2015;7:2891–2898. doi: 10.1021/am5082556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Adhikari M, Dhamane S, Hagstrom AE, Garvey G, Chen WH, Kourentzi K, Strych U, Willson RC. Analyst. 2013;138:5584–5587. doi: 10.1039/c3an00891f. [DOI] [PubMed] [Google Scholar]
- (25).Hagstrom AE, Garvey G, Paterson AS, Dhamane S, Adhikari M, Estes MK, Strych U, Kourentzi K, Atmar RL, Willson RC. PLoS One. 2015;10:e0126571. doi: 10.1371/journal.pone.0126571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Scholle MD, Collart FR, Kay BK. Protein Expr Purif. 2004;37:243–252. doi: 10.1016/j.pep.2004.05.012. [DOI] [PubMed] [Google Scholar]
- (27).Scholle MD, Kriplani U, Pabon A, Sishtla K, Glucksman MJ, Kay BK. Chembiochem. 2006;7:834–838. doi: 10.1002/cbic.200500427. [DOI] [PubMed] [Google Scholar]
- (28).Kehoe JW, Kay BK. Chem Rev. 2005;105:4056–4072. doi: 10.1021/cr000261r. [DOI] [PubMed] [Google Scholar]
- (29).Wiegand TW, Williams PB, Dreskin SC, Jouvin MH, Kinet JP, Tasset D. J Immunol. 1996;157:221–230. [PubMed] [Google Scholar]
- (30).Scholle MD, Kehoe JW, Kay BK. Comb Chem High Throughput Screen. 2005;8:545–551. doi: 10.2174/1386207054867337. [DOI] [PubMed] [Google Scholar]
- (31).Maniatis T, Fritsch EF, Sambrook J. Molecular cloning: a laboratory manual. Cold Spring Harbor; Cold Spring Harbor Laboratory, N.Y.: 1982. [Google Scholar]
- (32).Lee CM, Iorno N, Sierro F, Christ D. Nat Protoc. 2007;2:3001–3008. doi: 10.1038/nprot.2007.448. [DOI] [PubMed] [Google Scholar]
- (33).Tennico YH, Hutanu D, Koesdjojo MT, Bartel CM, Remcho VT. Anal Chem. 2010;82:5591–5597. doi: 10.1021/ac101269u. [DOI] [PubMed] [Google Scholar]
- (34).Tran DT, Vermeeren V, Grieten L, Wenmackers S, Wagner P, Pollet J, Janssen KP, Michiels L, Lammertyn J. Biosens Bioelectron. 2011;26:2987–2993. doi: 10.1016/j.bios.2010.11.053. [DOI] [PubMed] [Google Scholar]
- (35).Wei X, Li H, Li Z, Vuki M, Fan Y, Zhong W, Xu D. Anal Bioanal Chem. 2012;402:1057–1063. doi: 10.1007/s00216-011-5591-1. [DOI] [PubMed] [Google Scholar]
- (36).Xavier KA, Willson RC. Biophys J. 1998;74:2036–2045. doi: 10.1016/s0006-3495(98)77910-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Platts-Mills TA. Am J Respir Crit Care Med. 2001;164:S1–5. doi: 10.1164/ajrccm.164.supplement_1.2103024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.