

Abstract
The bacterial toxin RelE is a ribosome-dependent endoribonuclease. It is part of a type II toxin-antitoxin system that contributes to antibiotic resistance and biofilm formation. During amino acid starvation RelE cleaves mRNA in the ribosomal A-site, globally inhibiting protein translation. RelE is structurally similar to microbial RNases that employ general acid-base catalysis to facilitate RNA cleavage. The RelE active-site is atypical for acid-base catalysis, in that it is enriched for positively charged residues and lacks the prototypical histidine-glutamate catalytic pair, making the mechanism of mRNA cleavage unclear. In this study we use a single-turnover kinetic analysis to measure the effect of pH and phosphorothioate substitution on the rate constant for cleavage of mRNA by wild-type RelE and seven active-site mutants. Mutation and thio-effects indicate a major role for stabilization of increased negative change in the transition state by arginine 61. The wild-type RelE cleavage rate constant is pH-independent, but the reaction catalyzed by many of the mutants is strongly pH dependent, suggestive of general acid-base catalysis. pH-rate curves indicate that wild-type RelE operates with the pKa of at least one catalytic residue significantly downshifted by the local environment. Mutation of any single active-site residue is sufficient to disrupt this microenvironment and revert the shifted pKa back above neutrality. pH-rate curves are consistent with K54 functioning as a general base and R81 as a general acid. The capacity of RelE to effect a large pKa shift and facilitate a common catalytic mechanism by uncommon means furthers our understanding of other atypical enzymatic active sites.
The bacterial toxin RelE promotes sequence-specific cleavage of mRNA in a ribosome-dependent manner1–3. RelE and other type II bacterial toxins share common structural features with the RNase T1 family of endoribonucleases. All these enzymes cleave RNA phosphodiester bonds via a 2′,3′-cyclic phosphate intermediate4–9. Although RelE cannot cleave mRNA outside the ribosomal A-site and its low sequence homology with other RNases creates an enzyme active site significantly different from the long-studied RNases A and T1, it is hypothesized to employ the same catalytic mechanism2,4–6,8,10–13.
Toxin-antitoxin (TA) systems play a significant role in cell resistance to antibiotic challenge as well as biofilm formation and the bacterial stress response14–18. They are found widely in both bacteria and archaea and the number of TA systems within a bacterium may be linked to pathogenicity14,19. TA genetic loci typically code for two elements: a toxin, capable of interfering with cellular function and arresting growth, and an antitoxin, which inactivates the toxin 16,18. Type II TA systems are characterized by a tight protein-protein complex of toxin and antitoxin that eliminates toxin function. The endoribonuclease RelE, which is one of the most studied type II TA toxins, cleaves mRNA in the ribosomal A-site in response to amino acid starvation and arrests cell growth1,2,6,20–22. RelE shares structural features with the RNase T1 family of microbial RNases4–6,23,24 and, like many RNases, produces a 2′,3′-cyclic phosphate product upon cleavage of phosphodiester bonds6. It has been suggested that RelE, like many other nucleases, employs general acid-base catalysis to facilitate this reaction 6,13 but this has not been confirmed.
Despite the similar tertiary structure and reaction products of RelE and other RNases, a detailed characterization of RelE’s mechanism for RNase activity has been frustrated by a lack of sequence homology to other well-studied RNases4,10,11,25. Bond cleavage by many RNases is typically enabled by a glutamate-histidine pair that acts as the general base and general acid, respectively. This catalytic pair has been identified in many microbial RNases including members of the RNase T1 family that are structurally similar to RelE6,26–28. YoeB, another member of RelE type II bacterial toxin family, also has a glutamate-histidine pair located within its active site that correspond structurally with the catalytic pair in the RNase T1 family29. A second member of this type II TA family, HigB, has a conserved histidine in its active site30. However, RelE differs from these structurally similar RNases in its lack of conserved glutamate or histidine residues or a clear substitute for either residue within the active site5,6.
Identification of the RelE active site was confirmed by co-crystal structures of the enzyme bound to an mRNA substrate in the ribosomal A-site in both the pre- and post- cleavage states (Figure 1A,B)6. The crystal structures revealed a distorted mRNA configuration relative to the typical A-site path. The mRNA backbone is displaced by as much as 8 Å and the A-site bases are splayed apart, exposing the scissile phosphate and aligning the 2′-OH for nucleophilic attack on the adjacent phosphate6. Although distortion of base stacking and 2′-OH positioning are common structural features of metal-independent RNases9, the co-crystal structure revealed the atypical active site of RelE that lacks the glutamine-histidine catalytic pair and common alternatives. Instead, the active site showed a large number of positively charged amino acids. Many of the most conserved residues of bacterial RelE (R61, R81, Y87, K52, K54) are reasonably positioned for possible hydrogen bonding to the 2′-OH, the scissile phosphate, and the 5′-leaving group in wild-type enzyme6. However, the active site is sufficiently perturbed by the non-hydrolyzable substrate modifications that unambiguous assignment of enzyme-substrate hydrogen bonding interactions is difficult. Detailed analysis of single-turnover cleavage rates and binding effects for alanine deletions of these conserved residues confirmed their critical role in the chemical step of mRNA cleavage13.
Figure 1.

(A) The pre-cleavage structure of RelE (purple) and non-hydrolyzable mRNA substrate (2′-O-methyl, gray) in the ribosomal A-site. Critical residues are shown as sticks and elements colored blue (nitrogen), red (oxygen), and orange (sulfur). R81* is mutated to alanine in the crystal structure (PDB entry 3KIQ). (B) Post-cleavage crystal structure of A-site bound RelE, residues and atoms are colored in the same fashion as in A (PDB entry 4V7K). (C) Structural alignment of post-cleavage RelE and RNase T1 (cyan, PDB entry 1RGA) key residues for RNase T1 activity are shown as sticks, Scissile phosphate for both enzymes is show (orange spheres). (D) Overlay of RNase T1 (cyan) (4V7K) and pre-cleavage RelE (purple)(3KIQ), scissile phosphate is shown in orange.
The single-turnover kinetic data collected by Griffin et al. was consistent with rate defects of 103- to 106-fold that have been measured for deletion of acid-base side chains in other RNases27,31–33. This led to a revised mechanistic hypothesis for RelE cleavage. The Y87 is proposed to form stacking interactions that position the substrate. The positively charged microenvironment activates K52 to function as the general base, and R61 and K54 serve to stabilize the negative charge of the transition state, while R81 protonates the leaving group as the general acid. However, the similarity of rate effects among key mutants coupled with the atypical identity of active site residues leaves ambiguity as to the specific role for these residues in catalysis.
Here we extend the single-turnover kinetic analysis of the ribosome-dependent toxin RelE to directly measure the effect of pH and phosphorothioate substitution on the rate of chemistry for wild-type RelE and each of the active site mutants. These data are consistent with RelE’s reliance on transition state charge stabilization and atypical amino acid residues to cleave phosphodiester bonds by a general acid-base catalytic mechanism.
MATERIALS AND METHODS
Purification of RelE Protein
RelE wild-type and mutant protein were prepared as described previously13 with one modification: all RelBE complexes were expressed in E. coli BL21 (DE3)pLysS chemically competent cells. Briefly, the relBE locus from E. coli K-12 MG1655 with an N-terminal six-His tag was expressed in E. coli BL21 (DE3)pLysS13. Internal deletions of 3-9 amino acids starting at relB A19, described previously13, were retained to reduce the high affinity association of RelE and RelB, allowing for the subsequent purification of RelE by unfolding from RelB on a nickel column. The relBE E. coli cultures were grown in 100 μg/mL ampicillin and induced for 3 hours with 1 mM IPTG. Pelleted cells were lysed by sonication in at 4 °C in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 5 mM 2-mercaptoethanol, and 0.2 mg/mL lysozyme)22. Lysate was cleared by centrifugation and incubated with NTA-agarose resin for 1 hour at 4°C to bind the His6x-RelB:RelE complex. Resin was washed in lysis buffer of increased imidazole concentration (35 mM). RelE was selectively eluted by denaturation to disrupt contact with RelB in 9.8 M urea, 100 mM NaH2PO4, 10 mM Tris-HCl, 1 mM 2-mercaptoethanol (pH 8.0)22. Pure RelE fractions were dialyzed into 8 M urea, 50 mM Bicine (pH 8.4) overnight at room temperature. RelE protein was refolded overnight at 4 °C in 24 L of 50 mM Tris-HCl (pH 7.5), 70 mM NH4Cl, 300 mM KCl, 7 mM MgCl2, 1 M urea, and 1 mM dithiothreitol. Refolded protein was concentrated on ice under argon using a 10,000 molecular weight cutoff ultrafiltration membrane before dialysis into storage buffer (50 mM Tris-HCl pH 7.5, 70 mM NH4CL, 30 mM KCl, 7 mM MgCl2, 1 mM dithiothreitol, and 20% glycerol) at 4 °C overnight. RelE protein aliquots were flash frozen in liquid nitrogen and stored at −80 °C. Immediately prior to reaction, purified protein was buffer exchanged via centrifugation in a 10,000 molecular weight cutoff spin filter to pH-adjusted MMB buffer (50 mM MES 50 mM MOPS 50 mM Bicine, 70 mM NH4Cl, 30 mM KCl, 2 mM MgCl2, 1 mM dithiothreitol, pH adjusted to 5.5, 6.0, 6.5, 7.0, 7.5, 8.0, 8.5, 9.0).
Ribosome Complex Formation
E. coli 70S ribosomes were prepared as described previously34. mRNA oligonucleotides for fluorescence stopped flow reactions were ordered from Dharmacon with a 3′-C6 amino linker (5′- GGCAAGGAGGUAAAAAUGUAGAAAAACAAUN6-3′) and deprotected according to manufacturer’s instructions. Alexa Fluor 488 succinimidyl ester (Dharmacon) was dissolved in DMSO. Conjugation to the oligo was achieved by reacting 100 μg mRNA with 250 μg fluorophore in 75 μM Na2B4O7 (pH 8.4) overnight at room temperature with gentle shaking. Labeled mRNA was separated by polyacrylamide gel chromatography and purified by ethanol precipitation. 5′-32P-labeled mRNA was prepared as described previously35.
Ribosome complex (RC) was formed at 300 nM 70S ribosomes with 400 nM tRNAfMet and 100 nM Alexa Fluor labeled mRNA in formation buffer (50 mM Tris-HCl pH 7.5, 70 mM NH4Cl, 30 mM KCl, 7 mM MgCl2, 1 mM dithiothreitol) at 37 °C for 30 minutes. RCs for benchtop reactions contained 100 nM mRNA and trace 5′-32P mRNA in place of fluorescent RNA. Ribosome complex was then pelleted by ultracentrifugation at 120K RPM for 1hr (Beckman Optima Max TL, TLA120.1 rotor). Pelleted ribosome complex was resuspended in MMB buffer to a final ribosome concentration of 120 nM. MMB buffer for resuspension was pre-adjusted to specific pH values at half-unit increments from 5.5 – 9.0. Ribosome complexes containing the phosphorothioate mRNA substrates were not pelleted and remain in formation buffer for reactions. The mRNA containing the phosphorothioate linkage (5′-GGCAAGGAGGUAAAAAUGUAPSGAAAAACAAU-3′) was prepared and 5′-32P radiolabeled as previously described13.
Single Turnover Kinetic Reactions
Single-turnover kinetic measurements were taken for wild-type RelE and the K52A, K54A, and Y87F mutants using a fluorescence stopped-flow apparatus (Applied Photophysics SX.20) at 20±0.1 °C. Reactions were initiated by mixing an equal volume of RelE (final concentration > 4 μM) to the Alexa Fluor-containing reaction complex in MMB buffer (final ribosome concentration of 60 nM). Fluorescence was monitored at a 90° angle to incident light through a 515 nm long-pass colored glass filter with an excitation wavelength of 482 nm. At least three time courses were measured for each reaction trial and three trials were performed for each pH condition. Increase in fluorescence intensity was fit to equation 1 to determine the rate constants kfast and kslow. Throughout this paper kfast is referred to as kobs. pH-rate curves for kslow (not shown) show the same relationships as the kfast reported here. The remaining mutants (R81A, R61A, Y87A, K52A/Y87F) were measured using manual bench top quench methods13. Bench top reactions are exclusively composed of 5′-32P mRNA-containing reaction complex. The reactions were also initiated by addition of equal volume RelE and reaction complex (final concentrations >4 μM and 60 nM). Reactions were stopped by the addition of excess chemical quench (80% formamide, 50 mM Tris-MES pH 6.5, 65 mM EDTA, 0.2 mg/ML)13. As a control for mRNA stability, reactions without ribosomes and without RelE were measured for up to 48 hours. The extent of mRNA cleavage for all reactions was determined by separating RNA substrates and products by 15% denaturing PAGE and visualizing with a STORM phosphorimager (Molecular Dynamics). Individual band intensities were quantified with ImageQuant (GE Healthcare) and fraction reacted was determined as the product mRNA over the sum of product mRNA and remaining substrate mRNA. At least three trials were conducted for each mutant at each pH value.
Individual time courses collected by fluorescence stopped-flow and radiolabeled cleavage were fit to the double-exponential function in equation 1:
| (1) |
where Afast and Aslow are the amplitudes of the two phases, kfast and kslow are the rate constants for the two phases (sec−1), t is the reaction time in seconds, and Y0 is the initial fraction reacted. Because of the slow reaction rates for some mutants and low pH values it was not possible to fully fit kslow for all conditions. In these circumstances, reaction curves were fit to the single exponential equation 2:
| (2) |
where A is the amplitude, k is the rate constant of cleavage (sec−1), t is the reaction time in seconds, and Y0 is the initial fraction reacted.
Simulation of pH-Rate Curves
pH-rate curves were simulated with the assumption of a single general acid and a single general base that do not influence one another. Also, it is assumed that the enzyme is only able to react when both the acid and the base are in their functional states and they do so at a rate of k1. Given these assumptions, the partition function is given as equation 336:
| (3) |
where pKa,A and pKa,B are the pKas of the conjugate acid and base, respectively. Equation 3 allows for the calculation of the fractions of both the acid and the base in their active states with equations 4 and 536:
| (4) |
| (5) |
Where fHa+ and fB− are the fractions of the general acid and the general base in the functional state. The fraction of the enzymes in the reaction in which both the acid and the base are found in their active state is the product of the two fractions fHA+ and fB− and is given by equation 6.
| (6) |
fR(1) is the fraction of the enzyme in the functional state. The value of kobs is the product of the functional enzyme fraction and the reaction rate given by equation 736:
| (7) |
In order to approximate the pKa values for the RelE mutants, we substituted for fR(1) in equation 7 and fit the pH-rate data to equation 8:
| (8) |
where k1 is independent of pH and, in the context of the pH-rate curves, is equal to the maximum kobs.
RESULTS
pH Dependence of RelE Cleavage
We employed a single-turnover assay to measure the pH dependence of the RelE cleavage reaction. The RelE enzyme was present in excess of the substrate ribosomal complex (RC) comprised of 70S E. coli ribosomes, fluorescently labeled mRNA containing a UAG codon in the A-site, and the initiator tRNA bound to the P-site. The cleavage rate constant for wild-type RelE was determined by exponential fitting to the increase in fluorescence signal observed upon release of mRNA product from the ribosome complex using a stopped flow instrument (Figure 2). In the absence of RelE, a lack of change in fluorescence signal indicates either no cleavage or the rate of mRNA cleavage was below the observable limit (Figure 2). At high RelE concentrations (>3μM), kobs reaches a plateau13. All kinetic characterizations described below were performed at saturating enzyme concentrations (>4 μM). Using this system, we measured kobs of 390 ± 120 sec−1 for the wild-type RelE toxin. The previously published rate for wild-type RelE cleavage, determined using a radiolabeled substrate, was 380 ± 25 sec−1 13. The good agreement in these values indicates that product release is not rate limiting in our fluorescence assay and that kobs for the fluorescence assay is a measure of kchem as it was for the radiolabeled assay13.
Figure 2.
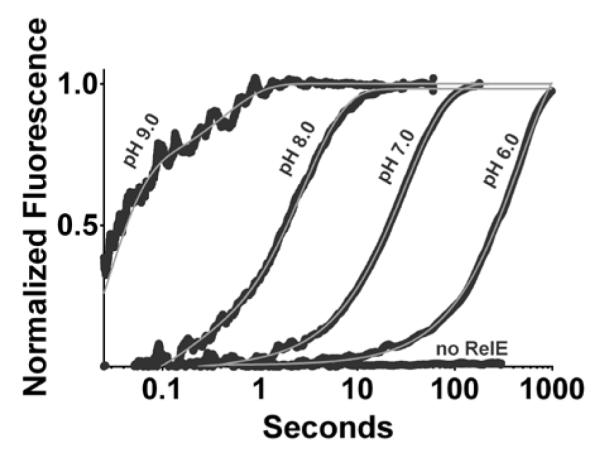
Stopped flow measurement of fluorescent mRNA cleavage by RelE. Four illustrative reaction time courses are shown in the presence of the K52A RelE mutant each at a different pH: 9.0, 8.0, 7.0, and 6.0. No increase in fluorescence is detected for mRNA-ribosome complex in the absence of RelE. All reaction curves are fit to equation 1 (grey lines), and voltage is normalized to the amplitude of the fit. The no-RelE voltage is normalized to the pH 7.0 fit.
We measured the rate constant for cleavage by RelE across a range of pH values from 5.5 to 9.0 and observed an essentially uniform kobs over the pH range (Figure 3A, black). One interpretation of the pH-rate independence for the wild-type enzyme is that it employs a general acid-base mechanism in which the pKa of the base and the pKa of the acid are widely separated36. In that scenario, our data indicate that the pKa of either the RelE general base or general acid is less than 6, while the pKa of the other is greater than 10. A more specific pKa determination is beyond the measureable range of the single-turnover assay as the ribosomal-mRNA complex inactivates at pH values above 9.0 and below 5.5. Alternative scenarios, in which kobs is not limited by the rate of chemistry or catalysis does not depend on titratable groups, are also consistent with a lack of pH dependence.
Figure 3.

pH-rate profiles for RelE and active site mutants. pH-rate profile for (A) wild-type (black), K54A (green), R81A (red), R61A (purple) and (B) wild-type (black), Y87F (blue), K52A (orange), Y87A (brown), K52A/Y87F (pink). Average vales of at least three independent trials with SEM error bars are plotted for each mutant. Trends are least squares fit to equation 8.
pH Dependence for the Active Site Mutants
To ascertain the contribution of RelE active-site residues to a proposed acid-base catalytic mechanism, we determined the pH-rate profiles for the Y87F, K52A, K54A, R81A, Y87A, K52A/Y87F, and R61A mutants. Cleavage rate constants greater than 0.1 sec−1 (Y87F, K52A, K54A) were determined by monitoring an increase in fluorescence signal observed by stopped flow as for the wild type (Figure 2). Cleavage rate constants for mutants with major rate defects (R81A, Y87A, K52A/Y87F, and R61A) were determined by the quantification of radiolabeled mRNA cleavage product separated by gel shift assay, as described previously13.
In contrast to wild-type RelE, many active-site mutants exhibited rate constants dependent on pH (Figure 3, Table S1). The variation in rate of observed mRNA cleavage by the mutant K52A was the most dramatic, with a log-linear pH-rate curve of slope equal to ~1 spanning four orders of magnitude in kobs (Figure 3B, orange). By contrast the pH-rate dependence of the K54A mutant resembles the wild-type enzyme, staying essentially flat across the testable range (Figure 3A, green). The pH-rate curves of the remaining mutants exhibit various inflection points at which the pH-rate dependence transitions from a slope of ~1 to ~0. The transitions in rate dependence for these mutants occur at pH 7.4 (Y87F), 8.5 (Y87A), 8.1 (R61A), and 8.7 (R81A) (Figure 3, blue, brown, purple, red). The shape of the pH-rate curves for these four active site mutants may indicate a shifted pKa of the general acid or general base36. However, pH-rate curves are not sufficient in themselves to conclude that the mutant rate dependencies are not merely the result of a change in the rate-limiting step of the cleavage reaction.
Phosphorothioate Effects for Mutant RelE Enzymes
In addition to the pH-rate curves, we examined the impact of a phosphorothioate substitution at the mRNA scissile phosphate on the rate constant for cleavage by RelE mutants. Substitution of a sulfur atom for either of the non-bridging phosphate oxygens of the mRNA substrate serves two purposes. First, observation of a “thio-effect” (calculated as the fold loss in kobs for the phosphorothioate-substituted substrate compared to the natural substrate) indicates that the reaction rate is at least partially limited by chemistry37. Second, the magnitude of the thio-effect provides information regarding the role of the non-bridging oxygen in the reaction mechanism. Importantly for this study, the change in thio-effect upon mutation to alanine can indicate an interaction between the amino acid side chain and the phosphate. Small thio-effects of at least 4- to 11- fold, consistent with effects seen in non-enzymatic reactions, are expected for reactions in which chemistry is the rate limiting step37. Larger effects can also indicate transition-state interactions between the enzyme and the non-bridging phosphate oxygen38–40. Our data indicate that all RelE mutant cleavage reactions are at least partially limited by the chemical step at pH 7.5. The thio-effect resulting from phosphorothioate substitution for the wild-type RelE was previously measured as 39- and 900-fold for the RP and SP stereoisomers, respectively13. Thio-effects for several active-site mutants of RelE using both of these substrates range from 2.3-fold to 2800-fold (Table 1). Only R81A retained a large thio-effects for both the RP (100-fold) and SP (320-fold) substitutions. Alanine mutations of K52 and K54 effectively eliminated the RP thio-effects (2.8-fold and 3-fold) while retaining the large SP thio-effect (390-fold & 2800-fold). The R61A mutation significantly impacted both the RP and SP thio-effects, reducing the RP and SP effects to 6.7-fold and 21-fold, respectively.
Table 1.
Effects of mRNA Phosphorothioate Substitution on RelE Cleavage Rate Constantsa
| O | S (RP) | S (SP) | |||||
|---|---|---|---|---|---|---|---|
| RelE | k(s−1) | k(s−1)b | Thio- effect (fold)c |
Apparent interaction energy (kcal/mol)e |
k(s−1)b | Thio- effect (fold)c |
Apparent interaction energy (kcal/mol)e |
|
|
|
|
|||||
| wild typed | 3.8 ± 0.2 ×102 | 9.7 ± 0.1 | 39 | 4 ± 2 ×10−1 | 900 | ||
| K52A | 1.4 ± 0.3 ×10−1 | 5.1 ± 0.4 ×10−2 | 2.8 | −1.5 | 3.7 ± 0.5 ×10−4 | 390 | −0.5 |
| K54A | 1.7 ± 0.2 ×10−1 | 5 ± 1 ×10−2 | 3 | −1.4 | 6.0 ± 0.2 ×10−5 | 2800 | −0.6 |
| R81A | 5.3 ± 0.6 ×10−3 | 5.02 ± 0.02 ×10−5 | 100 | −0.6 | 1.6 ± 0.5 ×10−5 | 320 | −0.6 |
| R61A | 3.1 ± 0.8 ×10−4 | 4.62 ± 0.07 ×10−5 | 6.7 | −1.0 | 1.5 ± 0.2 ×10−5 | 21 | −2.2 |
| Y87Ad | 2.1 ± 0.2 ×10−3 | 2.7 ± 0.6 ×10−4 | 8 | −0.9 | 4 ± 1 ×10−4 | 6 | −2.9 |
O, unsubstituted mRNA; S (RP) or (SP), sulfur substitution at the nonbridging oxygen between mRNA nucleotides 20 and 21.
Means ± SEM from at least two independent determinations of the rate constant at saturating enzyme concentrations.
Calculated as the change in cleavage rate constant for sulfur-substituted mRNA substrates relative to that of the unsubstituted substrate.
Wild-type and Y87A enzyme O and S reaction rate constants taken from Griffin, Davis and Strobel 2013.
Apparent interaction energy (kcal/mol), calculated as RT ln(WTthio-effect / Mutantthio-effect), negative values indicate a favorable interaction.
For wild-type and mutant RelE, similar thio-effects were observed at both pH 7.5 and pH 9.0 (Table S2). Large thio-effects for at least one of the stereoisomers (WT, K52A, K54A, R81A), indicates that chemistry is rate-limiting and that the inflection points in pH-rate curves observed for some mutants at high pH are not due to a change in rate-limiting step. Therefore the observed pKas can be interpreted as the effects of titratable groups on chemistry.
DISCUSSION
The highly conserved residues surrounding the mRNA scissile phosphate, revealed by the RelE co-crystal structure6, are atypical for acid-base catalysis and suggest the potential for an unusual reaction mechanism. Cleavage of phosphodiester bonds is often accelerated by four catalytic strategies: alignment of the nucleophile and leaving group, deprotonation of the nucleophile, stabilization of the developing negative charge on the phosphate in the transition state, and protonation of the leaving group. In the classic acid-base reaction mechanism, the general base deprotonates the nucleophile and the general acid protonates the leaving group9,36,38. Because all of the RelE active-site residues have native pKa values greater than 10, for RelE to function by acid-base catalysis a significant pKa shift is required for at least one residue. Our pH-rate dependence and phosphorothioate substrate data provide insight about how RelE performs this common enzymatic reaction within an uncommon active site, exclusively in the context of the ribosome.
A possible reaction pathway consistent with our data for mRNA cleavage via acid-base catalysis by RelE is described in figure 5. The rationale of specific residues assignments to catalytic roles is discussed in detail below. The 2′-OH of A20 is deprotonated by the general base, K54, whose pKa is reduced by the charged K52, R81, and R61 residues. A pentavalent intermediate is formed by inline nucleophilic attack on the phosphorus by the activated 2′-OH. Negative charge buildup on the non-bridging oxygen in the transition state is stabilized by a strong interaction with R61 and the positively charged active site, including K52 and K54. R81 acts as the general acid and protonates the 5′-leaving group. The pKa of either the general base or the general acid is lowered as much as 4 to 6 units by the positively charged active site. All of our data are consistent with a pKa shift, but we are unable to definitively assign whether the acid or the base is affected. These effects complement the distortion of the mRNA substrate from its normal path through the ribosome, which orients the nucleophile and leaving group for cleavage6.
Figure 5.
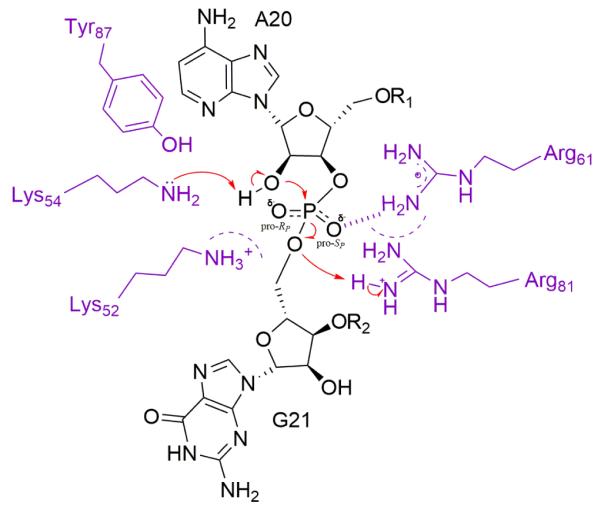
Possible reaction pathway for cleavage of mRNA (black) by RelE (purple). The 2′-OH of A20 is deprotonated by K54, its pKa reduced by the charged K52, R81, and R61 residues (whose influence is depicted by dashed semi-circles). Negative charge buildup on the non-bridging oxygen in the transition state is stabilized by a strong interaction with R61 (hashed line) and the positively charged active site including K52, K54, and R61. K52 and K54 help to stabilize the RP oxygen negative charge and interact minimally with the SP oxygen. R81 acts as the general acid and protonates the 5′-leaving group.
Charge Stabilization in the Transition State
As nucleophilic attack by the 2′-OH proceeds, the negative charge on the scissile phosphate increases. Our data support a role for the positively charged residues of the active site, and R61 especially, in stabilizing the transition state by neutralizing this buildup of negative charge. Furthermore, R61A’s impact on the rate constant for cleavage (106-fold) exceeds any other mutant, indicating a paramount role for charge stabilization in the RelE cleavage mechanism. Stabilization of negative charge in the transition state plays a large role in the proposed mechanism of RNase T1, Sa, and Sa2 among others28,39,41,42.
One way to investigate the specific enzymatic interaction with the transition state is to study the rate effects of phosphorothioate substitution on the cleavage reaction38,40,43. Small thio-effects of 4- to 11- fold are observed for phosphodiesterase cleavage in solution37, while enzymatic effects can range from 2-fold (RNase T2)44 to greater than 80,000-fold (RNase T1)39. Thio-effects greater than 10-fold are often attributed to either transition state interactions with the enzyme or protonation of a non-bridging oxygen during catalysis38–40. The observed 39-fold and 900-fold RP and SP thio-effects of wild-type RelE are consistent with significant stabilization of the negatively charged transition state by RelE. Protonation of a non-bridging oxygen, as has been proposed for RNase T1, would likely result in much larger thio-effects for both the RP and SP stereoisomers38,39,45
We measured the effect of phosphorothioate-substituted mRNA on cleavage rate constants for the K52A, K54A, R81A, and R61A mutants to investigate the degree to which each residue interacts with the mRNA non-bridging oxygen during the transition state. If a favorable interaction between a RelE residue and the phosphate contributes to the wild-type thio-effect, then the effect will be reduced when the residue is mutated to alanine. Alternatively, a reduced thio-effect could be derived from a change in mechanism upon phosphorothioate substitution; however, the retention of thio-effects similar to wild type for some of the mutants indicates the mechanism is unchanged.
R61A is the only mutant we tested that exhibits thio-effects much smaller than the wild type in both RP and SP cleavage rates, supporting a possible role for R61 as the residue primarily responsible for charge stabilization in the transition state. The SP thio-effect drops from 900-fold to 21-fold between wild-type and R61A RelE. This smaller adverse rate effect indicates a strong positive interaction between R61 and the SP oxygen. The RP thio-effect of R61A (7-fold) is also lower than the 39-fold effect of wild type, also indicating a favorable interaction. The absolute values of both thio-effects for R61A are similar to the range of 4- to 11-fold observed in the uncatalyzed reaction suggesting that the chemical step remains rate limiting37.
The R61 residue has previously been proposed as a key component in transition state stabilization for RelE in structural6 and kinetic studies13. The appropriate positioning of R61 for transition state stabilization was noted in the structure of pre-cleavage RelE6. Furthermore, R61 from RelE roughly aligns with R67 and R71 when superimposed on the structure of RNase Sa2. These residues are thought to be jointly responsible for charge stabilization28. Kinetically, the R61A rate effect on mRNA cleavage measured at pH 7.5 (106-fold) was larger than any other mutant, indicating a major role in catalysis. In this model, we expect the positively charged R61 guanidinium group of wild-type RelE to neutralize a partial negative charge that builds on the non-bridging oxygen during the transition state46.
The Y87A mutant thio-effects are also substantially reduced (8-fold and 6-fold for RP and SP, respectively13. However, the simultaneous loss of the phenyl ring and its proposed stacking interaction with A20 is likely to be responsible for the reduced thio-effect, either by decreasing the rate constant for the mRNA conformational change or altering the RelE active-site conformation sufficiently to disrupt the wild-type R61 charge interaction.
Large pKa Shift for the General Base or General Acid
The rate constant for wild-type RelE cleavage is independent of pH from 5.5 to 9.0 (Figure 3A, black), data that appear to be inconsistent with two previous proposals of acid-base-catalyzed cleavage6,13. However, a wide span of apparent pH-rate independence is possible in acid-base mechanisms where the pKa of the acid and the base are widely separated36. Because the native pKas of the conserved RelE active-site residues range from 10.1 (tyrosine) to 12.5 (arginine), a wide pKa separation requires the downshift of at least one residue. To evaluate whether a general acid-base mechanism could lead to the observed pH-rate dependence of mRNA cleavage in our experiments, we simulated pH-rate curves for a downshifted general-base pKa (model 1, Figure 4A) and a downshifted general-acid pKa (model 2, Figure 4E). Simulated pH-rate curves are flat across the testable range in either case. While our data cannot definitively distinguish between these two models we find precedence in the literature that leads us to prefer model 1, a shift in the general-base pKa, for reasons discussed in detail below.
Figure 4.

Simulated pH-rate curves and acid-base active fractions. Simulated pH-rate curves (green) and the fraction of general-acid (red) and general-base (blue) in the catalytically capable functional states for a simulated enzyme. Model 1 (A-D) assumes a downshifted general-base pKa and native general-acid pKa. Model 2 (E-H) assumes a downshifted general-acid pKa and a native general-base pKa. Curves simulated according to equations 4,5,7 and assume no influence on rate beyond acid and base protonation states.
These simulations describe the shape, but not the absolute values, of the pH-rate curve and are not applicable to reactions that do not proceed by general acid-base catalysis. However, the simulated pH-rate dependence of acid-base catalytic residue deletions (Figure 4B-D&F-H) provide useful predictions for RelE active-site-mutant pH curves (Figure 3).
What in the microenvironment of the RelE active site would lead to a pKa shift of this magnitude? Enzymes shift the pKa of critical residues primarily by charge-charge interactions, charge-dipole interactions (such as hydrogen bonding) and desolvation effects47. Charge-charge interactions strive to balance the electrostatic repulsion between groups of like charge inside the enzyme active site. Protonated groups will tend to decrease the pKas of adjacent resides until one is no longer protonated. As a result, the net charge of the active site moves closer to zero. The collection of positively charged active-site residues in RelE creates unfavorable charge-charge interactions, which are reduced by deprotonation of the general base. pH-rate data for each mutant was indeed supportive of roles in affecting a reduction in general-base pKa(see below). This RelE interaction is similar to the process used by acetoacetate decarboxylase to reduce the pKa of its catalytic lysine, K116. In this extensively studied enzyme, charge-charge interactions with the positively charged active site, and the adjacent K115 in particular, reduce the pKa of K116 by 4.4 units47–50.
Leaving Group Protonation
Protonation of the phosphodiester 5′-oxygen by a general acid can contribute significantly to the reaction mechanism. The pH-rate data, with structural and mutational data for RelE and similar RNases, support a role for R81 as the general acid responsible for leaving group protonation. The crystal structure of ribosome bound RelE in the pre-cleavage state shows that R81 and R61 are both positioned to potentially protonate the mRNA 5′-leaving group6. Furthermore, R81 aligns with the general-acid of RNase T1 (H92) when the two structures are superimposed13. The large rate effect (104-fold) of the R81A mutant indicates a role of this residue in catalysis and is consistent with rate effects observed in the mutation of the general acid residue of RNase A51. While both RNase Sa2 and T1 employ histidine as the general acid, there is precedent for general-acid activity mediated by arginine in X-family DNA polymerases and fumarate reductase9,47,52,53.
The pH-rate dependence of R81A (Figure 3A, red) is log-linear with a slope of ~1, which is consistent with deletion of the general acid and titration of a general base with a high pKa (Figure 4G). We have ascribed the existence of a low general-base pKa to a downshift of the native value by the RelE active-site microenvironment. Disruption of this microenvironment can revert the general-base pKa toward its native value of 10.4 for K54. If the general-acid deletion also disrupts the active-site microenvironment, a log-linear mutant pH-rate curve with slope of 1 is anticipated. We observe this positive slope pH-rate curve for R81A, among other mutants, supportive of its role as the general acid and a key contributor to the RelE microenvironment responsible for the large pKa shift.
An additive thio-effect, log-linear pH-rate relationship, and a major rate defect is not unique to R81A among the mutants we tested. K52A, for example, also retains an SP thio-effect, shows a log-linear rate dependence on pH and a similar (103-fold) rate defect at pH 7.5. But, unlike R81, neither K52 nor R61 align with the general-acids of RNase Sa2 and T1 in active-site structural alignments6. Our data, then, taken in context of known structures, argue for R81 to play the major role in leaving group protonation as the general acid.
Nucleophile Deprotonation
Ribonucleases typically enhance cleavage rates by abstracting a proton from the ribose 2′-OH, activating the oxygen nucleophile for attack on the adjacent 3′-phosphate. The pH-rate dependence data are consistent with K54 serving as the general base to activate the 2′-OH nucleophile. In RNases Sa2 and T1, proton abstraction is facilitated by glutamic acid (E56 and E58, respectively)26,28. Among RelE residues, K52 most closely corresponds to the general-base of Sa2 and T1 in an alignment of active sites6,13,23,28. In previous studies, both Y87 and K52 have been proposed as the sole RelE general-base6,13. The difficulty in assignment of the general base is due in part to distance of K52 from the 2′-OH in the pre-cleavage crystal structure. Both K52 and K54 are slightly farther from the 2′-OH than expected for hydrogen bonding. But this distance may be a result of the 2′-O-methyl chemical modification or R81A mutation introduced to prevent mRNA cleavage and not their inability to function as catalytic residues. Our pH-rate dependence data indicates that K52 is not the sole general base and supports a role for both K52 and Y87 in effecting the major pKa shift expected for the actual general base or general acid.
Mutation to inactivate the general base results in a rate dependence determined exclusively by the active fraction of the general acid. If the pKa of that acid is high, then the resulting mutant has a flat pH-rate curve (Figure 4B). This simulated pH-rate curve is a strong match for the observed dependence of K54A alone among the tested mutants, supportive of K54’s role as the general base.
We also conclude that K52 is not the sole general base as the positively sloped pH-rate curve of K52A (Figure 3B, orange) is not consistent with general acid-exclusive catalysis, whether the general acid’s pKa is downshifted by the active site (Figure 4F) or at its native value (Figure 4B). One possible alternative that we do not prefer, but cannot exclude, is that K52 is one of several residues that can function as a general base. Functional compensation by these residues could then prevent us from ever observing a pH-rate dependence wholly dependent on the general acid (Figure 4F). In this model, the active site may rely on contributions from K52 and Y87 to deprotonate the 2′-OH. It is even possible that the localized positive charge of the amino acids reduces the ribose 2′-OH pKa sufficiently that the moiety is frequently deprotonated and proton abstraction by a specific RelE residue is not required. Examples of partial rate compensation for general base mutation has been previously noted in RNase T1, α-sarcin, and the type II toxin YoeB29,54–56.
If the pKa of the general base is downshifted by as much as 4 units, as discussed above, the pH-rate curves for K52A, Y87A, Y87F, and R61A are all consistent with these residues collectively lowering the apparent pKa of K54 from lysine’s usual 10.4 to less than 6. The apparent pKa of the reaction for each mutant is informative of the relative contribution each residue makes to lowering K54’s pKa (Figure 4D). For example, Y87F demonstrates an apparent pKa of 7.5 for the general base, indicating a modest reversion toward the native pKa. The general-base pKa of the K52A mutant is greater than 9 and outside the testable range of the cleavage assay. The total reversion of the general acid to near its native pKa suggests a major role for K52 in maintaining the charge environment necessary to lower K54’s pKa at least 4 units in wild-type enzyme. Each of the other active site mutants exhibits a partial reversion of the general acid pKa somewhere between the impact of Y87F and K52A, indicating a role for all the active site residues in maintaining the pKa downshift.
An alternative model that shifts the general-acid pKa
An alternative explanation for the pH-rate profiles, including the pH independence of the wild-type reaction, is described in model 2 (Figure 4E-H). The model is still consistent with R81 and K54 playing roles in general acid-base catalysis, but in this model the pKa of the general acid, R81, is reduced rather than the general base, K54. The positively sloped R81A pH-rate curve is consistent with a general-acid deletion (Figure 4G). The negatively sloped pH-rate curve anticipated from a general-base deletion (Figure 4F) is not observed because mutation of the general-base, K54, also reverses the pKa shift of the acid due to disruption of the active-site microenvironment. This could result in the pH dependence shown in figure 4B, which matches the observed data for K54A (Figure 3A, green). Model 2 is also consistent with the interpretation of pH-rate curves for K52A, Y87F, Y87A, R61A and R81A as indicative of partially restoring the native pKa of the downshifted general-acid. Finally, both simulated models of the pH-rate dependencies are wholly consistent with our interpretation of the phosphorothioate data indicating a major role for charge stabilization in the transition state by R61.
While the pH-rate data does not definitively confirm one model over the other, we prefer model 1, which invokes a reduced pKa for the general-base K54. There is greater literature precedence for large pKa shifts for lysine than for arginine47,50,57,58. Also, the magnitude of the pKa shift for lysine is smaller, approximately 4 units, than the 6 units required for arginine. pKa reductions of this magnitude have been reported previously in enzyme active sites47, e.g. a reduction of 6.6 units for cysteine in the cysteine protease ficin59 or 4.4 units for lysine in acetoacetate decarboxylase50. We are not aware of any large pKa shifts for an arginine residue. In fact, specific studies of arginine protonation in enzymes and membranes have indicated that it is resistant to large reductions in pKa57,60.
RelE’s ribosome dependence
The specific microenvironment required by RelE to sufficiently lower the pKa of K54 for efficient cleavage may be a feature of the toxin’s ribosome dependence. Other bacterial toxins that can perhaps cleave on the ribosome, such as MqsR and MazF, are enriched for both arginine and lysine in their active sites at the expense of the canonical acid-base residues of glutamate and histidine61,62. However, unlike RelE, these two toxins are not exclusively ribosome dependent62,63. Not only is the positive charge state of the RelE active site conducive to shifting the pKa of its general base, but it may also support the enzymatic charge stabilization in the transition state. The preponderance of positively charged RelE residues may be partially responsible for reinforcing R61’s role in reducing this effect. Bacterial toxins are highly effective inhibitors of cellular translation that must be carefully regulated to ensure cell survival. The unusual active site of RelE and similar toxins appears to be an additional level of regulation employed by the cell, in addition to the anti-toxin system.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Kathryn D. Smith for helpful discussions and critical commentary on the manuscript and Dr. Meghan A. Griffin, Dr. Wenxiang Cao, and Emily Wong for experimental support. This work was supported by NIH grant GM054839 to SAS.
Abbreviations
- TA
Toxin-Antitoxin
- RNase
Ribonuclease
- RC
Ribosome Complex
- MMB
MES-MOPS-Bicine
- PAGE
Polyacrylamide gel electrophoresis
- SEM
Standard error of the mean
Footnotes
Complete table of cleavage rate constants for wild-type and all mutants at pH 5.5 through pH 9.0 and expanded phosphorothioate data at pH 7.5 and pH 9.0.
REFERENCES
- (1).Pedersen K, Zavialov AV, Pavlov MY, Elf J, Gerdes K, Ehrenberg M. The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell. 2003;112:131–140. doi: 10.1016/s0092-8674(02)01248-5. [DOI] [PubMed] [Google Scholar]
- (2).Goeders N, Drèze P-L, Van Melderen L. Relaxed Cleavage Specificity within the RelE Toxin Family. J. Bacteriol. 2013;195:2541–2549. doi: 10.1128/JB.02266-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Yamaguchi Y, Inouye M. mRNA Interferases, Sequence-Specific Endoribonucleases from the Toxin–Antitoxin Systems. Prog. Mol. Biol. Transl. Sci. 2009;85:467–500. doi: 10.1016/S0079-6603(08)00812-X. [DOI] [PubMed] [Google Scholar]
- (4).Takagi H, Kakuta Y, Okada T, Yao M, Tanaka I, Kimura M. Crystal structure of archaeal toxin-antitoxin RelE–RelB complex with implications for toxin activity and antitoxin effects. Nat. Struct. Mol. Biol. 2005;12:327–331. doi: 10.1038/nsmb911. [DOI] [PubMed] [Google Scholar]
- (5).Li G-Y, Zhang Y, Inouye M, Ikura M. Inhibitory mechanism of Escherichia coli RelE-RelB toxin-antitoxin module involves a helix displacement near an mRNA interferase active site. J. Biol. Chem. 2009;284:14628–14636. doi: 10.1074/jbc.M809656200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Neubauer C, Gao Y-G, Andersen KR, Dunham CM, Kelley AC, Hentschel J, Gerdes K, Ramakrishnan V, Brodersen DE. The Structural Basis for mRNA Recognition and Cleavage by the Ribosome-Dependent Endonuclease RelE. Cell. 2009;139:1084–1095. doi: 10.1016/j.cell.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Buckle AM, Fersht AR. Subsite Binding in an RNase: Structure of a Barnase-Tetranucleotide Complex at 1.76-. ANG. Resolution. Biochemistry (Mosc.) 1994;33:1644–1653. doi: 10.1021/bi00173a005. [DOI] [PubMed] [Google Scholar]
- (8).Yoshida H. The ribonuclease T1 family. Methods Enzymol. 2001;341:28. doi: 10.1016/s0076-6879(01)41143-8. [DOI] [PubMed] [Google Scholar]
- (9).Yang W. Nucleases: diversity of structure, function and mechanism. Q. Rev. Biophys. 2011;44:1–93. doi: 10.1017/S0033583510000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Pedersen K, Christensen SK, Gerdes K. Rapid induction and reversal of a bacteriostatic condition by controlled expression of toxins and antitoxins. Mol. Microbiol. 2002;45:501–510. doi: 10.1046/j.1365-2958.2002.03027.x. [DOI] [PubMed] [Google Scholar]
- (11).Gerdes K. Toxin-Antitoxin Modules May Regulate Synthesis of Macromolecules during Nutritional Stress. J. Bacteriol. 2000;182:561–572. doi: 10.1128/jb.182.3.561-572.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Raines RT. Ribonuclease a. Chem. Rev. 1998;98:1045–1066. doi: 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]
- (13).Griffin MA, Davis JH, Strobel SA. Bacterial Toxin RelE: A Highly Efficient Ribonuclease with Exquisite Substrate Specificity Using Atypical Catalytic Residues. Biochemistry (Mosc.) 2013;52:8633–8642. doi: 10.1021/bi401325c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Yamaguchi Y, Park J-H, Inouye M. Toxin-Antitoxin Systems in Bacteria and Archaea. Annu. Rev. Genet. 2011;45:61–79. doi: 10.1146/annurev-genet-110410-132412. [DOI] [PubMed] [Google Scholar]
- (15).Wang X, Wood TK. Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 2011;77:5577–5583. doi: 10.1128/AEM.05068-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Gerdes K, Maisonneuve E. Bacterial Persistence and Toxin-Antitoxin Loci. Annu. Rev. Microbiol. 2012;66:103–123. doi: 10.1146/annurev-micro-092611-150159. [DOI] [PubMed] [Google Scholar]
- (17).Karimi S, Ghafourian S, Kalani MT, Jalilian FA, Hemati S, Sadeghifard N. Association Between Toxin-Antitoxin Systems and Biofilm Formation. Jundishapur J. Microbiol. 2015;8(1):e14540. doi: 10.5812/jjm.14540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Schuster CF, Bertram R. Toxin–antitoxin systems are ubiquitous and versatile modulators of prokaryotic cell fate. FEMS Microbiol. Lett. 2013;340:73–85. doi: 10.1111/1574-6968.12074. [DOI] [PubMed] [Google Scholar]
- (19).Pandey DP, Gerdes K. Toxin–antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005;33:966–976. doi: 10.1093/nar/gki201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Christensen SK, Mikkelsen M, Pedersen K, Gerdes K. RelE, a global inhibitor of translation, is activated during nutritional stress. Proc. Natl. Acad. Sci. 2001;98:14328–14333. doi: 10.1073/pnas.251327898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Gotfredsen M, Gerdes K. The Escherichia coli relBE genes belong to a new toxin–antitoxin gene family. Mol. Microbiol. 1998;29:1065–1076. doi: 10.1046/j.1365-2958.1998.00993.x. [DOI] [PubMed] [Google Scholar]
- (22).Cherny I, Overgaard M, Borch J, Bram Y, Gerdes K, Gazit E. Structural and thermodynamic characterization of the Escherichia coli RelBE toxin-antitoxin system: indication for a functional role of differential stability. Biochemistry (Mosc.) 2007;46:12152–12163. doi: 10.1021/bi701037e. [DOI] [PubMed] [Google Scholar]
- (23).Zegers I, Haikal AF, Palmer R, Wyns L. Crystal structure of RNase T1 with 3′-guanylic acid and guanosine. J. Biol. Chem. 1994;269:127–133. [PubMed] [Google Scholar]
- (24).Overgaard M, Borch J, Gerdes K. RelB and RelE of Escherichia coli form a tight complex that represses transcription via the ribbon–helix–helix motif in RelB. J. Mol. Biol. 2009;394:183–196. doi: 10.1016/j.jmb.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Guglielmini J, Van Melderen L. Bacterial toxin-antitoxin systems: Translation inhibitors everywhere. Mob. Genet. Elem. 2011;1:283–306. doi: 10.4161/mge.18477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Heinemann U, Saenger W. Crystallographic Study of Mechanism of Ribonuclease Ti-Catalysed Specific RNA Hydrolysis. J. Biomol. Struct. Dyn. 1983;1:523–538. doi: 10.1080/07391102.1983.10507459. [DOI] [PubMed] [Google Scholar]
- (27).Mossakowska DE, Nyberg K, Fersht AR. Kinetic characterization of the recombinant ribonuclease from Bacillus amyloliquefaciens (barnase) and investigation of key residues in catalysis by site-directed mutagenesis. Biochemistry (Mosc.) 1989;28:3843–3850. doi: 10.1021/bi00435a033. [DOI] [PubMed] [Google Scholar]
- (28).Bauerová-Hlinková V, Dvorský R, Perečko D, Považanec F, Ševčík J. Structure of RNase Sa2 complexes with mononucleotides – new aspects of catalytic reaction and substrate recognition. FEBS J. 2009;276:4156–4168. doi: 10.1111/j.1742-4658.2009.07125.x. [DOI] [PubMed] [Google Scholar]
- (29).Kamada K, Hanaoka F. Conformational Change in the Catalytic Site of the Ribonuclease YoeB Toxin by YefM Antitoxin. Mol. Cell. 2005;19:497–509. doi: 10.1016/j.molcel.2005.07.004. [DOI] [PubMed] [Google Scholar]
- (30).Hurley JM, Woychik NA. Bacterial Toxin HigB Associates with Ribosomes and Mediates Translation-dependent mRNA Cleavage at A-rich Sites. J. Biol. Chem. 2009;284:18605–18613. doi: 10.1074/jbc.M109.008763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Meiering EM, Serrano L, Fersht AR. Effect of active site residues in barnase on activity and stability. J. Mol. Biol. 1992;225:585–589. doi: 10.1016/0022-2836(92)90387-y. [DOI] [PubMed] [Google Scholar]
- (32).Yakovlev GI, Mitkevich VA, Shaw KL, Trevino S, Newsom S, Pace CN, Makarov AA. Contribution of active site residues to the activity and thermal stability of ribonuclease Sa. Protein Sci. 2003;12:2367–2373. doi: 10.1110/ps.03176803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Shapiro R, Fox EA, Riordan JF. Role of lysines in human angiogenin: chemical modification and site-directed mutagenesis. Biochemistry (Mosc.) 1989;28:1726–1732. doi: 10.1021/bi00430a045. [DOI] [PubMed] [Google Scholar]
- (34).Rodnina MV, Fricke R, Kuhn L, Wintermeyer W. Codon-dependent conformational change of elongation factor Tu preceding GTP hydrolysis on the ribosome. EMBO J. 1995;14:2613. doi: 10.1002/j.1460-2075.1995.tb07259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ryder SP, Strobel SA. Nucleotide analog interference mapping. Methods. 1999;18:38–50. doi: 10.1006/meth.1999.0755. [DOI] [PubMed] [Google Scholar]
- (36).Bevilacqua PC. Mechanistic Considerations for General Acid–Base Catalysis by RNA: Revisiting the Mechanism of the Hairpin Ribozyme†. Biochemistry (Mosc.) 2003;42:2259–2265. doi: 10.1021/bi027273m. [DOI] [PubMed] [Google Scholar]
- (37).Herschlag D, Piccirilli JA, Cech TR. Ribozyme-catalyzed and nonenzymic reactions of phosphate diesters: rate effects upon substitution of sulfur for a nonbridging phosphoryl oxygen atom. Biochemistry (Mosc.) 1991;30:4844–4854. doi: 10.1021/bi00234a003. [DOI] [PubMed] [Google Scholar]
- (38).Herschlag D. Ribonuclease Revisited: Catalysis via the Classical General Acid-Base Mechanism or a Triester-like Mechanism? J. Am. Chem. Soc. 1994;116:11631–11635. [Google Scholar]
- (39).Loverix S, Winqvist A, Strömberg R, Steyaert J. Mechanism of RNase T1: concerted triester-like phosphoryl transfer via a catalytic three-centered hydrogen bond. Chem. Biol. 2000;7:651–658. doi: 10.1016/s1074-5521(00)00005-3. [DOI] [PubMed] [Google Scholar]
- (40).Stivers JT, Nagarajan R. Probing Enzyme Phosphoester Interactions by Combining Mutagenesis and Chemical Modification of Phosphate Ester Oxygens. Chem. Rev. 2006;106:3443–3467. doi: 10.1021/cr050317n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Sevcik J, Dodson EJ, Dodson GG. Determination and restrained least-squares refinement of the structures of ribonuclease Sa and its complex with 3′-guanylic acid at 1.8 Å resolution. Acta Crystallogr. Sect. B. 1991;47:240–253. [PubMed] [Google Scholar]
- (42).Steyaert J, Wyns L. Functional Interactions among the His40, Glu58 and His92 Catalysts of Ribonuclease T1 as Studied by Double and Triple Mutants. J. Mol. Biol. 1993;229:770–781. doi: 10.1006/jmbi.1993.1078. [DOI] [PubMed] [Google Scholar]
- (43).Milligan JF, Uhlenbeck OC. Determination of RNA-protein contacts using thiophosphate substitutions. Biochemistry (Mosc.) 1989;28:2849–2855. doi: 10.1021/bi00433a016. [DOI] [PubMed] [Google Scholar]
- (44).Burgers PMJ, Eckstein F. Diastereomers of 5′-O-adenosyl 3′-O-uridyl phosphorothioate: chemical synthesis and enzymic properties. Biochemistry (Mosc.) 1979;18:592–596. doi: 10.1021/bi00571a007. [DOI] [PubMed] [Google Scholar]
- (45).Breslow R, Xu R. Recognition and catalysis in nucleic acid chemistry. Proc. Natl. Acad. Sci. 1993;90:1201–1207. doi: 10.1073/pnas.90.4.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Gu H, Zhang S, Wong K-Y, Radak BK, Dissanayake T, Kellerman DL, Dai Q, Miyagi M, Anderson VE, York DM, Piccirilli JA, Harris ME. Experimental and computational analysis of the transition state for ribonuclease A-catalyzed RNA 2′-O-transphosphorylation. Proc. Natl. Acad. Sci. U. S. A. 2013;110:13002–13007. doi: 10.1073/pnas.1215086110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Harris TK, Turner GJ. Structural Basis of Perturbed pKa Values of Catalytic Groups in Enzyme Active Sites. IUBMB Life. 2002;53:85–98. doi: 10.1080/15216540211468. [DOI] [PubMed] [Google Scholar]
- (48).Kokesh FC, Westheimer FH. Reporter group at the active site of acetoacetate decarboxylase. II. Ionization constant of the amino group. J. Am. Chem. Soc. 1971;93:7270–7274. doi: 10.1021/ja00755a025. [DOI] [PubMed] [Google Scholar]
- (49).Frey PA, Kokesh FC, Westheimer FH. Reporter group at the active site of acetoacetate decarboxylase. I. Ionization constant of the nitrophenol. J. Am. Chem. Soc. 1971;93:7266–7269. doi: 10.1021/ja00755a024. [DOI] [PubMed] [Google Scholar]
- (50).Highbarger LA, Gerlt JA, Kenyon GL. Mechanism of the Reaction Catalyzed by Acetoacetate Decarboxylase. Importance of Lysine 116 in Determining the pKa of Active-Site Lysine 115. Biochemistry (Mosc.) 1996;35:41–46. doi: 10.1021/bi9518306. [DOI] [PubMed] [Google Scholar]
- (51).Thompson JE, Raines RT. Value of General Acid–Base Catalysis to Ribonuclease A. J. Am. Chem. Soc. 1994;116:5467–5468. doi: 10.1021/ja00091a060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Mowat CG, Moysey R, Miles CS, Leys D, Doherty MK, Taylor P, Walkinshaw MD, Reid GA, Chapman SK. Kinetic and Crystallographic Analysis of the Key Active Site Acid/Base Arginine in a Soluble Fumarate Reductase†. Biochemistry (Mosc.) 2001;40:12292–12298. doi: 10.1021/bi011360h. [DOI] [PubMed] [Google Scholar]
- (53).Doherty MK, Pealing SL, Miles CS, Moysey R, Taylor P, Walkinshaw MD, Reid GA, Chapman SK. Identification of the Active Site Acid/Base Catalyst in a Bacterial Fumarate Reductase: A Kinetic and Crystallographic Study†. Biochemistry (Mosc.) 2000;39:10695–10701. doi: 10.1021/bi000871l. [DOI] [PubMed] [Google Scholar]
- (54).Grunert H-P, ZOUNI A, BEINEKE M, QUAAS R, GEORGALIS Y, SAENGER W, HAHN U. Studies on RNase T1 mutants affecting enzyme catalysis. Eur. J. Biochem. 1991;197:203–207. doi: 10.1111/j.1432-1033.1991.tb15900.x. [DOI] [PubMed] [Google Scholar]
- (55).Nishikawa S, Morioka H, Kim HJ, Fuchimura K, Tanaka T, Uesugi S, Hakoshima T, Tomita K, Ohtsuka E, Ikehara M. Two histidine residues are essential for ribonuclease T1 activity as is the case for ribonuclease A. Biochemistry (Mosc.) 1987;26:8620–8624. doi: 10.1021/bi00400a019. [DOI] [PubMed] [Google Scholar]
- (56).Sylvester ID, Roberts LM, Lord JM. Characterization of prokaryotic recombinant Aspergillus ribotoxin α-sarcin. Biochim. Biophys. Acta BBA - Mol. Cell Res. 1997;1358:53–60. doi: 10.1016/s0167-4889(97)00048-7. [DOI] [PubMed] [Google Scholar]
- (57).Harms MJ, Schlessman JL, Sue GR, García-Moreno E. B. Arginine residues at internal positions in a protein are always charged. Proc. Natl. Acad. Sci. U. S. A. 2011;108:18954–18959. doi: 10.1073/pnas.1104808108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Isom DG, Castañeda CA, Cannon BR, García-Moreno E. B. Large shifts in pK(a) values of lysine residues buried inside a protein. Proc. Natl. Acad. Sci. U. S. A. 2011;108:5260–5265. doi: 10.1073/pnas.1010750108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Pinitglang S, Watts AB, Patel M, Reid JD, Noble MA, Gul S, Bokth A, Naeem A, Patel H, Thomas EW, Sreedharan SK, Verma C, Brocklehurst K. A Classical Enzyme Active Center Motif Lacks Catalytic Competence until Modulated Electrostatically. Biochemistry (Mosc.) 1997;36:9968–9982. doi: 10.1021/bi9705974. [DOI] [PubMed] [Google Scholar]
- (60).Li L, Vorobyov I, MacKerell AD, Allen TW. Is arginine charged in a membrane? Biophys. J. 2008;94:L11–L13. doi: 10.1529/biophysj.107.121566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Brown BL, Grigoriu S, Kim Y, Arruda JM, Davenport A, Wood TK, Peti W, Page R. Three Dimensional Structure of the MqsR:MqsA Complex: A Novel TA Pair Comprised of a Toxin Homologous to RelE and an Antitoxin with Unique Properties. PLoS Pathog. 2009;5:e1000706. doi: 10.1371/journal.ppat.1000706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Zhang Y, Zhang J, Hara H, Kato I, Inouye M. Insights into the mRNA Cleavage Mechanism by MazF, an mRNA Interferase. J. Biol. Chem. 2005;280:3143–3150. doi: 10.1074/jbc.M411811200. [DOI] [PubMed] [Google Scholar]
- (63).Yamaguchi Y, Park J-H, Inouye M. MqsR, a Crucial Regulator for Quorum Sensing and Biofilm Formation, Is a GCU-specific mRNA Interferase in Escherichia coli. J. Biol. Chem. 2009;284:28746–28753. doi: 10.1074/jbc.M109.032904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.