

Abstract
Design, synthesis, biological and X-ray crystallographic studies of a series of potent HIV-1 protease inhibitors are described. Various polar functionalities have been incorporated on the tetrahydropyranyl-tetrahydrofuran-derived P2 ligand to interact with the backbone atoms in the S2 subsite. The majority of the inhibitors showed very potent enzyme inhibitory and antiviral activity. Two high-resolution X-ray structures of 30b- and 30j-bound HIV-1 protease provide insight into ligand-binding site interactions. In particular, the polar functionalities on the P2 ligand appear to form unique hydrogen bonds with Gly48 amide NH and amide carbonyl groups in the flap region.
Keywords: HIV-1 protease inhibitors, antiviral, darunavir, multidrug-resistant, design, synthesis, X-ray crystal structure, backbone binding
Introduction
The development of human immunodeficiency virus type 1 (HIV-1) protease inhibitors (PIs) as part of the current active antiretroviral therapy has significantly reduced the mortality of HIV/AIDS patients.1,2 However, one of the major drawbacks of many approved protease inhibitors is the emergence of drug resistant HIV-1 strains which may severely limit long-term treatment options.3,4 Therefore, design and development of protease inhibitors with broad-spectrum activity against multidrug-resistant HIV-1 variants are critically important. As part of our continuing interest to develop novel PIs, we reported a variety of novel inhibitors that are potent against HIV-1 variants resistant to currently approved PIs.5,6 One of these PIs, darunavir (1, Figure 1) which contains a structure-based designed nonpeptidic P2 ligand, 3(R),3a(S),6a(R)-bis-tetrahydrofuranyl urethane (bis-THF), was approved as a first-line therapy for the treatment of HIV/AIDS patients.7–10
Figure 1.

Structure of PIs 1–4 and 30f,j
The bis-THF ligand in darunavir is responsible for its superb activity against multidrug resistant HIV-1 strains.5,7 In an effort to further optimize the structural feature of bis-THF, we have designed a furopyranol or tetrahydropyranyl tetrahydorfuran (Tp-THF) ligand.11,12 The X-ray structure of inhibitors 1 and 2 with HIV-1 protease revealed that both ring oxygens are involved in strong hydrogen bonding interactions with the backbone NHs of Asp29 and Asp30 in the S2 subsite. Also, the bicyclic ring system fills in the hydrophobic pocket in the S2 site. We have recently incorporated substituents on the bis-THF and Cp-THF rings (inhibitor 3) to effect specific ligand-binding site interactions in the S2-subsite of HIV-1 protease.13–16 Hohlfeld and co-workers have also reported potent HIV-1 protease inhibitors incorporating substituted bis-THF ligands.17,18 To further optimize ligand-binding site interactions, we speculated that a larger tetrahydropyranyl ring in place of the tri-substituted THF ring may improve hydrogen bonding with the active site aspartate backbone NHs due to an increase in the dihedral angle of the bicyclic acetal bearing the tetrahydropyranyl tetrahydrofuran ring system. Furthermore, a larger THP-ring in the fused THP-THF (Tp-THF) ligand may enhance van der Waals interactions. Tp-THF ligand-derived inhibitor 4 exhibited high enzyme affinity and antiviral activity. Also, inhibitor 4 has been shown to retain high potency against a variety of multi-drug resistant clinical HIV-1 strains with EC50 values in low nanomolar range. In an effort to further improve backbone ligand-binding site interactions, based upon the X-ray structure of 4-bound HIV-1 protease, we have incorporated various polar substituents at the C5-position of Tp-THF ligand. We speculated that a stereochemically defined hydroxyl group, alkoxy group or amine derivative could form a hydrogen bond with the backbone atoms of Gly48 located in the flap region of HIV-1 protease. Also, the corresponding alkyl chain may enhance additional van der Waals interactions in the active site. Herein we report our studies on the design, synthesis, biological evaluation and X-ray structural studies of C5-substituted tetrahydropyranyl tetrahydrofuran ligands. A number of inhibitors exhibited very high enzyme inhibitory and antiviral activity. For the synthesis of the tetrahydropyranyl tetrahydrofuran ring system P2-ligands in this study, we developed a highly diastereoselective tetrahydropyranyl tetrahydrofuran ring system using a [2,3]-sigmatropic rearrangement as the key reaction.
Results and discussion
Synthesis of the fused tetrahydropyranyl-tetrahydrofuran ligand is shown in Scheme 1. Methyl ester 5 is obtained in four steps from L-malic acid as described by Solladié and coworkers.19 Dibal-H reduction of 5 provided the corresponding aldehyde. Wittig olefination of the resulting aldehyde in MeOH at 0 °C afforded α,β-unsaturated ester 6 in 70% yield as a mixture (8:1) of cis/trans isomers. Dibal-H reduction of 6 provided the corresponding cis-alcohol. Cesium hydroxide promoted O-alkylation with t-butyl bromoacetate gave the desired alkylated product 7 in 95% yield over 2 steps. Sigmatropic rearrangement of 7 using LiHMDS at −65 °C resulted in a mixture of rearrangement products 9 and 10 in 77% yield (diastereomeric ratio 8.5:1).16,20 The absolute stereochemical configuration of the major isomer 9 was determined through the use of 2D COSY and 1D NOE after conversion to the subsequent ligand derivatives.
Scheme 1.

Reagents and conditions: (a) Dibal-H, CH2Cl2, −78 °C, 1 h; (b) MeOH, Ph3P=CHCO2Et, 0 °C, 70%; (c) Dibal-H, Ch2Cl2, −78 °C; (d) t-butyl bromoacetate, CsOH·H2O, TBAI, CH3CN, 12 h, 95% 2 steps; (e) LiHMDS, THF, −65 °C −20 °C, 2 h, 77% overall, major/minor (8.5:1); (f) MsCl, Et3N, THF, 0 °C, 1 h; (g) LAH, THF, 23 °C, 24 h, 70% 2 steps; (h) O3, DMS, −78 °C, then p-TsOH, 23 °C, 18 h, 70%.
The stereochemical outcome of the [2,3]-sigmatropic rearrangement can be rationalized based upon the transition state model 8 as described in the literature. 21,22 The observed selectivity is somewhat lower as compared to the isopropylidene derivative. This is possibly due to the greater flexibility of the 6-membered dioxane ring in substrate 7. Mesylation of compound 9 followed by lithium aluminum hydride reduction gave alcohol 11 in 70 % yield over 2 steps. Ozonolysis and subsequent acid catalyzed cyclization resulted in the desired Tp-THF ligand 12 in 70 % yield. The route provided Tp-THF ligand 12 in 25% overall yield from 5. This ligand was converted to the desired inhibitor as described previously.14,23 The sigmatropic rearrangement product 9 is an effective intermediate for the synthesis of substituted P2-ligands. As shown in Scheme 2, the hydroxyl group was converted to methyl ether 13 using NaH and methyl iodide in THF. Reduction of 13 by LiAlH4, ozonolytic cleavage of the double bond, and subsequent treatment of the resulting aldehyde with p-TsOH in THF provided the corresponding C3-methoxy substituted ligand 14.
Scheme 2.

Reagents and conditions: (a) NaH, MeI, THF, 0 °C – 23 °C, 90%; (e) PPh3, DIAD, p-nitrobenzoic acid, DCM, 0 °C, 18 h, 81%; (b) LAH; (c) O3, DMS; (d), THF, p-TsOH, 30%, 2 steps; (f) K2CO3/MeOH, 0 °C, 1 h, 98%; (g) MeI, NaH, THF, 0 °C – 23 °C, or, BnBr, TBAI, NaH, THF, 0 °C to 23 °C 2 h; (h) LAH, THF, 0 °C, 1 h; (i) O3, DMS, then, THF, p-TsOH.
Rearrangement product 9 was subjected to Mitsunobu inversion with p-nitrobenzoic acid to provide benzoate 15.24 Selective deprotection of the p-nitrobenzoate in the presence of the t-butyl ester was accomplished in K2CO3 and MeOH at 0 °C over 1 h. Alkylation of the resulting alcohol using NaH and respective alkyl halide provided the corresponding alkylated product in high yield (88 – 96%). Ozonolysis of the O-alkylated products 16–18, followed by acid catalyzed cyclization, gave various C3 O-alkylated Tp-THF ligands 19–21 in 60 – 70 % yield.
The synthesis of various C3-amine derivatives was accomplished utilizing hydroxy ester 9. As shown in Scheme 3, reduction of 9 with LAH provided the corresponding diol. Protection of the primary alcohol with trityl chloride and Et3N afforded the trityl derivative 22 over 92% yield in two-steps. Alcohol 22 was subjected to Mitsunobu azidation using diphenyl phosphorazidate (DPPA), triphenyl phosphine and diethyl azodicarboxylate (DEAD) to provide the corresponding azide derivative in 70% yield. Ozonolysis of the resulting olefin followed by treatment with p-TsOH in CH2Cl2 afforded bicyclic azide derivative 23 in 74% yield in two-steps. Catalytic hydrogenation of 23 with 10% Pd/C under a hydrogen-filled balloon provided the corresponding amine in near quantitative yield. The resulting amine was converted to various benzyl derivatives as follows: reductive amination with benzyldehyde in the presence of NaBH3CN in MeOH affording the corresponding benzylamine, which was then subjected to further reductive amination with formaldehyde, acetaldehyde or propionaldehyde to provide substituted amines 24 – 26 in 45–81% yield. This double alkylation was carried out in a single pot operation.25
Scheme 3.

Reagents and conditions: (a) LAH, THF, 0 °C – 23 °C, 1h; (b) TrCl, Et3N, DCM, 23 °C, 18 h, 92% 2 steps; (c) PPh3, DPPA, DEAD, THF, 0 °C – 23 °C, 24 h, 70%; (d) O3, DMS, −78 °C, then p-TsOH, 23 °C, 18h, 74%; (e) Pd/C, H2, MeOH, 1h; (f) PhCHO, NaBH3CN, MeOH, then RCHO, 24 h, 45 – 81%.
For the synthesis of C3-substituted Tp-THF-derived inhibitors, various selected alcohol derivatives were converted to the corresponding mixed activated carbonate derivatives 27a–h. As shown in Scheme 4, optically active ligand alcohols (14, 19–21, 24–26) were reacted with 4-nitrophenyl chloroformate and pyridine in CH2Cl2 at 0 °C to 23 °C for 12 h, furnishing mixed carbonates 27a–h in 24–90% yields.26 The synthesis of inhibitors with hydroxyethylsulfonamide isosteres with p-methoxysulfonamide and p-aminosulfonamide as the P2′-ligand is shown in Scheme 5. Treatment of amines 28 and 29 with mixed activated carbonates 27a–e in CH3CN in the presence of diisopropylethylamine (DIPEA) provided inhibitors 30a–e, g. For the synthesis of inhibitor 30f, inhibitor 30e was subjected to catalytic hydrogenation using Pearlman’s Catalyst in MeOH at 60 psi hydrogen pressure for 12 h. This has resulted in inhibitor 30f in 38% yield over two steps. Similarly, hydrogenation of azide 30g over 10% Pd-C in ethyl acetate under a hydrogen-filled balloon for 1 h provided inhibitor 30h in 85% yield.
Scheme 4.

Reagents and conditions: (a) pyridine, CH2Cl2, 0 °C – 23 °C, 12 h (32–90%).
Scheme 5.

Reagents and conditions: (a) DIPEA, CH3CN, 23 °C, 24 h (30–85%); (b) H2, Pd(OH)2, MeOH, 60 psi; (c) H2, 10% Pd-C, EtOAc; (d) H2, Pd(OH)2, 5% NH3 in MeOH; (e) NaBH3CN, HCHO, MeOH,
For synthesis of inhibitors 30i–l, amines 28 and 29 were reacted with active mixed carbonate 27f–h in the presence of DIEPA to provide corresponding urethanes. Catalytic hydrogenation of the resulting urethanes provided inhibitors 30i–l. For the synthesis of inhibitor 30m, methylamine derivative 30i was subjected to reductive amination using Na(CN)BH3 in MeOH in the presence of paraformaldehyde in CH2Cl2 to provide 30m in 67% yield.
As stated earlier, our main objective is to investigate the effect of additional inhibitor-HIV-1 protease interactions in the S2 subsite. In particular, we planned to target the Gly48 backbone atoms in the flap region of HIV-1 protease. We first investigated the effect of the hydroxyl group and its alkyl ethers at the C4 position of the Tp-THF ligand. The structure and activity of these inhibitors are shown in Table 1. Enzyme inhibitory potency was evaluated using assay developed by Toth and Marshall.27 Inhibitors 30a and 30b were designed to examine the importance of the R/S configuration of the C5-methoxy group. As it turned out, compound 30b with a 5-(R)-configuration has shown over 300-fold better enzymatic Ki compared to compound 30a with a 5-(S)-methoxy group. Antiviral activity of selected compounds were evaluated using protocol described previously.28 Consistent with potent enzyme inhibitory activity of 30b, it showed an antiviral IC50 value of 0.2 nM, which is over 85-fold better than the antiviral activity of 30a. Based upon our preliminary model, we presume that C5-(R)-configuration is optimum for hydrogen bonding interactions with Gly48 backbone atoms. We subsequently investigated other C5-substituents. Compound 30c with 4-aminosulfonamide as the P2′-ligand showed reduction of potency compared to 30b. Compound 30d with 5-(R)-ethoxy substituent showed comparable enzyme inhibitory activity. Benzyl ether derivative 30e also showed similar antiviral potency as compound 30b. Compound 30f with a 5-(R)-hydroxy group also showed very potent enzyme inhibitory and antiviral activity, however it was 10-fold less potent than compound 30b. Interestingly, inhibitor 30f maintained full antiviral activity against HIVDRVRP20 drug resistant strains of HIV (IC50 = 3.3 nM). Another objective of our investigation is to incorporate a basic amine functionality on the P2-ligand. Particularly, incorporation of an alkyl amine may serve the purpose of hydrogen bonding interaction with Gly48 backbone atoms through the amine NH. Also, a suitable alkyl group may fill in the hydrophobic pocket in the S2-subsite. Towards this goal, we have incorporated various amine and substituted amine derivatives at the C5-position of the Tp-THF ligand. Based upon previous observation of the preference for the C5-(R)-configuration, we have investigated derivatives containing 5-(R)-configuration. The structures and activity of amine derivatives are shown in Table 2. As can be seen, compounds 30g and 30h with C5-azide and C5-amine functionalities are significantly less potent than the corresponding methoxy and hydroxyl derivatives 30b and 30f respectively. Compound 30i with a methylamine substituted is very potent. Compound 30j with an ethylamine functionality showed very potent antiviral activity with an IC50 value of 0.8 nM. The corresponding 4-aminosulfonamide derivative 30k, however, showed significant reduction in antiviral potency. The propyl amine derivative 30l, with a sterically demanding alkyl side chain, showed nearly 3-fold reduction in antiviral potency compared to ethylamine derivative 30j. Compound 30m with a dimethylamine functionality showed reduction in enzyme Ki, however it showed 3-fold improved antiviral activity over its methyl derivative 30i.
Table 1.
Structure and activity of O-substituted inhibitors
| Entry | Inhibitor | Ki(nM)a | IC50(nM)a,b |
|---|---|---|---|
| 1. |
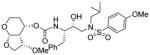
|
1.38 | 47 |
| 2. |
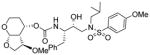
|
0.0045 | 0.2 |
| 3. |
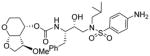
|
0.037 | 15 |
| 4. |
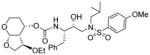
|
0.0033 | nt |
| 5. |
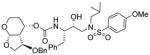
|
0.012 | 0.45 |
| 6. |
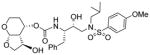
|
0.06 | 3.15 |
Ki values represents at least 5 data points. Standard error in all cases less than 7%. Darunavir (control) exhibited Ki = 16 pM.
IC50 values were determined using the MTT assay. Values are means of at least three experiments. Standard error in all cases less than 5%. Darunavir exhibited IC50 = 1.6 nM. nt, not tested.
Table 2.
Structure and activity of N-substituted inhibitors
| Entry | Inhibitor | Ki(nM)a | IC50(nM)b |
|---|---|---|---|
| 1. |
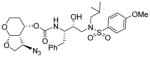
|
0.043 | 44 |
| 2. |
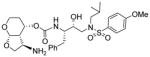
|
21.4 | 520 |
| 3. |
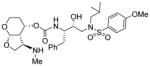
|
0.008 | 27 |
| 4. |
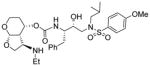
|
0.009 | 0.8 |
| 5. |
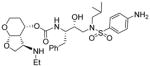
|
0.0058 | 22 |
| 6 |
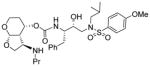
|
0.0073 | 2.8 |
| 7. |
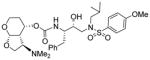
|
0.023 | 0.8 |
Ki values represents at least 5 data points. Standard error in all cases less than 7%. Darunavir (control) exhibited Ki = 16 pM.
IC50 values were determined using the MTT assay. Values are means of at least three experiments. Standard error in all cases less than 5%. Darunavir exhibited IC50 = 1.6 nM.
In order to obtain molecular insight into the HIV-1 protease-inhibitor interactions, we have determined X-ray structures of HIV-1 protease complexed with compounds 30b and 30j. A stereoview of 30b-bound HIV-1 protease structure is shown in Figure 2. The crystal structure of wild type HIV-1 protease with the inhibitor 30b was determined and refined at 1.22 Å resolution. This crystal structure contains the protease dimer and the inhibitor bound in two orientations related by a 180° rotation with 55/45% relative occupancies. The overall structure is similar to the structure of HIV-1 protease with darunavir29 with root mean square differences of 0.13 Å for Cα atoms. Inhibitor 30b has a tetrahydropyrano-tetrahydrofuran (Tp-THF) group at P2, which bears a methoxy group. The Tp-THF ring shows a slight bend compared to the complex with the related bis-THF inhibitor bis-THF derived (pdb code: 3QAA).30 As shown, the methoxy group on the Tp-THF ligand forms a water-mediated interaction with the amide NH of Gly 48. These interactions likely stabilize the flexible flap region of the active site cavity. The new interactions with the flap region may be responsible for its robust antiviral activity.
Figure 2.

Stereoview of the X-ray structure of inhibitor 30b (turquoise color)-bound to the active site of wild-type HIV-1 protease (PDB code: 5DGU). All strong hydrogen bonding interactions are shown as dotted lines.
We have also determined the X-ray crystal structure of 30j-bound HIV-1 protease. A stereoview of the active site is shown in Figure 3. This structure was determined and refined at 1.62Å resolution. Similar to structure for 30b, the structure contains the protease dimer and inhibitor 30j is bound in two orientations related to 180 ° rotation with 55/45 relative occupancies. The structure is related to 30b-bound HIV-1 protease. The major difference is in the binding of the ethylamino substituent on the Tp-THF ring. As it turns out, the amine NH on the Tp-THF ring of the inhibitor forms a water-mediated hydrogen bonding interaction with the backbone amide NH of Gly48. In addition, the amine NH forms a direct hydrogen bond with the carbonyl oxygen of Gly48. This interaction is lacking for inhibitor 30b. The ethylamino nitrogen atom in inhibitor 30j shifts its position to enhance the interactions with the wild-type HIV-1 protease. These interactions are likely to stabilize the flexible flap region of the active site. This may help maintain antiviral activity of the inhibitor against multidrug-resistant HIV-1 variants.
Figure 3.

Stereoview of the X-ray structure of inhibitor 30j (green color)-bound to the active site of wild-type HIV-1 protease (PDB code: 5DGW). All strong hydrogen bonding interactions are shown as dotted lines.
Conclusion
In summary, we have designed and synthesized alkoxy- and amine-substituted derivatives of a tetrahydropyran-tetrahydrofuranyl ligand. These molecules are specifically designed to make hydrogen bonding interactions with the flap region of HIV-1 protease. In addition, the alkyl substituents are expected to make hydrophobic interactions in the S2 subsite. The syntheses of the substituted ligands were carried out in a stereoselective manner using a [2,3]-sigmatropic rearrangement as the key step. A number of inhibitors with alkoxy and alkylamine substituents on the P2 ligands showed excellent enzyme Ki and antiviral IC50 values. Inhibitors with a 5-(R)-configuration showed improved potency over inhibitors with a 5-(S)-configuration. Both inhibitors 30b and 30j with a methoxy and ethylamine functionalities, respectively displayed subnanomolar antiviral activity. Our preliminary assessment of inhibitor 30f showed that it maintained full antiviral activity against the HIVDRVP20 resistant HIV-1 variant. We have determined high resolution X-ray crystal structures of 30b- and 30j-bound HIV-1 protease. The structure of 30b-bound HIV-1 protease revealed that the methoxy oxygen forms a strong water-mediated hydrogen bond with the amide NH of Gly48 located in the flap region of the HIV-1 protease active site. The ethylamino group in inhibitor 30j also forms a water-mediated interaction with Gly48. In addition, the amine NH forms a direct hydrogen bond with Gly48 carbonyl oxygen. The ethyl side chain also nicely packed in the hydrophobic pocket in the S2-subsite. The basic amine functionality on the inhibitor may be important for improving pharmacological properties. More detailed drug-resistance studies as well as further optimization of enzyme-inhibitor interactions are currently in progress.
Experimental Section
All reactions were carried out under an inert atmosphere, either N2 or Ar, using magnetic stirring and oven-dried glassware. All solvents were anhydrous and distilled prior to use. Dichloromethane and triethylamine were distilled from calcium hydride. Tetrahydrofuran, diethyl ether, and benzene were distilled from sodium/benzophenone. All other solvents were HPLC grade or better. Flash column chromatography was performed using EM Science 60–200 mesh silica gel. Thin-layer chromatography was performed using 60 F-254 E. Merck silica gel plates. 1H- and 13C-NMR were recorded using Bruker AV-400 MHz, Avance DRX-500, Varian Mercury-Vx-300, and Gemini-2300 spectrometers and use Me4Si as an internal standard. Optical rotations were recorded on a Perkin-Elmer 341 polarimeter. A Thermo Finnigan LCQ Classic mass was used for MS analyses. The purity of test compounds was determined by HRMS and HPLC analysis. All test compounds showed ≥95% purity.
(S,Z)-Ethyl 3-(2,2-dimethyl-1,3-dioxan-4-yl)acrylate (6)
To a solution of (S)-methyl 2,2-dimethyl-1,3-dioxane-4-carboxylate (5) (3.0g, 17.2 mmol ) in CH2Cl2 (100 mL) at −78 °C was added Dibal-H (1M in CH2Cl2, 19.0 mL). The solution was allowed to warm slowly to −50 °C over 1 h. Upon completion, methanol (100 mL) was added to the reaction followed by Ph3PCHCO2Et (4.20 g, 12.1 mmol) and the reaction was allowed to stir for 1 h at 0 °C. The reaction was concentrated under vacuum and the solid was washed with CH2Cl2 (3×20 mL) and concentrated under vacuum. The crude mixture was purified on silica gel using 5– 10% ethyl acetate/hexanes. The desired product 6 was obtained as a separable mixture of cis/trans (8:1) isomers (2.6 g, 70 % yield). cis: Rf = 0.38 (10% ethyl acetate/hexanes). [α]23D = −12.5 (c 1.01, CHCl3). 1H NMR (300 MHz, Chloroform-d) δ 6.17 (dd, J = 11.4 Hz, 7.1 Hz, 1H), 5.76 (d, J = 11.7 Hz, 1H), 5.52 (q, J = 7.1 Hz, 1H), 4.24 – 4.10 (q, J = 7.5 Hz, 2H), 4.10 – 3.97 (m, 1H), 3.90 – 3.76 (d, J = 16 Hz, 1H), 1.67 – 1.61 (m, 2H), 1.50 (s, 3H), 1.39 (s, 3H), 1.28 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 165.7, 149.7, 119.2, 98.3, 66.8, 60.2, 59.5, 30.0, 29.4, 19.3, 14.2. trans: Rf = 0.20 (10% ethyl acetate/hexanes). 1H NMR (400 MHz, Chloroform-d) δ 6.86 (dd, J = 15.7 Hz, 7.1 Hz, 1H), 6.04 (d, J = 11.8 Hz, 1H), 4.27 – 4.08 (q, J = 7.5 Hz, 2H), 4.01 (ddt, J = 14.9 Hz, 8.3 Hz, 2.7 Hz, 1H), 3.90 – 3.76 (m, 1H), 3.76 – 3.59 (m, 1H), 1.82 – 1.60 (m, 1H), 1.60 – 1.50 (m, 1H), 1.48 (s, 3H), 1.41 (s, 3H), 1.28 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 166.4, 147.0, 120.3, 108.6, 98.5, 67.8, 60.3, 59.5, 30.4, 29.7, 19.0, 14.1; LRMS-CI (m/z): 215 (m + H)+.
(S,Z)-t-butyl-2-(3-(2,2-dimethyl-1,3-dioxan-4-yl)allyloxy)-acetate (7)
Diisobutyl aluminum hydride (1 M in CH2Cl2, 27.5 mL, 27.5 mmol,) was slowly added to a cold solution (−78 °C) of 6 (12.0 g, 56.0 mmol) in dichloromethane (200 mL). The solution was allowed to stir for 15 min at −78 °C. A saturated solution of Rochelle’s salt (50 mL) was added and the reaction mixture was warmed to room temperature. The reaction was stirred until both layers were clear. The organic layer was separated and the aqueous layer was extracted with dichloromethane (3 × 15 mL). The organic layers were combined, washed with brine and dried over MgSO4. The solid was filtered out and the organic layer was concentrated under vacuum. The crude mixture was purified on silica gel using 20% ethyl acetate/hexanes to obtain the desired allyl alcohol (9.4 g, 98% yield) as a colorless oil. Rf = 0.27 (40% ethyl acetate/hexanes). 1H NMR (300 MHz, Chloroform-d) δ 5.78 – 5.74 (m, 1H), 5.50 (ddt, J = 11.2 Hz, 7.0 Hz, 1.3 Hz, 1H), 4.78 – 4.62 (m, 1H), 4.24 (dd, J = 12.9 Hz, 7.0 Hz, 1H), 4.20 – 4.08 (m, 1H), 4.00 (td, J = 12.2 Hz, 2.8 Hz, 1H), 3.83 (ddd, J = 11.8 Hz, 5.4 Hz, 1.5 Hz, 1H), 2.47 – 2.27 (m, 1H), 1.86 – 1.63 (m, 1H), 1.55 (s, 3H), 1.48 – 1.45 (m, 1H), 1.39 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 132.3, 131.5, 98.5, 65.7, 59.6, 58.8, 31.2, 29.9, 19.1
To a round bottom flask charged with activated molecular sieves (6.0 g) was added a solution of substrate, (S,Z)-3-(2,2-dimethyl-1,3-dioxan-4-yl)prop-2-en-1-ol (9.0 g, 52.0 mmol) in acetonitrile (150 mL), followed by t-butylbromoacetate (9.90 mL, 67.2 mmol), tetrabutylammonium iodide (2.0 g, 5.2 mmol) and cesium hydroxide monohydrate (13.1 g, 78.0 mmol) at room temperature. The reaction was allowed to stir for 6 h. The solid was filtered out and the solvent was concentrated under vacuum; the residue was purified by flash column chromatography (5% ethyl acetate/hexanes) to afford 7 (14.4 g, 97% yield) as a colorless oil. Rf = 0.57 (30% ethyl acetate/hexanes). [α]23D = + 22.9 (c 1.29, CHCl3) 1H NMR (300 MHz, Chloroform-d) δ 5.72 – 5.51 (m, 2H), 4.77 – 4.65 (m, 1H), 4.26 – 4.09 (m, 2H), 4.01 (dd, J = 17.3, 2.6 Hz, 1H), 3.93 (d, J = 3.6 Hz, 2H) 3.82 (ddd, J = 11.8 Hz, 5.4 Hz, 1.4 Hz, 1H), 1.79 – 1.65 (m, 1H), 1.48 (s, 3H), 1.46 (s, 10H), 1.37 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 169.5, 134.1, 127.7, 98.3, 81.5, 67.6, 66.8, 65.5, 59.5, 31.1, 29.9, 28.0, 19.1; LRMS-ESI (m/z): 438.4 (m + Na)+.
(2S,3S)-t-Butyl 3-((S)-2,2-dimethyl-1,3-dioxan-4-yl)-2-hydroxypent-4-enoate (9)
A solution of LiHMDS (84.0 mL 1.0 M in THF, 84.0 mmol) was added to a cold solution (−60 °C) of 7 (12.0 g, 42.0 mmol) in THF (250 mL) (LiHMDS was added at a rate that did not exceed −50 °C). The reaction mixture was allowed to warm slowly to −20 °C over 2 h. The reaction was quenched with saturated ammonium chloride (10 mL) extracted with ethyl acetate (3×20 mL) after warming to room temperature. The organic layers were combined washed with brine, dry over anhydrous MgSO4 and reduce under vacuum. The residue was purified with a 5 – 10 % gradient of ethyl acetate/hexanes. The desired 9 (9.0 g, 77% yield, 8.5:1 mixture of diastereomers) was obtained as a colorless oil. Rf = 0.30 (20% ethyl acetate/hexanes). [α]23D = + 5.8 (c 1.02, CHCl3); 1H NMR (300 MHz, Chloroform-d) δ 5.86 – 5.74 (m, 1H), 5.26 – 5.01 (dd, J = 12.1 Hz, 2H), 4.23 (t, J = 4.23 Hz, 1H), 4.12 (m, 1H), 3.99 (td, J = 11.9 Hz, 2.8 Hz, 1H), 3.86 (dd, J = 11.1 Hz, 4.9 Hz, 1H), 3.15 (d, J = 4.2 Hz, 1H), 2.48 – 2.45 (m, 1H), 1.90 – 1.64 (m, 1H), 1.45 (d, J = 4.7 Hz, 13H), 1.35 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 172.8, 133.1, 119.3, 98.5, 82.5, 71.0, 69.3, 59.8, 52.2, 29.8, 28.6, 28.1, 19.2; LRMS-ESI (m/z); 309.3 (M− + Na)+.
Minor isomer 10:
Rf = 0.37 (20% ethyl acetate/hexanes) [α]23D = −17.5 (c 1.8, CHCl3) 1H NMR (300 MHz, Chloroform-d) δ 5.69 – 5.51 (m, 1H), 5.20 – 5.04 (m, 2H), 4.46 (dd, J = 5.1 Hz, 2.2 Hz, 1H), 4.08 – 3.80 (m, 3H), 2.94 (d, J = 5.2 Hz, 1H), 2.40 (td, J = 9.8 Hz, 2.2 Hz, 1H), 1.53 – 1.34 (m, 17H). 13C NMR (75 MHz, CDCl3) δ 174.1, 132.1, 119.9, 98.6, 82.4, 69.0, 67.0, 60.1, 53.9, 30.0, 29.9 28.1, 19.2; LRMS-ESI (m/z): 309.3 (M− + Na)+.
(R)-3-((S)-2,2-dimethyl-1,3-dioxan-4-yl)pent-4-en-1-ol (11)
To a cold (0 °C) THF (25 mL) solution of 9 (0.98 g, 3.42 mmol) was added triethyl amine (0.57 mL, 4.10 mmol) followed by MsCl (0.32 mL, 4.1 mmol). The reaction was stirred for 2 h at 0 °C. The reaction was quenched with saturated ammonium chloride (10 mL) then extracted with ethyl acetate (3×15 mL). The organic layers were combined and dried over anhydrous MgSO4. The solvent was concentrated under vacuum and the crude mixture was purified using 10% ethyl acetate/hexanes to obtain the desired mesylated compound (1.2 g, 99 % yield) as a colorless oil. Rf = 0.37 (20% ethyl acetate/hexanes). 1H NMR (400 MHz, Chloroform-d) δ 5.77 (dt, J = 17.2 Hz, 9.9 Hz, 1H), 5.20 (dd, J = 21.0 Hz, 12.9 Hz, 2H), 5.06 (d, J = 4.1 Hz, 1H), 4.07 – 4.01 (m, 1H), 4.01 – 3.92 (m, 1H), 3.89 – 3.81 (m, 1H), 3.15 (s, 3H), 2.66 – 2.61 (m, 1H), 1.74 (qd, J = 12.3 Hz, 5.5 Hz, 1H), 1.48 (s, 10H), 1.43 (s, 3H), 1.36 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 167.2, 132.0, 120.4, 98.6, 83.4, 78.0, 68.1, 59.7, 51.0, 39.4, 29.7, 28.2, 28.0, 19.0.
The mesylated compound above was dissolved (1.23 g, 3.39 mmol) in THF (20 mL). At 0 °C lithium aluminum hydride (0.54 g, 14.2 mmol) was added and the reaction was stirred for 1 h at 0 °C then stirred for 6 h at room temperature. A small aliquot of the reaction was quenched and checked by NMR to determine the reaction’s progress. The reaction was cooled to 0 °C and diluted with ethyl acetate. This was followed by the stepwise addition of 3N NaOH (0.5 mL) and H2O (1 mL). The reaction was stirred until a white precipitate formed. MgSO4 was added to the solution and the white solid was filtered out. The solvent was removed under vacuum and the crude was purified by flash column chromatography (gradient 10% – 20% ethyl acetate/hexanes) to give 11 (0.46 g, 70% yield, 2 steps) as a colorless oil. Rf = 0.45 (50% ethyl acetate/hexanes). [α]23D = −29.2 (c 1.4, CHCl3); 1H NMR (400 MHz, Chloroform-d) δ 5.75 (m, 1H), 5.20 – 5.00 (m, 2H), 4.01 – 3.86 (m, 2H), 3.97 – 3.87 (m, 1H), 3.85 – 3.80 (m, 1H), 3.69 – 3.57 (m, 1H), 2.36 – 2.22 (m, 1H), 2.13 (bs, 1H), 1.86 – 1.59 (m, 2H), 1.44 (s, 3H), 1.37 (s, 3H), 1.34 – 1.21 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 138.1, 117.0, 98.4, 71.4, 60.8, 59.87, 46.0, 33.5, 29.8, 27.5, 19.1; LRMS-CI (m/z): 201.0 (M + H)+.
(3aS,4S,7aR)-hexahydro-2H-furo[2,3-b]pyran-4-ol (12)
Into a cold a solution of 11 (0.45 g, 2.25 mmol) in CH2Cl3/MeOH (20 mL, 4:1) at −78 °C was bubbled a stream of ozone until a blue color persisted. The ozone stream was stopped and a stream of argon was bubbled through the reaction mixture to remove the excess ozone. Dimethyl sulfide (0.50 mL, 6.9 mmol) was added to the reaction and the mixture was warmed to room temperature and stirred an additional 3 h. p-TsOH (10.0 mg, 5 mol %) was added and the reaction mixture was stirred for 16 h. The reaction was again carefully concentrated and the residue was purified on silica gel (20% ether/hexanes to 40% ether/hexanes) to afford compound 12, (0.22, 70 % yield) as a white solid. Rf = 0.23 (70% ethyl acetate/hexanes). [α]23D = −34.2 (c 1.4, CHCl3); 1H NMR (400 MHz, Chloroform-d) δ 4.97 (d, J = 3.4 Hz, 1H), 4.26 – 4.09 (m, 2H), 4.01 – 3.82 (m, 2H), 3.42 – 3.20 (m, 1H), 2.49 (m, 1H), 2.20 (bs, 1H), 2.09 – 1.98 (m, 1H), 1.94 – 1.82 (m, 1H), 1.82 – 1.58 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 101.3, 68.4, 67.3, 61.1, 46.3, 29.3, 21.8
(2S,3R)-t-Butyl 3-((S)-2,2-dimethyl-1,3-dioxan-4-yl)-2-methoxypent-4-enoate (13)
To a cold (0 °C) solution of 11 (0.18 g, 0.63 mmol) and methyl iodide (50 μL, 0.75 mmol) in THF (5 mL) was added sodium hydride (20 mg, 0.8 mmol). The reaction was allowed to stir for 2 h at 23 °C then quenched with saturated ammonium chloride (5 mL). The reaction mixture was extracted with ethyl acetate (3×10 mL). The organic layers were combined, washed with brine and dried over anhydrous sodium sulfate. The solvent was reduced under vacuum and the residue was purified on silica gel to obtain 13 (0.17 g, 90% yield) as a colorless oil. Rf = 0.54 (20% ethyl acetate/hexanes. 1H NMR (400 MHz, Chloroform-d) δ 5.78 (dt J = 17.2 Hz, 9.5 Hz, 1H), 5.21 – 5.00 (m, 2H), 4.03 – 3.88 (m, 2H), 3.84 (ddd, J = 11.7 Hz, 5.5 Hz, 1.7 Hz, 1H), 3.79 (d, J = 4.7 Hz, 1H), 3.36 (s, 3H), 2.45 (dt, J = 10.1 Hz, 5.3 Hz, 1H), 1.82 – 1.69 (m, 1H), 1.47 (m, 9H), 1.42 (s, 3H),1.35 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 170.6, 134.0, 118.8, 98.4, 81.5, 80.4, 68.5, 59.9, 58.2, 52.0, 29.8, 28.5, 28.2, 19.13.
(2R,3R)-1-t-Butoxy-3-((S)-2,2-dimethyl-1,3-dioxan-4-yl)-1-oxopent-4-en-2-yl4-nitrobenzoate (15)
Into a cold (0 °C) solution of 9 (0.15 g, 0.52 mmol) in THF (10 mL) was added triphenylphosphine (0.55 g, 2.1 mmol) p-nitrophenylbenzoic acid (0.35 g, 2.1 mmol) and diethyl azodicarboxylate (0.95 μL, 2.1 mmol). The reaction was allowed to stir 36 h. The reaction was diluted with ethyl acetate (10 mL) and quenched with a saturated solution of sodium bicarbonate (10 mL). The reaction was extracted with ethyl acetate (3×15 ml). The organic layers were combined, washed with brine and dried over anhydrous sodium sulfate. The solvent was concentrated under vacuum and the crude mixture was purified on silica gel using 5% ethyl acetate/hexanes to obtain 15 (0.21 g, 91%) as a pale yellow solid. Rf = 0.36 (10% ethyl acetate/hexanes). [α]23D = −0.27 (c 1.1, CHCl3). 1H NMR (300 MHz, Chloroform-d) δ 8.36 – 8.21 (m, 4H), 5.86 (dt, J = 17.2 Hz, 10.0 Hz, 1H), 5.32 – 5.09 (m, 2H), 4.24 (dt, J = 11.8 Hz, 2.7 Hz, 1H), 3.97 (td, J = 12.0 Hz, 2.8 Hz, 1H), 3.89 – 3.73 (m, 1H), 2.65 (td, J = 9.5 Hz, 2.9 Hz, 1H), 1.93 – 1.74 (m, 1H), 1.44 (s, 9H), 1.34 (s, 3H), 1.27 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 168.3, 163.8, 150.68, 134.9, 132.3, 130.8, 123.6, 120.4, 98.4, 82.8, 73.6, 66.6, 59.7, 50.9, 29.6, 28.4, 28.0, 18.8; LRMS-ESI (m/z); 481.5 (M + Na)+.
(2R,3R)-t-Butyl 3-((S)-2,2-dimethyl-1,3-dioxan-4-yl)-2-methoxypent-4-enoate (16)
To a cold (0 °C) solution of 15 (0.42 g, 0.96 mmol) in methanol was added potassium carbonate (0.16 g, 1.16 mmol). The reaction was allowed to stir for 0.5 h. The reaction was quenched with a saturated ammonium chloride (5 mL) and the methanol was removed under vacuum. The solution was extracted with ethyl acetate (3×10 mL) and the combined organic layer was combined, washed with brine and dried over anhydrous sodium sulfate. The solvent was removed under vacuum and the residue was purified on silica gel using 10% ethyl acetate/hexanes to obtain the free secondary alcohol (0.28 mg, 99% yield) as an amorphous solid. Rf = 0.38 (30% ethyl acetate/hexanes). [α]23D = −21.6 (c 1.1, CHCl3). 1H NMR (400 MHz, Chloroform-d) δ 5.92 (dt, J = 17.2 Hz, 10.0 Hz, 1H), 5.29 – 5.03 (m, 2H), 4.22 (dt, J = 11.8 Hz, 2.8 Hz, 1H), 4.11 (dd, J = 8.9 Hz, 6.2 Hz, 1H), 3.95 (td, J = 12.1 Hz, 2.8 Hz, 1H), 3.80 (ddd, J = 11.7, 5.4, 1.6 Hz, 1H), 3.28 (d, J = 9.1 Hz, 1H), 2.32 – 2.27 (m, 1H), 1.85 – 1.74 (m, 1H), 1.46 (s, 9H), 1.44 (s, 3H), 1.35 (s, 3H), 1.19 (d, J = 13.0 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 173.1, 134.1, 118.8, 98.5, 82.1, 73.0, 68.6, 59.7, 52.5, 29.7, 28.4, 28.1, 18.9
To a cold (0 °C) solution of (2R,3S)-t-Butyl 3-((S)-2,2-dimethyl-1,3-dioxan-4-yl)-2-hydroxypent-4-enoate (0.12 g, 0.40 mmol) in THF (5 mL) was added sodium hydride (20.0 mg, 0.8 mmol) followed by methyl iodide (50 μL, 0.8 mmol). The reaction was allowed to stir for 2 h at 23 °C then quenched with saturated ammonium chloride (5 mL). The reaction mixture was extracted with ethyl acetate (3×10 mL). The organic layers were combined, washed with brine and dried over anhydrous sodium sulfate. The solvent was reduced under vacuum and the residue was purified on silica gel to obtain 16 (0.12 g, 96% yield) as a colorless oil. Rf = 0.54 (20% ethyl acetate/hexanes. 1H NMR (300 MHz, Chloroform-d) δ 5.74 (dt, J = 17.2 Hz, 10.1 Hz, 1H), 5.22 – 4.93 (m, 2H), 4.29 (d, J = 11.9 Hz, 1H), 3.95 (td, J = 12.0 Hz, 2.8 Hz, 1H), 3.83 – 3.73 (m, 2H), 3.35 (s, 3H), 2.26 (td, J = 9.9 Hz, 2.2 Hz, 1H), 1.79 – 1.71 (m, 1H), 1.43 (s, 9H), 1.41 (s, 3H), 1.35 (s, 3H), 1.19 – 1.07 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 171.3, 132.9, 119.8, 98.2, 81.5, 80.5, 65.9, 60.0, 58.1, 52.3, 29.8, 28.4, 28.1, 19.1
(2R,3R)-t-Butyl 2-(benzyloxy)-3-((S)-2,2-dimethyl-1,3-dioxan-4-yl)pent-4-enoate (18)
Compound 18 was prepared by following the procedure outlined for o-alkylation outlined above. Compound 18 was obtained in 88% yield. Rf = 0.70 (20% ethyl acetate/hexanes). 1H NMR (300 MHz, Chloroform-d) δ 7.43 – 7.27 (m, 5H), 5.76 (dt, J = 17.2 Hz, 10.1 Hz, 1H), 5.24 – 4.94 (m, 2H), 4.58 (d, J = 11.3 Hz, 1H), 4.48 – 4.28 (m, 2H), 3.99 – 3.89 (m, 2H), 3.86 – 3.70 (m, 1H), 2.37 (td, J = 10.0 Hz, 2.1 Hz, 1H), 1.82 −1.68 (m, 1H), 1.44 (s, 9H), 1.37 (s, 3H), 1.33 (s, 3H) 1.15 (d, J = 12.9, 1H). 13C NMR (75 MHz, CDCl3) δ 171.2, 137.4, 132.8, 128.3, 127.9, 119.9, 98.2, 81.4, 78.4, 72.5, 65.8, 59.9, 52.4, 29.8, 28.3, 28.1, 19.1.
(3S,3aS,4S,7aS)-3-Methoxyhexahydro-2H-furo[2,3-b]pyran-4-ol (14)
To a cold (0 °C) solution of 13 (0.14 mg, 0.47 mmol) in THF (5 mL) was added lithium aluminum hydride (41 mg, 1.03 mmol). The reaction was allowed to stir for 1 h at 23 °C after which the reaction was cooled to 0 °C and quenched by adding excess ethyl acetate, 1 N NaOH (0.5 mL), H2O (0.5 mL). After a white precipitate formed magnesium sulfate was added and stirred for 15 min. The reaction mixture was filtered and concentrated under vacuum to provide the corresponding diol. 1H NMR (400 MHz, Chloroform-d) δ 5.87 (dt, J = 17.3 Hz, 9.6 Hz, 1H), 5.19 (dd, J = 10.3 Hz, 2.0 Hz, 1H), 5.10 (ddd, J = 17.2 Hz, 2.0, 0.6 Hz, 1H), 4.13 (dt, J = 11.9 Hz, 2.6 Hz, 1H), 3.95 (td, J = 12.1 Hz, 2.8 Hz, 1H), 3.79 (ddd, J = 11.7 Hz, 5.4 Hz, 1.6 Hz, 1H), 3.74 – 3.55 (m, 2H), 3.38 (s, 3H), 3.31 – 3.28 (m, 1H), 2.71 (bs, 1H), 2.37 – 2.32 (m, 1H), 1.80 (qd, J = 12.5 Hz, 5.5 Hz, 1H), 1.44 (s, 3H), 1.34 (s, 3H), 1.22 (m, J = 13.2 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 134.8, 118.8, 98.3, 82.7, 67.0, 61.0, 59.9, 57.7, 50.4, 29.7, 28.6, 19.0
The crude diol. mixture was taken up in CHCl3/MeOH (4:1) and a stream of O3 was bubble through the solution at −78 °C until a blue color persisted. Argon was bubbled through the blue solution until the solution became clear. Dimethyl sulfide (0.13 mL, 5.0 eq) was added to the reaction and the mixture was warmed to room temperature and stirred an additional 3 h. To the reaction mixture was added p-TsOH (10 mol %) the mixture was stirred for 18 h at room temperature. The reaction was carefully concentrated and the residue was purified on silica gel (20% ether/hexanes to 50% ether/hexanes) to afford 14, (24 mg, 30 % yield 2 steps) as a colorless oil. Rf = 0.20 (60% ethyl acetate/hexanes). [α]23D = −111.7 (c 1.2, CHCl3); 1H NMR (400 MHz, Chloroform-d) δ 4.99 (d, J = 4.1 Hz, 1H), 4.24 – 4.17 (m, 1H), 4.17 – 4.14 (m, 2H), 4.14 – 4.02 (m, 1H), 4.01 – 3.96 (m, 1H), 3.43 – 3.37 (d, J = 13.6 Hz, 1H), 3.32 (s, 3H), 2.74 (d, J = 10.1 Hz, 1H), 2.53 – 2.48 (m, 1H), 2.00 – 1.85 (m, 1H), 1.80 – 1.74 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 101.5, 80.7, 73.3, 67.3, 60.7, 57.7, 47.5, 33.0.
(3R,3aS,4S,7aS)-3-Methoxyhexahydro-2H-furo[2,3-b]pyran-4-ol (19)
Methyl ether derivative 19 was obtained by following the two-step procedures outlined above for the formation of compound 14. Methyl ether 19 was obtained in 45 % yield. Rf = 0.20 (70% ethyl acetate/hexanes). 1H NMR (300 MHz, Chloroform-d) δ 5.06 (d, J = 3.8 Hz, 1H), 4.38 – 4.22 (m, 2H), 4.21 – 4.13 (m, 1H), 3.94 – 3.86 (m, 3H), 3.33 (s, 3H), 2.53 – 2.47 (m, 2H), 1.92 – 1.76 (m, 1H), 1.76 – 1.54 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 101.4, 78.9, 72.2, 66.3, 60.5, 57.9, 51.1, 30.2.
(3R,3aS,4S,7aS)-3-Ethoxyhexahydro-2H-furo[2,3-b]pyran-4-ol (20)
Ethyl ether derivative 20 was prepared by following the two-step procedures described for compound 19. Ethyl ether 20 was obtained in 60 % yield. Rf = 0.20 (50% ethyl acetate/hexanes). 1H NMR (400 MHz, Chloroform-d) δ 5.05 (d, J = 3.8 Hz, 1H), 4.45 – 4.26 (m, 2H), 4.26 – 4.07 (m, 1H), 4.01 – 3.87 (m, 1H), 3.85 (dd, J = 8.6 Hz, 4.7 Hz, 1H), 3.59 – 3.41 (m, 2H), 3.32 (td, J = 11.9 Hz, 2.3 Hz, 1H), 2.64 – 2.41 (m, 2H), 1.91 – 1.78 (m, 1H), 1.78 – 1.64 (m, 1H), 1.19 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 101.3, 77.1, 72.6, 66.6, 66.0, 60.7, 50.8, 30.2, 15.5.
(3R,3aS,4S,7aS)-3-(Benzyloxy)hexahydro-2H-furo[2,3-b]pyran-4-ol (21)
Benzyl ether derivative 21 was prepared by following the two-step procedures as described for compound 19. Benzyl ether 21 was obtained in 88 % yield. [α]23D = +45 (c 1.1, CHCl3); Rf = 0.22 (60% ethyl acetate/hexanes). 1H NMR (300 MHz, Chloroform-d) δ 7.47 – 7.18 (m, 5H), 5.05 (d, J = 3.7 Hz, 1H), 4.51 (d, J = 1.6 Hz, 2H), 4.49 – 4.36 (m, 1H), 4.26 (dd, J = 9.0 Hz, 6.8 Hz, 1H), 4.21 – 4.06 (m, 1H), 3.93 – 3.85 (m, 2H), 3.31 (td, J = 11.8 Hz, 2.4 Hz, 1H), 2.66 – 2.54 (m, 1H), 2.49 (bs, 1H), 1.93 – 1.75 (m, 1H), 1.75 – 1.47 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 137.4, 128.5, 128.0, 128.0, 101.3, 72.7, 72.5, 66.4, 60.6, 51.2, 30.1.
(2S,3S)-3-((S)-2,2-Dimethyl-1,3-dioxan-4-yl)-1-(trityloxy)pent-4-en-2-ol (22)
To a cold (0 °C) solution of 9 (1.50 g, 5.30 mmol) in THF (30 mL) was added lithium aluminum hydride (0.45 g, 11.7 mmol). The reaction was allowed to stir for 1 h at 23 °C after which the reaction was cooled to 0 °C and quenched by adding excess ethyl acetate, 1 N NaOH (0.5 mL), H2O (0.5 mL). After a white precipitate formed magnesium sulfate was added and stirred for 15 min. The reaction mixture was filtered and concentrated under vacuum.
The crude 1,2-diol was dissolved in CH2Cl2 (20.0 mL). To that mixture was added triethyl amine (1.6 mL, 11.1 mmol) and triphenylmethyl chloride (1.6 g, 5.83 mmol). The reaction was allowed to stirr for 24 h. Upon completion the reaction was concentrated under vacuum and purified on silica gel to obtain the 22. (2.20 g, 92% yield) as a white solid. Rf = 0.30 (20% ethyl acetate/hexanes). [α]23D = −1.86 (c 1.5, CHCl3). 1H NMR (400 MHz, Chloroform-d) δ 7.48 – 7.39 (m, 6H), 7.35 – 7.18 (m, 10H), 5.85 (dt, J = 17.4 Hz, 10.1 Hz, 1H), 5.16 (dd, J = 10.3 Hz, 2.1 Hz, 1H), 4.99 (dd, J = 17.3 Hz, 2.1 Hz, 1H), 4.17 – 3.98 (m, 2H), 3.93 (td, J = 12.1 Hz, 2.7 Hz, 1H), 3.79 (dd, J = 11.2 Hz, 4.8 Hz, 1H), 3.21 – 3.06 (m, 2H), 3.04 (d, J = 1.1 Hz, 1H), 2.30 (dt, J = 9.8 Hz, 3.3 Hz, 1H), 1.80 (dd, J = 12.5 Hz, 5.3 Hz, 1H), 1.30 (d, J = 5.5 Hz, 6H), 1.29 – 1.15 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 144.0, 128.7, 127.8, 127.0, 119.8, 98.2, 86.5, 73.0, 71.2, 64.9, 59.8, 50.9, 29.8, 28.7, 19.0; LRMS-ESI (m/z); 481.5 (M + Na)+.
(3R,3aS,4S,7aS)-3-Azidohexahydro-2H-furo[2,3-b]pyran-4-ol (23)
To a solution of 22 (2.2 g, 4.80 mmol) in THF (50.0 mL) at 0 °C was added triphenyl phosphine (5.0 g, 19.2 mmol), diethylazodicarboxylate (3.3 g, 19.2 mmol), Diphenylphosphoryl azide (2.5 g, 9.6 mmol) sequentially. The reaction mixture was stirred at room temperature for 24 h after which it was concentrated under vacuum and purified on silica gel to get the desired azide (1.6 g, 70 % yield). Rf = 0.35 (10% ethyl acetate/hexanes). [α]23D = + 16.3 (c 1.6, CHCl3). 1H NMR (400 MHz, Chloroform-d) δ 7.56 – 7.39 (m, 6H), 7.38 – 7.13 (m, 10H), 5.48 (dt, J = 17.3 Hz, 10.1 Hz, 1H), 4.96 (dd, J = 10.3 Hz, 1.9 Hz, 1H), 4.84 (dd, J = 17.3 Hz, 1.8 Hz, 1H), 4.30 (dt, J = 11.9 Hz, 2.4 Hz, 1H), 3.98 (td, J = 12.1 Hz, 2.7 Hz, 1H), 3.88 – 3.71 (m, 1H), 3.69 – 3.64 (m, 1H), 3.38 (dd, J = 10.1 Hz, 2.6 Hz, 1H), 3.07 (dd, J = 10.0 Hz, 7.9 Hz, 1H), 1.98 (td, J = 10.2 Hz, 2.2 Hz, 1H), 1.81 – 1.59 (m, 1H), 1.49 (s, 3H), 1.35 (s, 3H), 1.20 – 1.03 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 143.8, 133.6, 128.6, 127.8, 127.0, 119.6, 98.4, 87.0, 67.2, 65.4, 62.3, 59.9, 50.5, 29.7, 28.4, 18.9.
(S)-4-((3S,4R)-4-azido-5-(trityloxy)pent-1-en-3-yl)-2,2-dimethyl-1,3-dioxane (.08 g, 0.17 mmol) was taken up in CH2Cl3/MeOH (20 ml, 4:1) and a stream of O3 was bubble through the solution at −78 °C until a blue color persisted. Argon was bubbled through the blue solution until the solution became clear. Dimethyl sulfide (0.5 mL) was added to the reaction and the mixture was warmed to room temperature and stirred an additional 3 h. To the reaction mixture was added p-TsOH (10 mol %) the mixture was stirred for 18 h at room temperature. The reaction was carefully concentrated and the residue was purified on silica gel (20% ether/hexanes to 50% ether/hexanes) to afford 23, (23 mg, 74 % yield) as a white solid. Rf = 0.33 (50% ethyl acetate/hexanes). [α]23D = −20.4 (c 1.0, CHCl3). 1H NMR (400 MHz, Chloroform-d) δ 5.07 (d, J = 3.7 Hz, 1H), 4.42 – 4.27 (m, 2H), 4.21 (dt, J = 10.9 Hz, 5.7 Hz, 1H), 3.90 (ddd, J = 12.2 Hz, 4.3 Hz, 2.3 Hz, 1H), 3.83 – 3.71 (m, 1H), 3.32 (td, J = 12.0 Hz, 2.0 Hz, 1H), 2.63 – 2.45 (m, 2H), 1.79 (ddd, J = 13.2 Hz, 3.8 Hz, 1.6 Hz, 1H), 1.69 (td, J = 11.6 Hz, 4.5 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 101.7, 72.3, 65.5, 60.8, 59.0, 52.01, 29.8; LRMS-CI (m/z); 186.0 (M + H)+.
General Procedure for the Synthesis of N-alkylated Ligands
(3R,3aS,4S,7aS)-3-azidohexahydro-2H-furo[2,3-b]pyran-4-ol was dissolved in a solution methanol and placed under argon. 10 % Palladium on carbon (10 mol%) was added and the mixture was stirred under a hydrogen balloon for 1 h. Upon completion the reaction was filtered through a plug of silica. The solvent was removed under vacuum and the product was used without further purification.
The amino alcohol obtained from the previous step was dissolved in methanol and treated with benzaldehyde (1.0 eq) and sodium triacetoxyborohydride (1.2 eq). The reaction was allowed to stir for 18 h (or until the starting material was consumed). The desired aldehyde (1.5 eq) was added followed by additional sodium triacetoxyborohydride (1.5 eq) and the reaction was allowed to continue for an additional 12 h to give the desired dialkylated amino-Tp-THF ligand.
(3R,3aS,4S,7aS)-3-(Benzyl(methyl)amino)hexahydro-2H-furo[2,3-b]pyran-4-ol (24)
Tertiary amine 24 was prepared following the general procedure outlined above. Amine 24 was obtained in 81 % yield. Rf = 0.61 (10% MeOH/DCM). 1H NMR (400 MHz, Chloroform-d) δ 7.46 – 7.10 (m, 5H), 4.93 (d, J = 3.7 Hz, 1H), 4.19 (q, J = 11.4 Hz, 1H), 4.07 – 3.94 (m, 3H), 3.84 (ddd, J = 12.3 Hz, 4.5 Hz, 1.9 Hz, 1H), 3.76 (d, J = 12.9 Hz, 1H), 3.54 (d, J = 12.9 Hz, 1H), 3.25 (td, J = 12.5 Hz, 1.8 Hz, 1H), 2.72 – 2.65 (m, 1H) 2.32 (s, 3H), 1.81 (dd, J = 13.4 Hz, 5.6 Hz, 1H), 1.68 – 1.52 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 128.9, 128.6, 127.7, 100.6, 67.9, 62.8, 61.3, 60.3, 60.2, 43.5, 36.0, 30.5; LRMS-ESI (m/z): 286.3 (M + Na)+.
(3R,3aS,4S,7aS)-3-(Benzyl(ethyl)amino)hexahydro-2H-furo[2,3-b]pyran-4-ol (25)
Tertiary amine 25 was obtained following the general procedure outlined above. Amine 25 was obtained in 82 % yield. Rf = 0.34 (5% MeOH/DCM). [α]23D = +74.6 (c 1.23, CHCl3). 1H NMR (400 MHz, Chloroform-d) δ 7.48 – 7.12 (m, 5H), 4.91 (d, J = 3.6 Hz, 1H), 4.16 (q, J = 11.3 Hz, 1H), 4.09 – 4.04 (m, 2H), 4.03 – 3.86 (m, 2H), 3.85 – 3.75 (m, 1H), 3.33 (d, J = 13.3 Hz, 1H), 3.23 (td, J = 12.4 Hz, 1.6 Hz, 1H), 2.80 (dq, J = 14.7 Hz, 7.4 Hz, 1H), 2.66 – 2.63 (m, 1H), 2.43 (dq, J = 13.6 Hz, 6.9 Hz, 1H), 1.76 (dd, J = 13.3 Hz, 5.6 Hz, 1H), 1.57 – 1.42 (m, 1H), 1.12 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 137.7, 128.9, 128.6, 127.5, 100.4, 67.9, 64.1, 61.2, 57.9, 54.3, 44.1, 43.9, 30.6, 13.0
(3R,3aS,4S,7aS)-3-(Benzyl(propyl)amino)hexahydro-2H-furo[2,3-b]pyran-4-ol (26)
Tertiary amine 26 was obtained following the general procedure outlined above. Amine 26 was obtained in 45 % yield. Rf = 0.48 (5% MeOH/DCM). 1H NMR (400 MHz, Chloroform-d) δ 7.40 – 7.22 (m, 5H), 4.91 (d, J = 3.6 Hz, 1H), 4.16 (q, J = 11.5 Hz, 1H), 4.08 – 4.01 (m, 2H), 3.98 (d, J = 13.3 Hz, 1H), 3.94 – 3.87 (m, 1H), 3.81 (ddd, J = 12.2 Hz, 4.4 Hz, 1.8 Hz, 1H), 3.33 (d, J = 13.3 Hz, 1H), 3.23 (td, J = 12.5 Hz, 1.8 Hz, 1H), 2.69 – 2.62 (m, 2H), 2.41 – 2.29 (m, 1H), 1.75 (dd, J = 13.3 Hz, 5.6 Hz, 1H), 1.70 – 1.56 (m, 1H), 1.56 – 1.38 (m, 2H), 0.87 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 137.7, 129.0, 128.5, 127.5, 100.5, 67.9, 64.0, 61.2, 58.0, 55.4, 51.8, 43.9, 30.5, 21.0, 11.5; LRMS-ESI (m/z): 314.5 (M + Na)+.
General Procedure for the preparation of activated carbonates from polycyclic P2-ligands
To a solution of the desired Tp-THF alcohol in dry CH2Cl2 was added pyridine (2.3 equivalents). The resulting mixture was cooled to 0 °C under argon and 4-nitrophenylchloroformate (2.2 equivalents) was added in one portion. The resulting mixture was stirred at 0 °C until completion. The reaction mixture was evaporated to dryness and the residue was purified by flash column chromatography on silica gel using a gradient of 20–40% ethyl acetate/hexanes to afford the desired mixed carbonate.
(3S,3aR,4S,7aS)-3-Methoxyhexahydro-2H-furo[2,3-b]pyran-4-yl 4-nitrophenyl carbonate (27a)
Following the general procedure outlined above, activated carbonate 27a was obtained in 81 % yield. Rf = 0.55 (60% ethyl acetate/hexanes). 1H NMR (400 MHz, Chloroform-d) δ 8.29 (d, J = 9.2 Hz, 2H), 7.39 (d, J = 9.2 Hz, 2H), 5.26 (dt, J = 11.2 Hz, 6.3 Hz, 1H), 5.10 (d, J = 4.0 Hz, 1H), 4.27 (dd, J = 9.7 Hz, 1.6 Hz, 1H), 4.15 – 4.08 (m, 1H), 4.05 (m, 1H), 3.50 – 3.41 (m, 2H), 3.40 (s, 3H), 2.58 – 2.54 (m, 1H), 2.39 (qd, J = 11.9 Hz, 4.8 Hz, 1H), 1.95 – 1.86 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 155.4, 152.0, 145.4, 125.3, 121.7, 101.4, 80.1, 74.7, 74.6, 60.2, 58.5, 45.4, 28.2; LRMS-ESI (m/z): 362.4 (M + Na)+.
(3R,3aR,4S,7aS)-3-Methoxyhexahydro-2H-furo[2,3-b]pyran-4-yl 4-nitrophenyl carbonate (27b)
Following the general procedure outlined above, activated carbonate 27a was obtained in 90 % yield. Rf = 0.29 (40% ethyl acetate/hexanes).1H NMR (300 MHz, Chloroform-d) δ 8.27 (d, J = 9.1 Hz, 2H), 7.38 (d, J = 9.3 Hz, 2H), 5.22 – 4.97 (m, 2H), 4.36 (dd, J = 9.0 Hz, 6.8 Hz, 1H), 4.27 – 4.22 (m, 1H), 4.07 – 3.84 (m, 2H), 3.51 – 3.34 (m, 1H), 3.34 (s, 3H), 2.96 – 2.91 (m, 1H), 2.06 – 1.79 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 155.5, 151.5, 145.4, 125.3, 121.7, 101.4, 79.2, 73.7, 72.6, 60.1, 58.0, 48.2, 26.9; LRMS-ESI (m/z): 362.4 (M + Na)+.
(3R,3aR,4S,7aS)-3-Ethoxyhexahydro-2H-furo[2,3-b]pyran-4-yl 4-nitrophenyl carbonate (27c)
Following the general procedure outlined above, activated carbonate 27c was obtained in 48 % yield. Rf = 0.17 (30% ethyl acetate/hexanes). 1H NMR (400 MHz, Chloroform-d) δ 8.28 (m, J = 9.2 Hz, 2H), 7.40 (m, 2H), 5.24 – 5.15 (m, 1H), 5.14 (d, J = 3.8 Hz, 1H), 4.42 – 4.27 (m, 2H), 4.01 (dt, J = 12.3 Hz, 3.7 Hz, 1H), 3.97 – 3.87 (m, 1H), 3.50 (q, J = 7.0 Hz, 2H), 3.45 – 3.33 (m, 1H), 2.98 – 2.95 (m, 1H), 2.03 – 1.90 (m, 2H), 1.15 (t, J = 7.0 Hz, 3H).
(3R,3aR,4S,7aS)-3-(Benzyloxy)hexahydro-2H-furo[2,3-b]pyran-4-yl 4-nitrophenyl carbonate (27d)
Following the general procedure outlined above, activated carbonate 27d was obtained in 87 % yield. Rf = 0.35 (40% ethyl acetate/hexanes). 1H NMR (300 MHz, Chloroform-d) δ 8.11 (d, J = 7.1 Hz, 2H), 7.44 – 7.16 (m, 5H), 7.04 (d, J = 7.1 Hz, 2H), 5.27 – 5.07 (m, 2H), 4.59 – 4.42 (m, 3H), 4.33 (dd, J = 9.1 Hz, 6.9 Hz, 1H), 4.03 – 3.96 (m, 2H), 3.44 – 3.36 (m, 1H), 3.09 – 3.04 (m, 1H), 2.04 – 1.91 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 155.2, 151.5, 145.1, 137.4, 128.3, 127.9, 125.0, 121.6, 101.3, 77.3, 73.8, 72.9, 60.2, 48.3, 26.8; LRMS-ESI (m/z): 438.4 (M + Na)+.
(3R,3aS,4S,7aS)-3-Azidohexahydro-2H-furo[2,3-b]pyran-4-yl 4-nitrophenyl carbonate (27e)
Following the general procedure outlined above, activated carbonate 27e was obtained in 80 % yield. Rf = 0.25 (30% ethyl acetate/hexanes). 1H NMR (400 MHz, Chloroform-d) δ 8.28 (d, J = 9.2 Hz, 2H), 7.39 (d, J = 9.2 Hz, 2H), 5.29 – 5.22 (m, 1H), 5.15 (d, J = 3.6 Hz, 1H), 4.41 (q, J = 8.0 Hz, 1H), 4.41 – 4.37 (m, 1H), 4.02 (dd, J = 12.5 Hz, 4.6Hz, 1H), 3.87 (dd, J = 9.0 Hz, 4.9 Hz, 1H), 3.42 (td, J = 12.2 Hz, 2.0 Hz, 1H), 2.88 – 2.79 (m, 1H), 2.15 – 2.01 (m, 1H), 1.95 (td, J = 12.1 Hz, 4.7 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 155.2, 151.6, 145.5, 125.3, 121.7, 101.7, 73.5, 72.5, 60.4, 59.3, 48.9, 26.6; LRMS-ESI (m/z): 373.2 (M + Na)+.
(3R,3aS,4S,7aS)-3-(Benzyl(methyl)amino)hexahydro-2H-furo[2,3-b]pyran-4-yl 4-nitrophenyl carbonate (27f)
Following the general procedure outlined above, activated carbonate 27f was obtained in 36 % yield. Rf = 0.4 (40% ethyl acetate/hexanes). 1H NMR (400 MHz, Chloroform-d) δ 8.20 (d, J = 9.2 Hz, 2H), 7.34 – 7.09 (m, 7H), 5.25 – 5.07 (m, 2H), 4.10 (dd, J = 9.4 Hz, 4.4 Hz, 1H), 4.05 – 3.94 (m, 2H), 3.85 (td, J = 8.1 Hz, 4.4 Hz, 1H), 3.62 (d, J = 13.0 Hz, 1H), 3.50 (d, J = 13.1 Hz, 1H), 3.47 – 3.37 (m, 1H), 3.11 – 3.06 (m, 1H), 2.21 (s, 3H),1.95 (dt, J = 8.6, 4.5 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 155.4, 151.7, 145.3, 138.4, 129.2, 128.2, 127.2, 125.2, 121.8, 101.3, 74.6, 64.9, 62.0, 60.3, 59.6, 43.9, 36.4, 26.7.
(3R,3aS,4S,7aS)-3-(Benzyl(ethyl)amino)hexahydro-2H-furo[2,3-b]pyran-4-yl 4-nitrophenyl carbonate (27g)
Following the general procedure outlined above, activated carbonate 27g was obtained in 77 % yield. Rf = 0.6 (30% ethyl acetate/hexanes). 1H NMR (400 MHz, Chloroform-d) δ 8.23 (d, J = 9.2 Hz, 2H), 7.32 – 7.14 (m, 7H), 5.24 −5.19 (m, 1H), 5.17 (d, J = 3.9 Hz, 1H), 4.08 – 3.98 (m, 2H), 3.97 (t, J = 3.6 Hz, 1H), 3.87 (d, J = 4.5 Hz, 1H), 3.75 (d, J = 13.7 Hz, 1H), 3.49 – 3.36 (m, 2H), 3.03 – 2.98 (m, 1H), 2.62 (p, J = 7.2 Hz, 1H), 2.52 – 2.39 (p, J = 6.8 Hz, 1H), 1.98 – 1.85 (m, 2H), 1.01 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 155.4, 151.6, 145.3, 139.1, 131.0, 129.1, 128.1, 127.0, 125.2, 121.9, 101.1, 74.6, 66.1, 60.3, 59.1, 53.1, 45.8, 44.5, 26.9, 12.5.
(3R,3aS,4S,7aS)-3-(Benzyl(propyl)amino)hexahydro-2H-furo[2,3-b]pyran-4-yl 4-nitrophenyl carbonate (27h)
Following the general procedure outlined above, activated carbonate 27h was obtained in 32 % yield. Rf = 0.6 (40% ethyl acetate/hexanes. 1H NMR (400 MHz, Chloroform-d) δ 8.23 (d, J = 9.2 Hz, 2H), 7.32 – 7.13 (m, 7H), 5.23 – 5.18 (m, 1H), 5.17 (d, J = 3.9 Hz, 1H), 4.03 (d, J = 5.6 Hz, 2H), 4.01 – 3.95, (m, 1H), 3.85 (m, 1H), 3.76 (d, J = 13.8 Hz, 1H), 3.48 (d, J = 13.8 Hz, 1H), 3.42 (td, J = 12.2 Hz, 11.8 Hz, 3.3 Hz, 1H), 2.98 – 2.93 (m, 1H), 2.53 – 2.46 (m, 1H), 2.41 – 2.30 (m, 1H), 1.97 – 1.82 (m, 2H), 1.65 – 1.48 (m, 1H), 1.41 – 1.23 (m, 1H), 0.82 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 155.5, 151.7, 145.3, 139.2, 129.0, 128.2, 127.0, 125.3, 121.8, 101.3, 74.7, 66.6, 60.4, 59.4, 54.1, 52.6, 45.7, 26.9, 20.6, 11.7.
(3S,3aR,4S,7aS)-3-Methoxyhexahydro-2H-furo[2,3-b]pyran-4-yl (2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-yl carbamate (30a)
To a stirred solution of amine 28 in CH3CN at cooled to 0 °C, DIPEA (5 equivalent) followed by activated ligand 27a. The resulting solution was stirred at 23 °C until the reaction was complete. The solution was evaporated to dryness and the crude residue purified by flash column chromatography on silica gel to yield the desired inhibitor. The titled inhibitor was synthesized using the general procedure outlined above. The desired inhibitor was obtained as a white solid (15 % yield). Rf = 0.25 (60% ethyl acetate/hexanes). 1H NMR (400 MHz, Chloroform-d) δ 7.72 (d, J = 8.8 Hz, 2H), 7.35 – 7.16 (m, 5H), 6.98 (d, J = 8.9 Hz, 2H), 5.02 – 4.96 (m, 2H), 4.89 (d, J = 8.8 Hz, 1H), 4.16 (d, J = 10.0 Hz, 1H), 4.03 – 3.92 (m, 3H), 3.92 – 3.84 (s, 4H), 3.78 (bs, 1H), 3.46 – 3.27 (m, 3H), 3.19 (s, 3H), 3.16 – 3.08 (m, 1H), 3.10 – 2.94 (m, 3H), 2.81 (dd, J = 13.4 Hz, 6.7 Hz, 2H), 2.31 (d, J = 4.4 Hz, 1H), 2.18 (dd, J = 12.1 Hz, 4.4 Hz, 1H), 1.90 – 1.80 (m, 1H), 0.93 (d, J = 6.6 Hz, 3H), 0.87 (t, J = 7.6 Hz, 3H). Mass: HRMS (ESI), Calcd for C30H42N2O9S: m/z 629.2509 (M+Na), found m/z 629.2505 (M+Na).
(3R,3aR,4S,7aS)-3-Methoxyhexahydro-2H-furo[2,3-b]pyran-4-yl (2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-ylcarbamate (30b)
The titled inhibitor was synthesized using the general procedure outlined above. Inhibitor 30b was obtained in 61 % yield. Rf = 0.38 (60% ethyl acetate/hexanes). 1H NMR (300 MHz, Chloroform-d) δ 7.70 (d, J = 8.6 Hz, 2H), 7.43 – 7.13 (m, 5H), 6.97 (d, J = 8.7 Hz, 2H), 5.12 (d, J = 3.7 Hz, 1H), 5.09 – 4.96 (m, 1H), 4.93 (d, J = 8.5 Hz, 1H), 4.17 – 4.12 (m, 1H), 3.87 (s, 9H), 3.47 – 3.25 (m, 1H), 3.12 (s, 3H), 2.96 (dd, J = 13.9 Hz, 7.4 Hz, 4H), 2.78 (dd, J = 13.3 Hz, 6.7 Hz, 1H), 2.55 (d, J = 4.5 Hz, 1H), 1.94 – 1.72 (m, 2H), 1.62 (dd, J = 16.9 Hz, 7.9 Hz, 1H), 0.91 (d, J = 6.5 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H). Mass: HRMS (ESI), Calcd for C30H42N2O9S: m/z 629.2509 (M+Na), found m/z 629.2505 (M+Na).
(3R,3aR,4S,7aS)-3-Methoxyhexahydro-2H-furo[2,3-b]pyran-4-yl (2S,3R)-4-(4-amino-N-isobutylphenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate (30c)
The titled inhibitor was synthesized from amine 29 and activated carbonate 27b using the general procedure outlined above. Inhibitor 30c was obtained in 24% yield. 1H NMR (300 MHz, Chloroform-d) δ 7.54 (d, J = 8.5 Hz, 2H), 7.41 – 7.00 (m, 5H), 6.67 (d, J = 8.6 Hz, 2H), 5.13 (d, J = 3.8 Hz, 1H), 5.03 (dd, J = 10.2 Hz, 5.2 Hz, 1H), 4.96 (d, J = 8.2 Hz, 1H), 4.39 – 3.99 (m, 3H), 3.99 – 3.70 (m, 6H), 3.48 – 3.24 (m, 2H), 3.12 (s, 3H), 3.02 – 2.83 (m, 4H), 2.75 (dd, J = 13.3 Hz, 6.3 Hz, 1H), 2.54 (d, J = 4.7 Hz, 1H), 1.89 – 1.64 (m, 3H), 0.91 (d, J = 6.5 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H). Mass: LRMS: m/z 592.8 (M+H). Mass: HRMS (ESI), Calcd for C29H41N3O8S: m/z 614.2513 (M+Na), found m/z 614.2502 (M+Na).
(3R,3aS,4S,7aS)-3-Hydroxyhexahydro-2H-furo[2,3-b]pyran-4-yl (2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenyl-sulfonamido)-1-phenylbutan-2-ylcarbamate (30f)
For the synthesis of inhibitor 30f, compound 30e was treated with Pd(OH)2 (10 mol %) in methanol (2.0 mL) at 60 Psi for 12 h. The reaction was filtered through a plug of Celite and concentrated under vacuum. The crude product was purified by silica gel chromatography to give the desired inhibitor 30f as an amorphous solid. (38 % yield, 2 steps). Rf = 0.22, (50 % ethyl acetate/hexanes). 1H NMR (300 MHz, Chloroform-d) δ 7.72 (d, J = 7.1 Hz, 2H), 7.30 – 7.22 (m, 5H), 6.99 (d, J = 8.9 Hz, 2H), 5.17 (s, 1H), 5.04 (d, J = 3.7 Hz, 2H), 4.57 (s, 1H), 4.32 (t, J = 8.4 Hz, 1H), 3.88 (s, 7H), 3.73 (dd, J = 9.2 Hz, 5.0 Hz, 1H), 3.41 – 3.08 (m, 2H), 3.02 – 2.90 (m, 4H), 2.79 (dd, J = 13.5 Hz, 6.8 Hz, 1H), 2.47 (s, 1H), 1.93 – 1.52 (m, 3H), 0.88 (dd, J = 13.2 Hz, 6.4 Hz, 6H). Mass: HRMS (ESI), Calcd for C29H40N2O9S: m/z 593.2532 (M+H) and 615.2352 (M+Na), found m/z 593.2520 (M+H) and 615.2330 (M+Na).
(3R,3aR,4S,7aS)-3-Ethoxyhexahydro-2H-furo[2,3-b]pyran-4-yl (2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenyl-sulfonamido)-1-phenylbutan-2-ylcarbamate (30d)
The titled inhibitor 30d was synthesized using the general procedure outlined above. Inhibitor 30d was obtained in 47 % yield. Rf = 0.20 (40% ethyl acetate/hexanes). 1H NMR (400 MHz, Chloroform-d) δ 7.69 (d, J = 8.3 Hz, 2H), 7.41 – 7.12 (m, 5H), 6.97 (d, J = 8.9 Hz, 2H), 5.14 (s, 1H), 5.04 (bs, 1H), 4.28 – 4.05 (m, 1H), 3.87 (s, 8H), 3.34 (d, J = 7.0 Hz, 3H), 3.10 – 2.91 (d, 5H), 2.86 – 2.65 (m, 1H), 2.54 (bs, 1H), 1.75 – 1.63 (m, 3H), 1.14 – 1.09 (m, 3H), 0.98 – 0.85 (m, 6H). Mass: LRMS: m/z 621.83 (M+H). Mass: HRMS (ESI), Calcd for C31H44N2O9S: m/z 643.2665 (M+Na), found m/z 643.2660 (M+Na).
(3R,3aR,4S,7aS)-3-(Benzyloxy)hexahydro-2H-furo[2,3-b]pyran-4-yl (2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxy-phenylsulfonamido)-1-phenylbutan-2-ylcarbamate (30e)
The titled inhibitor 30e was synthesized using the general procedure outlined above. Inhibitor 30e was obtained in 82 % yield. Rf = 0.6, (50 % ethyl acetate/hexanes).1H NMR (300 MHz, Chloroform-d) δ 7.69 (d, J = 8.9 Hz, 2H), 7.39 – 7.10 (m, 10H), 6.96 (d, J = 8.9 Hz, 2H), 5.13 (d, J = 3.7 Hz, 1H), 5.05 (d, J = 4.3 Hz, 1H), 4.82 (d, J = 8.4 Hz, 1H), 4.37 (s, 2H), 4.23 – 3.99 (m, 2H), 3.99 – 3.73 (m, 8H), 3.37 (t, J = 10.2 Hz, 1H), 3.09 (dd, J = 15.1 Hz, 8.4 Hz, 1H), 2.95 – 2.95 (m, 4H), 2.78 (dd, J = 13.5 Hz, 6.7 Hz, 1H), 2.64 – 2.59 (m, 1H), 1.79 (d, J = 7.4 Hz, 2H), 1.71 – 1.53 (m, 1H), 0.88 (dd, J = 14.5 Hz, 7.0 Hz, 6H). Mass: LRMS: m/z 683.9 (M+H). Mass: HRMS (ESI), Calcd for C36H46N2O9S: m/z 705.2822 (M+Na), found m/z 705.2816 (M+Na).
(3R,3aS,4S,7aS)-3-Azidohexahydro-2H-furo[2,3-b]pyran-4-yl (2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenyl-sulfonamido)-1-phenylbutan-2-ylcarbamate (30g)
The titled inhibitor 30g was synthesized using the general procedure outlined above. The desired inhibitor was obtained as an amorphous solid in 75% yield. Rf = 0.23 (40% ethyl acetate/hexanes). 1H NMR (400 MHz, CD3OD ) δ 7.75 (d, J = 8.8 Hz, 2H), 7.24 (d, J = 4.4 Hz, 4H), 7.19– 7.15 (m, 1H), 7.11 – 7.01 (d, J = 8.8 Hz, 2H), 5.09 (d, J = 3.7 Hz, 1H), 5.06 – 5.01 (m, 1H), 4.26 – 4.21 (m, 1H), 4.23 – 4.14 (m, 1H), 3.86 (s, 4H), 3.81 (bs, 2H), 3.67 (dd, J = 8.9 Hz, 4.2 Hz, 1H), 3.39 (dd, J = 10.8 Hz, 4.1 Hz, 2H), 3.12 – 3.00 (m, 3H), 2.90 – 2.85 (m, 1H), 2.72 – 2.60 (m, 1H), 2.48 – 2.41 (m, 1H), 2.04 – 1.97 (m, 1H), 1.83 – 1.62 (m, 2H), 0.97 – 0.80 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 163.0, 155.4, 137.4, 129.7, 129.4, 128.4, 126.6, 114.3, 99.3, 72.4, 72.3, 71.6, 59.4, 58.7, 55.5, 55.3, 55.0, 53.5, 44.3, 35.3, 28.2, 27.2, 20.0, 19.8. Mass: HRMS (ESI), calcd for (C29H39N5O8S): m/z 618.2598 (M+H), found m/z 618.2597.
(3R,3aS,4S,7aS)-3-Aminohexahydro-2H-furo[2,3-b]pyran-4-yl (2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-ylcarbamate (30h)
Inhibitor 30g was dissolved in ethyl acetate and 10% Pd/C (10 % by weight) was added. The resulting mixture was stirred under a hydrogen-filled balloon for 1 h. The mixture was filtered through a plug of Celite and concentrated under vacuum. The crude product was purified by silica gel chromatography to obtain inhibitor 30h as an amorphous solid in 85 % yield. Rf = 0.18 (5% (5% NH3/MeOH)/dichloromethane). 1H NMR (400 MHz, Chloroform-d) δ 7.71 (d, J = 8.8 Hz, 2H), 7.37 – 7.14 (m, 5H), 6.97 (d, J = 8.7 Hz, 2H), 5.53 (s, 1H), 5.34 – 5.13 (m, 2H), 4.07 (dd, J = 9.8 Hz, 7.0 Hz, 1H), 3.98 – 3.73 (m, 8H), 3.67 (dd, J = 9.6 Hz, 4.9 Hz, 1H), 3.36 – 3.27 (m, 1H), 3.17 (dd, J = 14.9 Hz, 8.9 Hz, 1H), 3.04 – 2.68 (m, 5H), 2.66 – 2.16 (bs, 2H), 2.10 – 1.95 (m, 1H), 1.82 (dd, J = 14.1, 6.9 Hz, 2H), 1.46 – 1.28 (m, 1H), 0.89 (d, J = 6.5 Hz, 3H), 0.85 (d, J = 6.5 Hz, 3H). Mass: LRMS: m/z 592.8 (M+H). Mass: HRMS (ESI), Calcd for C29H41N3O8S: m/z 592.2693 (M+H), found m/z 592.2686 (M+H).
(3R,3aS,4S,7aS)-3-(Methylamino)hexahydro-2H-furo[2,3-b]pyran-4-yl (2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-yl carbamate (30i)
To a stirred solution of amine 28 and activated carbonate 27f in CH3CN, DIPEA (5 equivalent) was added and the resulting mixture was stirred for 24 h. Standard work up and purification as described for inhibitor 30a provided the corresponding carbamate derivative. Above carbamate was dissolved in 5% ammonia in MeOH (2 mL) and 10% Pd(OH)2 was added. The mixture was stirred under a hydrogen-filled balloon for 4 h. The mixture was filtered through a plug of Celite and the filtrate was concentrated under reduced pressure. The residue was chromatographed on a silica gel column to provide inhibitor 30i in 76% yield for two-steps. Rf = 0.20 (10% methanol/DCM).1H NMR (400 MHz, CD3OD) δ 7.77 (d, J = 8.9 Hz, 2H), 7.32 – 7.21 (m, 4H), 7.20 – 7.17 (m, 1H), 7.07 (d, J = 8.7 Hz, 2H), 5.08 (d, J = 3.7 Hz, 1H), 5.00 – 4.91 (m, 1H), 4.21 (t, J = 8.4 Hz,1H), 3.87 (s, 3H), 3.86 – 3.65 (m, 4H), 3.50 – 3.34 (m, 3H), 3.15 (dd, J = 14.0 Hz, 3.6 Hz, 1H), 3.06 (dd, J = 13.7 Hz, 8.2 Hz, 1H), 2.95 (dd, J = 14.9 Hz, 8.1 Hz, 1H), 2.86 (dd, J = 13.6 Hz, 6.9 Hz, 1H), 2.60 (dd, J = 14.0 Hz, 10.7 Hz, 1H), 2.38 – 2.26 (m, 2H), 2.24 (s, 3H), 2.10 – 1.92 (m, 1H), 1.85 – 1.74 (m, 1H), 1.75 – 1.58 (m, 1H), 0.92 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 164.5, 157.4, 140.2, 132.1, 130.6, 130.3, 129.2, 127.3, 115.4, 103.3, 74.2, 73.8, 70.5, 61.2, 59.0, 58.9, 57.1, 56.2, 53.9, 36.6, 34.5, 28.5, 28.1, 20.5. Mass: HRMS (ESI), Calcd for C30H43N3O8S: m/z 606.2849 (M+H), found m/z 606.2840 (M+Na).
(3R,3aS,4S,7aS)-3-(Ethylamino)hexahydro-2H-furo[2,3-b]pyran-4-yl (2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-ylcarbamate (30j)
The titled inhibitor was synthesized using the general procedure outlined above. The desired inhibitor 30j was obtained as an amorphous solid in 50 % yield for the 2 steps. Rf = 0.25 (10% methanol/DCM). 1H NMR (400 MHz, CD3OD ) δ 7.76 (d, J = 8.8 Hz, 2H), 7.36 – 7.13 (m, 5H), 7.13 – 7.02 (d, J = 9.2 Hz, 2H), 5.09 (d, J = 3.8 Hz, 1H), 5.01 (dq, J = 10.7 Hz, 5.3 Hz, 4.7 Hz, 1H), 4.25 – 4.14 (t, J = 8.8 Hz, 1H), 3.87 (s, 3H), 3.86 – 3.79 (m, 3H), 3.78 – 3.72 (m, 1H), 3.70 (dd, J = 8.7 Hz, 5.1 Hz, 1H), 3.50 (td, J = 7.3 Hz, 5.3 Hz, 1H), 3.46 – 3.35 (m, 2H), 3.15 (dd, J = 14.0 Hz, 3.7 Hz, 1H), 3.05 (dd, J = 13.6 Hz, 8.1 Hz, 1H), 2.96 (dd, J = 14.9 Hz, 8.3 Hz, 1H), 2.87 (dd, J = 13.6 Hz, 6.9 Hz, 1H), 2.65 – 2.51 (m, 2H), 2.50 – 2.41 (m, 1H), 2.29 (td, J = 6.8 Hz, 4.0 Hz, 1H), 2.07 – 1.96 (m, 1H), 1.82 – 1.60 (m, 2H), 1.10 (t, J = 7.1 Hz, 3H), 0.91 (d, J = 6.6 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H). 13C NMR (100 MHz, CD3OD) δ 164.5, 157.4, 140.2, 132.2, 130.6, 130.3, 129.3, 127.3, 115.4, 103.1, 74.3, 74.1, 70.4, 60.9, 58.8, 57.4, 57.2, 56.2, 53.9, 43.5, 36.7, 28.6, 28.1, 20.5, 15.0. Mass: HRMS (ESI), Calcd for C31H45N3O8S: m/z 620.3005 (M+Na), found m/z 620.3000 (M+Na).
(3R,3aS,4S,7aS)-3-(Ethylamino)hexahydro-2H-furo[2,3-b]pyran-4-yl (2S,3R)-4-(4-amino-N-isobutylphenylsulfonamido)-3-hydroxy-1-phenylbutan-2-ylcarbamate (30k)
The titled inhibitor 30k was synthesized following the general procedure outlined above. The desired inhibitor was obtained as an amorphous solid in 61 % yield for the 2 steps. 1H NMR (400 MHz, CD3OD ) δ 7.47 (d, J = 8.8 Hz, 2H), 7.32 – 7.10 (m, 5H), 6.68 (d, J = 9.1 Hz, 2H), 5.09 (d, J = 3.7 Hz, 1H), 5.04 – 4.92 (m, 2H), 4.21 (t, J = 8.0 Hz, 1H), 3.95 – 3.79 (m, 2H), 3.90 – 3.79 (m, 2H), 3.53 – 3.48 (m,1H), 3.44 – 3.33 (m, 3H), 3.17 (dd, J = 14.0 Hz, 3.7 Hz, 2H), 2.99 (dd, J = 13.6 Hz, 8.2 Hz, 1H), 2.89 (dd, J = 14.8 Hz, 8.4 Hz, 1H), 2.80 (dd, J = 13.5 Hz, 6.8 Hz, 1H), 2.65 – 2.51 (m, 2H), 2.50 – 2.43 (m, 1H), 2.31 – 2.26 (m, 1H), 2.03 – 1.98 (m, 1H), 1.78 – 1.63 (d, 2H), 1.10 (t, J = 7.1 Hz, 3H), 0.92 (d, J = 6.5 Hz, 3H), 0.89 – 0.80 (d, J = 6.5 Hz, 3H). Mass: LRMS: m/z 605.8 (M+H). Mass: HRMS (ESI), Calcd for C30H44N4O7S: m/z 605.3009 (M+H), found m/z 605.3005 (M+H).
(3R,3aS,4S,7aS)-3-(Propylamino)hexahydro-2H-furo[2,3-b]pyran-4-yl (2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-ylcarbamate (30l)
The titled inhibitor 30l was synthesized following the general procedure outlined above. Inhibitor 30l was obtained as an amorphous solid in 60% yield. 1H NMR (400 MHz, CD3OD) δ 7.76 (d, J = 8.9 Hz, 2H), 7.35 – 7.14 (m, 5H), 7.08 (d, J = 8.9 Hz, 2H), 5.10 (d, J = 3.8 Hz, 1H), 5.04 – 4.99 (m, 1H), 4.21 (t, J = 6.8 Hz, 1H), 3.88 (s, 4H), 3.86 – 3.79 (m, 1H), 3.79 – 3.68 (m, 2H), 3.53 – 3.48 (m, 1H), 3.47 – 3.35 (m, 3H), 3.26 – 3.24 (m, 1H), 3.18 – 3.11 (dd, J = 14.0 Hz, 3.6 Hz, 1H), 3.05 (dd, J = 13.4 Hz, 8.4 Hz, 1H), 2.96 (dd, J = 14.9 Hz, 8.3 Hz, 1H), 2.86 (dd, J = 14.9 Hz, 8.3 Hz, 1H), 2.62 (dd, J = 14.0 Hz, 10.7 Hz, 1H), 2.55 – 2.45 (m, 1H), 2.45 – 2.35 (m, 1H), 2.34 – 2.30 (m, 1H), 2.07 – 1.94 (m, 1H), 1.83 – 1.62 (m, 2H), 1.55 – 1.45 (m, 1H), 0.97 – 0.89 (m, 6H), 0.86 (d, J = 6.7 Hz, 3H). Mass: LRMS: m/z 634. 9 (M+H). Mass: HRMS (ESI), Calcd for C32H47N3O8S: m/z 634.3163 (M+H), found m/z 634.3156 (M+H).
(3R,3aS,4S,7aS)-3-(Dimethylamino)hexahydro-2H-furo[2,3-b]pyran-4-yl (2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-ylcarbamate (30m)
Compound 30h was dissolved in 1,2-dichloroethane (2 mL). Powdered paraformaldehyde (3 equivalent) and sodium triacetoxy borohydride (3 equivalent) were added and the mixture was stirred for 24 h. The crude inhibitor was purified by silca gel chromatography to obtain the desired inhibitor 30m as an amorphous solid in 67 % yield. Rf = 0.25 (10% methanol/DCM). 1H NMR (400 MHz, CD3OD ) δ 7.77 (d, J = 8.8 Hz, 2H), 7.29 – 7.18 (m, 5H), 7.08 (d, J = 8.8 Hz, 2H), 5.18 (d, J = 4.3 Hz, 1H), 5.01 – 4.98 (m, 1H), 4.18 – 4.01 (m, 1H), 3.96 – 3.83 (m, 5H), 3.84 – 3.76 (m, 2H), 3.50 – 3.42 (m, 3H), 3.15 (dd, J = 13.8 Hz, 2.5 Hz, 1H), 3.06 (dd, J = 13.7 Hz, 8.1 Hz, 1H), 2.96 (dd, J = 14.8 Hz, 7.8 Hz, 1H), 2.87 (dd, J = 13.6 Hz, 6.9 Hz, 1H), 2.67 – 2.50 (m, 1H), 2.23 (s, 3H), 1.97 (s, 3H), 1.81 – 1.71 (m, 1H), 1.70 – 1.61 (m, 1H), 0.92 (d, J = 6.6 Hz, 3H), 0.87 (d, J = 6.6 Hz, 3H). Mass: LRMS: m/z 620.8 (M+H). Mass: HRMS (ESI), Calcd for C31H45N3O8S: m/z 620.3006 (M+H), found m/z 620.2998 (M+H).
Methods: Determination of X-ray structures of HIV-1 protease-inhibitor complexes
The optimized HIV-1 protease was expressed and purified as described.31 The protease-inhibitor complex was crystallized by the hanging drop vapor diffusion method with well solutions of 1.18 M NaCl, 0.1 M Sodium Acetate buffer (pH 4.6) for the complex with inhibitor 30b and 1.3M NaCl, 0.1M Sodium Acetate buffer (pH 5.5) for the complex with inhibitor 30j respectively. X-ray diffraction data were collected on a single crystal cooling to 90 K at SER-CAT (22-BM beamline), Advanced Photon Source, Argonne National Lab (Chicago, USA) with X-ray wavelength of 1.0 Å, and processed by HKL-200032 with an Rmerge of 6.8% and 5.7%, respectively, for the 30b and 30j complexes. Using the isomorphous structure33, the crystal structures were solved by PHASER34 in CCP4i Suite35, 36 and refined by SHELX-9737 to 1.22 Å and 1.62 Å resolution, respectively. COOT38 was used for visual modification of the structures. PRODRG-239 was used to construct the inhibitor and the restraints for refinement. Alternative conformations were modeled. Anisotropic atomic displacement parameters (B factors) were applied for all atoms, including solvent molecules in the higher resolution complex of HIV protease with inhibitor 30b. The final refined solvent structure comprised two sodium ions, three chloride ions, one acetate ion and 186 water molecules for HIV protease with inhibitor 30b and one sodium ion, three chloride ions and 138 water molecules for HIV protease with inhibitor 30j. The crystallographic statistics are listed in Table 2 (ESI). The coordinates and structure factors of the HIV-1 protease complexes with inhibitor 30b and inhibitor 30j have been deposited in the Protein Data Bank with accession codes 5DGU and 5DGW.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (Grant GM53386, AKG and Grant GM 62920, IW). X-ray data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) beamline 22BM at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the US Department of Energy, Basic Energy Sciences, Office of Science, under Contract No. W-31–109-Eng-38. This work was also supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health, and in part by a Grant-in-Aid for Scientific Research (Priority Areas) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Monbu Kagakusho), a Grant for Promotion of AIDS Research from the Ministry of Health, Welfare, and Labor of Japan, and the Grant to the Cooperative Research Project on Clinical and Epidemiological Studies of Emerging and Reemerging Infectious Diseases (Renkei Jigyo) of Monbu-Kagakusho. The authors would like to thank the Purdue University Center for Cancer Research, which supports the shared NMR and mass spectrometry facilities.
Footnotes
Electronic Supplementary Information (ESI) available: Crystallographic data Collection, Refinement Statisticsand NMR spectra of compounds are available. See DOI:
References
- 1.Edmonds A, Yotebieng M, Lusiama J, Matumona Y, Kitetele F, Napravnik S, Cole SR, Van Rie A, Behets F. Med PLoS. 2011:8e1001044. doi: 10.1371/journal.pmed.1001044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mitsuya H, Maeda K, Das D, Ghosh AK. Adv Pharmacol. 2008;56:169–197. doi: 10.1016/S1054-3589(07)56006-0. [DOI] [PubMed] [Google Scholar]
- 3.Hue S, Gifford RJ, Dunn D, Fernhill E, Pillay D. J Virol. 2009;83:2645–2654. doi: 10.1128/JVI.01556-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conway B. Future Virol. 2009;4:39–41. [Google Scholar]
- 5.Ghosh AK, Anderson DD, Weber IT, Mitsuya H. Angew Chem Int Ed. 2012;51:1778–1802. doi: 10.1002/anie.201102762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghosh AK, Chapsal BD, Weber IT, Mitsuya H. Acc Chem Res. 2008;41:78–86. doi: 10.1021/ar7001232. [DOI] [PubMed] [Google Scholar]
- 7.Ghosh AK, Chapsal B, Mitsuya H. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2010. pp. 205–243. [Google Scholar]
- 8.Ghosh AK, Dawson ZL, Mitsuya H. Bioorg Med Chem. 2007;15:7576–7580. doi: 10.1016/j.bmc.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghosh AK, Sridhar PR, Kumaragurubaran N, Koh Y, Weber IT, Mitsuya H. ChemMedChem. 2006;1:939–950. doi: 10.1002/cmdc.200600103. [DOI] [PubMed] [Google Scholar]
- 10.Ghosh AK, Xu CX, Rao KV, Baldridge A, Agniswamy J, Wang YF, Weber IT, Aoki M, Miguel SGP, Amano M, Mitsuya H. ChemMedChem. 2010;5:1850–1854. doi: 10.1002/cmdc.201000318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghosh AK, Chapsal BD, Baldridge A, Steffy MP, Walters DE, Koh Y, Amano M, Mitsuya H. J Med Chem. 2011;54:622–634. doi: 10.1021/jm1012787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ide K, Aoki M, Amano M, Koh Y, Yedidi RS, Das D, Leschenko S, Chapsal B, Ghosh AK, Mitsuya H. Antimicrob Agents Chemother. 2001;55:1717–1727. doi: 10.1128/AAC.01540-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghosh AK, Martyr CD, Steffey M, Wang YF, Agniswamy J, Amano M, Weber IT, Mitsuya H. ACS Med Chem. 2011;2:298–302. doi: 10.1021/ml100289m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh AK, Chapsal BD, Parham GL, Steffey M, Agniswamy J, Wang YF, Amano M, Weber IT, Mitsuya H. J Med Chem. 2011;54:5890–5901. doi: 10.1021/jm200649p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ghosh AK, Chapsal BD, Steffey M, Agniswamy J, Wang YF, Amano M, Weber IT, Mitsuya H. Bioorg Med Chem Lett. 2012;22:2308–2311. doi: 10.1016/j.bmcl.2012.01.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh AK, Martyr CD, Osswald HL, Sheri VR, Kassekert LA, Chen S, Agniswamy J, Wang YF, Hayashi H, Aoki M, Weber IT, Mitsuya H. J Med Chem. 2015;58:6994–7006. doi: 10.1021/acs.jmedchem.5b00900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hohlfeld K, Wegner JK, Kesteleyn B, Linclau B, Unge J. J Med Chem. 2015;58:4029–4038. doi: 10.1021/acs.jmedchem.5b00358. [DOI] [PubMed] [Google Scholar]
- 18.Hohlfeld K, Tomass C, Wegner JK, Kesteleyn B, Linclau B. ACS Med Chem Lett. 2011;2:461–465. doi: 10.1021/ml2000356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Solladié G, Arce E, Bauder C, Carreno MC. J Org Chem. 1998;63:2332–2337. [Google Scholar]
- 20.Briickner R. Chem Ber. 1989;122:703–710. [Google Scholar]
- 21.Briickner R, Priepke H. Angew Chem. 1988;27:278–280. [Google Scholar]
- 22.White KN, Konopelski JP. Org Lett. doi: 10.1021/ol051441w. [DOI] [PubMed] [Google Scholar]; Ghosh AK, Parham GL, Martyr CD, Nyalapatla PR, Osswald HL, Agniswamy J, Wang Y-F, Amano M, Weber IT, Mitsuya H. J Med Chem. 2013;56:6792–6802. doi: 10.1021/jm400768f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghosh AK, Parham GL, Martyr CD, Nyalapatla PR, Osswald HL, Agniswamy J, Wang YF, Amano M, Weber IT, Mitsuya H. J Med Chem. 2013;56:6792–6802. doi: 10.1021/jm400768f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumara Swamy KC, Bhuvan Kumar NN, Balaraman E, Pavan Kumar KVP. Chem Rev. 2009;109:2551–2651. doi: 10.1021/cr800278z. [DOI] [PubMed] [Google Scholar]
- 25.Ghosh AK, Martyr CD. Modern Drug Synthesis. 2010:29–44. [Google Scholar]
- 26.Ghosh AK, McKee SP, Lee HY, Thompson WT. J Chem Soc Chem Comm. 1992;3:273–274. doi: 10.1039/C39920000273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Toth MV, Marshall GR. J Pept Protein Res. 1990;36:544–550. doi: 10.1111/j.1399-3011.1990.tb00994.x. [DOI] [PubMed] [Google Scholar]
- 28.Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang YF, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. Antimicrob Agents Chemother. 2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tie Y, Boross PI, Wang YF, Gaddis L, Hussain AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. J Mol Biol. 2004;338:341–352. doi: 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]
- 30.Ghosh AK, Martyr CD, Steffey M, Wang YF, Agniswamy J, Miguel S, Amano M, Weber IT, Mitsuya H. ACS Med Chem Lett. 2011;2:298–302. doi: 10.1021/ml100289m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mahalingam B, Louis JM, Hung J, Harrison RW, Weber IT. Proteins. 2001;43:455–464. doi: 10.1002/prot.1057. [DOI] [PubMed] [Google Scholar]
- 32.Otwinowski Z, Minor W. In: Macromolecular Crystallography, Part A. Carter CW Jr, Sweet RM, editors. 1997. pp. 307–326. [Google Scholar]
- 33.Shen CH, Wang YF, Kovalevsky AY, Harrison RW, Weber IT. Febs J. 2010;277:3699–3714. doi: 10.1111/j.1742-4658.2010.07771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNichols SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Acta Crystallogr Sect D: Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Potterton E, Briggs P, Turkenburg M, Dodson E. Acta Crystallogr, Sect D: Biol Crystallogr. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 37.Sheldrick GM. Acta Crystallogr Sect A: Found Crystallogr. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 38.Emsley P, Lohkamp B, Scott WG, Cowtan K. Acta Crystallogr Sect D: Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schuettelkopf AW, van Aalten DMF. Acta Crystallogr Sect D: Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.