

Abstract
Cell and protein arrays have demonstrated remarkable utility in the high-throughput evaluation of biological responses; however, they lack the complexity of native tissue and organs. Here, we describe tissue extracellular matrix (ECM) arrays for screening biological outputs and systems analysis. We spotted processed tissue ECM particles as two-dimensional arrays or incorporated them with cells to generate three-dimensional cell-matrix microtissue arrays. We then investigated the response of human stem, cancer, and immune cells to tissue ECM arrays originating from 11 different tissues, and validated the 2D and 3D arrays as representative of the in vivo microenvironment through quantitative analysis of tissue-specific cellular responses, including matrix production, adhesion and proliferation, and morphological changes following culture. The biological outputs correlated with tissue proteomics, and network analysis identified several proteins linked to cell function. Our methodology enables broad screening of ECMs to connect tissue-specific composition with biological activity, providing a new resource for biomaterials research and translation.
INTRODUCTION
Tissues and organs in the body are composed of cells and their surrounding extracellular matrix (ECM) generated by self-assembly and cellular processing1. Tissue specificity is created by the unique composition—from hundreds of different biomolecules—and the supramolecular structures that interact physically, chemically, and biologically with cells to regulate cellular-level functions2–5. Ongoing research continues to elucidate how the structural and compositional properties of the ECM influence resident cells6, 7. Despite the use of tissue-derived materials in the clinic, detailed mechanistic information on how tissue ECMs directly influence cell behavior or repair processes is largely unknown, likely owing to the complex chemical and physical cues that cannot be untangled or reduced to a single component.
Microarray-based strategies allow high-throughput in vitro screening of cellular functions and biological outputs on diverse substrates8–12. Although DNA, RNA, and single-protein microarrays are commonplace, more complex biomaterial arrays have yet to reach their full potential. To investigate cell-microenvironment interactions, purified ECM proteins, such as collagen, or synthetic biomaterials that mimic the ECM have been studied in an array format. For example, two-dimensional (2D) microarray libraries of synthetic polymers delineated optimal scaffold composition for lineage-specific stem cell differentiation8, 9. ECM proteins have been integrated with synthetic hydrogels to identify combinations that stimulate stem cell osteogenesis in 3D13–15. Individual and combinatorial screening of purified proteins in microarray formats has suggested mechanisms of cell-protein interactions10 and identified candidate cell-protein interactions that correlate with cancer metastasis11. All of these previous arrays started with simple building blocks, such as polymers or proteins that can be tested in a combinatorial manner. However, cells in the body exist within tissues and organs with a complex ECM that includes hundreds of different molecules organized with a hierarchy ranging from nanometer fibrils to micrometer units that can modulate cell behavior16.
Tissue ECMs have been used for regenerative medicine and wound healing in humans23–25, typically matching “like with like”19–22; for instance, stem cells cultured on liver ECM to create new liver tissue17. However, broader screening of tissue ECM properties may elucidate more general biological functions and novel therapeutic entities. To advance the understanding and use of tissue-derived biomaterials, high-throughput screening tools are needed to probe variability in ECM composition and complex cell-matrix interactions in vitro. In doing so, the intricate mechanisms of cell-material response and repair processes can be teased apart to understand how these materials can be used, in turn, to influence cell behavior in vivo.
Here, we developed 2D and 3D tissue ECM arrays for screening biological responses to tissue specific scaffold microenvironments. To generate a biomaterial scaffold from tissues, various chemical and mechanical processing conditions can be employed18, ranging from mild conditions (minimal disruption of ECM composition) to harsh conditions (complete removal of cellular debris and remnants). We processed tissues to remove soluble tissue components, and then mechanically fragmented the matrix to create tissue ECM microparticles that retained the proteomic complexity of the natural ECM in a medium compatible with array fabrication (spotting). We characterized a range of cell-matrix interactions at cellular and functional levels, including mineralization, cell adhesion and proliferation, gene expression, and changes in cell morphology, using stem cells, cancer cells and macrophages. We then correlated tissue ECM array outputs with proteomic composition to build networks that highlight candidate proteins responsible for tissue-specific differences in cell function. Using this array platform and systems biology, we can ultimately understand the interplay between cells and complex matrices, with the hope of developing novel tissue engineering strategies based on tissue ECMs.
RESULTS
Fabrication of 2D and 3D tissue ECM arrays
We established a physiologically broad tissue ECM data set by harvesting 11 different porcine tissues and organs (spleen, small intestine, bladder, bone, brain, cartilage, cardiac, kidney, liver, lung, and adipose) and chemically treating the tissues using a combination (unless noted otherwise) of acid, detergent and DNAse19, and then lyophilizing them to mechanically break down into microparticles using a cryomill (Fig. 1a). The tissue ECM particles contained numerous intact proteins reminiscent of the original tissue (Supplementary Fig. 1a)—a feature that is lost during alternative methods of enzymatic digestion.
Figure 1. Tissue and organ model ECM arrays in 2D and 3D.
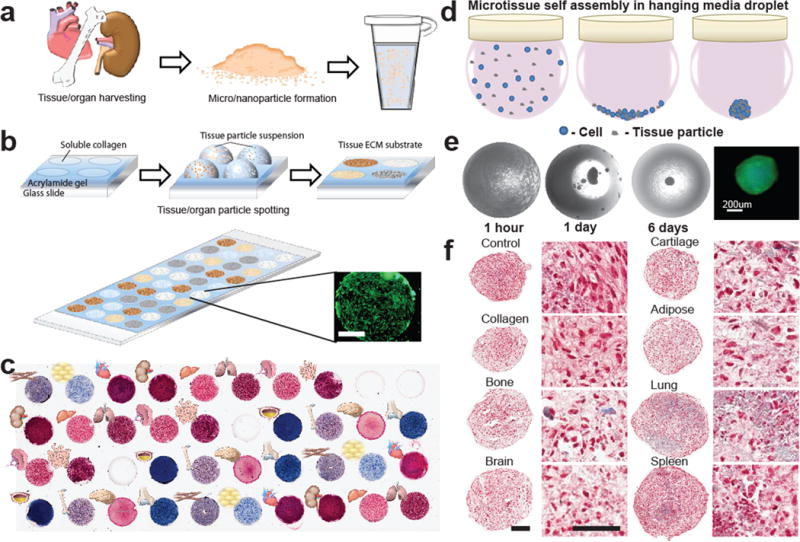
(a) Fresh porcine tissue was chemically treated, mechanically processed into particles, and suspended in water to render into a form compatible with microarray spotting techniques. (b) Microarrays were defined by a silicone gasket placed on acrylamide-coated coverslips. Each well was spotted with a particle suspension and dried to form circular tissue substrates on the chip before gasket removal. Human adipose-derived stem cells (hASCs) stained with calcein AM remained confined to the adhesive tissue spots. Scale bar=1 mm. (c) Masson’s trichrome staining, which stains collagen blue, highlights the variation in biochemical composition of 11 different tissues and 2 different collagen controls (n=3 per microarray chip). (d) 3D microtissues were formed in hanging drop culture from cell/tissue particle suspension. Cells and tissue particles settled at the bottom of the droplet, where they subsequently self-assembled into a cell-tissue spheroid. (e) Brightfield images of hanging droplets show the progressive formation of a cell-tissue spheroid containing ASC cells and processed cartilage particles over 6 days of culture. Calcein AM staining confirmed the viability of cells contained in the spheroids. (f) Cells and tissue particles distributed throughout histological sections of the spheroid microtissues were stained with Masson’s trichrome (day 17, Cell=red, Collagen=blue).
Array spots (3-mm diameter) consisted of tissue matrix particles spotted on acrylamide-coated glass (Fig. 1b) with a layer of purified soluble type I collagen to promote homogeneous particle binding, as confirmed by SEM (Supplementary Fig. 1b). Collagen controls included acid-soluble type I collagen protein without the addition of tissue ECM particles and insoluble collagen particles that match the structure of processed tissue ECM particles. We prepared up to 40 spots of tissue ECM per array (Fig. 1c and Supplementary Fig. 2a) and then seeded cells uniformly on the array using a confined flow device (Supplementary Fig. 2b), which limited cell spot seeding variability to 13% of the mean per row and column (n=10 rows, n=4 columns per 40 spot array) (Supplementary Fig. 2c). Further, spot locations and seeding direction were organized so that replicates of each tissue were distributed evenly throughout the seeding area to limit the effects of flow patterns within the seeding device (Supplementary Fig. 2a).
Three-dimensional (3D) systems provide an additional method to evaluate and predict cell-tissue interactions in an environment that may better predict some in vivo behaviors. To this end, we also developed hanging droplet arrays of 3D tissue ECM spheroids where each spheroid contained 10,000 – 20,000 cells and ECM particles at a concentration of 0–10 ng/cell in 40 μL culture medium (Fig. 1d). Spontaneous cell-matrix assembly resulted in formation of large agglomerations after 24 hrs in culture, and continued to self-assemble over the course of 2–6 days (Fig. 1e). Tissue particle-to-cell ratios were optimized to maximize tissue ECM content without disrupting compact spheroid formation or cell viability (Supplementary Fig. 3a–c). Compact spheroid formation and microtissue size was consistent across all ECM types tested at concentrations of 2 ng/cell or less after 6 days of culture. Cells were viable at ECM concentrations up to 2 ng/cell, but decreased at higher particle concentrations for some tissues.
To form 3D spheroids, we seeded human adipose-derived stem cells (hASCs) with ~17,000 cells and 16 μg of tissue particles to create compact spheroids with a uniform diameter ~460 μm (+/− 40, n=8) despite different tissue particle composition (purified type I collagen particles, bone, brain, cartilage, adipose, lung, spleen). To enable high-throughput morphological, histological and immunohistochemical analyses of the 3D microtissue arrays, we developed a method similar to tissue microarray (TMA) technology used in tumor pathology20. We covered cell-tissue spheroids that were arranged in a microarray mold with agarose gel, to fix the location of the microtissues and make possible sectioning of the spheroids. Microtissue cross-sections revealed a relatively uniform distribution of cells and tissue particles throughout each spheroid (Fig. 1f).
Characterization of tissue ECM arrays
Having established both 2D and 3D approaches to fabricate arrays, we first characterized the physical and biochemical properties of the tissue-specific ECMs to understand tissue-specific attributes and to confirm reproducibility in array fabrication.
Physical properties
After complete drying, spot surfaces for brain, bladder, and small intestine were generally smooth; but other tissues, such as spleen, bone, and liver, had more variation in roughness and texture (Supplementary Fig. 1b). The elastic moduli of the dry tissue spots were two orders of magnitude higher and demonstrated less variation between tissues types than stiffness reported for fresh hydrated tissue21 (Supplementary Fig. 1c). For example, after processing into particles and spotting in 2D, cardiac tissue spots were two-fold stiffer than brain; by comparison, there is an estimated ~20-fold difference between fresh heart and brain tissues21.
Reproducibility
Masson’s Trichrome stain was able to contrast the collagen composition between tissue spots. Spots derived from connective tissues, cartilage, bone, and bladder contained qualitatively more collagen than solid organ spots, such as liver and muscle (Fig. 1c). We demonstrated differences in collagen composition between the ECMs from different tissue sources as well as between healthy and diseased tissues (cancer and muscular dystrophy), where ECM changes in composition and structure often occur during pathogenesis (Supplementary Fig. 4). The variability in staining intensity of the most prominent color in tissue spot replicates remained below 25% (coefficient of variation, n=3 spots/tissue type) for 10 of the 11 tissue types analyzed with ImageJ (Supplementary Fig. 5a–b). We confirmed reproducibility of tissue particle composition using mass spectrometry and proteomic analysis comparing replicates of small intestine (Supplementary Fig. 5c) with similar peptide abundances in triplicate runs. Approximately 95% of proteins identified by at least 2 peptide spectral matches (PSMs) were found in all small intestine samples, whereas only 42% of less abundant proteins (1 PSM) were identified across all 3 runs (Supplementary Fig. 5d). Taken together, there was low tissue inter-spot variability and high tissue specificity within tissue array chips.
We demonstrated broad utility of our tissue array with a range of tissue preparation methods by exposing two model tissues, cardiac and lung, to a range of chemical processing conditions that produced various molecular weights of ECMs (Supplementary Fig. 6a). Spots from the same tissues processed with different methods were clearly differentiated by their collagen content (Supplementary Fig. 6b). Using an automated algorithm, we also looked for any changes in morphology of primary mouse bone marrow-derived macrophages (BMDMs) seeded on the variably processed tissue ECMs (Supplementary Fig. 7a). Lung and cardiac tissues (and combinations of the two) that underwent different types of preparation produced distinct cell morphologies (Supplementary Fig. 7b,c), with lung tissue processing changes yielding the greatest effect on cell morphology (Supplementary Fig. 7d). Thus, the arrays can be used with multiple tissue processing methods and, importantly, variable cell response to the arrays can capture differences in tissue processing and, thus, the tissues’ resulting compositions.
Proteomic analysis
The tissue particles used to fabricate the 2D and 3D arrays retained a complex protein composition that was representative of the native tissues from which they were derived Nearly 4,000 unique proteins were identified across the 11 processed porcine tissue types, of which 111 were ECM and 98 were ECM-associated. To understand the effect of processing on tissue composition in the primary arrays, we compared several processed tissues in this study to native tissues from a recently published draft map of the human proteome6. Processed tissue particles showed strong similarities in ECM-specific proteins compared to the native tissues; however, processing reduced protein diversity of non-ECM proteins (Fig. 2a). Total protein abundance of ECM and non-ECM proteins varied by tissue type, although at similar ratios, with the exception of bone and cartilage, which had lower amounts of non-ECM proteins (Fig. 2b).
Figure 2. Tissue ECM protein composition and analysis.
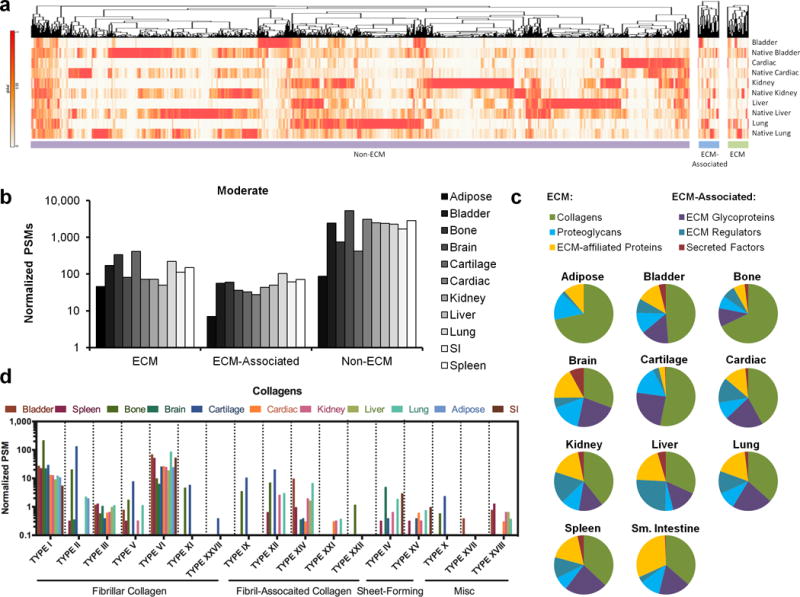
(a) Mass spectrometry and proteomics analysis comparing processed tissues to native human tissues from a draft map of the human proteome. Hierarchical clustering analysis was conducted for proteins mapped to ECM, ECM-associated, and Non-ECM genes. (b) Protein composition of moderately processed tissues and organs (peracetic acid, Triton X-100 & DNAse) were determined with mass spectrometry and proteomic analysis. Protein abundance of ECM, ECM-associated, and non-ECM proteins was determined from total normalized peptide spectrum matches (PSMs) mapped to gene level annotation, and differed in each tissue from a single mass spectrometry run. (c) The relative proportions of different categories of ECM and ECM-associated proteins varied between tissue types. (d) The abundances of fibrillar, fibril-associated, sheet-forming, and other collagen types were identified by proteomic analysis in processed tissues and organs.
Sub-classification of ECM and ECM-associated proteins in tissue particles revealed tissue-specific differences. Proteoglycans were enriched in adipose, cartilage, and brain tissues, whereas secreted factors (including growth factors) were miniscule in most tissues except for brain, where they comprised 8% of ECM total protein (Fig. 2c). Collagen subtypes accounted for the largest ECM fraction in all tissues except for brain and heart, and represented greater than 50% of the ECM in bladder, bone, and cartilage (Fig. 2c). Detailed analysis of collagen composition revealed expected tissue specificity (Fig. 2d); for example, type X collagen is associated with endochondral ossification, and was only identified in bone and cartilage.
Human stem cell response to 2D and 3D tissue ECM arrays
Having established optimal processing conditions as well as assay reproducibility, we used both 2D and 3D tissue arrays to study stem cell interactions with different ECMs. We seeded hASCs on 2D tissue ECM arrays or cultured them in 3D cell-ECM spheroids, in both instances in either control or osteogenic medium. The percent area of calcified matrix on each spot or microtissue represented osteogenic differentiation. hASCs cultured on bone, bladder, and lung 2D tissue ECMs demonstrated nearly 100% matrix deposition, whereas other tissue ECMs (adipose, collagen control) induced minimal osteogenic differentiation after only 6 days of culture (Fig. 3a–c). In 3D cell-ECM spheroids, osteogenic medium induced stem cell differentiation predominantly in bone and lung, with minimal matrix production observed in cartilage, adipose tissue, and spleen after 11 days of culture (Fig. 3d–f).
Figure 3. Stem cell-tissue ECM interactions in 2D (a–c) and 3D (d–f). (a–c).
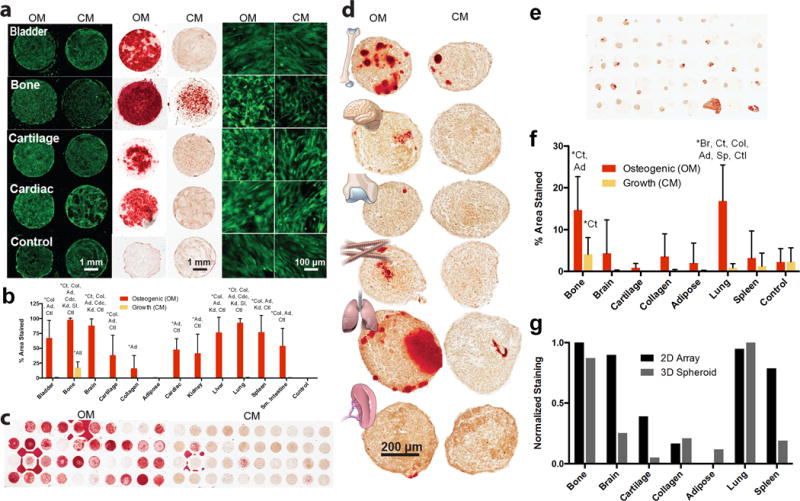
Human adipose derived stem cells (hASCs) were cultured on 2D tissue microarray substrates for 6 days in osteogenic (OM) or control/growth (CM) media. (a) hASC cells are stained with calcein AM (green, left and right). Alizarin Red staining (red) labels calcified matrix deposition (middle) (images representative of n=3). (b) Osteogenic differentiation, as quantified by the percent area of each spot that stained positively with Alizarin Red. Data are mean±s.d. (n=9). (c) Chips incubated in CM and OM are compared (images representative of n=3). (d–f) hASCs were cultured with different types of tissue particles in 3D microtissue spheroids for 11 days in OM or CM. (d) Histological sections of individual spheroids (images representative of n=5) were stained with Alizarin Red (e) Spheroid microarrays contained up to 36 spheroids on a single slide (image representative of n=2). (f) Quantification of percent area stained by Alizarin Red. Data are mean±s.d. (n=5 spheroids). (g) Normalized staining comparison between tissue types in 2D and 3D culture conditions. Spearman’s rank correlation coefficient=0.79 (P<0.05). One-way ANOVA with a post-hoc Tukey test was used for statistical analysis (b & d). Statistically significant increases (P<0.05) are labeled with (*). Abbreviations are: Bl=bladder, Bo=bone, Br=brain, Ct=Cartilage, Col=purified collagen I particles, Ad=adipose Cdc=cardiac, Kd=kidney, Lv=liver, Lg=lung, Sp=spleen, SI=small intestine, Ctl=control soluble collagen only, or All=all other tissues.
Overall osteogenic differentiation was similar in the 2D and 3D tissue arrays (Spearman’s rank coefficient 0.79 (p<0.05), but the amount of calcified matrix present in the 3D cellular microtissues was less than in the 2D arrays (Fig. 3g). As expected, bone tissue matrix supported and stimulated osteogenesis. However, newly formed calcified matrix was also abundant on lung ECM in both array systems so differentiation was confirmed in bone and lung spheroids as confirmed by increased osteocalcin gene expression, a late-stage marker of osteogenesis (Supplementary Fig. 8a).
Cancer and immune cell behavior on tissue ECM arrays
After using tissue ECM arrays to evaluate stem cell differentiation, we then demonstrated that the arrays are broadly applicable to cancer and immune cell types and their relevant biological outputs. For proof-of-concept, we screened human cancer cell lines in 2D and 3D to measure cell adhesion and proliferation, and mouse primary macrophages on the 2D tissue ECM arrays to quantify cell morphology and phenotype changes.
Cancer cell adhesion and proliferation
The tissue microenvironment plays a major role in tumor progression and metastasis22, and using tissue-specific ECMs could provide insights into how the local environment impacts tumor growth or metastatic preferences. To this end, we screened the adhesion of three different human cancer cell lines (lung, breast and skin) after two hours on 2D ECM microarrays. Cell adhesion on different tissue matrices varied by up to 10-fold for all cell types (Fig. 4a,b and Supplementary Table 1), but notably the cancer cells exhibited minimal adhesion to kidney- and liver-derived ECMs, and adhered to all of the mesenchymal tissues (adipose, bone, cardiac and cartilage) except spleen.
Figure 4. Cancer cell and macrophage interactions with tissue microarrays.
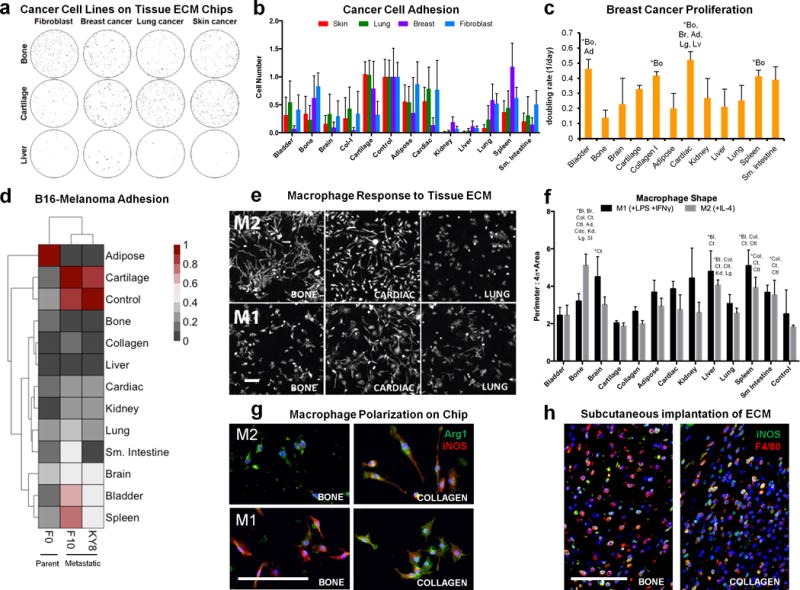
(a) Adhesion of three different human cancer cell lines and a control murine fibroblast line to different tissue ECM types was assessed on tissue microarrays. Cells (black) were stained with calcein-AM and counted with ImageJ software (representative of n=6 array spots). (b) Cell number was counted and normalized to the average number of cells on soluble collagen control spots located on the same microarray. Data are mean±s.d. (n=6 array spots). (c) Breast cancer cell doubling rate was calculated from cell number after 1, 2, and 7 days of culture on each tissue microarray spot. Data are mean±s.d. (n=3 array spots). (d) Heat map representation and hierarchial clustering of normalized (0–1) B16 murine melanoma cell adhesion between F0 (parent) and the metastatic KY8 (liver) and F10 (lung) cell lines seeded on the tissue array (n=6 tissue array spots and n=4 soluble collagen control spots). (e–f) The immunological properties of different tissue array spots were screened by seeding murine bone marrow-derived macrophages that were chemically polarized toward M1 or M2 phenotypes on tissue arrays. (e) Cell morphology was visualized after 24 hours with actin phalloidin staining (n=3 array spots, scale bar=200 μm). (f) Cell morphology was quantified as the convex hull area ratio. Data are mean±s.d. (n=3 array spots). (g) Macrophage phenotype on bone and collagen array spots after 24 hours in polarizing conditions was determined by immunofluorescence staining for Arginase-1 (green) and iNOS (red) (representative of n=3 array spots, scale bar=100 μm). (h) Bone tissue ECM and collagen were hydrated and injected subcutaneously into wild type C57BL/6 mice. Implants were harvested after one week for immunofluorescence staining for iNOS (green) and F4/80 (red) (representative of 4 images from n=2 animals). Scale bars=100 μm. The effect of array spot tissue type was analyzed by one-way ANOVA with a post-hoc Tukey test. Statistically significant increases (P<0.05) are signified with (*) and the abbreviations of the tissues they are greater than: Bl=bladder, Bo=bone, Br=brain, Ct=Cartilage, Col=purified collagen I particles, Ad= adipose Cdc=cardiac, Kd=kidney, Lv=liver, Lg=lung, Sp=spleen, SI=small intestine, Ctl=control soluble collagen only, or All=different from all other tissues. Statistical comparisons of cancer cell adhesion on different tissue spots are provided in Supplementary Table 1.
We next compared human breast cancer cell proliferation on 2D and 3D tissue ECMs. In 2D, bone ECM suppressed cell proliferation, whereas cardiac tissue amplified proliferation (Fig. 4c & Supplementary Fig. 8b). However, proliferation within 3D cell-ECM spheroids over 7 days was similar between bone, kidney, and cardiac ECMs (Supplementary Fig. 8c). We also compared the adhesion of three types of murine B16 melanoma cells was to evaluate tissue preferences of primary and metastatic tumor lines: the B16-F0 parent cell line, the B16-F10 line associated with lung metastasis, and the B16-KY8 line associated with liver metastasis. F0 cells demonstrated the highest overall attachment and KY8 the lowest, as expected from their known in vivo behavior. Cartilage and spleen tissues promoted adhesion of all three melanoma cell types; conversely, liver, kidney, and collagen control did not support attachment of melanoma cells, which was similar to the response of human breast cancer cells. The most substantial difference between cell lines was observed for attachment to adipose tissue. Cluster analysis revealed similarities in tissue adhesion between the two metastatic melanoma lines (F10 and KY8) compared to the primary cells, despite the fact that the metastatic lines have distinct metastatic profiles (Fig. 4d and Supplementary Table 1).
Immune cell morphology and polarization
Macrophages play a key role in normal tissue repair and in response to disease. For example, the tissue regenerative response has been linked to macrophage skewing toward a remodeling phenotype called M2; conversely, the M2 phenotype is associated with negative outcomes in a tumor environment23. Recent studies have also demonstrated the importance of the local tissue environment on macrophage phenotype5. We used our 2D tissue array method to probe M2 and M1 (pro-inflammatory) macrophage responses to specific tissue microenvironments (Fig. 4e). We skewed murine BMDMs towards an M1 phenotype (interferon-γ [IFNγ] + E. coli lipopolysaccharide [LPS]) or an M2 phenotype (interleukin-4 [IL-4]) and then quantified cell morphology, as a surrogate marker of immune polarization, on tissue ECMs. Macrophage shape (convex hull area ratio) varied up to 40% across the tissue matrix types in both cytokine environments, suggesting that tissue-specific matrix environments can modulate cell response (Fig. 4e–f and Supplementary Fig. 9). Macrophages changed morphology dramatically when cultured on bone tissue ECM, with M1-stimulated cells elongated and spread compared to the more compact M2-stimulated cells (Fig. 4e–f).
We further investigated macrophage polarization on the 2D tissue array by immunostaining M1 and M2 cytokine-stimulated BMDMs for the expression of the M1 marker inducible nitric oxide synthase (iNOS/Nos2) and the M2 marker Arginase-1 (Arg-1) on both bone ECM and control collagen (insoluble particles) spots. Macrophages on the bone ECM spots increased Arg-1 staining in M2 conditions and increased iNOS in M1 conditions compared to collagen, suggesting enhanced polarization and responsiveness to either M1 or M2 conditions (Fig. 4g). To provide a correlation between in vitro and in vivo results and demonstrate a predictive value of in vitro results on in vivo responses, we implanted the bone ECM and control collagen matrices subcutaneously in C57BL/6 mice. Both scaffolds showed substantial mononuclear cell infiltration following implantation, many of which were F4/80+ macrophages expressing iNOS (Fig. 4h). Similar to the increased macrophage polarization of the bone ECM observed in vitro, more macrophages expressed iNOS in the bone ECM implants than in collagen in vivo.
Lastly, we evaluated the effect of 2D tissue substrates on macrophage polarization via expression of M1- and M2-associated genes (Supplementary Fig. 10). Macrophage gene expression was dependent on source tissue and cytokine stimulation with a number of statistically significant differences between tissues ECM sources (Supplementary Table 1). Bone ECM induced extensive M1 gene expression in M0 cytokine conditions, whereas liver did not alter phenotype compared to uncoated wells. In contrast, all tissues resulted in decreased M2 gene expression in M2 cytokine conditions, and did not substantially alter M1 expression in M1 conditions, although cardiac tissue increased pro-infllammatory IL10 expression.
Systems analysis of tissue composition and outcomes
An advantage of the tissue arrays is the ability to perform multiplexed assays using a variety of cell types and matrix compositions. For example, to identify potential mediators of in vitro biologic function, the proteomic composition of tissue array spots was integrated with the outputs of the various in vitro assays. Heat map representation of correlation coefficients showed that different ECM and ECM-related proteins clustered with in vitro osteogenesis, cancer cell adhesion, and macrophage polarization—the three outputs we investigated in this study (Fig. 5a). We selected the most statistically significant correlations (n=28 proteins with P<0.05 for both original and rank transformed data, Supplementary Table 2) for network analysis, to further probe common proteins that may influence cell behavior. Several ECM components, including S100A9, SERPINB10, CTSB, HAPLN3, and PRSS2, were strongly linked with multiple in vitro assay outcomes (Fig. 5b). The S100A family, for instance, correlated with both osteogenic and immunomodulatory assay outcomes. Some of the more abundant ECM proteins, such as collagens, did not distinguish different in vitro outcomes between tissues. Conversely, ECM glycoproteins correlated with several biological assay outputs in the network analysis; specifically, positive correlations to cancer cell adhesion and osteogenesis and negative correlations to macrphage morphology.
Figure 5. Systems biology analysis of tissue proteomic composition and in vitro function.
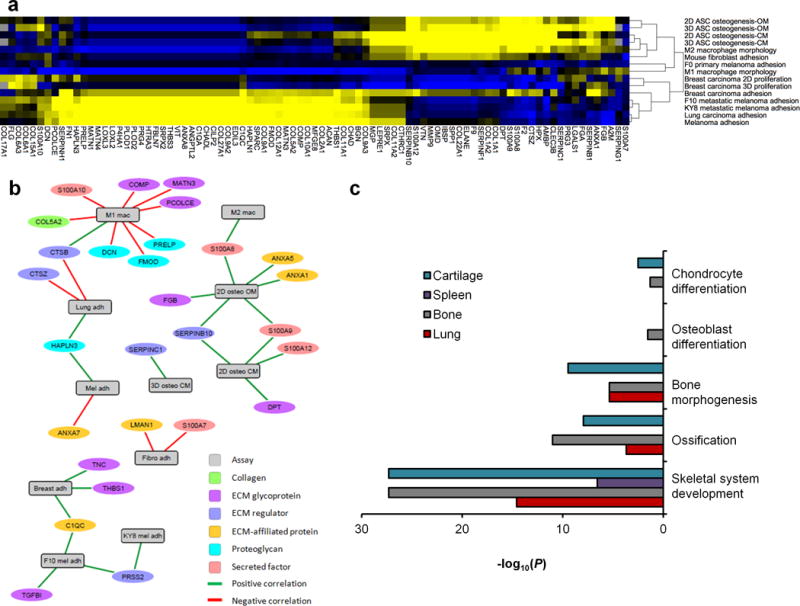
(a) Correlation analysis was conducted between the proteomics data and cell responses to 11 porcine tissues across 15 in vitro assays. Hierarchical clustering and heat map representation of the correlation coefficients between ECM proteins and each in vitro assay outcome (portion of heatmap shown; full dataset available in Supplementary Table 2). Clusters of positive (yellow) and negative (blue) correlations showed an association of osteogenic assays with M2 macrophage morphology and cancer cell adhesion with M1 macrophage morphology. (b) Network analysis showing the statistically significant correlations (edges) between in vitro outcomes and ECM proteins (vertices). Statistically significant correlations were defined as P<0.05 for both the original assay and rank-transformed assay results. (c) Gene ontology enrichment was performed on ECM proteins in tissues that promoted in vitro osteogenesis (bone and lung) and compared to those did not (cartilage and spleen). All identified ECM and ECM-associated proteins were entered into enrichment analysis for biological processes gene ontology, and terms related to osteogenesis or skeletal development were selected with corresponding P-values (list of ECM proteins identified in each GO term provided in Supplementary Table 3).
To further investigate the unexpected stem cell osteogenesis that we observed on lung tissue in 2D and 3D (Fig 3), we probed the ECM and ECM-associated proteomic composition using gene ontology (GO) enrichment; specifically, terms that related to skeletal development or bone formation. Lung ECM components exhibited significant enrichment for these terms in lung compared to spleen (Fig. 5c, including ossification (GO:0001503) and bone morphogenesis (GO:0060349), suggesting that factors were retained in lung following processing that individually or in combination may have promoted osteogenesis (Supplementary Table 3). Cartilage was also enriched in these pro-osteogenic factors to a similar extent as bone, although it did not show any in vitro osteogenic potential, indicating that GO enrichment analysis alone is not sufficient to explain this behavior, or that there are inhibitors of osteogenesis present within processed cartilage. Correlation networks and GO analysis revealed individual molecules and groups of molecules, respectively, in the complex tissue ECMs.
DISCUSSION
Synthetic approaches to recreating the ECM use a reductionist approach that incorporates select components required to modulate cell behavior24, 25. However, ECM isolated directly from mammalian tissues can be used to achieve the complexity of the native tissue in a biomaterial scaffold. Such tissue-derived scaffolds have been under investigation for over 25 years26, and during that time, have been implanted in over 1 million patients27. Although starting as simple sheets for surgical repair (i.e. dermis and intestine), recent advances have shown the capability to retain the structure and composition of whole organs. However, the biochemical complexity of tissue- and organ-derived matrices renders it difficult to fully understand the material composition, specific interactions with cells, and therapeutic action. High-throughput bottom-up approaches—studying single or paired protein combinations—have been unable to mimic the highly complex composition of healthy and diseased tissues. ECM, if treated properly—without enzymes or chemicals to solubilize—can retain bulk tissue and organ properties on the micro scale, more effectively preserving the physical and biochemical properties of a given tissue. We therefore created tissue arrays using mechanically processed ECM, allowing us to formulate 11 different tissue ECMs as particles, which could then be spotted in 2D or combined with cells in 3D “microtissues” for further characterization and modulation.
Tissues and organs are processed using a wide range of mechanical, chemical, and/or enzymatic treatments18. The specific treatment regimen, as well as the tissue to which it is applied, dictates the level of disruption to the native matrix components. Frequently used processing agents (often-termed “decellularization agents”) were employed to produce tissue spots that were composed of both ECM and non-ECM components, indicating minimal tissue disruption. It may not be possible to completely remove all cell-associated components from a tissue matrix, and full decellularization is not an appropriate terminology for most tissue products that may rather be considered as a decellularization spectrum or simply processed tissues. Moreover, cell-associated proteins, both intracellular and secreted, may be relevant for the biological and even therapeutic outcomes of these materials, and should therefore be preserved. Tissue ECMs from different sources exhibited relatively minor differences in mechanical and structural properties, but large differences in composition, which is likely the primary influence in cell response.
Using our ECM array methodology, we found that the effects of specific tissues on stem cell osteogenesis were consistent in both 2D and 3D, although the 2D assays demonstrated an estimated 50-fold lower ratio of tissue matrix to cells. Bone tissue ECM enhanced osteogenesis, which is expected because demineralized bone materials are used clinically to stimulate bone healing. However, lung tissue ECM unexpectedly enhanced osteogenesis compared to most other tissues. These unexpected results may lead to further study and understanding of lung tumor calcification28. Gene ontology analysis revealed that processed lung ECM was enriched for components associated with osteogenesis and chondrogenesis compared to other tissues, including ECM proteins such as type XII and II collagen, matrilins, thrombospondin 1, and asporin. (Supplementary Table 3).
Correlating in vitro assay outcomes with in vivo behavior will be an important link between our method and biology, with the hope that specific components of the tissue matrix can be linked to cell signaling pathways. To this end, we performed an extensive network analysis to correlate proteomics and in vitro functional outcomes. Correlation network analysis is ideal for identifying individual ECM candidates and for discovering potentially overlapping mechanisms across different cell processes. Conversely, gene ontology enrichment is well suited for deriving complex combinations of ECM components for further investigation using previously defined annotations. Both analysis methods are complimentary and easily applied to datasets generated by these arrays to discover drivers of the biological responses. We identified several tissue ECM components with strong correlations (both positive and negative) to multiple in vitro assay outcomes, including S100A9, SERPINB10, CTSB, HAPLN3, and PRSS2 indicating different roles depending on cell type and functional assay outcome. Of particular interest, S100A8 and S100A9, which were most abundant in bone, spleen, and lung, are DAMPs that possess immunoregulatory activity via interaction with the receptor for advanced glycation end products (RAGE). Other members of the S100 family that were found in bone and lung, such as S100A12, have recently been shown to activate osteogenic gene pathways29 and are considered biomarkers of osteoarthritis. The dual immunoregulatory and osteogenic functions of S100 proteins may plausibly contribute to the connection between the M2 macrophage morphology assay (which express RAGE) and stem cell osteogenesis described by the array. ECM correlation networks applied to the cancer cells found that both metastatic melanoma lines, though not the parent, are positively correlated with PRSS2, a serine protease that was enriched in bladder, spleen, and brain tissue arrays. PRSS2 and other enzymes have long been associated with cancer and promote metastasis by cleaving cell adhesion molecules and ECM barriers. However, our data suggest that PRSS2 has a direct effect on metastatic cancer adhesion, and may promote initial metastasis to these tissues through mechanisms such as shedding of other membrane-bound growth factors or enzymes30. Although these data do not define mechanisms of the tissue ECM modulation of processes such as osteogenic differentiation, cancer metastasis or immune modulation, the array screening and resulting analysis were able to establish unique correlations and provide promising candidate molecules for further investigation, either isolated components with novel, non-canonical functions or complex combinations of proteins that are not otherwise apparent.
The 2D and 3D arrays each present advantages and may complement each other; 2D microarrays are compatible with high-throughput microscope slide scanning and are simple to culture, whereas 3D microarrays better represent the natural microenvironment, do not have sample crosstalk, can be sectioned multiple times, and the ratio of cells to tissue matrix can be varied to measure dose dependence. Three-dimensional microtissues are also compatible with metabolic assays, single cell analysis or sorting, and gene expression analysis. Each method, however, offers the unprecedented ability to probe mechanisms of cell interactions with complex tissue microenvironments, support discovery of new therapeutic targets and applications, and ultimately transform the therapeutic potential of ECM-based therapeutics.
METHODS
Tissue processing
Porcine tissues were harvested from 6-month-old market weight pigs weighing approximately 100 kg (Wagner’s Meats, Mt. Airy, MD). Murine tissues were harvested from normal mammary gland tissue of 6–10 week old female wild type C57BL/6 mice and early stage (diameter ~ 9 mm) and late stage tumor (diameter ~ 16 mm) tissue from MMTV-PyMT mutant mice (Jackson Laboratories). Skeletal muscle was harvested from the quadriceps of 26–32 week old female Dmdmdx-5Cv mutant mice (Jackson Laboratories) and wild type control. Whole tissues and organs were cut into approximately 100 mm3 sized pieces and rinsed several times with phosphate buffered saline (PBS). Bone tissue required an addition decalcification preparation in 10% formic acid for 18 hours at room temperature and fat was mechanically pressed to reduce lipid content before processing. Unless otherwise noted, tissue was processed by incubation with three different solutions with thorough washing in PBS between each step 19: (1) 3% peracetic acid for 3 hours at 37 °C, (2) 1% Triton™ X-100 containing 2 mM EDTA for 18 hrs at 37 °C and (3) 600 U/mL DNAse (Sigma) containing 10 mM MgCl2 for 18 hrs at 37 °C. After the final treatment, the tissue was washed thoroughly with PBS followed by distilled water, and then lyophilized.
For experiments comparing multiple processing protocols, the following modifications were used: (Fresh) - tissues were not treated with acid, detergent or DNAse; (Mild) - tissues were incubated in (1) 3% peracetic acid for 10 min at 37 °C and (2) 600 U/mL DNAse containing 10 mM MgCl2 for 18 hrs at 37 °C; (Moderate) -tissues were incubated in (1) 3% peracetic acid for 3 hours at 37 °C, (2) 1% Triton™ X-100 containing 2 mM EDTA for 18 hrs at 37 °C and (3) 600 U/mL DNAse containing 10 mM MgCl2 for 18 hrs at 37 °C; (Harsh) - tissues were incubated in: (1) 3% peracetic acid for 3 hours at 37 °C, (2) 1% Triton™ X-100 containing 2 mM EDTA for 18 hrs at 37 °C and (3) 4% SDS for 16 hr at 37°C (4) 600 U/mL DNAse containing 10 mM MgCl2 for 18 hrs at 37 °C; (Digest) – ECM particles treated using the moderate protocol were further digested in a 1 mg/ml solution of porcine pepsin in 0.01NHCL for 72 hr.
Tissue particle fabrication
Lyophilized processed tissue was cryogenically pulverized in a cryomill (SPEX 6770, SPEX SamplePrep®) at −195 °C under liquid nitrogen. Approximately 300–500 mg of sample was processed in each batch. Cryomill settings were 8–15 one-minute cycles at 10 cycles per second with 3 minute cooling periods between runs. The resulting powder was suspended in distilled water or DMEM media at 10 mg/mL and sonicated with a probe sonicator (GE 130PB, Cole-Parmer) at an output power of 10–15 W two times for 30 s in an ice bath. Water suspensions were centrifuged at 4000 rpm for 10 min and resuspended in fresh DI water. Sonication was repeated and the suspension was filtered through a 40-μm cell sieve. The final concentrations of solutions were determined from the mass of lyophilized aliquots.
Microarray chip fabrication
Glass coverslips (22 × 60 mm) were cleaned and functionalized with methacrylate groups using a silane reaction, as previously described 10. Acrylamide was mixed with bis-acrylamide and dissolved in DI water at a concentration of 10.55% and 0.55% wt/v, respectively. A photoinitiator solution of Irgacure (I2959) dissolved in methanol at 200 mg/mL was added to the acrylamide solution at a concentration of 10% v/v. A 20-uL drop of working solution was pipetted onto the functionalized 22 × 60-mm coverslip and an untreated 22 × 50-mm glass slide was carefully placed on top of the liquid to form a thin layer estimated to be 36-μm thick. The solution was polymerized with UV light (~2 mW/cm2) for 10 min and the 22 × 50-mm coverslip was removed after incubation in DI water for 30 min. Gel-coated slides were soaked in DI water overnight and dried on a hot plate at 40 °C for 45 min.
Silicon gaskets with arrays of 3-mm diameter wells (CWCS-50R, Grace Bio-Labs, Inc.) were placed on the dry gel-coated slide with 40 wells in full contact. Collagen (C7661, Sigma-Aldrich) dissolved at 0.25 mg/mL in 0.1M acetic acid was pipetted (9 μL) in each chamber and allowed to dry overnight. Next, 10 μL of tissue particle suspension was spotted in each of the collagen-coated wells. The working tissue particle concentration was 2 mg/mL for suspensions of liver, lung, spleen and small intestine, and 3 mg/mL for all other tissue types. Selected concentration was determined visually as the amount required to form a complete monolayer spot without visualization of the underlying acrylamide gel surface. Mixed cardiac and lung ECM solutions were combined at ratios of 1:3, 3:1, and 1:1 v/v and spotted at a total concentration of 3 mg/ml to form composite ECM substrates. Spotted chips were left to dry overnight in a cell culture hood at room temperature and the gaskets were removed. Chips were sterilized in a culture hood with UV light for 30 min on each side.
Tissue spot variability
The reproducibility of the spotting techniques were validated by histochemical staining and proteomic analysis. Dried substrates spotted from 3 mg/ml ECM particle suspensions of 11 different tissues and collagen controls were stained using Masson’s Trichrome. Three replicates of each tissue type were visually compared for color content and distribution. The overall staining variation between tissues was quantitatively compared using a hue filter in ImageJ to threshold the percent of total spot area contained the hue ranges: 151–159, 209–229, 237–255, 188–238, 227–234. Standard deviation and coefficient of variation were calculated for each tissue (n=3) at each hue range.
Protein size characterization
The effect of different processing methods on protein size was compared for lung and cardiac tissues by boiling in Laemmli buffer and running on an SDS-PAGE gel at 120 V for 1.5 hours. Changes in protein size following pepsin digestion were compared between tissue ECM particles from bladder, cartilage, lung, spleen, and purified collagen. Undigested tissue was either prepared in Laemmli buffer as above, or digested in 1 mg/mL porcine pepsin (Sigma) in 0.01 N HCL. After 72 hr of agitation in pepsin solution, pH was neutralized and a sample was run on an SDS-PAGE gel to compare changes in protein size distribution versus control undigested tissue particles.
Proteomics analysis
Tissue samples were reduced with dithiothreitol (DTT) and alkylated with iodoacetamide, then digested in solution with trypsin (Promega) at 37 °C overnight. Digested peptides were acidified and dried. The peptides were reconstituted in 40 uL 2% acetonitrile (ACN)/0.1% formic acid and 6 μL injected (15% of total volume). Protein identification by liquid chromatography tandem mass spectrometry (LCMS/MS) analysis of peptides was performed using a Q-Exactive interfaced with a Thermo Easy-nLC 1000 system (Thermo Scientific) or a Velos Orbitrap (Thermo Scientific) interfaced with a NanoACQUITY UPLC system (Waters Corp.). Peptides were fractionated by reverse-phase HPLC on a 75 um × 12 cm column with a 15-um emitter tip (New Objective, Woburn, MA) in-house packed with Magic C18AQ (5 μm, 120 Å, Michrom Bioresources) using 0–90% ACN/0.1% formic acid gradient over 90 min at 300 nL/min. Eluting peptides were sprayed directly into the Q-Exactive or Velos at 2.0 kV. Q-Exactive survey scans were acquired from 350–1800 m/z with up to 15 peptide masses (precursor ions) individually isolated with a 2.0 Da window and fragmented (MS/MS) using a collision energy of 27 and 30s dynamic exclusion. Precursor and fragment ions were analyzed at 70,000 and 17,500 resolution, respectively. Velos survey scans were acquired from 350–1800 m/z with up to 8 peptide masses (precursor ions) individually isolated with a 1.9 Da window and fragmented (MS/MS) using a collision energy of 35 and 30s dynamic exclusion. Precursor and fragment ions were analyzed at 30,000 and 15,000 resolution, respectively.
The mass spectrometry derived data was searched against a combined human and porcine RefSeq protein database (Version 65 with common contaminants added) using the SEQUEST HT search algorithm through Proteome Discoverer (version 1.4.1.14, Thermo Scientific). Search parameters included a maximum of one missed trypsin cleavage, cysteine carbamidomethylation as a fixed modification, and methionine oxidation as a variable modification. The precursor mass tolerance was 20 ppm and fragment mass tolerance 0.05 Da, and the maximum peptide length was specified as 7 amino acids. Peptides that passed the 1% FDR threshold were used for protein identification. Protein inference was based on rules of parsimony as employed by Proteome Discoverer software. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium31 via the PRIDE partner repository with the dataset identifier PXD002571. Gene-level measurements were obtained by summing the peptide spectral matches (PSMs) for all proteins corresponding to a given gene. The summed spectral counts in each experiment were normalized by dividing by the total acquired tandem mass spectra as previously described6. Small intestine spectra from each experiment were compared for repeatability and averaged prior to normalization. The identified genes were then categorized as ECM (subclassified as a Collagen, proteoglycan, or glycoprotein), ECM-associated (subclassified as ECM affiliated protein, ECM regulator, or secreted factor), or non-ECM as previously described32. The proteomic composition of processed tissues was compared to the native tissues from a recent draft map of the human proteome6 (online resource available at: http://www.humanproteomemap.org/). Proteins found in both processed tissues and the corresponding native tissues were normalized between 0–1, and summarized as separate heat maps for ECM, ECM-associated, and non-ECM components using Gene-E software (http://www.broadinstitute.org/cancer/software/GENE-E/). Hierarchal clustering for each protein was performed across all processed and native tissues in each heat map. The total abundance of ECM and non-ECM components in each tissue was determined from the sum of the normalized PSMs for all proteins in each category.
The variation in proteomic composition between replicate spots in a fabricated microarray was evaluated from three separate aliquots of a representative 3 mg/ml small intestine particle suspension. The total normalized PSMs and the corresponding number of identified proteins in ECM and non-ECM categories was compared for each run. Protein identification consistency was evaluated for rare proteins (identified by 1 PSM) and more abundant proteins (identified by 2 or more PSMs) across the three runs and defined as: (No Overlap) identified in one of three samples, (2 of 3) identified in 2 of three samples, or (3 of 3) identified in all samples.
Cell seeding methods
Uniform cell distribution on 2D culture plate can be enhanced with a confined chamber. In order to limit variation in initial cell seeding density on each tissue array spot,a custom confined flow chamber was designed. The flow chambers were made from Polydimethylsiloxane (PDMS) such that the depth of media over the microarray was approximately 1 mm. The tissue microarrays were placed on a tissue culture plate (1 well rectangular, NUNC) and their edges were sealed with parafilm to stabilize and prevent media from flowing under the tissue microarray chip. The PDMS flow chamber was pressed over the array to seal the chamber and cell suspension was slowly injected through the inlet. The location of tissue spot replicates were randomized within the microarrays so that each type was evenly distributed across rows and columns to minimize systematic error associated with flow patterns during cell seeding (Supplementary Fig 2a).
A cell adhesion assay was performed on control microarrays to quantitatively investigate the experimental error associated with cell seeding and washing procedures. Control microarrays were fabricated where all spots were collagen control. Breast cancer cells were seeded at 15,000 cells/cm2 in serum free media and allowed 1.5 hr to attach. The PDMS flow chambers were removed and the microarrays were washed 4x in PBS and incubated in media with 10% FBS for 30min, stained with calcein AM (3 μg/ml) for 30 min, and imaged. Total cell number per spot was counted with ImageJ.
The assay was run in duplicate and the total error was quantified as the coefficient of variation for all spots in the microarray. Systematic error was analyzed by quantifying the average cells/spot in each row and each column. The coefficient of variation between rows or columns was calculated for each experiment. Final coefficient of variation in rows or columns was calculated by averaging the row and column coefficients of variation of duplicate tissue microarray chip.
3D microtissue optimization
Different ratios of cells to tissue ECM weight were screened to optimize parameters for formation of breast cancer-tissue ECM 3D spheroids. Cells were suspended at 500,000 cells/mL and added to tissue ECM suspended at 4−0.25 mg/ml concentrations at a 1:1 v/v ratio. 40 uL of the cell/tissue suspension was pipetted into a 96 well hanging drop culture plate (inSphero) to form a hanging drop. Seven different tissue particle types (bone, cardiac, kidney, liver, lung, spleen, and collagen) were mixed at 0, 0.25, 0.5, 1, 2, and 4 mg/ml with breast cancer cells (250k cells/ml final concentration) in hanging drops. Spheroids were imaged at 250x magnification in bright field at days 1, 2, 4, and 6 to capture spheroid formation kinetics over time. Relative cell viability/proliferation at day 6 was compared between each group with Alamar Blue assay (Life Technologies).
Spheroid microarray fabrication
We formed the microtissue array by first preparing an array of 1 mm wells in a plastic mold.. The wells were filled with water and cleared of air bubbles. Cell-tissue particle spheroids were transferred from a GravityTRAP (InSphero AG) culture dish into the wells of the plastic mold. After tissues had settled to the bottom of the wells, the mold was infiltrated with a 2% agarose solution in water at 70 °C and allowed to cool and form a gel. The agarose diffused into the water-filled chambers and embedded the microtissues. After cooling, the agarose block was removed, dehydrated and infiltrated with paraffin similar to previously described 33. Dehydration was performed with a graded ethanol solutions (100 mL: 30%, 50%, 70%, 80%, 95% × 2, 100% × 2) for 3 hrs each and 100% again overnight. Ethanol solutions were cleared with HistoClear II (100 mL) 3 times for 2 hours and once overnight and infiltrated with paraffin (100 mL, 60 °C, 4× 2hrs) and cast in paraffin.
Macrophage cell culture and morphological analysis
Bone marrow-derived macrophage (BMDM) progenitors were isolated from the femurs of wild type C57BL/6 mice and were differentiated toward a macrophage phenotype in a mixture of 80% DMEM/F12 (supplemented with 10% FBS and 1%P/S) and 20% fibroblast (L929) conditioned media (10% FBS, 100-mm L-Glutamine, and 1% P/S). Macrophage progenitors were differentiated for 7 days with media changed at day 4. Resulting BMDMs were suspended in M1 or M2 macrophage polarization media (2.5 × 105 cells in 1.5 mL) on the tissue microarrays and the surrounding acrylamide. The M1 polarization medium contained 200 ng/mL lipopolysaccharide (LPS, 055:B5, Sigma) and 20 ng/mL interferon ɣ (IFNγ, PeproTech), and M2 polarization medium contained 20 ng/mL interleukin-4 (IL-4, Peprotech). Cells were incubated for 24 hours in their respective cytokine environments before being fixed, stained for actin and imaged and analyzed with the Cellomics platform. BMDMs were suspended in unsupplemented macrophage differentiation media for seeding on tissue spots with different degrees of processing.
Cancer cell culture and adhesion analysis
Human skin cancer cell line (A-375, ATCC: CRL-1619) and human lung cancer cell line (A549, ATCC: CCL-185) were cultured in growth media. Human breast cancer cell line (Hs 578T, ATCC: HTB-126) was cultured in growth media supplemented with 0.01 mg/mL bovine insulin. Melanoma derived mouse cancer cell lines (B16-F0 [ATCC: CRL-6322], B16-F10 [ATCC: CRL-6475], B16-KY834) and L929 fibroblasts were cultured in growth media. Cell line identity was verified by referencing the ICLAC database of cross-contaminated or misidentified cell lines. Cell lines were not tested for mycoplasma contamination.
Cancer cells and L929 fibroblasts were seeded in serum free media at a final concentration of 15,000 cells/cm2 and allowed to attach for 2 hours. Cells were washed gently 4 times with PBS and incubated in media with 10% serum for 45 min and then washed again with PBS. Cells were stained with calcein AM at (3uL/mL from 1mg/mL stock) for 30 minutes at 37°C and imaged at 50x magnification. Total cells per spot were counted with ImageJ. Two chips with different tissue spot patterns (Supplementary Fig 2a) were analyzed for each cell types for a total of six spot of each tissue type (n=6). Cell adhesion was for each tissue was quantified by normalizing to the solubilized collagen coated control spots.
Cancer cell proliferation analysis
Chips were placed in one well rectangular culture dishes (Nunc) surrounded by parafilm and seeded with labeled breast cancer cells using the PDMS gasket method. Cells were pre-stained in a T-75 flask with CellTracker Green dye CMFDA (5-chloromethylfluorescein diacetate, Invitrogen) according to manufacturer instructions. A suspension of dyed cells was seeded onto the chip with a concentration of 5,000 cells/cm2 in 10% FBS media. Cells were allowed to attach overnight. The next day the gasket was removed and the media was changed. Cells were imaged at 50x magnification at day 1 and day 2 after seeding. By day 3 the tracker dyes signal had become too weak for segmentation imaging. At day 7 cells were incubated with calcein AM at (3 μg/ml) for 30 minutes at 37°C and imaged and viable cell number per spot was quantified using ImageJ. Autofluorescence of liver ECM made it impossible to resolve cell staining at day 2 so this data was not available. Cells formed an over confluent monolayer and peeled off of the soluble collagen control spot at day 7 so this data was not available. Cell number over time was used to estimate the doubling rate when seeded on different tissue spots using an exponential growth model:
Cell attachment was confirmed at day 1, and a growth lag phase assumed between day 1 and 2. Thus “t” represents a 5 day time span, the day 2 and day 7 cell counts represent the initial and final cell counts, respectively, and the doubling rate calculated as “k”.
Viability/analysis of 3D cancer cell/tissue ECM microtissues was conducted with the Alamar Blue assay. Relative cell viability was compared between each group by repeated sampling from wells incubated with Alamar Blue reagent (Life Technologies) at days 1, 3 and 6 (n=3).
hASC culture and analysis
hASCs were isolated, as previously described 35 and passaged at 90% confluence in growth media (GM) with media changes every 3 days. hASC differentiation was induced with osteogenic differentiation media (OM, DMEM, 10% FBS, 1% P/S, 100 nM dexamethasone, 50 μM ascorbic acid-2-phosphate, 10 mM β-glycerophosphate).
2D microarray
For culture on microarray chips, hASCs were seeded on tissue microarrays (NUNC) at 6000 cells/cm2 as described above. hASCs were cultured on microarrays for 5 days in GM to reach confluency, media replaced with either osteogenic media or growth media, and cultured for an additional 6 days before fixation and staining with Alizarin Red.
3D microtissues
Microtissues were formed by self assembly of cells and tissue particles into spheroids in hanging drop culture 36. Tissue particles suspensions were diluted to 0.8 mg/mL in serum-free DMEM culture media and sterilized with UV light (~1.5 mW/cm2 for 20 min). hASCs were suspended in GM at 850,000 cells/mL with a 1:1 mixture of ECM particle suspension for microtissue formation as described above. Microtissue medium was changed with GM every 2 days by replacing half of the hanging drop volume with fresh media twice. Microtissues were cultured for 6 days in GM and then transferred to 96-well GravityTRAP (InSphero) plates with control growth media or osteogenic media with media changes every 3 days. After 11 days of culture, microtissues were fixed, embedded in well molds as described above, sectioned and stained with Masson’s Trichrome or Alizarin Red.
Macrophage and hASC PCR
Changes in mouse macrophage and hASC gene expression following culture with tissue ECM was determined in 2D and 3D microenvironments, respectively.
2D macrophage microarrays
Bone, cardiac, liver, lung, and spleen tissues were processed using a modified procedure. Tissues were mechanically fragmented using a knife-mill processor (Retsch, Germany) with particle sizes no larger than 10 mm3, and rinsed thoroughly distilled water until blood was fully cleared from samples. Bone samples were decalcified by incubation in 10% formic acid (Sigma) for 3 days, verified by a colorimetric calcium test (STANBIO Laboratory). All tissues were then incubated in 3.0% peracetic acid (Sigma) with agitation at 37°C for 4 hours, changing to fresh solution after 1 hour. The pH was adjusted to 7 with thorough extensive rinsing with water and PBS. Samples were transferred to a 1% Triton-X100 (Sigma) + 2 mM sodium EDTA (Sigma) solution with agitation at room temperature for 3 days, changing to fresh solution daily. Tissues were rinsed thoroughly with distilled water until and incubated in 600 U/ml DNase I (Roche Diagnostics) + 10 mM MgCl2 (Sigma) + 10% Antifungal-Antimycotic (Gibco) at 37°C for 24 hours. Tissues were rinsed thoroughly with distilled water, frozen at −80°C, and lyophilized. Tissue particles were prepared from lyophilized samples as described earlier. Tissue particle suspensions in distilled water were adjusted to 4–5 mg/ml for coating six-well plates with 1 ml of solution followed by air drying. After drying plates were sterilized under UV light for 1 hour, then rinsed to remove any non-adhered particles prior to seeding with macrophages. Macrophages were cultured as described earlier in M1, M2, or non-polarizing M0 media conditions for 24 hours (n=3).
Macrophage RNA was extracted for PCR analysis using TRIzol reagent (Life Technologies), and RNA was purified using RNeasy Mini columns (Qiagen). cDNA was synthesized through the use of SuperScript Reverse Transcriptase III (Life Technologies) as per manufacturer’s instructions. RT-PCR was conducted on an Applied Biosystems Real Time PCR Machine using SYBR Green (Life Technologies) as a reporter. Macrophage polarization was evaluated by expression of genes associated with M1 polarization (Interleukin-1 beta [IL1β], inducible nitric oxide synthase [iNOS], and tumor necrosis factor alpha [TNFα]) and M2 polarization (Arginase-1 [Arg1], Fizz1, and Interleukin-10 [IL10]). Expression was calculated relative to non-polarized M0 macrophages cultured in uncoated control wells using β2 microglobulin (B2M) as the reference gene via the 2−ΔΔCt method, and the following primers: B2M forward CTC GGT GAC CCT GGT CTT TC, B2M reverse GGA TTT CAA TGT GAG GCG GG; TNFα forward GTC CAT TCC TGA GTT CTG, TNFα reverse GAA AGG TCT GAA GGT AGG; IL1β forward GTA TGG GCT GGA CTG TTT C, IL1β reverse GCT GTC TGC TCA TTC ACG; iNOS forward GAC GAG ACG GAT AGG CAG AG, iNOS reverse GTG GGG TTG TTG CTG AAC TT; Arg1 forward CAG AAG AAT GGA AGA GTC AG, Arg1 reverse CAG ATA TGC AGG GAG TCA CC; Fizz1 forward CTT TCC TGA GAT TCT GCC CCA G, Fizz1 reverse CAC AAG CAC ACC CAG TAG CA; IL10 forward TCT CAC CCA GGG AAT TCA AA, IL10 reverse AAG TGA TGC CCC AGG CA (Integrated DNA Technologies). Fold change expression for each polarizing media condition was displayed as a heat map with hierarchial clustering by tissue ECM coating.
3D hASC microtissues
Osteogenic differentiation was determined in bone and lung microtissues using qRT-PCR. Microtissues were formed as described above for 7 days in GM followed by an additional 14 days in osteogenic media. A total of 3 microspheres per treatment group were pooled and homogenized for RNA extraction using TRIzol Reagent per manufacturer’s instructions. RNA was then reverse transcribed to cDNA using Superscript III reverse transcriptase (Life Technologies) following the manufacturer’s instructions. Real-time PCR was carried out using a StepOnePlus Real-time PCR System (Applied Biosystems). Osteocalcin gene expression at day 21 for each tissue relative to day 7 was calculated using GAPDH as the reference gene via the 2-ΔΔCt method and the following primers: Osteocalcin forward CCTCACACTCCTCGCCCTAT, Osteocalcin reverse CTTGGACACAAAGGCTGCAC; GAPDH forward AGGAGCGAGATCCCTCCAAA, GAPDH reverse AAATGAGCCCCAGCCTTCTC.
Histology
Microarray chips were fixed in 4% paraformaldehyde (PFA) prior to staining procedures. 3D microtissues were fixed in PFA for 1 hr prior to embedding in microarrays, as described above. Paraffin blocks containing microtissue arrays were sectioned at 5 μm and stained as described below.
Actin and nuclear staining
Cells were permeabilized in 1% Triton X-100 and stained for actin with Alexa Fluor-647 conjugated Phalloidin (Life Technologies) at a 1:25 dilution in 0.1% BSA (Sigma) in PBS for 30 min at 37 °C. Nuclei were stained with DAPI (100 ng/mL, Life Technologies) for 30 min.
In vitro macrophage immunofluorescence
Macrophages seeded on tissue arrays in M1 or M2 polarizing cytokines as described above were evaluated for expression of the M2 and M1 markers Arginase-1 and iNOS. After 24 hours, macrophages were fixed for 10 min in 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 in TBS for 10 min. Nonspecific protein interactions were blocked with 1% bovine serum albumin, 2% horse serum in 0.05% Tween-20. Tissue microarrays were then incubated with primary antibodies against Arginase-1 (rabbit polyclonal diluted 1:100, GTX109242, GeneTex) and iNOS (mouse monoclonal [4E5] diluted 1:300, ab129372, abcam) diluted in 1% bovine serum albumin in TBS overnight at 4°C. Arrays were washed and probed with FITC goat anti-rabbit IgG (1:250, Jackson Immuno) and Alexa Fluor-568 goat anti-mouse IgG (1:250, Invitrogen) secondary antibodies diluted in 1% bovine serum albumin for 1 hour. ECM autofluorescence was then blocked by treatment with 0.1% Sudan Black B in 70% ethanol for 20 min. Arrays were washed, coverslipped, and imaged. Variations in background autofluorescence were corrected by subtracting the mean background intensity of acellular regions of each tissue array spot using ImageJ software.
Subcutaneous tissue ECM implantation and immunofluorescence
Animal experiments were conducted in accordance with guidelines set by the Johns Hopkins University Animal Care and Use Committee. Bone and collagen tissue ECM particulate was hydrated with saline (100 mg dry wt/0.2 ml) and injected subcutaneously on the dorsum of 6–8 week old female C57BL/6 mice. Animals were sacrificed after 1 week, implants explanted, fixed in formalin, and embedded in paraffin for sectioning. Sections were then deparaffinized, rehydrated, and immunolabeled for the pan-macrophage marker F4/80 and the M1 marker iNOS. Antigen retrieval was conducted in citrate (10 mM, pH 6) for 30 min in a vegetable steamer, and rinsed with TBS in 0.05% Tween-20. Sections were blocked with 1% bovine serum albumin, 2% goat serum, and 0.05% Tween-20 for 1 hour. Sections were then incubated with primary antibodies against F4/80 (rat monoclonal [BM8] diluted 1:100, ab16911, abcam) and iNOS (mouse monoclonal [4E5] diluted 1:200, ab129372, abcam) overnight at 4°C in blocking solution. Sections were washed and probed with FITC goat anti-rabbit IgG (1:250, Jackson Immuno) and Alexa Fluor-594 goat anti-rat IgG (1:250, Invitrogen) secondary antibodies diluted in blocking solution for 1.5 hours. ECM autofluorescence was then blocked by treatment with 0.1% Sudan Black B in 70% ethanol for 20 min. Sections were counterstained with DAPI for 5 min, washed, coverslipped, and imaged.
Alizarin red staining
Calcified matrix staining was performed on microarray chips with Alizarin Red (40 mM, pH 4.1) for 25 min. Chips were briefly rinsed with water 3 times and then a fourth time for 5 min, before rapid dehydration and clearing in acetone, acetone:xylene (50:50) and xylene, followed by coverslipping. Sectioned slides containing microtissue arrays were deparaffinized and rehydrated, stained with Alizarin Red for 5 min, washed, dehydrated and cleared rapidly through acetone, 1:1 acetone-xylene, xylene, and coverslipped.
Electron microscopy
Dry tissue microarrays were placed in a desiccator overnight to dry completely, attached to aluminum stubs via carbon sticky tabs (Ted Pella, Inc.), and coated with 20 nm of AuPd with a Denton Vacuum Desk III sputter coater (Denton Vacuum, LLC). Stubs were viewed and digital images captured at a 60° tilt on a LEO 1530 Field Emission Scanning Electron Microscope (FESEM) operating at 1 kV.
Slide scanning and analysis
Cell counts and morphology on microarray chips were imaged and quantified using a Cellomics high content scanner (Cellomics) with ArrayScan VTI software (Thermo Fisher). Microarray chips were adhered to 4-well rectangular culture plates (NUNC). Cell counts were quantified from 50x images (2 fields per spot) with calcein AM staining. Morphological quantification was performed following phalloidin staining as described above. The object identification parameters were adjusted to outline the perimeters of individual cells in each image that were subsequently counted and analyzed for morphology using the convex hull area ratio. 2D Microarrays stained with Alizarin red and histological slides containing 3D spheroid microarrays were scanned with a ScanScope AT (Aperio) at 200x magnification and viewed with ImageScope software (Aperio).
Image processing
Cell counts for adhesion studies were analyzed from 50x images using ImageJ (NIH, Bethesda, MD). Cell number for each spot (n=6) was normalized control spots (soluble collagen) on the same microarray. Color images of 2D and 3D Alizarin red stained microarrays were analyzed with ImageJ to quantify the percent area of Alizarin red staining. Regions of interest (ROI) were traced around the perimeter of each individual sample in the array to measure total area. Microarrays were threshold color filtered in the RGB color space to select areas of positive red staining and the filtered image was converted to binary. The percent area of positive staining was calculated using the measure command in the ROI manager.
Mechanical Testing
The elastic modulus and hardness of dried tissue spots were measured using a Nano Indentor XP (MTS Systems Corporation) and Nanosuite software which supports measurements of complex, thin materials. Samples were approached with a velocity of 10 nm/s and samples were indented with a Berkovich tip to a depth of 400 nm at a strain rate of 0.05 with a peak hold time of 10 s and 90% unload. To minimize the influence of the underlying glass coverslip upon array mechanics, sample thickness was increased by double spotting. First, 10 μL of particle suspension was spotted and dried as described previously, and followed by an additional 10μL of particle suspension dried on top of the original spot. Since each test required several hours, measurements were made in a dried state to avoid confounding factors of evaporation. Six different tissue spots (bladder, brain, cardiac, liver, lung, and spleen) were indented in 8–10 different locations per sample. Outlier measurements were excluded from analysis as determined by Grubbs’ test (1 measurement each was excluded for bladder, brain, lung, and spleen) as described in the statistical methods.
Proteomics network analysis
The abundance of extracellular proteins (identified at the gene level) in each tissue were correlated with in vitro assay results. In some cases, assay results were not available for all tissues, resulting in fewer than 11 tissues for a given assay. Correlation coefficients for each of the 3879 identified proteins in the 15 in vitro assays were calculated using the assay results directly and by replacing each assay result with its rank-ordered value. The P-value and t-statistic for each protein/assay correlation was calculated from the linear model for both the original and rank-transformed assay data. The assay/protein results were visualized as a heatmap showing the correlation coefficients for all genes/assays (Cluster 3.0) and by creating an assay/protein network (Cytoscape v3.2.1). Proteins and assays are displayed as vertices, and edges connect proteins to assays when they are strongly correlated (p < 0.05 for both the original assay and the rank-transformed assay results). The full results of these analyses are provided in the Supplementary Table 2.
Gene set enrichment analysis was conducted to determine whether defined groups of proteins improved predictive value for each assay. This was applied to Bone, Cartilage, Lung, and Spleen tissues using the Biological Processes Gene Ontology database. ECM proteins from each of these tissues were evaluated for enrichment terms related to osteogenesis, and the corresponding p-values for enrichment compared. Terms related to osteogenesis include: skeletal system development (GO: GO:0001501), ossification (GO:0001503), bone morphogenesis (GO:0060349), osteoblast differentiation (GO:0001649), and chondrocyte differentiation (GO:0002062). Identified ECM proteins annotated to these processes are listed in Supplementary Table 3.
Statistical Analysis
Statistical significance for quantitative in vitro cell assays were determined by a one-way analysis of variance (ANOVA) with post-hoc Tukey test (Graphpad Prism v6, GraphPad Software Inc.). The effect of different processing methods on macrophage morphology was determined by a student’s t-test with the Bonferroni correction. Statistical significance was defined as a P-value<0.05. Differences between tissue spots were represented as tissue symbols above each bar (representing the mean ± s.d.), or attached in the Supplementary Information (Supplementary Table 1). Statistical outliers for nanoindentation measurements were observed from Tukey box plots as values beyond 1.5 times the interquartile range of the quartile borders, and confirmed via Grubbs’ outlier test using alpha=0.05 (Graphpad Prism v6). These outlier measurements were excluded from analysis of tissue spot elastic modulus and hardness. Power analysis was not conducted to determine sample size and investigators were not blinded.
Supplementary Material
Acknowledgments
The authors wish to acknowledge Maryland Stem Cell Investigator-Initiated Grant, the Department of Defense Grant W81XWH-l1-2-0022 and Armed Forces Institute of Regenerative Medicine, and the Jules Stein Chair from the Research to Prevent Blindness for funding. The authors thank M. Frisk for carefully editing the manuscript and providing insightful suggestions. VB thanks the Hartwell Foundation Postdoctoral Fellowship. AP is supported by NCI’s Clinical Proteomic Tumor Analysis Consortium initiative (U24CA160036). JE would like to acknowledge the Johns Hopkins Tilghman Fellowship Sabbatical Grant and the Swiss National Foundation Visiting Professor Grant along with M. Swartz and the Swartz lab for hosting a semester sabbatical. We acknowledge A. Ewald for discussions on the systems biology analysis and for providing mammary tumors for processing and array spotting. We thank R. Cole and L. DeVine of the Johns Hopkins School of Medicine Mass Spectrometry and Proteomics Facility for mass spectrometry preparation and analysis.
Footnotes
Author Contributions
VB and JE conceived idea. VB made 2D and 3D tissue arrays. VB performed stem cell studies. MB and HJ developed seeding chambers and performed cancer adhesion and proliferation studies. KS and MW performed macrophage studies. KS performed gel electrophoresis work and collagen ECM classification. SM, VB, MW, and AP performed proteomic analysis. DP provided melanoma lines and guidance for cancer studies. MW and JB performed systems analysis. VB, MW, and JE evaluated all data and wrote the manuscript.
There is no competing financial interest for the array technologies. JE has financial interest in a specific tissue ECM technology, acellular adipose tissue.
References
- 1.Hay ED. Extracellular matrix. J Cell Biol. 1981;91:205s–223s. doi: 10.1083/jcb.91.3.205s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Watt FM, Huck WTS. Role of the extracellular matrix in regulating stem cell fate. Nat Rev Mol Cell Biol. 2013;14:467–473. doi: 10.1038/nrm3620. [DOI] [PubMed] [Google Scholar]
- 3.Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15:786–801. doi: 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee GY, Kenny PA, Lee EH, Bissell MJ. Three-dimensional culture models of normal and malignant breast epithelial cells. Nat Methods. 2007;4:359–365. doi: 10.1038/nmeth1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lavin Y, et al. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell. 2014;159:1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim MS, et al. A draft map of the human proteome. Nature. 2014;509:575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uhlen M, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 8.Anderson DG, Levenberg S, Langer R. Nanoliter-scale synthesis of arrayed biomaterials and application to human embryonic stem cells. Nat Biotechnol. 2004;22:863–866. doi: 10.1038/nbt981. [DOI] [PubMed] [Google Scholar]
- 9.Mei Y, et al. Combinatorial development of biomaterials for clonal growth of human pluripotent stem cells. Nat Mater. 2010;9:768–778. doi: 10.1038/nmat2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flaim CJ, Chien S, Bhatia SN. An extracellular matrix microarray for probing cellular differentiation. Nat Methods. 2005;2:119–125. doi: 10.1038/nmeth736. [DOI] [PubMed] [Google Scholar]
- 11.Reticker-Flynn NE, et al. A combinatorial extracellular matrix platform identifies cell-extracellular matrix interactions that correlate with metastasis. Nat Commun. 2012;3:1122. doi: 10.1038/ncomms2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gobaa S, et al. Artificial niche microarrays for probing single stem cell fate in high throughput. Nat Methods. 2011;8:949–955. doi: 10.1038/nmeth.1732. [DOI] [PubMed] [Google Scholar]
- 13.Dolatshahi-Pirouz A, et al. A combinatorial cell-laden gel microarray for inducing osteogenic differentiation of human mesenchymal stem cells. Sci Rep. 2014;4:3896. doi: 10.1038/srep03896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woodruff K, Fidalgo LM, Gobaa S, Lutolf MP, Maerkl SJ. Live mammalian cell arrays. Nat Methods. 2013;10:550–552. doi: 10.1038/nmeth.2473. [DOI] [PubMed] [Google Scholar]
- 15.Ranga A, et al. 3D niche microarrays for systems-level analyses of cell fate. Nat Commun. 2014;5:4324. doi: 10.1038/ncomms5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mouw JK, Ou G, Weaver VM. Extracellular matrix assembly: a multiscale deconstruction. Nat Rev Mol Cell Biol. 2014;15:771–785. doi: 10.1038/nrm3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uygun BE, et al. Organ reengineering through development of a transplantable recellularized liver graft using decellularized liver matrix. Nat Med. 2010;16:814–820. doi: 10.1038/nm.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crapo PM, Gilbert TW, Badylak SF. An overview of tissue and whole organ decellularization processes. Biomaterials. 2011;32:3233–3243. doi: 10.1016/j.biomaterials.2011.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu I, Nahas Z, Kimmerling KA, Rosson GD, Elisseeff JH. An injectable adipose matrix for soft-tissue reconstruction. Plast Reconstr Surg. 2012;129:1247–1257. doi: 10.1097/PRS.0b013e31824ec3dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kononen J, et al. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med. 1998;4:844–847. doi: 10.1038/nm0798-844. [DOI] [PubMed] [Google Scholar]
- 21.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 22.Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011;17:320–329. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Badylak SF, Valentin JE, Ravindra AK, McCabe GP, Stewart-Akers AM. Macrophage phenotype as a determinant of biologic scaffold remodeling. Tissue Eng Part A. 2008;14:1835–1842. doi: 10.1089/ten.tea.2007.0264. [DOI] [PubMed] [Google Scholar]
- 24.DeForest CA, Polizzotti BD, Anseth KS. Sequential click reactions for synthesizing and patterning three-dimensional cell microenvironments. Nat Mater. 2009;8:659–664. doi: 10.1038/nmat2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tibbitt MW, Anseth KS. Dynamic microenvironments: the fourth dimension. Science translational medicine. 2012;4:160ps124. doi: 10.1126/scitranslmed.3004804. [DOI] [PubMed] [Google Scholar]
- 26.Badylak SF, Lantz GC, Coffey A, Geddes LA. Small intestinal submucosa as a large diameter vascular graft in the dog. J Surg Res. 1989;47:74–80. doi: 10.1016/0022-4804(89)90050-4. [DOI] [PubMed] [Google Scholar]
- 27.Nerem RM. Regenerative medicine: the emergence of an industry. Journal of the Royal Society, Interface/the Royal Society. 2010;7(Suppl 6):S771–775. doi: 10.1098/rsif.2010.0348.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuribayashi H, et al. Clinicopathological analysis of primary lung carcinoma with heterotopic ossification. Lung Cancer. 2009;64:160–165. doi: 10.1016/j.lungcan.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 29.Averill MM, Kerkhoff C, Bornfeldt KE. S100A8 and S100A9 in cardiovascular biology and disease. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:223–229. doi: 10.1161/ATVBAHA.111.236927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vilen ST, et al. Trypsin-2 enhances carcinoma invasion by processing tight junctions and activating ProMT1-MMP. Cancer investigation. 2012;30:583–592. doi: 10.3109/07357907.2012.716467. [DOI] [PubMed] [Google Scholar]
- 31.Vizcaino JA, et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol. 2014;32:223–226. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naba A, et al. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics. 2012;11 doi: 10.1074/mcp.M111.014647. M111014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan P, Seelentag W, Bachmann A, Bosman FT. An agarose matrix facilitates sectioning of tissue microarray blocks. J Histochem Cytochem. 2007;55:21–24. doi: 10.1369/jhc.6A6987.2006. [DOI] [PubMed] [Google Scholar]
- 34.Yoshimura K, et al. Integrin alpha2 mediates selective metastasis to the liver. Cancer research. 2009;69:7320–7328. doi: 10.1158/0008-5472.CAN-09-0315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mitchell JB, et al. Immunophenotype of human adipose-derived cells: temporal changes in stromal-associated and stem cell-associated markers. Stem Cells. 2006;24:376–385. doi: 10.1634/stemcells.2005-0234. [DOI] [PubMed] [Google Scholar]
- 36.Bratt-Leal AM, Carpenedo RL, Ungrin MD, Zandstra PW, McDevitt TC. Incorporation of biomaterials in multicellular aggregates modulates pluripotent stem cell differentiation. Biomaterials. 2011;32:48–56. doi: 10.1016/j.biomaterials.2010.08.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.