

Abstract
Epithelial sheets fold into complex topographies that contribute to their function in vivo. Cells can sense and respond to substrate topography in their immediate vicinity by modulating their interfacial mechanics, but the extent to which these mechanical properties contribute to their ability to sense substrate topography across length scales larger than a single cell has not been explored in detail. To study the relationship between the interfacial mechanics of single cells and their collective behavior as tissues, we grew cell-sheets on substrates engraved with surface features spanning macroscopic length-scales. We found that many epithelial cell-types sense and respond to substrate topography, even when it is locally nearly planar. Cells clear or detach from regions of local negative curvature, but not from regions with positive or no curvature. We investigated this phenomenon using a finite element model where substrate topography is coupled to epithelial response through a balance of tissue contractility and adhesive forces. The model correctly predicts the focal sites of cell-clearing and epithelial detachment. Furthermore, the model predicts that local tissue response to substrate curvature is a function of the surrounding topography of the substrate across long distances. Analysis of cell-cell and cell-substrate contact angle suggests a relationship between these single-cell interfacial properties, epithelial interfacial properties, and collective epithelial response to substrate topography. Finally, we show that contact angles change upon activation of oncogenes or inhibition of cell-contractility, and that these changes correlate with collective epithelial response. Our results demonstrate that in mechanically integrated epithelial sheets, cell contractility can be transmitted through multiple cells and focused by substrate topography to affect a behavioral response at distant sites.
Introduction
Sheets of epithelial cells possess topographies – the folding of a tissue perpendicular to the sheet – that are neither wholly planar nor tubular. Tissues like intestinal villi, gastric pits, the choroid plexus, dermal-epidermal interface, or the corneal limbus exist as convoluted, corrugated, or ruffled sheets. In these topographically rich microenvironments, each individual cell experiences a nearly planar substrate even while the tissue can contain considerable heterogeneity in substrate curvature over a larger length scale.1 How cells sense and respond to the topography of their substrate may inform why epithelial tissues feature cells whose behavior is a function of their position within complex microenvironments.2-4 To date, numerous studies have explored the mechanisms through which single cells sense the local and microscale topography of their substrates.5-7 Studies mimicking the structure of intestinal villi have also confirmed that substrate geometry can influence the behavior of cells and disease processes.8,9 However, less is known about how cells sense the topography of the surrounding epithelial sheet over much larger length scales.
Pioneering work relating tissue geometry to cell behavior demonstrated that the lateral 2D shape of a 3D tissue can influence morphogenesis by converting global tissue contractility to local differences in tissue tension.10,11 Other studies have investigated the impact of microtopography (or micro- and nanogrooves) of culture substrates on cell behaviors, finding that microgrooves affect cell morphology, alignment, adhesion, and motility.5-7 In these cases, substrate topography varied dramatically over the diameter of a single cell, allowing each cell to sense and individually respond to the topography of its immediate microenvironment. Few studies, however, have explored topographic features that are significantly larger than individual cells. Under such conditions, the immediate topographic microenvironment of a cell would appear planar. Therefore, cells would have to coordinate their individual behaviors if they were to sense and respond to their geometry collectively. Given a role for cell contractility in sensing both organ size and local tissue geometry, we reasoned that a coordinated epithelial response to substrate topography across tens to hundreds of microns might also occur through a mechanisms involving cell contractility.
To draw a link between the contractile properties of single cells and their ability to sense substrate topographical cues at the multicellular level, we developed a technique to prepare substrates that are composed of regularly arrayed, smoothly contoured, and large-scale topographical features. By manipulating the width and spacing of these channels we reveal tissue-level responses to changes in substrate topography that correlate with single-cell measurements such as cell-cell and cell-substrate contact angles. Contact angles reflect the balance of tensions acting at the cell-cell, cell-medium, and cell substrate interfaces, and these tensions converge at sites of cell-cell or cell-substrate adhesion.12,13 We further investigated whether the tensions accumulating at cell-cell, cell-substrate and cell-medium interfaces are coupled in epithelial sheets across long distances, thereby allowing them to sense and respond collectively to substrate topography that spans many cell diameters.
Experimental methods
Cell lines and cell culture
MDCK cells were kindly provided by Professor Keith Mostov. HUVEC cells were purchased from Lonza, and Caco-2 cells were purchased from ATCC. All cell lines were cultured according to standard practices listed with ATCC or Lonza.
Antibiodies, and inhibitors
The following antibodies were used for immunofluorescence: E-cadherin from BD Biosciences, YAP (63.7) from Santa Cruz Biotechnology, and Phospho-Myosin Light Chain 2 (Thr18/Ser19) and cleaved caspase-3 from Cell Signaling Technology). Secondary staining was performed using Alexa Fluor 488- and 568-conjugated goat anti-mouse, anti-rat, and anti-rabbit antibodies (Invitrogen). F-actin was stained with Alexa Fluor 488- or 568-conjugated phalloidin (Invitrogen). Y-27632, blebbistatin, ML-7, and PF-573228 were obtained from Tocris Bioscience. Nocodazole was purchased from Sigma-Aldrich, PD-0325901 was purchased from R&D Systems, and PIK-90 was purchased from EMD Millipore.
Preparation of structured PDMS substrates
Freshly cleaned Si wafers were coated with SU-8 2005 and cured according to manufacturer protocol (MicroChem Corp) to provide an adhesive surface. Positive photoresist AZ9620 (Microchemicals GmbH) was then deposited in two coatings to a thickness of 50 um. After exposure at 3200 mJ/cm2 at 365 nm, wafers were developed in AZ 400K (Microchemicals GmbH), and baked at 115 °C for 45 min to melt square patterns into desired curved geometry.
Preparation of structured PDMS for cell culture
PDMS (Dow Corning SYLGARD 184, 10:1 base:cure) was mixed and bubbles were removed by centrifugation and vacuum. Patterned wafers were then coated in uncured PDMS (~12 mL) and cured at 70 °C overnight. PDMS was then cut and stored or used directly for culture experiments. Immediately preceding use in cell culture, PDMS was cleaned with tape and exposed for 35 s to an air plasma (Plasma Etch, PE-50), then soaked for 1 h in EtOH, washed with PBS, and then soaked in 0.1 mg/mL Collagen I (PureCol) in PBS for 3 h at RT.
Cell culture on structured substrates
Cells were freshly lifted, counted, and seeded onto substrates at 7.5 × 105 cell/mL. In experiments using inhibitors, cells were incubated with inhibitors at 37 °C for 10 min immediately prior to seeding. Inhibitors were also included in culture media for the duration of the experiment. After the desired time period (typically 48 h), cells were fixed using 4% formaldehyde in PBS for 15 min at RT, then permeabilized and blocked using PFS (0.7 % fish skin gelatin, 0.025% saponin, 0.02% NaN3 in PBS) for 20 min at RT. Samples were stored in PFS at 4 °C.
Image acquisition
All confocal microscopy images were acquired using a spinning disk confocal microscope: a Zeiss Cell Observer Z1 equipped with a Yokagawa spinning disk, and Photometrics Evolve EM-CCD camera. Illumination was provided by LED lasers at 405, 488, 561, and 640 nm. The instrument was controlled using Zeiss Zen Software and images were analyzed using FIJI14 and Wolfram Mathematica.
Cell-Cell contact angle measurements
MDCK cells were centrifuged into small rectangular agarose microwells (20 μm × 40 μm × 15 μm) fabricated using the photolithography and micromolding methods described above. Wells were designed to accommodate two cells. After 4 h in the wells, the cell doublets were imaged at 40X magnification and analyzed in FIJI in order to determine the contact angle at the cell-cell interface. For these experiments we visually screened for microwells that contained cell doublets that were alive and contained 2 cells of comparable size.
Cell-ECM contact angle measurements
Glass coverslips were coated with a thin layer of PDMS, which was cured overnight at 70 °C. After curing, coverslips were soaked in 0.1 mg/mL Collagen I for 3 h at RT. The coverslips were then rinsed with PBS and incubated for 3 h with a solution of 4 nM QD605 (Invitrogen) to assist in visualizing the interface. Coverslips were then washed with media and used as substrates for the spreading of MDCK cells. Cells were seeded at low density (~103 cells/cm2) and, after 1 h incubation at 37 °C, were fixed in 4% paraformaldehyde (PFA). Cells were then treated with DAPI, Phalloidin-AF488, and WGA-AF555 to stain nuclei, actin, and membrane, respectively. For these experiments we visually screened for cells that appeared to be alive and that were not interacting with nearby cells. To measure the contact angle at the cell-substrate interface, we analyzed 63X confocal Z-stacks using a custom FIJI script (Scripts S5-6, ESI).
Results and Discussion
Cell-sheets on curved substrates detach and clear
To study the effects of substrate topography, we first developed a reproducible method to microfabricate locally smooth substrates with curved topologies over tens to hundreds of microns. Silicon wafers were coated with a thick positive photoresist, exposed to high-intensity UV light, and developed to form square-profiled structures. These structures were then heated above the glass-transition temperature of the photoresist to allow thermal relaxation.5 The resulting curved structures could then be cast in PDMS to yield suitable substrates for cell culture (Fig. 1B). Feature dimensions deviated by less than 4% across a 1 cm square, allowing for the consistent observation of tissue-scale behaviors (Fig. S1, ESI). Madine-Darby Canine Kidney (MDCK) cells were chosen as a model due to their ability to establish true apico-basal polarity, retain barrier function, and form growth arrested and lumenized 3D cysts in matrigel and collagen.15-18 Substrates were coated in collagen and cells were seeded such that they were confluent within 12 h. The resulting epithelial sheets were found to conform to the shape of the channels as a monolayer, with actin and E-cadherin (eCad) forming belts at the apical cell-cell and cell-medium interface (Fig. 1C). The local variation of substrate height across the length of an individual cell was at most 1 μm, 5% the monolayer height (Fig. S2, ESI). Within the first 12 h, actin can be observed to align with the channels, especially within the channels (Fig. S3, ESI). After 24-48 h, we observed the formation of discontinuities in the confluent cell sheet. Discontinuities initially appeared at the base of the curved regions of the structured substrates and expanded as time progressed (Fig. 1D,E). This phenomenon increased over time up to 96 h, at which time nearly all channel space was devoid of cells. Discontinuities were not observed on unstructured PDMS (Fig. S4, ESI), indicating that topographical features, and not the substrates themselves, were driving this phenomenon. Because this behavior was clearly observable by 36 h, future experiments were carried out for 48 h. Clearance from substrate channels was also observed in other non-malignant human epithelial, and endothelial cell types including human umbilical vein endothelial cells (HUVEC) and Caco-2 (Fig. 1F,G). However, MDCK cells transformed with a constitutively active mutant of the oncogene H-Ras did not clear from the structured regions of the substrates.
Figure 1.

Epithelial tissues clear from large-scale curvature. (A) Epithelial sheets having large-scale curvature can be nearly flat on the length scale of a single cell. (B) Cross-section of a PDMS substrate. (C) XY and XZ cross-sectional view of a confluent monolayer of MDCK on a curved PDMS substrate taken 12 h after seeding of cells. Average intensity of actin (green) and eCad (red) staining relative to cell nuclei (blue). (D) Time course showing cells clearing selectively from channels. Channel location indicated by gray schematic (right) (E) Quantitation of fluorescence from the indicated region showing lifting of tissues coincident with clearing. (F) 48 h fluorescence micrographs of MDCK, HUVEC, and Caco2, and Ras-transformed MDCK. (G) Quantitation of proportion adhered, lifted, and cleared tissue for each cell type. All scale bars: 100 μm.
Epithelial clearing is not driven by differences in proliferation or apoptosis
To investigate the mechanism of channel clearing, we first explored the roles of localized cell proliferation and cell death. A recent study suggested that epithelial bridges lose integrity in part due to mitosis.19 To measure localized changes in the rate of mitosis prior to cell clearing or detachment, MDCK were seeded onto the structured substrates in media containing 5-ethynyl-2′-deoxyuridine (EdU) and fixed at 12 h. Fluorescent staining of ethynyl groups using click chemistry revealed actively proliferating cells (Fig. 2A). Significantly fewer EdU-positive cells were observed in channels as compared to plateau regions (Fig. 2B). Although reduced relative to cells on plateaus, over 40% of cells in channels were proliferating. To determine if mitosis in channels contributed to clearing and detachment, we treated cultures with mitosis inhibitors aphidicolin or mimosine. Neither drug had an appreciable effect on channel clearance, excluding the possibility that clearing and detachment were caused by differential proliferation or mitosis (Fig. S5, ESI). We also stained for cleaved caspase 3 (Casp3) by immunofluorescence, but observed no significant difference in the proportion of Casp3-positive cells between channels and plateau regions. Although we observed slightly higher levels of activated Casp3 in nascent discontinuities (Fig. 2A, circled), we observed similarly high levels in areas that would not go on to form discontinuities. These experiments suggest that while apoptosis may contribute to selective clearance of channels, it is unlikely to be the sole or proximate cause.
Figure 2.

Clearing involves mechanical stress, and does not require differential proliferation or apoptosis. (A) Multichannel fluorescence imaging of EdU uptake and cleaved caspase-3 staining in channels and in nascent tissue discontinuities (circled). Scale bar 100 μm. (B) Percentage of EdU and Casp3 positive cells in channel and plateau regions. (C) Cell density compared between channel and plateau regions. (D) Per-cell quantification of nuclear YAP localization. Channel edges are masked to avoid double-counting cells (E-F) High-magnification images of lifted tissues at channels or ridges shows cells lifted from regions of negative curvature with anchoring cells remaining at the edges of the discontinuity. Note: dehydration by mounting medium causes reduction of tissue thickness. Schematic arrows indicate direction and locus of lifting. (G) Histogram of tear centroid positions relative to their local channel center (n = 56). Scale bars: 20 μm. All error bars are 95% confidence intervals.
Previous reports suggest that high cell density in intestinal villi and the zebrafish epidermis can trigger cell delamination.20 We therefore measured cell density in channel regions prior to cell detachment and clearly. We found that cell density was actually slightly reduced in channels relative to plateaus by 12 h (Fig. 2C). This observation suggests that cell clearing and detachment from channels may occur through a different mechanism than occurs in the epidermis and intestinal epithelium.
Epithelial sheets sense substrate curvature through a contractile mechanism
Epithelial sheets must balance the mechanical forces acting at cell-cell, cell-medium, and cell substrate interfaces so as to maintain their architecture and mechanical equilibrium. We hypothesized that curved substrates might put these forces in conflict, driving the accumulation of differential stresses on the epithelial sheets either inside or outside of the channels. Previous studies have implicated the Yorkie-homologue proteins YAP and Taz as important signaling relays for mechanosensation,21 and we reasoned that differential mechanical stresses would be reported by positional differences in YAP activation. We therefore stained for YAP-related mechanotransduction and calculated the ratio of nuclear to cytoplasmic YAP within each cell (Fig S6, ESI). To aid analysis, cells were computationally segmented. To control for potential errors introduced by this method, measurements were validated against a subset of manually segmented cells (Figure S7, ESI). The ratio of nuclear to cytoplasmic YAP levels were on average 56% higher within the channel regions of the substrates compared to plateaus (Fig 2D and Fig. S8, ESI). Statistical analysis confirms that there is a highly significant difference in the distribution of YAP activation between these two spatial locations (Kruskall-Wallis, p < 10−75). The differences in nuclear YAP staining persisted throughout the process of cell clearing and detachment. These findings are consistent with a model wherein cells experience unique mechanical microenvironments inside and outside the channels that are reflected in their signaling states.
A close inspection of cells in the process of lifting and clearing revealed some common features (Fig. 2E). First, cells tended to form discontinuities in the sheet by clearing from the center of channels and expanding outward to the edges of the channel. They also lifted from the substrate as the discontinuities in the tissue expanded (Movie S1, ESI). Staining for actin and phosphorylated myosin light chain (pMLC) revealed regions of intense signal at the edges of discontinuities, similar in appearance to contractile actomyosin “purse-strings” observed in epithelial wound healing.22,23 Cumulative projections in the XZ plane revealed portions of the tissue that were out of contact with substrate. In contrast to channels, clearance from inverted channels (i.e. convex “ridges”) followed a different pattern (Fig. 2F). Rather than occurring at the region of greatest positive curvature (the ridge apex), clearance occured predominantly near the region having the greatest concavity (negative curvature). In both cases, the vast majority of detachments appeared to be centered at the areas of greatest concavity, and those detachments that were not tended to be directly next to these areas (Figure 2G and S9, ESI). Together, these observations are consistent with a contractile mechanism where tissue failure occurs at regions of increased stress (within channels, where nuclear YAP is more prevalent). At these positions of high concavity, a component of the lateral contractile force generated by the surrounding tissue is projected normal to the substrate, potentially putting it out of balance with substrate adhesive forces. When tissue failure occurs at these sites, contractile forces could then expand the discontinuities, and peel the tissue away from its substrate.
Perturbing contractility affects epithelial clearing and detachment
We further investigated a role for tissue contractility by perturbation with small molecule inhibitors (Fig. 3). The ROCK inhibitor Y-27632 completely blocked cell clearing and detachment. ML-7 and blebbistatin act downstream of ROCK by inhibiting myosin light chain kinase phosphorylation and the association of myosin and actin, respectively. Both drugs also effectively blocked the clearance and lifting phenotypes. Blocking of cell division with mimosine and aphidocolin also excludes the possibility that contact remodeling during mitosis plays significant role in clearance (see above).24 Together, these data indicate that cell contractility is necessary for cells to respond to the topography of their substrate. We therefore attempted to increase net cell contractility by treating cell monolayers with Nocodazole. Nocodazole triggers microtubule depolymerization, releasing a variety of microtubule-bound Rho GEFs into the cytosol that increase cell contractility.25 Nocodazole treatement increased the frequency and severity of cells clearing from channels, although cell clearance was also observed on plateaus under these conditions. Focal adhesion kinase (FAK) has been previously shown to have an important effect in collective cell behaviors regulated by cell-ECM interactions.26-28 The FAK inhibitor PF-573228 was therefore used to probe the role of focal adhesion remodeling in the collective sensation of epithelial substrate topography. Detachment frequency was not affected by FAK inhibition, however, suggesting that focal adhesion remodeling is not necessary for epithelial clearing. Instead, clearing was more severe, implying that focal adhesion remodeling may work to help inhibit detachment. Taken together, these findings suggest that epithelial cells are able to sense and respond to their topographical microenvironment through regulation of cell contractility, and implicate focal adhesion remodeling as opposing the process of epithelial tearing and lifting.
Figure 3.
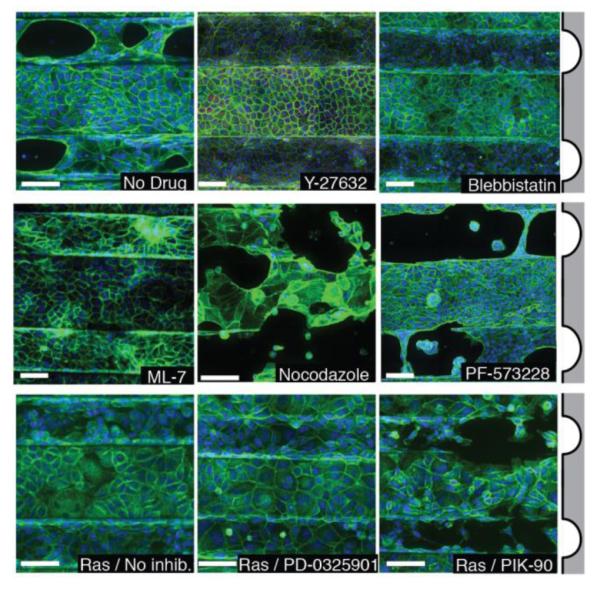
Treatment of cells with small-molecule drugs that modulate cell physicochemical properties affects their response to topography. MDCK cells treated with Y-27632, blebbistatin, or ML-7 do not clear, while untreated cells and those treated with Nocodazole or PF-573228 do clear (top two rows). MDCK-Ras cells do not clear, nor do MDCK-Ras cells treated with PD-0325901, but treatment with PIK-90 restores clearing (bottom row). Scale bars: 100 μm.
Unlike wild-type MDCK cells, lines overexpressing activated H-Ras did not clear of detach from channels suggesting that chronic stimulation of pathways downstream of Ras alter cell contractility and topography sensing (Fig. 1F and Fig. 3). Ras activation stimulates several downstream pathways, most notably the Ras-Raf-MEK-ERK pathway and the Ras-PI3K pathway.29,30 Inhibition of MEK with PD-0325901 did not rescue the ability for Ras mutants to sense and respond to substrate topography, but inhibition of PI3K with PIK-90 did.31 Because inhibition of PI3K rescues the clearing phenotype in cells that express oncogenic Ras, we concluded that chronic signaling through PI3K, but not MEK, is responsible for the diminished capacity of those cells to sense and respond to their substrate topography.
A finite-element model predicts the site of epithelial detachment on curved substrates
The experiments above imply that cell clearing and detachment occur as a result of epithelial contractility. We hypothesized that on curved substrates, epithelial contractility in the plane of the sheet generates a lifting force that is normal to the substrate. To explore this hypothesis, we generated a model using finite element method calculations. Tissues were modeled as continuous contractile materials bound to curved substrates by adhesive forces. Qualitatively, this model recapitulated the observation that cells lift from their substrate at the point of greatest negative curvature (Fig. 4A). Importantly, removing surface-adhesion from the model resulted in incorrectly predicting the location of clearance: lifting was predicted to initiate from the base of each channel, but also the peak of each ridge (Fig. S10, ESI). In contrast, increasing the adhesive forces between the substrate and tissue decreases the degree of tissue lifting, further supporting a coupling between epithelial contractility and substrate adhesion in the model (Fig. 4B). Interestingly, this analysis also indicated that tension along the apical face (cell-medium interface) of the tissue is necessary for channel clearing, as contraction solely along the basal surface of the sheet led to an altered geometry of tissue lifting (Fig. S10, ESI). The importance of apical tension suggested by modeling is reflected in the observation that actin and phosphorylated myosin are localized apically in intact monolayers and forming discontinuities (Fig. 1C, 2D,E, S11, ESI). Interestingly, when the model tissue is blocked from lifting, tension accumulates at the apical face, and especially at the base of the channel, matching experimental observations of tissues prior to lifting (Fig. S10, ESI). Even though the model does not account for factors like tearing, proliferation, or cell death, these qualitative observations correlate with our experimental observations of tissue lifting and tearing.
Figure 4.

FEM modeling supports a mechanism where apical tension is transmitted to basal cell-ECM adhesions. (A) Model output, with tissues treated as a contractile continuum adhered to a substrate. (B) Plot showing the modeled extent of lifting as a function of adhesion strength between the tissue and substrate. (C) Plot showing the modeled extent of lifting as a function of plateau width between channels. (D) Cells grown on substrates with increasing plateau width. Lifting can be seen in the XY views (top) and clearing can be seen in XY projections (bottom) in tissues that lift. (E) Quantification of lifting and tearing as a function of plateau width between channels.
Long-range topographical cues affect epithelial detachment
An important final implication of this contractile model is that forces are conducted between mechanically coupled groups of cells. Even if there were no variation in the magnitude of lateral forces, those cells that lie on the flat regions between channels (plateaus) would be expected to produce a greater integrated lateral force than those on curved regions, whose lateral forces run tangent to their underlying substrate curvature. Thus, in a ‘tug-of-war’ between the two plateaus surrounding a channel, the tissue inside the channel will experience unbalanced forces that result in clearing, lifting, or tissue failure (Fig S12, ESI). Finite element modeling of variable plateau width supports this outcome (Fig. 4C). As plateau width is increased the qualitative degree of tissue lifting also increases, suggesting increased tension in the channel. This led to the following hypothesis: if the plateau regions on a substrate are made small enough, they should not be able to generate sufficient force in the channels to trigger cell clearance. We therefore prepared substrates with 100 μm wide channels as in previous experiments, but with 25, 50, or 75 μm intervening plateaus (Fig 4D,E). As predicted, decreasing the plateau width to 25 μm resulted in fully intact tissues with no lifting or cell clearing. As plateau width increased, we observed progressively more pronounced lifting and clearing of cells from channels. Together, these experiments suggest that tension is transmitted across many cell diameters, ultimately allowing the properties of the plateau regions to contribute to the degree of cell clearing at the base of the valleys.
Single-cell interfacial properties correlate with collective epithelial clearing and detachment
The above computational modeling supports the notion that cell contractility in the plane of an epithelial monolayer can generate a lifting force normal to the monolayer in nearby regions of curved substrate. This lifting force would arise as a consequence of cell or tissue contractility along the apical face of the tissue (cell-medium interface) between the walls of the curved channel. Cell-substrate adhesion at the base of the channel must therefore act in the opposite direction in order to maintain a conformal relationship between the epithelial sheet and the substrate. This reasoning suggests that for a given lifting force, the balance of forces projected along cell-cell interfaces and normal to the substrate would correlate with the propensity of a tissue to clear from a channel or remain intact. As these forces arise from the combined action of many single cells, we hypothesized that the adhesive and contractile properties of single cells or cell pairs might be diagnostic of the collective properties of those cells in sheets. We explored this notion using cell-cell and cell-substrate contact angles as a simple measure of the balance of forces acting at cell-cell, cell-medium, and cell-substrate interfaces.12,13,32,33 Assuming a constant cell-medium interfacial tension, we extracted the projection of forces acting normal to the substrate (analogous to the lifting force and adhesive force) using the cosine and sine of the cell-cell and cell-substrate contact angles, respectively (Fig S14, ESI). These forces would be projected in opposite directions, and while not identical to the forces acting within intact epithelial sheets, they could nonetheless be diagnostic of the properties of epithelial sheets as a whole.
Cell-substrate contact angles (θ) were measured 1 h after seeding cells at low density onto a flat PDMS substrate (Fig. 5B). Because cells were not uniformly round, θ measurements were made for each of 60 radial slices per cell and for >30 cells (Fig. S13, ESI). Relative to controls, blebbistatin-treated cells were observed to have increased average contact angles and became spread out and morphologically dendritic within 1 h (Fig. 5C and S15, ESI). In contrast, cells exposed to the Y-27632 had significantly decreased affinity for their surface. Compared to control MDCK cells, Ras expressing MDCK cells also had decreased cell-substrate contact angles, and this effect was exacerbated by inhibition of the PI3K pathway.
Figure 5.

Single cell morphometric analysis is diagnostic of the capacity to sense and respond to large-scale substrate topography. (A) Diagram indicating the balance of forces acting within contracting monolayers on curved substrates. Blue represents tension at the apical surface; purple indicates tension at the cell-cell interface; red indicates tension across cell-substrate adhesions. (B) Representative single radial slice indicating the contact angle (θ) at the cell-substrate interface. (C) Average cell-substrate contact angle measured under the indicated conditions. Light gray: cells detach or clear, dark gray: no detachment. (D) The contact angle at the cell-cell interface (φ) was measured 4 h after seeding into agarose microwells. (E) Overlay of phase contrast images and fluorescence micrographs of actin or eCad stained cells showing polarization of doublets. (F) Average cell-cell contact angle measured under the indicated conditions. (G) The ratio of cos φ to sin θ from 5D and 5E under the indicated conditions. Red qualitatively highlights ratios that do not detach; green highlights those that do. All error bars are 95% confidence intervals. All scale bars: 10 μm.
The contact angle between cell-cell doublets provides an expedient method for quantifying the ratio of cell-cell and cell-medium interfacial tension. Although cells in growth arrested epithelial sheets have a different structure compared to cell doublets, we and others have found that, like epithelial sheets, eCad in cell doublets concentrates at cell-cell interfaces, and actin is enriched at the cell-medium interface (see below).34,35 Thus, the cell-cell and cell-medium interfaces in cell doublets are, at a minimum, topologically similar to the cell-cell and cell-medium interfaces in epithelial sheets. Cell-cell contact angles (Φ) were measured by seeding cell pairs into small and non-adhesive agarose wells and imaging live after 4 h (Fig. 5D). As expected, actin localized to the apical (cell-medium) interface, and eCad localized to the basolateral (cell-cell) interface (Fig. 5E). Pairs that were observed to lie flat within the wells were measured for their cell-cell contact angle, Φ (n>30, Fig. 5F). Cells treated with either blebbistatin or Y-27632 had decreased cell-cell contact angle, relative to untreated cells, likely attributable to decreased cell contractility at the cell-medium interface.36,37 RasG12V-expressing cells also had a reduced contact angle. Although cell-cell contact angle was partially restored by treatment with PI3K inhibitors, it was not statistically distinguishable from blebbistatin treated MDCK cells (p=0.08).
To extract a correlate to the interfacial forces directed parallel to the cell-cell interface and normal to the cell-substrate interface we used the average of the sine and cosine of the cell-substrate and cell-cell contact angles, respectively. While the ratio of these two values is a fundamental property of isolated cells, it is not necessarily a measure of the actual ratio of forces acting along the same interfaces in intact epithelial sheets. Nevertheless, we reasoned that this property of single cells and cell doublets might be diagnostic of the relative strength of the lifting and adhesive forces in intact epithelial sheets (Fig. S14, ESI). Consistent with our reasoning, we observed that perturbations effecting tissue clearing produced markedly lower values of this ratio than those that did not affect tissue clearing (Fig 5G). For example, Ras transformed cells had reduced contact angle at both the cell-cell and cell-substrate interfaces, suggesting decreases in both cell-medium interfacial tension as well as cell-substrate adhesion. However, the proportionally higher decrease in cell-cell contact angle predicts their inability to sense and respond to the tested geometries. The inhibition of the PI3K pathway in Ras transformed cells results in a corresponding increase in the cell-cell contact angle, and a decrease in cell-substrate contact angle. This changes the overall ratio of forces acting normal to the tissue substrate, and rescues the ability of the epithelium to sense and respond to substrate curvature. These experiments suggest that the ratio of forces acting at the interfaces in isolated cell pairs is diagnostic of tissue clearing. However, additional experiments would be necessary to directly measure and quantify the balance of forces acting within intact epithelial sheets, but at the cell-medium, cell-cell, and cell-substrate interfaces.
Conclusions
Epithelial tissues can exist in topographically rich microenvironments in vivo.38-40 Here we have shown that cells can sense the topographical features of their environment collectively and over long distances by coupling their contractile apparatus to their cell-cell and cell-substrate adhesion machinery. This capacity to sense substrate topography is inherently multicellular, as changing the intervening space between features can cause drastic differences in the way that cells respond as an epithelial sheet, even though the locus of response is multiple cell diameters away. Our results suggest that apical contractility in mechanically integrated epithelial sheets leads to the accumulation of tension that can be transmitted through multiple cells to affect a response at a distant site. The topography of a substrate can focus components of the accumulating tension toward the basal adhesion apparatus, suggesting an important coupling between these adhesion complexes in topographically heterogeneous microenvironments. We find that the properties of single cells can be predictive of a given epithelium’s response to topographical heterogeneity. Specifically, the ratio of the cosine and sine of cell-cell and cell-substrate contact angles correlates with epithelial clearing and detachment of substrate channels.
Morphologically similar cells can be molecularly distinct when in different topographical microenvironments.2-4 When cells are grown in channels, these distinct mechanical microenvironments are reflected by differences in the nuclear localization of YAP. However, increased YAP signaling in channels did not correlate with Edu incorporation, contrary to previous reports where cells were strained isotropically and from their basal surface using an isotropic strain array.41 This discrepancy may be a consequence of how stresses are applied to the epithelial sheets in different contexts. In curved channels, it appears that cell-cycle entry is not induced by the YAP-associated stresses that accumulate gradually and concomitantly with establishment of confluence in channels. In contrast, instantaneously applied strain to confluent monolayers on flat substrates does induce YAP-associated proliferation. Additional studies will be necessary to reveal how cellular response to YAP is regulated by mechanical microenvironment, but our results suggest that while YAP signaling may be necessary for cell cycle entry, it may not be sufficient.
Although it was reasonable to expect that the topography of a tissue might also affect cell behavior, it was nonetheless surprising to find that the context of the topography could play such a significant role. For example, we found that when the width of the intervening plateaus between channels was decreased, epithelia ceased to clear and detach, even when the topography of the channels were identical. Therefore, this sensation of topography must be a multicellular phenomenon. Finite element modeling suggests that at the base of channels, the epithelial sheet experiences a different mechanical microenvironment on substrates with wide plateaus when compared to substrates with narrow plateaus.
Given the inherently multicellular nature of topography sensation, it is intriguing that the physical properties of individual cells, such as contact angles, can be diagnostic of their behaviors as collectives. Contact angles at the interface of isolated cells arise as a consequence of the balance of forces between the cell-medium, cell-cell, and cell-substrate interfaces. It is a similar balance of forces in the mechanical model that generate responses qualitatively similar to epithelial sheets. While the forces acting on isolated cells are likely distinct from those acting in intact epithelial sheets, the balance of forces acting at the cell-cell and cell medium interfaces in isolated cell and cell pairs nevertheless correlates with the collective behavior of the cells in intact sheets. We and others recently found that cell-cell12 and cell-substrate13 contact angles can also be diagnostic of cell sorting in heterotypic tissues spanning many cell diameters, suggesting similarities between the channel clearing phenomenon observed here and cell sorting by differential interfacial tension.42,43 Thus, parallel mechanisms may contribute to the mechanical coupling within epithelial sheets during morphogenesis and homeostasis,44,45 and to the long range response of epithelial sheets to substrate topography.
The relationship between long-range substrate topography, cell contractility, and its integration with substrate adhesion may have implications for human disease. Heritable blistering diseases of the skin, for example, involve the physical detachment of the epidermis from the convoluted upper layer of the dermis. Intriguingly, these diseases are associated with mutations in key stress bearing components of tissues: the intermediate filaments and basal adhesion machinery.46,47 These studies may also have implications for tumor progression. A characteristic common to many solid tumors is the dysregulation of tissue structure and improper boundary formation.48,49 We find that cells with transforming mutations in Ras, among the most common mutations in cancer,50 cannot sense the structure of the plateau regions in a manner analogous to wild type cells. This observation would suggest that Ras transformed cells have lost the ability to sense tissue structure extending beyond their immediate microenvironment. While it is now well accepted that capacity to sense the structure of a cell’s immediate microenvironment is essential to suppress tumorigenesis,11,48 our work adds to this notion in two important ways. First, the structure of a cell’s microenvironment even hundreds of microns away may also be important for suppressing tumorigenesis; second, the capacity of a cell to sense the structure of the surrounding tissue can become altered by certain tumor-associated mutations. Indeed, a variety of mutations and microenvironmental changes can differentially affect the relative strength of cell-cell and cell-substrate interactions. For example, stiffening of substrates is known to strengthen cell-substrate association and contribute to tumorigenesis.51,52 Additionally, cells undergoing EMT possess decreased cell-cell affinity and have also been observed to increase cell substrate interactions.53,54 Future work will be needed to investigate whether mutations affecting the capacity of epithelial cells to sense their long-range substrate topography are a common feature of aggressive tumors.
Supplementary Material
{kind=link}
Acknowledgments
We would like to thank Nathan Nguyen for experimental assistance and Justin Farlow for help using Mathematica. This work was supported by the National Institute of Health and the National Science Foundation (DP2 HD080351-01 and MCB-1330864 to Z.J.G). ZJG is supported by the UCSF Center for Systems and Synthetic Biology (NIGMS Systems Biology Center grant P50 GM081879) K.E.B. was supported by the US National Institutes of Health (NRSA NCI 5F32CA165620). A.E.C was supported by the US Department of Defense through a National Defense Science and Engineering Graduate (NDSEG) Fellowship.
Notes and references
- 1.Gibson WT, Gibson MC. Curr. Top. Dev. Biol. 2009;89:87–114. doi: 10.1016/S0070-2153(09)89004-2. [DOI] [PubMed] [Google Scholar]
- 2.Eberwein P, Reinhard T. Stem Cells. 2015;33:916–924. doi: 10.1002/stem.1886. [DOI] [PubMed] [Google Scholar]
- 3.Bullen TF, Forrest S, Campbell F, Dodson AR, Hershman MJ, Pritchard DM, Turner JR, Montrose MH, Watson AJM. Lab. Invest. 2006;86:1052–1063. doi: 10.1038/labinvest.3700464. [DOI] [PubMed] [Google Scholar]
- 4.Kouznetsova I, Kalinski T, Meyer F, Hoffmann W. Mol Biosyst. 2011;7:1105–1112. doi: 10.1039/c0mb00233j. [DOI] [PubMed] [Google Scholar]
- 5.Mata A, Boehm C, Fleischman AJ, Muschler GF, Roy S. International journal of nanomedicine. 2007;2:389–406. [PMC free article] [PubMed] [Google Scholar]
- 6.Bettinger CJ, Langer R, Borenstein JT. Angew. Chem. Int. Ed. Engl. 2009;48:5406–5415. doi: 10.1002/anie.200805179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schindler M, Nur-E-Kamal A, Ahmed I, Kamal J, Liu H-Y, Amor N, Ponery AS, Crockett DP, Grafe TH, Chung HY, Weik T, Jones E, Meiners S. Cell Biochem. Biophys. 2006;45:215–227. doi: 10.1385/CBB:45:2:215. [DOI] [PubMed] [Google Scholar]
- 8.Costello CM, Sorna RM, Goh Y-L, Cengic I, Jain NK, March JC. Mol. Pharm. 2014;11:2030–2039. doi: 10.1021/mp5001422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim SH, Chi M, Yi B, Kim SH, Oh S, Kim Y, Park S, Sung JH. Integr. Biol. 2014;6:1122–1131. doi: 10.1039/c4ib00157e. [DOI] [PubMed] [Google Scholar]
- 10.Nelson CM, Inman JL, Bissell MJ. Nat Protoc. 2008;3:674–678. doi: 10.1038/nprot.2008.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gjorevski N, Nelson CM. Integr Biol (Camb) 2010;2:424–434. doi: 10.1039/c0ib00040j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maître J-L, Berthoumieux H, Krens SFG, Salbreux G, Jülicher F, Paluch E, Heisenberg C-P. Science. 2012;338:253–256. doi: 10.1126/science.1225399. [DOI] [PubMed] [Google Scholar]
- 13.Cerchiari AE, Garbe JC, Jee NY, Todhunter ME, Broaders KE, Peehl DM, Desai TA, LaBarge MA, Thomson M, Gartner ZJ. Proc. Natl. Acad. Sci. U.S.A. 2015;112:2287–2292. doi: 10.1073/pnas.1410776112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elia N, Lippincott-Schwartz J. Curr Protoc Cell Biol. 2009 doi: 10.1002/0471143030.cb0422s43. Chapter 4, Unit 4.22–4.22.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Debnath J, Brugge JS. Nat. Rev. Cancer. 2005;5:675–688. doi: 10.1038/nrc1695. [DOI] [PubMed] [Google Scholar]
- 17.O'Brien LE, Zegers MMP, Mostov KE. Nat. Rev. Mol. Cell Biol. 2002;3:531–537. doi: 10.1038/nrm859. [DOI] [PubMed] [Google Scholar]
- 18.Cerchiari A, Garbe JC, Todhunter ME, Jee NY, Pinney JR, LaBarge MA, Desai TA, Gartner ZJ. Tissue Eng Part C Methods. 2014;21:150127064141009–547. doi: 10.1089/ten.tec.2014.0450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vedula SRK, Hirata H, Nai MH, Brugués A, Toyama Y, Trepat X, Lim CT, Ladoux B. Nat Mater. 2013;13:87–96. doi: 10.1038/nmat3814. [DOI] [PubMed] [Google Scholar]
- 20.Eisenhoffer GT, Loftus PD, Yoshigi M, Otsuna H, Chien C-B, Morcos PA, Rosenblatt J. Nature. 2012;484:546–549. doi: 10.1038/nature10999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 22.Bement WM. The Journal of Cell Biology. 1993;121:565–578. doi: 10.1083/jcb.121.3.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abreu-Blanco MT, Verboon JM, Liu R, Watts JJ, Parkhurst SM. J Cell Sci. 2012;125:5984–5997. doi: 10.1242/jcs.109066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thery M, Bornens M. HFSP journal. 2008;2:65–71. doi: 10.2976/1.2895661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang Y-C, Nalbant P, Birkenfeld J, Chang Z-F, Bokoch GM. Mol. Biol. Cell. 2008;19:2147–2153. doi: 10.1091/mbc.E07-12-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boghaert E, Gleghorn JP, Lee K, Gjorevski N, Radisky DC, Nelson CM. Proc. Natl. Acad. Sci. U.S.A. 2012;109:19632–19637. doi: 10.1073/pnas.1118872109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schober M, Raghavan S, Nikolova M, Polak L, Pasolli HA, Beggs HE, Reichardt LF, Fuchs E. The Journal of Cell Biology. 2007;176:667–680. doi: 10.1083/jcb.200608010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Provenzano PP, Inman DR, Eliceiri KW, Beggs HE, Keely PJ. Am. J. Pathol. 2008;173:1551–1565. doi: 10.2353/ajpath.2008.080308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Steelman LS, Chappell WH, Abrams SL, Kempf RC, Long J, Laidler P, Mijatovic S, Maksimovic-Ivanic D, Stivala F, Mazzarino MC, Donia M, Fagone P, Malaponte G, Nicoletti F, Libra M, Milella M, Tafuri A, Bonati A, Bäsecke J, Cocco L, Evangelisti C, Martelli AM, Montalto G, Cervello M, McCubrey JA. Aging (Albany NY) 2011;3:192–222. doi: 10.18632/aging.100296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tönges L, Koch J-C, Bähr M, Lingor P. Front Mol Neurosci. 2011;4:39. doi: 10.3389/fnmol.2011.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu JS, Farlow JT, Paulson AK, LaBarge MA, Gartner ZJ. Cell Rep. 2012;2:1461–1470. doi: 10.1016/j.celrep.2012.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fagotto F. Development. 2014;141:3303–3318. doi: 10.1242/dev.090332. [DOI] [PubMed] [Google Scholar]
- 33.Gonzalez-Rodriguez D, Guevorkian K, Douezan S, Brochard-Wyart F. Science. 2012;338:910–917. doi: 10.1126/science.1226418. [DOI] [PubMed] [Google Scholar]
- 34.Bryant DM, Datta A, Rodríguez-Fraticelli AE, Peränen J, Martín-Belmonte F, Mostov KE. Nat Cell Biol. 2010;12:1035–1045. doi: 10.1038/ncb2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Engl W, Arasi B, Yap LL, Thiery JP, Viasnoff V. Nat Cell Biol. 2014;16:587–594. doi: 10.1038/ncb2973. [DOI] [PubMed] [Google Scholar]
- 36.Kovács M, Tóth J, Hetényi C, Málnási-Csizmadia A, Sellers JR. J. Biol. Chem. 2004;279:35557–35563. doi: 10.1074/jbc.M405319200. [DOI] [PubMed] [Google Scholar]
- 37.Narumiya S, Ishizaki T, Uehata M. Meth. Enzymol. 2000;325:273–284. doi: 10.1016/s0076-6879(00)25449-9. [DOI] [PubMed] [Google Scholar]
- 38.Ewald AJ, Brenot A, Duong M, Chan BS, Werb Z. Dev. Cell. 2008;14:570–581. doi: 10.1016/j.devcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miura T. J. Biochem. 2015;157:121–127. doi: 10.1093/jb/mvu087. [DOI] [PubMed] [Google Scholar]
- 40.Menco BP, Jackson JE. J. Comp. Neurol. 1997;388:293–306. [PubMed] [Google Scholar]
- 41.Benham-Pyle BW, Pruitt BL, Nelson WJ. Science. 2015;348:1024–1027. doi: 10.1126/science.aaa4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Foty RA, Steinberg MS. Dev. Biol. 2005;278:255–263. doi: 10.1016/j.ydbio.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 43.Brodland GW. J Biomech Eng. 2002;124:188–197. doi: 10.1115/1.1449491. [DOI] [PubMed] [Google Scholar]
- 44.Heisenberg C-P, Bellaïche Y. Cell. 2013;153:948–962. doi: 10.1016/j.cell.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 45.Mertz AF, Che Y, Banerjee S, Goldstein JM, Rosowski KA, Revilla SF, Niessen CM, Marchetti MC, Dufresne ER, Horsley V. Proc. Natl. Acad. Sci. U.S.A. 2013;110:842–847. doi: 10.1073/pnas.1217279110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bruckner-Tuderman L. Biochem. Cell Biol. 1996;74:729–736. doi: 10.1139/o96-079. [DOI] [PubMed] [Google Scholar]
- 47.Uitto J, Pulkkinen L. Arch Dermatol. 2001;137:1458–1461. doi: 10.1001/archderm.137.11.1458. [DOI] [PubMed] [Google Scholar]
- 48.Bissell MJ, Hines WC. Nat. Med. 2011;17:320–329. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bissell MJ, Radisky D. Nat. Rev. Cancer. 2001;1:46–54. doi: 10.1038/35094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. Nat. Rev. Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu H, Mouw JK, Weaver VM. Trends Cell Biol. 2011;21:47–56. doi: 10.1016/j.tcb.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yeung T, Georges PC, Flanagan LA, Marg B, Ortiz M, Funaki M, Zahir N, Ming W, Weaver V, Janmey PA. Cell Motil. Cytoskeleton. 2005;60:24–34. doi: 10.1002/cm.20041. [DOI] [PubMed] [Google Scholar]
- 53.Shamir ER, Pappalardo E, Jorgens DM, Coutinho K, Tsai W-T, Aziz K, Auer M, Tran PT, Bader JS, Ewald AJ. The Journal of Cell Biology. 2014;204:839–856. doi: 10.1083/jcb.201306088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lamouille S, Xu J, Derynck R. Nat. Rev. Mol. Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.