

Abstract
The oligodendrocyte transcription factor Olig1 is critical for both oligodendrocyte development and remyelination in mice. Nuclear to cytoplasmic translocation of Olig1 protein occurs during brain development and in multiple sclerosis, but the detailed molecular mechanism of this translocation remains elusive. Here, we report that Olig1 acetylation and deacetylation drive its active translocation between the nucleus and the cytoplasm in both mouse and rat oligodendrocytes. We identified three functional nuclear export sequences (NES) localized in the basic helix-loop-helix domain and one specific acetylation site at Lys 150 (human Olig1) in NES1. Olig1 acetylation and deacetylation are regulated by the acetyltransferase CREB-binding protein and the histone deacetylases HDAC1, HDAC3, and HDAC10. Acetylation of Olig1 decreased its chromatin association, increased its interaction with inhibitor of DNA binding 2 and facilitated its retention in the cytoplasm of mature oligodendrocytes. These studies establish that acetylation of Olig1 regulates its chromatin dissociation and subsequent translocation to the cytoplasm and is required for its function in oligodendrocyte maturation.
SIGNIFICANCE STATEMENT The nuclear to cytoplasmic translocation of Olig1 protein has been observed during mouse and human brain development and in multiple sclerosis in several studies, but the detailed molecular mechanism of this translocation remains elusive. Here, we provide insight into the mechanism by which acetylation of Olig1 regulates its unique nuclear-cytoplasmic shuttling during oligodendrocyte development and how the acetylation status of Olig1 modulates its distinct function in the nucleus versus the cytoplasm. The current study provides a unique example of a lineage-specific transcription factor that is actively translocated from the nucleus to the cytoplasm as the cell differentiates. Importantly, we demonstrate that this process is tightly controlled by acetylation at a single lysine.
Keywords: acetylation, nuclear export, Olig1
Introduction
Myelin ensheathment of axons provides valuable trophic support to axons and it is physiologically critical to increase electrical conduction dramatically (Bullock et al., 1984). Human demyelinating diseases in the CNS, such as multiple sclerosis (MS), result in oligodendrocyte cell death, axonal degeneration, and cognitive and motor deficits (Trapp and Nave, 2008; Franklin and ffrench-Constant, 2008). Oligodendrocyte progenitor cells (OPCs) are present in MS lesions, suggesting that demyelinated lesions should be able to remyelinate (Chang et al., 2000). Nevertheless, only minimal remyelination occurs and it appears that OPC maturation is particularly vulnerable in MS (Wolswijk, 1998, 2000; Chang et al., 2002; Kuhlmann et al., 2008). The molecular mechanisms of oligodendrocyte differentiation and myelination during development and after demyelination are being actively investigated and the current studies address one element of this program.
Two closely related basic helix-loop-helix (bHLH) transcription factors, Olig1 and Olig2, function in the well established transcriptional regulatory network of oligodendrocyte lineage progression (Emery, 2010a,b) and are critical for oligodendrocyte specification and differentiation (Lu et al., 2002; Zhou and Anderson, 2002; Mei et al., 2013). However, the function of Olig1 during oligodendrocyte development has been unclear, with differing evidence based on the generation of different Olig1-null lines (Lu et al., 2002; Xin et al., 2005; Paes de Faria et al., 2014). Our recent analysis of brain development in Olig1-null mice reveals an essential role for Olig1 in OPC differentiation in brain, but less so in spinal cord (Dai et al., 2015).The current studies focus on elements of the regulation of Olig1 function in oligodendrocyte development in brain.
Subcellular translocation of Olig1 during both oligodendrocyte development and remyelination is unique to Olig1 relative to Olig2, which is only found in the nucleus. Olig1 is present in OPC nuclei and translocates progressively to the cytoplasm as oligodendrocytes mature in the developing mouse and human brain (Arnett et al., 2004; Jakovcevski and Zecevic, 2005; Kitada and Rowitch, 2006; Niu et al., 2012). Interestingly, during remyelination, Olig1 relocates back to the nucleus, suggesting a nuclear function for Olig1 that appears essential for remyelination (Arnett et al., 2004). The mechanism regulating this intriguing Olig1 translocation has not been established. Posttranslational modification of proteins, particularly phosphorylation and acetylation, regulates the subcellular location of several transcription factors (Krämer et al., 2009; Meek and Anderson, 2009) and a recent in vitro study indicates that Olig1 phosphorylation at Ser149 (Ser138, mouse Olig1) can regulate its cytoplasmic location and cell membrane extension (Niu et al., 2012).
In the current study, we focused on the role of acetylation in regulating Olig1 localization. We established that Olig1 is acetylated by CREB-binding protein (CBP) on lysine150 (Lys139, mouse Olig1) as oligodendrocytes mature both in vitro and in vivo. Acetylation of Olig1 increased its binding with inhibitor of DNA binding 2 (Id2), decreased its association with chromatin, and facilitated its nuclear to cytoplasmic relocation during oligodendrocyte development. Our data show that Olig1 acetylation is important for its function in promoting oligodendrocyte lineage progression and morphological differentiation and reveal the mechanism by which its acetylation regulates its nuclear to cytoplasmic translocation.
Materials and Methods
Animal procedures and primary cell culture.
All animal procedures were conducted in accordance with the University of Colorado Institutional Animal Care and Use Committee and the National Institutes of Health guidelines for the care and use of laboratory animals. Mouse neural progenitor cells were isolated from neocortex of male and female embryonic day 12.5 (E12.5) to E14.5 embryos of wild-type C57BL/6J or Olig1-null mice [B6;129S4-Olig1tm1(cre)Rth/J strain; Jackson Laboratories, catalog # 011105] and converted to OPCs as described previously (Pedraza et al., 2008). Mixed glial cultures were generated from male and female postnatal day 0 (P0) to 3-d-old Sprague Dawley rat pups, as described previously (Dai et al., 2014).
Plasmid vectors.
The open reading frame of human Olig1 was amplified from a human embryonic cDNA library (Origene) and subcloned into pCMV-myc vector (Clontech), pEGFP-N1/C3 vectors (Clontech), pGEX-KG vector (for GST tag protein purification), and HA-TAT vector (a gift from Dr. Steven Dowdy, University of California, for TAT tag fusion protein purification). The Olig1 and Olig2 mutant vectors used in this study were generated using Stratagene's QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies) following the manufacturer's protocol (detailed primers and PCR conditions provided upon request). The p300-myc and p300DY-myc vectors were kindly provided by Dr. Tso-Pang Yao (Duke University). The pEGFPC1-mId2 and pEGFPC1-mId4 vectors described previously (Kurooka and Yokota, 2005) were kind gifts from Dr. Hisanori Kurooka (University of Fukui, Japan). The HA-tagged human HDAC1, HDAC 2, HDAC3, HDAC4, HDAC5, HDAC6, HDAC7, HDAC8, and HDAC10 vectors were from Drs. Minoru Yoshida and Akihisa Matsuyama (Chemical Genetics Laboratory, RIKEN, Japan).The Flag-Sirtuin1/2 vectors were from Dr. Eric Verdin (University of California). The pCMV5-Sox10 vector was kindly provided by Dr. Michael Wegner (Lehrstuhl für Biochemie und Pathobiochemie, Germany). The following vectors were obtained from Addgene: pRc/RSV-m CBP-HA (catalog #16701), pAdEasy Flag GCN5 (catalog #14106), pAd-Track Flag GCN5 Y621A/P622A (catalog #14425), pCI-flag-PCAF (catalog #8941), p3xFLAG-mE47 (catalog #34585), and Flag-hCRM1 (catalog #17647). The GCN5 and GCN5-Y621A/P622A cDNA were digested with KpnI and XhoI and subcloned into pcDNA3.1+ vector (Life Technologies). CBP shRNA was constructed by Genepharma into a lentivirus expression vector pGLVH1/GFP+Puro using the sequence 5′-gcagcagccagcattgata-3′ and 5′-ctgatgagctgatacccaatg-3′.
Antibodies.
Two rabbit anti-mouse Olig1 antibodies targeting different regions were generated by Abgent. The sequences of immunizing peptides were SLLPKPAREKAEAP (residue 55-68 of mOlig1, rabbit anti-Olig1-1) and TKYLSLALDEPPC (residue 206-218, rabbit anti-Olig1-2). The specificity of these two antibodies was validated by Western blot, immunoprecipitation, and immunocytochemistry (data not shown). Based on their performance, the Olig1-1 antibody was used for Western blots and Olig1-2 antibodies were used for immunoprecipitation studies. The K150 acetylation-site-specific antibody of Olig1, which is conserved among human, mouse, and rat Olig1 proteins, was generated against the modified peptide GAPGRKLS[Ac]KIAT (residues 142-153 of human Olig1/131-142 of mouse Olig1) by Abgent. The S149 phosphorylation-site-specific antibody of Olig1, which is conserved among human, mouse, and rat Olig1 proteins, was generated against the modified peptide GAPGRKL[pSer]KIAT (residues 142-153 of human Olig1/131-142 of mouse Olig1) by Abgent. The antiserum was first absorbed on a column containing nonacetylated or nonphosphorylated peptide and then affinity purified on an acetylpeptide or phosphopeptide column, respectively. The purified acetyl-Olig1 antibody and phospho-Olig1 antibody were validated and used at 1:250 for Western blot.
Primary antibodies used for Western blot, immunoprecipitation, or immunofluorescence were as follows: rabbit anti-Olig2 and Olig1 (a gift from Dr. Charles Stiles, Harvard University); mouse anti-Olig1 (Millipore, catalog #MAB5540); the following antibodies were purchased from Santa Cruz Biotechnology: mouse anti-Olig1 (sc-166257); rabbit anti-Id2 (sc-489); rabbit anti-CBP (sc-369); rabbit anti-p300 (sc-584); goat anti-Sox10 (sc17342); rabbit anti-Sox10 (Sigma-Aldrich, catalog #S8193); mouse anti-β-tubulin (Sigma-Aldrich, catalog #T8328); mouse anti-CC1 antibody (Calbiochem, catalog #OP80); rat anti-PDGFRα (BD PharMingen, catalog #55874); O4 hybridoma (gift from Dr. Rashmi Bansal, University of Connecticut Health Sciences Center); mouse anti-myelin basic protein (MBP; Covance, catalog #SMI-94); mouse anti-Flag M2 (Sigma-Aldrich, catalog #F3165); anti-Flag M2 affinity gel (Sigma-Aldrich, catalog #A2220); mouse anti-myc (9E10) supernatant (Developmental Studies Hybridoma Bank). The following antibodies were obtained from Cell Signaling Technology: rabbit anti-Id2 (catalog #3431); rabbit anti-CBP (catalog #7389); rabbit anti-GCN5 (catalog #3305); rabbit anti-GAPDH (catalog #2118); rabbit anti-LaminA/C (catalog #2032,); rabbit anti-acetylated lysine (catalog #9814); mouse anti-acetylated lysine(catalog #9681); rabbit anti-Histone H3 (catalog #4620); acetyl-histone H3 antibody sampler kit (catalog #9927); and mouse anti-GFP (catalog #2955).
Cell line culture and transfection.
HEK293T cells were cultured in DMEM high glucose (Life Technologies) supplemented with 10% (v/v) fetal bovine serum at 37°C with 5% (v/v) CO2. Plasmid transfection was performed using either Lipofectamine LTX and Plus reagent (Invitrogen, catalog #15338; if using three plasmids or more) or LipoD 293 DNA transfection reagent (SignaGen, catalog # SL100668; if using two plasmids or fewer) according to manufacturer's instructions. Total DNA concentrations were normalized with empty vector DNA when required. All of the biochemistry analyses of protein overexpression were done in the HEK293T cells.
OPC transfections.
Rat OPCs were collected after shaking and 5 × 106 cells were electroporated (Amaxa nucleofection apparatus) in 100 μl of nucleofection solution (Amaxa basic glial cells nucleofector kit #VPI-1006, Lonza) with Olig1 siRNAs (catalog #L100044-01, 10 μl of 20 μm) or siControl nontargeting siRNA pool (catalog #B002000-UB) from Dharmacon. After electroporation, cells were resuspended and seeded onto poly-d-lysine/Laminin (Sigma-Aldrich, catalog #I6758)-coated dishes or round, 12 mm coverslips in DMEM supplemented with N2 (Life Technologies, catalog #17502-048), fibroblast growth factor (FGF, 10 ng/ml), and platelet-derived growth factor (PDGF, 10 ng/ml), and allowed to recover for 24 h. The expression constructs (2 μg of each well of 24 well plate) were transfected into cultured OPCs using Xfect transfection reagent (Clontech, catalog #631318) according to the manufacturer's instructions.
Expression and purification of recombinant TAT and GST fusion proteins.
Plasmids for TAT-HA or GST fusion proteins were transformed into the Escherichia coli strain BL21 (DE3). Protein production was induced with 100 μm isopropyl β-d-1-thiogalactopyranoside (Sigma-Aldrich, catalog #I6758) at 16°C for 18 h, and protein was purified by TALON Metal Affinity Resin (Clontech, catalog #635501) as described previously (Mi et al., 2012) or Glutathione-Superflow Resin (Clontech, catalog #635607), respectively. The size and purity of all of the proteins were confirmed by Western blot using tag-specific and Olig1-specific antibodies. Apparent degradation of Olig1 protein was noticed under different purification conditions, likely because Olig1 protein is relatively unstable (unpublished observation). After dialysis and filtration, proteins were aliquoted and stored at −80°C.
In vitro histone acetyl transferase assay.
Recombinant GST-tagged human histone acetyl transferase (HAT) proteins were purchased from SignalChem: CBP (catalog #C07-31G, p300), E1A-binding protein (300 kDa; catalog #P07-31G), and GCN5 (KAT2; catalog #K311-381G). Their HAT activity was validated using histone H3 1-21 aa peptide as a substrate (data not shown). To perform the in vitro HAT peptide assay, varying amounts (0.1 ng to 1 μg) of nonacetylated peptide (GAPGRKLSKIAT; Abgent) were incubated with recombinant HATs (CBP, 500 ng; p300, 500 ng; GCN5, 200 ng) or 200 ng glutathione S-transferase (GST) in 20 μl of acetyltransferase assay buffer (250 mm Tris-HCl, pH 8.0, 0.5 mm EDTA, and 2 mm dithiothreitol), with 100 μm acetyl-CoA (Sigma-Aldrich, catalog #A2506) at room temperature (RT) for 1 h. The reactions were spotted on PVDF membrane to detect acetylation of Olig1 peptides with acetyl-K150-Olig1 antibody. To perform the acetylation assay on Olig1 protein, 40 pmol of purified Olig1-GST, Olig1-K150R-GST protein was incubated with recombinant HATs (CBP, 500 ng; p300, 500 ng; GCN5, 200 ng) in 40 μl of acetyltransferase assay buffer at RT for 1 h. The reactions were stopped by adding 5× Laemmli buffer and boiling for 5 min. The reaction product was analyzed by Western blot and the acetylation of Olig1 protein was detected by either general acetyl-lysine antibody or acetyl-K150-Olig1 antibody.
Western blots and immunoprecipitation.
After transfection or treatment, cultured cells or brain tissue were lysed in RIPA buffer (25 mm Tris-HCl, pH 7.5, 150 mm NaCl; 1 mm EDTA, 1% NP-40, 0.1% DOC, 0.1%SDS) supplemented with complete mini-protease inhibitor mixture (Roche Applied Science) and phosphatase inhibitor mixture set II (CalbioChem, catalog #564652). Samples were analyzed by Western blot and protein bands were detected with the LICOR Odyssey infrared scanner. Protein expression levels in scanned images were quantified using the Odyssey Scanner Software version 2.0. For immunoprecipitations, primary antibodies or control mouse IgG (Invitrogen, catalog #10400C) or rabbit IgG (Invitrogen, catalog #10500C) were incubated with protein A/G agarose (Pierce, catalog #20421) or Dynabeads protein A or G (Invitrogen, catalog #10004D) overnight. Antibodies were then cross-linked to beads using the Pierce immunoprecipitation kit (catalog #26147) following the manufacturer's instructions. Cell lysates were precleared by incubation with IgG beads for 2 h; antibody-crosslinked beads were then added and samples incubated overnight at 4°C while rotating. Beads were washed 3 times in RIPA buffer wash, resuspended in 30 μl of SDS gel sample buffer, boiled for 5 min, and subjected to SDS-PAGE, followed by Western blot.
Immunohistochemistry and immunocytochemistry.
Immunohistochemistry was performed as described previously (Trapp et al., 1997). Free-floating cortex sections (30 μm) were analyzed, with antigen retrieval in 10 mm sodium citrate, pH 6.0, at 65°C for 10 min as needed, using a Pelco Biowave Pro tissue processor (Ted Pella). For immunocytochemistry, oligodendrocytes were cultured on coverslips and fixed with 4% paraformaldehyde for 15 min at RT. Cells were permeabilized with 0.1% Triton X-100 for 10 min, blocked with 3% BSA in PBS for 60 min at RT, and incubated with primary antibodies overnight at 4°C. For detection of O4 cell surface antigens, O4 antibody was diluted with medium and incubated with live cells on coverslips for 1 h before fixation.
Images were taken on a Zeiss Axio Imager M2 or Leica SP5 confocal microscopy. A total of six microscopic fields per coverslip from two coverslips per condition were sampled in each experiment. The covered area of O4+ cells was analyzed with Imaris surfaces module (Bitplane).
Subcellular fractionation.
Subcellular fractionation of cells was performed as described previously (MacDonald et al., 2004). Cultured cells or brain tissue were suspended in hypotonic buffer (10 mm HEPES 7.9, 10 mm KCl, 2 mm MgCl2, 0.5% NP40, and 1 mm dithiothreitol with protease and phosphatase inhibitors). The cells were incubated on ice for 5 min and cytoplasmic and nuclear fractions were harvested by centrifugation at 1500 × g for 5 min. The isolated nuclei were then washed in hypotonic buffer, lysed in hypotonic buffer plus 500 mm NaCl, and incubated on ice for 30 min. The nuclear fraction was used for immunoprecipitations or Western blots or subsequent subnuclear fractionation was performed as described previously (Niida et al., 2007). The isolated nuclei were then washed in hypotonic buffer, lysed in chromatin extraction buffer (3 mm EDTA, 0.2 mm EGTA, 1 mm DTT), and incubated on ice for 30 min. The soluble nuclear and chromatin fractions were harvested by centrifugation at 1500 × g for 15 min. The isolated chromatin was then washed in chromatin extraction buffer, centrifuged at 10,000 × g for 1 min, resuspended in Laemmli sample buffer, and boiled for 5 min.
DNA pulldown assay.
DNA pulldown assays were performed as described previously (Bujalka et al., 2013) with slight modifications. Briefly, the nuclear fraction was collected as described above, lysates were clarified by centrifugation at 15,000 × g for 20 min at 4°C, and adjusted to a total protein concentration of 1 μg/μl using buffer H (100 mm KCl, 200 mm HEPES, pH 7.8, 20% glycerol, 1 mm DTT, 0.1% NP40). Five micrograms of biotinylated, annealed oligonucleotides (5′-BioTEG-AACAGCTGTGAACAGCTGTGAACAGCTGTGAACAGCTGTGAACAGCTGTGAACAGCTGTG-3′) were conjugated to Steptavidin magnetic beads (Roche, catalog #11636502103) following the manufacturer's instructions. The oligonucleotide-conjugated beads were washed twice with 500 μl buffer H and added to the cell lysate in the presence of 2 μg poly (dIdC; Sigma-Aldrich, catalog #4929). Beads were incubated with cell lysates overnight at 4°C with rotation and then washed three times in 500 μl of buffer H. Bound protein was eluted by boiling the beads in 1.5× Laemmli buffer and subjected to Western blot.
Results
Three nuclear export sequences of Olig1 regulate its export to the cytoplasm
Olig1 and Olig2 are highly conserved at the molecular and functional level, yet they have distinct subcellular localization during oligodendrocyte development (Arnett et al., 2004; Kitada and Rowitch, 2006; Niu et al., 2012). Therefore, we predicted that Olig1 must have nuclear export sequences (NES) not found in Olig2 and analyzed the Olig1 protein sequence for potential NES. Three consensus NES were identified either within or adjacent to the bHLH domain of Olig1 (Fig. 1A). The sequence of NES1 was fully conserved with Olig2, but there were two additional NES sequences in Olig1 (NES2 and NES3; Fig. 1A,B). The three Olig1 NES were conserved across different species from mouse to human, but not zebrafish (Danio rerio; Fig. 1B).
Figure 1.
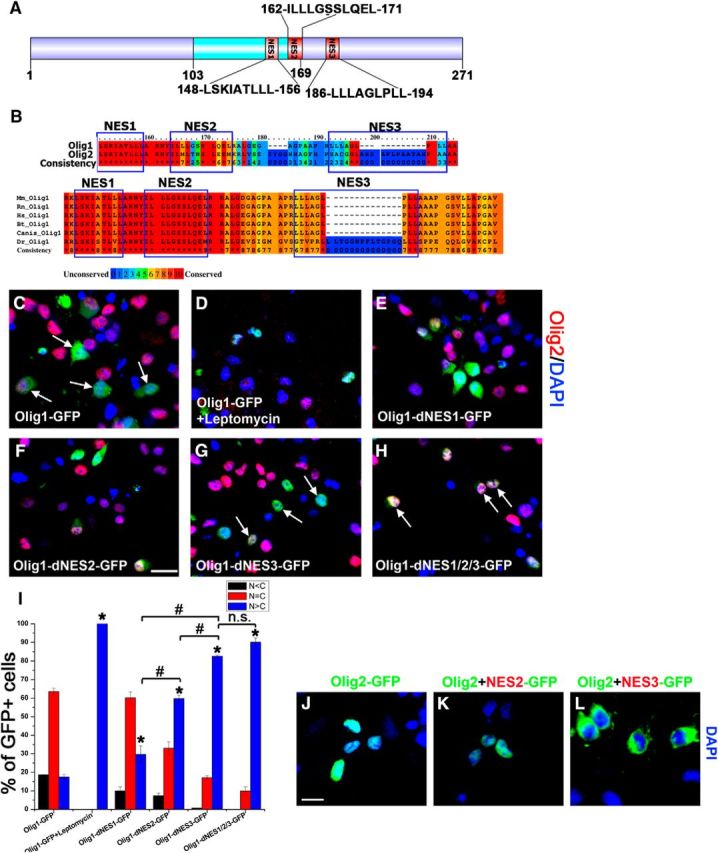
Three NESs of Olig1 regulate its export. A, Structural features of human Olig1 protein showing the conserved bHLH domain. Three predicted NESs, as determined by analysis with NetNES1.1, are highlighted in red. B, Alignment of the bHLH domain of human Olig1 and Olig2 (top). Note high conservation of NES1, with less for NES2 and NES3. Sequence of this Olig1 region among different species with the three NES domains boxed in blue (bottom). Note unique D. rerio (zebrafish) sequence in blue highlight that disrupts NES3 in that species. C–H, Subcellular location of GFP-tagged Olig1 or its mutants deleted for the indicated NES sequences (dNES) 48 h after transfection into cultured rat OPCs. C, Olig1-GFP, note cytoplasmic localization (arrow). D, Olig1-GFP plus leptomycin (20 nm); E, Olig1-dNES1-GFP. F, Olig1-dNES2-GFP. G, Olig1-dNES3-GFP, note nuclear localization (arrow). H, Olig1-dNES1/2/3-GFP, note nuclear localization (arrow). Costained with Olig2 (red), DAPI (blue). Scale bar, 15 μm. Arrow labels transfected OPCs. I, Quantitative analysis of the subcellular distribution of Olig1-GFP or mutants imaged in C–H. Data were quantified as N < C (black), N = C (red), or N > C (blue). Data represent the mean ± SEM from at least three independent experiments and, for each different construct in each experiment, at least 300 transfected cells were counted. The percentage of nuclear localization of indicated mutants was compared with that of WT Olig1(*p < 0.05) and between different mutants (#p < 0.05; one-way ANOVA). J, K, Impact of ectopic Olig1 nuclear export sequences on Olig2 subcellular location. Olig2-GFP (J), chimeric Olig2 with nuclear export sequence 2 derived from Olig1 (Olig2+NES2-GFP; K), and chimeric Olig2 with nuclear export sequence 3 derived from Olig1 (Olig2+NES3-GFP; L), note cytoplasmic localization. Scale bar, 10 μm.
To test the role of the conserved Olig1 NES in regulating nuclear export, GFP-tagged human Olig1 was transfected into primary cultured OPCs. In contrast to dominant nuclear location of Olig1 in OPCs in vivo, the majority of the Olig1-GFP (63.59%) was evenly distributed in the nucleus and cytoplasm (N = C) in transfected OPCs in vitro (Fig. 1C,I). To test active export of Olig1-GFP after overexpression in cultured OPCs, cells were treated with the nuclear exportin inhibitor leptomycin. In contrast to untreated transfected cells (Fig. 1C), all of the Olig1-GFP localization was nuclear after leptomycin treatment (N > C; Fig. 1D,I). To investigate the role of the three Olig1 NES, they were deleted individually or collectively. After NES1, NES2, or NES3 were individually deleted, nuclear Olig1 increased significantly to 29.5% (dNES1), 59.75% (dNES2), and 82.64% (dNES3), respectively, compared with 17.45% nuclear retention of wild-type Olig1 (Fig. 1E,F,G,I). Consistent with this, when all three Olig1 NES were collectively deleted, Olig1 cytoplasmic export was severely blocked, with 92.5% of Olig1 retained in the nucleus (Fig. 1H,I). These data suggest that the three predicted NES regulate Olig1 export together and eliminating any one alters Olig1 export out of the nucleus to a different extent. Among them, NES3 functions as the strongest export signal. Interestingly, NES3 is the least conserved between Olig1 and Olig2 (Fig. 1B). To test whether the extra NES2 and/or NES3 of Olig1 could explain its nuclear export, in contrast to the predominant nuclear location of Olig2, we introduced NES2 or NES3 sequences into the Olig2 protein to examine directly their impact on Olig2 subcellular location. GFP-tagged human Olig2 and chimeric Olig2 with NES2 or NES3 of Olig1 were transfected into primary cultured OPCs. Olig2-GFP was found predominantly in the nucleus, as is endogenous Olig2 (Fig. 1J). Surprisingly, adding the Olig1 NES2 sequence to Olig2 did not change its nuclear location (Fig. 1K), but inserting the NES3 sequence of Olig1 to Olig2 forced complete export of Olig2 to the cytoplasm (Fig. 1L). These data support the function of NES3 as a strong nuclear export signal because its insertion into Olig2 is sufficient to drive nuclear export of Olig2. However, despite the presence of the three NES sequences in Olig1, including the strong nuclear export signal NES3, the majority of Olig1 is not completely exported to cytoplasm in cultured OPCs. From this, we concluded that all three NES are necessary but not sufficient to drive the complete nuclear export of Olig1. Additional mechanisms must exist to regulate the active export of Olig1 from the nucleus.
Acetylation of Olig1 is correlated with its cytoplasmic localization
In the developing mouse brain, Olig1 localized only to the nucleus of OPCs that expressed PDGFRα at P1 (Fig. 2A). At P10, Olig1 was present in both the nucleus and the cytoplasm in maturing oligodendrocytes that expressed CC1 (Fig. 2B) and it was completely cytoplasmic in myelinating oligodendrocytes at 1 month (Fig. 2C). These results were consistent with previous findings (Arnett et al., 2004; Niu et al., 2012; Dai et al., 2015) and suggested that Olig1 translocation to the cytoplasm in maturing oligodendrocytes at P10 is an actively regulated process. The fact that, despite the presence of the three consensus nuclear export sequences in Olig1, its nuclear export occurs only in myelinating oligodendrocytes but not in OPCs suggests that additional modification of the Olig1 protein may regulate its translocation as oligodendrocytes mature.
Figure 2.
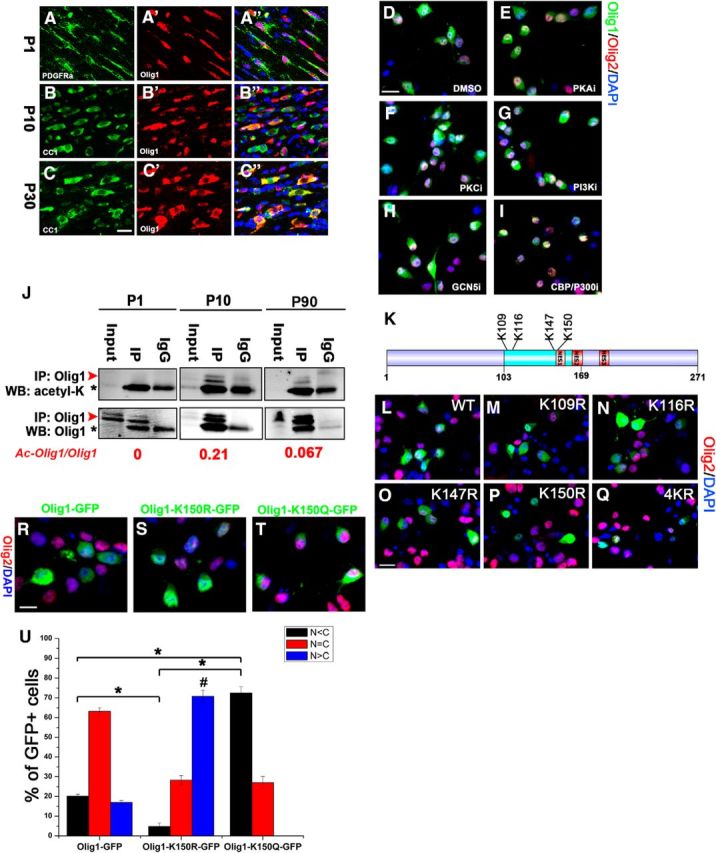
Acetylation of Olig1 is correlated with its cytoplasmic localization in developing mouse brain. A–C, Double immunostaining of Olig1 (red, A′, B′, C′) with PDGFRα (OPCs, green, A, A″) or CC1 (mature oligodendrocytes, green, B, C, B″, C″) in corpus callosum of P1 (A), P10 (B), or P30 (C) mice (blue in A″, B′, C″ is DAPI). Olig1 is in the nucleus of PDGFRα-positive OPCs at P1 (A), switches from the nucleus to cytoplasm of CC1-positive oligodendrocytes at P10 (B), and is exclusively localized to the cytoplasm in CC1-positive cells at P30 (C). Scale bar, 20 μm. D–I, Primary cultured rat OPCs were treated with DMSO (D); the kinase inhibitors protein kinase A inhibitor, 10 μm H89 (B), PKC inhibitor, 10 μm Gö6976 (F); PI3K inhibitor, 25 μm LY294002 (G); or different histone acetyltransferase inhibitors: CBP/P300 inhibitor, 20 μm C646 (H), GCN5 inhibitor, 100 μm MB-3 (I). Twenty-four hours after treatment, Olig1 subcellular locations were determined by Olig1 antibody staining (green) and costained with Olig2 (red) or DAPI (blue). Scale bar, 15 μm. J, Olig1 was acetylated in developing mouse brain. Endogenous Olig1 was immunoprecipitated from subcortical white matter homogenates of different postnatal ages as indicated and Olig1 acetylation was detected with general acetylated-lysine antibody. Arrowheads (red), Olig1; asterisks, rabbit IgG. Ratio of acetylated Olig1 to total Olig1 is indicated below in red. K, Schematic illustrating the predicted acetylated lysine sites in the Olig1 bHLH domain and NES sequences, including K109, K116, K147, and K150. L–Q, Subcellular location of deacetylated mimic of four different predicted lysine sites in cultured rat OPCs. WT Olig1-GFP (L), Olig1-K109R-GFP (M), Olig1-K116R-GFP (N), Olig1-K147R-GFP (O), Olig1-K150R-GFP (P), or Olig1-K109K116K147K150R(4KR)-GFP (Q) mutants were transfected into the rat OPCs and Olig1 subcellular location was determined 48 h later and costained with Olig2 (red) or DAPI (blue). Scale bar, 15 μm. R–T, Acetylation of lysine (K), 150 on Olig1 regulated its cytoplasmic localization in cultured rat OPCs. Olig1-GFP (R), Olig1-K150R-GFP (S), or Olig1-K150Q-GFP (T) mutants were transfected into the rat OPCs and Olig1 subcellular location was analyzed 48 h later in transfected OLs using Olig2 (red) and DAPI (blue) to localize nuclei. Scale bar, 10 μm. U, Quantitative analysis of the subcellular distribution of Olig1-GFP or its mutants imaged in R–T. Data were quantified as nuclear N < C (black), N = C (red), or N > C (blue). Data represent the mean ± SEM from at least three independent experiments and, for each different construct in each experiment, at least 300 transfected cells were counted. The percentage of cytoplasmic localization of indicated mutants was compared with each other (*p < 0.05) and nuclear localization of indicated mutants were compared with wild-type Olig1 (#p < 0.05; one-way ANOVA).
Phosphorylation and acetylation are well known posttranslational modifications that can regulate the nuclear-cytoplasmic shuttle of many transcription factors (di Bari et al., 2006; Krämer et al., 2009). To test the role of phosphorylation or acetylation in regulating Olig1 nuclear to cytoplasmic translocation, we applied a variety of kinase inhibitors (protein kinase A, protein kinase C or Akt) and HAT inhibitors (C646, MB-3) to primary cultured OPCs (Fig. 2D–I). Only C646, which inhibits CBP and P300, blocked the nuclear to cytoplasmic translocation of Olig1 (Fig. 2I). This suggested that Olig1 acetylation could regulate its nuclear to cytoplasmic translocation.
To determine whether acetylation played a role in Olig1 translocation in vivo, we investigated whether Olig1 translocation from the nucleus to the cytoplasm correlated with increased Olig1 acetylation in the brain. At P1, when Olig1 was in OPC nuclei (Fig. 2A), it was very minimally acetylated (Fig. 2J, red arrowhead, not asterisk; note endogenous mouse Olig1 migrates very near normal IgG and is often the minor band seen above the nonspecific IgG band in blots of immunoprecipitated mouse tissue). The peak of Olig1 acetylation was at P10, when the Olig1 nuclear to cytoplasmic translocation occurred (Fig. 2B,J). At P30, Olig1 acetylation was reduced compared with P10 (Fig. 2J). Therefore, Olig1 acetylation increased during oligodendrocyte maturation in vivo, particularly during the translocation period, which suggested that Olig1 acetylation could regulate its cytoplasmic translocation, but it was not required for maintenance in the adult oligodendrocyte cytoplasm.
Olig1 lysine (K) 150 acetylation regulated Olig1 cytoplasmic localization
The Olig1 protein sequence was analyzed for potential acetylated lysines using the Prediction of Acetylation on Internal Lysines (PAIL) program (Li et al., 2006), and four (K109, K116, K147, K150) potential acetylateable lysines were identified in the Olig1 bHLH domain (Fig. 2K, Table 1). Analysis of Olig1 protein sequences from different species revealed that the predicted lysine sites are conserved. Each lysine was mutated to arginine (R) to generate individual mutants that mimic the deacetylated status and a mutant Olig1 with all four R mutations at K109, K116, K147, and K150 (4KR) was generated. These deacetylation-mimicking mutants were transfected into primary cultured OPCs and their subcellular location was determined (Fig. 2L–Q). K150 was the only lysine in the Olig1 NES (Fig. 2K) and K150R was the only single mutation that showed nuclear retention (Fig. 2P).
Table 1.
Predicted Olig1 acetylation sites
| Peptide | Position | Score |
|---|---|---|
| TAPLLPKAAREKP | 67 | 1.15 |
| PKAAREKPEAPAE | 72 | 1.91 |
| QQQLRRKINSRER | 109 | 0.81 |
| INSRERKRMQDLN | 116 | 1.07 |
| QGAPGRKLSKIAT | 147 | 2.10 |
| PGRKLSKIATLLL | 150 | 2.23 |
| DALRPAKYLSLAL | 218 | 1.80 |
| CTCAVCKFPHLVP | 251 | 1.51 |
Potential acetylation sites of human Olig1 were predicted by the PAIL server (http://bdmpail.biocuckoo.org/prediction.php) with a 0.75 threshold cutoff.
To further evaluate the role of K150 acetylation in regulating Olig1 subcellular localization, the K150 was mutated to glutamine (Q) to mimic acetylation or to arginine to mimic the nonacetylated form. WT-Olig1-GFP, Olig1-K150R-GFP, and Olig1-K150Q-GFP were electroporated into primary cultured rat OPCs. WT Olig1 was present in both the nucleus and cytoplasm (Fig. 2R,U), which was similar to endogenous Olig1 in cultured OPCs (see Fig. 9A). Nuclear retention of Olig1-K150R was found in 78.8% of the transfected OPCs (Fig. 2S,U). Strikingly, the acetylated Olig1 mimic Olig1-K150Q was frequently found to be exclusively localized to rat OPC cytoplasm (Fig. 2T,U; 72.5% C > N).
Figure 9.
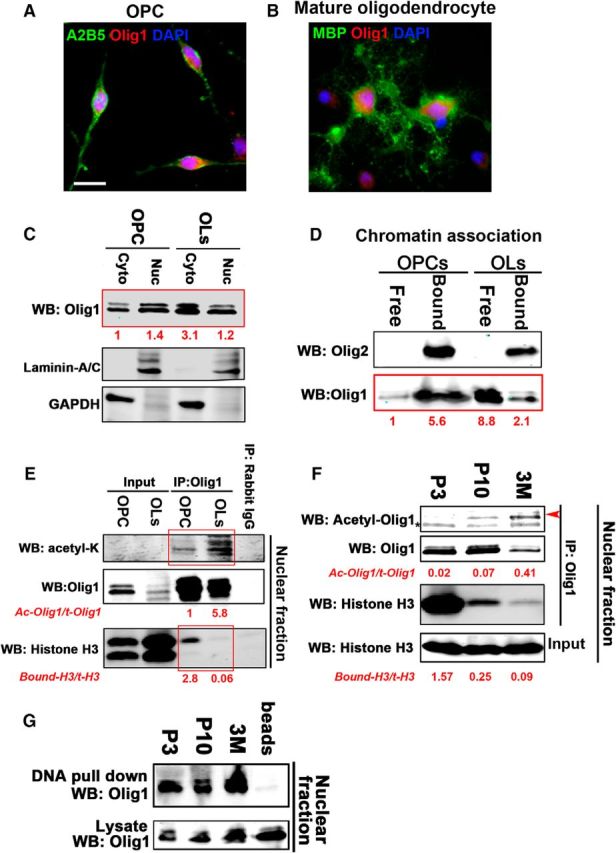
Increased acetylation of Olig1 decreased its chromatin binding as oligodendrocytes differentiated. A, B, Primary cultured rat OPCs were either maintained in proliferation media for 3 d (A) or differentiated for another 5 d and stained with Olig1 antibody (red). OPCs were detected with A2B5 (green) and mature oligodendrocytes were labeled with MBP. Scale bar, 15 μm. C, Some Olig1 was retained in the nucleus of mature oligodendrocytes in culture. Nuclear and cytoplasmic fractions were isolated from cultured OPCs or differentiated oligodendrocytes. The subcellular distribution of endogenous Olig1 was determined by Western blot. Laminin A/C and GAPDH were used as markers for nuclear and cytoplasmic fractions, respectively (bottom row). Quantification of the Olig1 level is indicated in red and normalized to that of the cytoplasmic OPC fraction. D, Olig1 dissociated from chromatin as oligodendrocytes differentiated in vitro. The chromatin and soluble nuclear fractions of cultured OPCs (PDGF/bFGF, 3 d) or differentiated oligodendrocytes (T3, 3 d) were isolated by subnuclear fractionation and the presence of Olig1 or Olig2 in either the soluble or pelleted chromatin fraction was determined by Western blot. Note the presence of Olig1 primarily in the chromatin pellet in OPCs, but primarily in the soluble fraction, dissociated from chromatin, in cultured oligodendrocytes. Quantification of the Olig1 level indicated in red and normalized to that from chromatin-free fraction of OPCs. E, Binding of Olig1 with Histone H3 decreased as Olig1 acetylation increased in cultured oligodendrocytes. Mouse OPCs were maintained in PDGF/bFGF for 3 d or differentiated for 5 d. Cell lysates were then immunoprecipitated with Olig1 antibody and immunoblotted for acetyl-lysine or histone H3 association (note boxed area). Quantification of the ratio of acetylated Olig1 to total Olig1 and indicated in red, normalized to that from OPCs, and Olig1-bound histone H3 is indicated in red. F, In developing mouse brain, Olig1 binding with histone H3 decreased as oligodendrocytes matured. Subcortical white matter from different postnatal ages was fractionated into nuclear and cytoplasmic fractions. Endogenous Olig1 was immunoprecipitated from the nuclear fraction with Olig1 antibody. Olig1 acetylation was detected with acetyl-Olig1-K150 antibody (top), and Olig1-bound histone H3 was detected with histone H3 antibody. Arrowheads, Acetyl-Olig1; asterisks, rabbit IgG. Note increase in acetylated Olig1 with age at the same time that the Olig1 association with histone decreased. Quantification of the ratio of acetylated Olig1 to total Olig1 and Olig1 bound histone H3 to total histone H3 indicated in red. G, DNA-binding affinity of Olig1 did not change as oligodendrocytes matured in the developing mouse brain. The same nuclear fractions as in E were incubated with a streptavidin-coupled tandem E-box consensus DNA probe. Endogenous Olig1 was pulled down with DNA probes and detected with Olig1 antibody. Note comparable association of Olig1 with DNA regardless of the age of the samples. All data represent the results from three independent experiments.
Interestingly, the lysine 150 site is located in the NES1 of Olig1 and this site is conserved in Olig2. To assess whether acetylation of K150 was unique to Olig1, the acetylation of Olig2 was studied in developing brain. An antibody was generated that specifically recognized acetylated K150 in Olig1 (see Fig. 6). That sequence is identical in Olig2 and, using that antibody on immunoprecipitation/Western blots of tissue samples, it was clear that Olig2 was acetylated at K148 (the equivalent lysine site in Olig2 for K150 in Olig1) in vivo (Fig. 3A). However, in contrast to Olig1, Olig2 K148 acetylation was found primarily at P1, likely in OPCs, and very little in myelinating oligodendrocytes at P10 or 3 months (Fig. 3A). This result indicates that, although Olig2 can be acetylated at the counterpart lysine site to that in Olig1, their acetylation is differently regulated. Clearly, acetylation of Olig2 is not sufficient to drive its nuclear export in OPCs in vivo. Furthermore, when an Olig2 lysine 148 acetylation mimic (Olig2-K148Q) was transfected into primary culture OPCs, it was predominantly localized in the nucleus of OPCs (Fig. 3B,C). Importantly, acetylation of K150 in Olig1 was also not sufficient to drive Olig1 nuclear export in the absence of NES3 (Fig. 3D–G). Therefore, Olig1 acetylation at lysine 150 is required for its cytoplasmic localization in primary cultured oligodendrocytes, but the active nuclear export of Olig1 also requires the presence of NES3.
Figure 6.
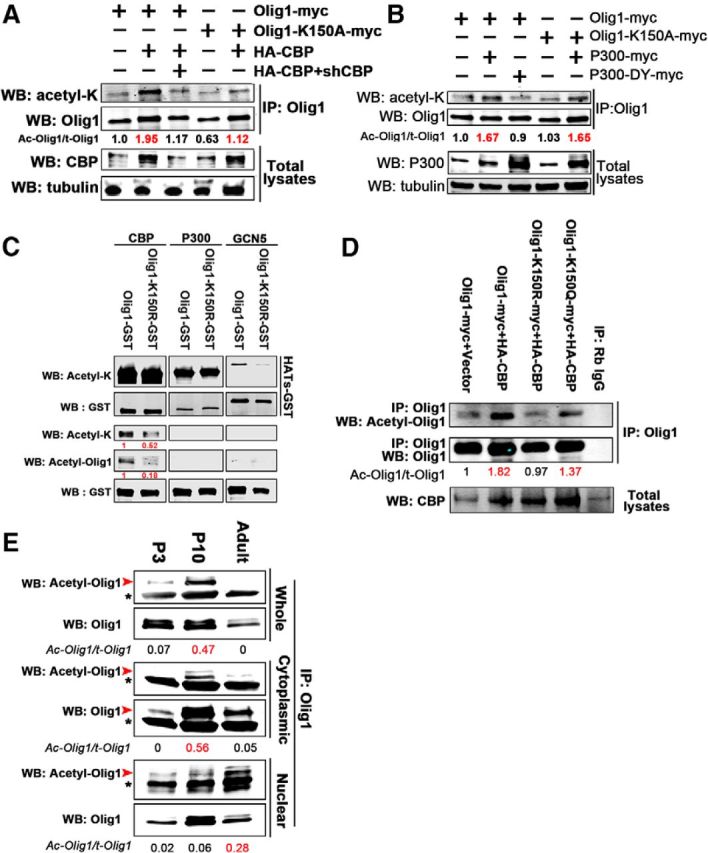
Olig1 is acetylated at lysine 150 in vivo. A, B, Olig1 is acetylated at lysine 150 by CBP in HEK293T cells. Olig1-myc or Olig1-K150A-myc was cotransfected with HA-CBP plus/minus CBP shRNA A, or with p300-myc or p300DY-myc (acetylase-deficient p300; B) into HEK293T cells. Note increased acetylation in the presence of HA-CBP, which is reduced for Olig1-K150A-myc (A) or by the presence of shCBP. However, increased acetylation by P300 is comparable for WT Olig1 and Olig1 K150A-myc (B). C, The acetyl-Olig1-K150 antibody specifically recognized recombinant human Olig1 protein acetylated on lysine 150. One microgram of recombinant WT Olig1 or acetylation-deficient Olig1 mutant protein (Olig1-K150R) was incubated in vitro with purified CBP, P300, or GCN5 and their acetylation was detected by immunoblotting with acetyl-Olig1-K150 antibody, acetylated lysine antibody, or GST antibody. The activity of the different HATs was indicated by auto-acetylation of the GST-acetyltransferase detected by acetylated lysine antibody (top rows). The GST-Olig1 proteins were detected by GST immunostaining (bottom rows) and quantification of the ratio of acetylated Olig1 to total Olig1 is presented in red, normalized to the WT control. D, Olig1 was acetylated at lysine 150 by CBP in HEK293T cells, as detected by acetyl-Olig1-K150 antibody. Olig1-myc, Olig1-K150R-myc, or Olig1-K150Q-myc was cotransfected with HA-CBP into HEK293T cells. Twenty-four hours later, cells were treated with 500 nm TSA for 10 h to inhibit deacetylation before the cell lysate was immunoprecipitated with Olig1 antibody and detected with acetyl-Olig1-K150 antibody. E, Olig1 was acetylated at lysine 150 in developing mouse brain. Subcortical white matter from P3, P10, or adult samples was fractionated and endogenous Olig1 was immunoprecipitated from whole lysates or cytoplasmic or nuclear fractions and immunoblotted with acetyl-Olig1-K150 antibody. Arrowheads (red), Olig1; asterisks (black), rabbit IgG. All data represent the results from three independent experiments. Quantification of the ratio of acetylated Olig1 to total Olig1 is indicated below in red.
Figure 3.
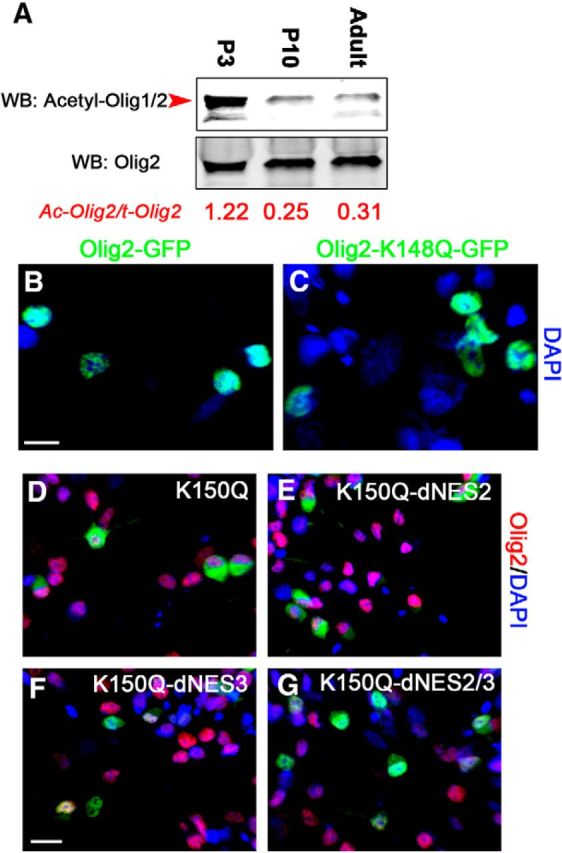
Acetylation of Olig2 does not alter its subcellular location. A, Olig2 was acetylated in developing mouse brain at P3. Endogenous Olig2 was immunoprecipitated from subcortical white matter homogenates of different postnatal ages as indicated and Olig2 acetylation was detected with acetyl-Olig1-K150 antibody that targets the acetylated K150 epitope that is conserved between Olig1 and Olig2. Arrowheads (red), Olig2. Ratio of acetylated Olig2:total Olig2 indicated below in red. Note it is high at P3. B, C, Acetylation of lysine (K)148 on Olig2 (equivalent to K150 of Olig1) did not induce nuclear export of Olig2 in cultured rat OPCs. Olig2-GFP (B) or Olig2-K150Q-GFP (C) mutants were transfected into the rat OPCs and Olig2 subcellular location was determined 48 h later using DAPI (blue) to localize nuclei. Scale bar, 10 μm. D–G, Acetylation of lysine (K)150 on Olig1 is insufficient to drive its nuclear export in cultured rat OPCs. Shown is the Olig1 K150 acetylation mimic (Olig1-K150Q-GFP) or Olig1-K150Q mutant with deletion of the indicated NES sequences (dNES) 48 h after transfection into cultured rat OPCs. Olig1-K150Q-GFP (D), Olig1-K150Q-dNES2-GFP (E), Olig1-K150Q-dNES3-GFP (D), and Olig1-K150Q-dNES2/3-GFP (D) using Olig2 (red) and DAPI (blue) to localize nuclei. Scale bar, 15 μm.
Acetylation of Olig1 is critical for its role in morphological differentiation and lineage progression
Our earlier studies (Dai et al., 2015) establish that Olig1 is critical for initial OPC differentiation both in culture and in mouse brain. Because Olig1 acetylation at K150 correlated with oligodendrocyte maturation both in vitro and in vivo, we examined whether Olig1 K150 acetylation had a direct impact on oligodendrocyte differentiation. To introduce the WT Olig1 or Olig1 lysine 150 mutants into differentiating OPCs, we treated cells with HA-tagged Olig1 protein linked to the cell-penetrating peptide TAT. TAT cell-penetrating peptide has been widely used to help transduce tagged protein into cells both in vitro and in vivo (Soane and Fiskum, 2005; Hotchkiss et al., 2006; Mi et al., 2012; O'Meara et al., 2013). The proteins were taken up by OPCs after 1 d of growth in differentiation medium plus purified TAT-tagged proteins, as demonstrated both by immunostaining and Western blotting (Fig. 4A,B). This allowed us to study overexpression of WT Olig1 or its lysine 150 mutants during oligodendrocyte differentiation. Control cells or Olig1-knock down cells were generated by electroporating OPCs with control siRNA (Fig. 5A–D) or Olig1 siRNA (Fig. 5E–H). Differentiation of control cells or Olig1-knock-down cells was then studied after incubation with TAT-tagged WT or mutant Olig1 proteins. Initial studies were done to analyze the effect of WT Olig1 overexpression in control cells that express endogenous Olig1. This overexpression did not influence their initial lineage progression, as measured by the number of O4-positive cells (Fig. 5A,B,I, cf. TAT-GFP-treated, TAT-Olig1-treated cells). However, WT Olig1 overexpression in control cells significantly increased their morphological differentiation, as measured by total covered area (Fig. 5A,B,J). The overexpression of the K150 acetylation mimic Olig1 K150Q in control cells significantly promoted both initial OPC differentiation to O4-positive cells (Fig. 5A,C,I) and their morphological differentiation (Fig. 5A,C,J). In contrast, overexpression of the K150 deacetylation mimic Olig1-K150R impaired OPC lineage progression and reduced the total covered area of O4-positive cells by 36%, despite the presence of endogenous Olig1 (Fig. 5A,D,I,J). These results suggested that Olig1 acetylation at K150 is critical for promoting oligodendrocyte lineage progression and for morphological differentiation.
Figure 4.
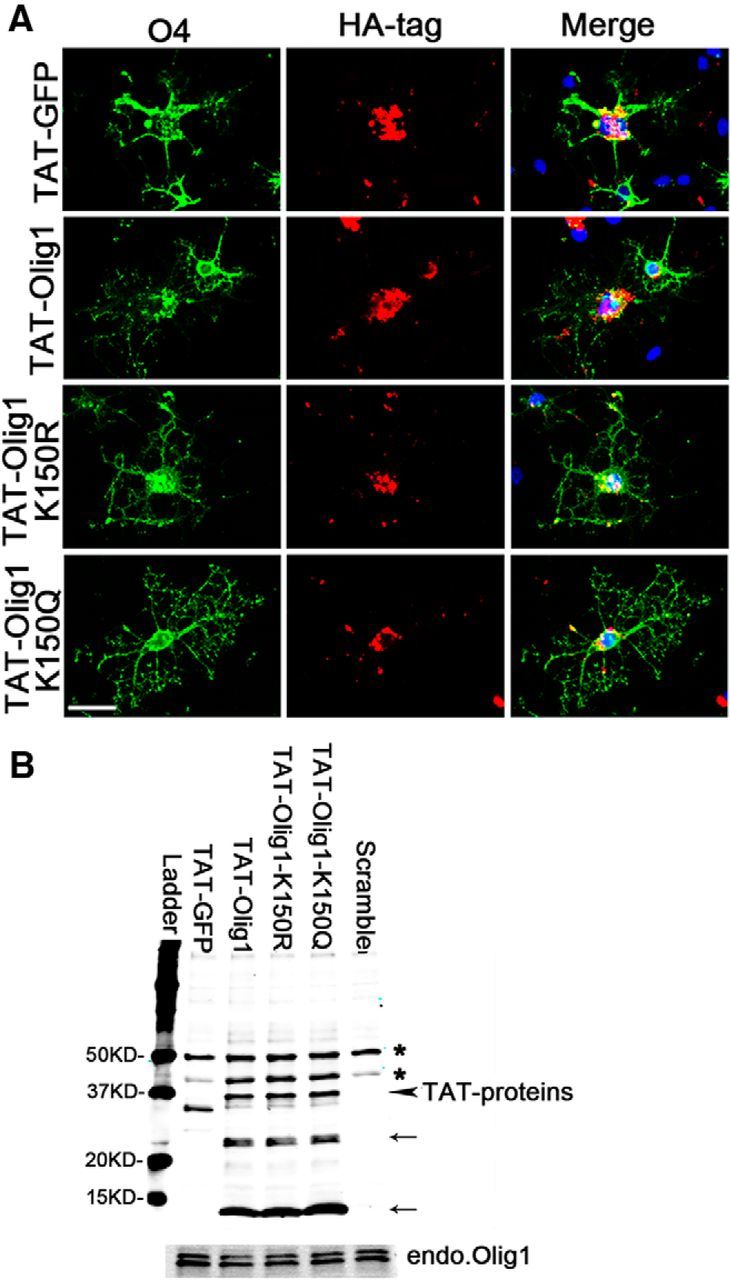
Transduction of primary oligodendrocyte with TAT-tagged purified Olig1 mutant proteins. A, Cultured rat oligodendrocytes were electroporated with smart pool control siRNA and recovered in proliferation media for 1 d and then shifted to differentiation medium with addition of either purified HA-tagged TAT-GFP, TAT-Olig1, TAT-Olig1-K150R, or TAT-Olig1-K150Q protein (0.4 μm). After differentiation for 1 d, the cells were costained with O4- and anti-HA-tagged antibodies. Scale bar, 50 μm. B, Cultured rat oligodendrocytes were grown and treated as in A, collected, and immunoblotted with HA antibody and Olig1 antibody. Arrows, Olig1 degradation products; arrowhead, TAT-Olig1 protein; asterisks, nonspecific bands.
Figure 5.
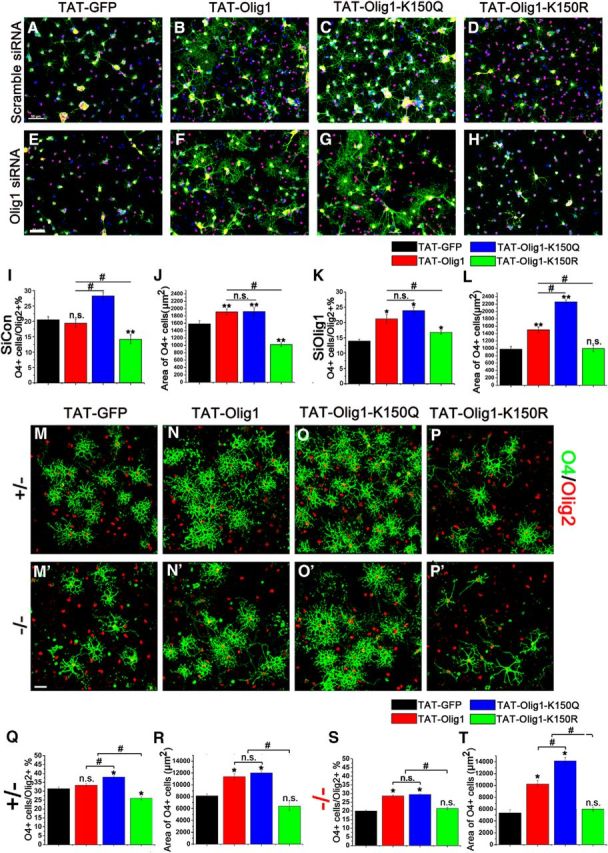
Acetylation of Olig1 is critical for its role in morphological differentiation and lineage progression. A–H, Cultured rat oligodendrocytes were electroporated with smart pool control siRNA (A–D) or Olig1 siRNA (E–H) and recovered in differentiation media for 1 d. Cells were then switched to differentiation medium and purified TAT-GFP (A, E), TAT-Olig1 (B, F), TAT-Olig1-K150Q (C, G), or TAT-Olig1-K150R (D, H) protein (0.4 μm) was added. After differentiation for 1 d, the cells were live stained with O4 antibody (green) and costained with Olig2 antibody (red). Scale bar, 50 μm. I–L, Quantitative analysis of the impact of TAT-GFP, TAT-Olig1, TAT-Olig1-K150R, or TAT-Olig1-K150Q protein transduction on OPC differentiation with control siRNA shown in A–D (I, J) or after Olig1 siRNA knock-down shown in E–H (K, L). The percentage of Olig2-positive cells that express O4 (I, K) and average covered area of O4-positive cells (J, L) were quantified. M–P, Cultured mouse oligodendrocytes derived from Olig1 heterozygous (+/−; M–P) or Olig1 null (−/−; M′–P′) mice were differentiated for 3 d together with purified TAT-GFP (M, M′), TAT-Olig1 (N, N′), TAT-Olig1-K150Q (O, O′), or TAT-Olig1-K150R (P, P′) protein (0.4 μm). Cells were live stained with O4 antibody (green) and costained with Olig2 antibody (red). Scale bar, 50 μm. Q–T, Quantitative analysis of the impact of TAT-GFP, TAT-Olig1, TAT-Olig1-K150R, or TAT-Olig1-K150Q protein transduction on differentiation of OPCs derived from either Olig1 heterzygous neurospheres shown in M–P (Q, R) or Olig1-null neurospheres shown in M′–P′ (S, T). The percentage of Olig2-positive cells that express O4 (Q, S) and average covered area of O4-positive cells (R, T) were quantified. For all experiments, the data represent the mean ± SEM from three independent experiments and, in each condition, at least 300 cells were examined visually and counted. Data of TAT-Olig1 or mutants treated group were compared with that of TAT-GFP treated group (*p < 0.05, **p < 0.005) and between different mutants (#p < 0.05; one-way ANOVA).
The role of Olig1 K150 acetylation in regulating oligodendrocyte differentiation was further examined in Olig1-knock down cells. As noted above, oligodendrocyte differentiation was significantly reduced upon Olig1 knock-down (cf. Fig. 5A,E,I,K). Lineage progression (the percentage of O4-positive cells) and morphological differentiation were rescued by reintroduction of either WT Olig1 (Fig. 5F,K,L) or the acetylation mimic Olig1-K150Q (Fig. 5G,K,L) and the impact of Olig1-K150Q on morphological differentiation was more dramatic than that of WT Olig1 (Fig. 5L). In contrast, the deacetylation mimic Olig1-K150R failed to rescue the morphological differentiation defect after Olig1 knock-down (Fig. 6H,L). Consistent with these results (Fig. 5M–T), the TAT-Olig1 and the TAT-Olig1-K150Q acetylation mimic were also able to rescue the differentiation deficit of OPCs derived from Olig1-null mice (Fig. 5N′,O′, S,T), but the deactylated mimic TAT-Olig1-K150R was not (Fig. 5P′,S,T). These results confirmed that K150 acetylation of Olig1 is critical for its role in oligodendrocyte differentiation and membrane expansion.
Olig1 is acetylated at lysine 150 in vivo
As noted above, to investigate the role of Olig1 K150 acetylation in vivo, antibodies specific to the acetylated lysine 150 of Olig1 were generated. We first started to identify the histone acetyltransferase (HAT) responsible for Olig1 acetylation at K150. Among the four major HATs, CBP (cAMP response element-binding protein [CREB]-binding protein) and P300 (E1A-binding protein, 300 kDa) appeared most likely to be the HATs responsible for Olig1 acetylation at K150, since Olig1 nuclear export was blocked by CBP/P300 inhibitor C646 (Fig. 2I). Indeed, both CBP and p300 overexpression increased Olig1 acetylation in 293T cells (Fig. 6A,B). Furthermore, Olig1 K150 acetylation by CBP overexpression, but not P300, was largely abolished by the single mutation at lysine 150 in Olig1 (Fig. 6A,B). These results confirm that CBP is the HAT responsible for Olig1 K150 acetylation.
Furthermore, the acetyl-Olig1-K150 (acetyl-Olig1) antibody specificity was confirmed by its ability to recognize purified WT Olig1 that was acetylated by CBP in vitro, but not the Olig1-K150R mutant that could not be acetylated at K150 (Fig. 6C). Overexpression of CBP in HEK293T cells increased WT Olig1 acetylation at K150, but not that of the K150 de-acetylated mimic Olig1-K150R (Fig. 6D).Therefore, Olig1 was acetylated by CBP at K150 both in vitro and in vivo and this acetylated K150 could be specifically recognized by the acetyl-Olig1 antibody.
To identify Olig1 K150 acetylation in developing mouse brain, endogenous Olig1 was immunoprecipitated from P3, P10, or 3 month subcortical white matter lysates or subcellular fractions. Consistent with our earlier results (Fig. 2J), studies with acetyl-Olig1 antibody demonstrated that Olig1 K150 acetylation peaked in the P10 fraction, when Olig1 translocation was occurring (Fig. 6E, red arrow). Furthermore, acetylated Olig1 K150 was enriched in the cytoplasmic fraction of the tissue, not the nuclear fraction. Acetylated Olig1 K150 was almost undetectable at P3 or adult in the cytoplasmic fraction, when Olig1 was either predominantly nuclear (P3) or cytoplasmic (adult), respectively. Weak Olig1 K150 acetylation was detected in the P10 nuclear fraction at the time Olig1 would be translocating to the cytoplasm but, interestingly, at 3 months, there was increased nuclear Olig1 K150 acetylation compared with P10 (Fig. 6E). Because Olig1 is not nuclear in mature oligodendrocytes in the adult, the acetylated nuclear Olig1 protein detected by Western blot is likely in adult OPCs. Overall, these studies suggest that acetylation of Olig1 at K150 is developmentally regulated and peaks when Olig1 is actively translocating from the nucleus to the cytoplasm.
Olig1 acetylation is regulated by preferentially interacting with CBP and HDAC1
Olig1 acetylation increased as OPCs differentiated to oligodendrocytes in vitro (Fig. 7A) and the interaction between Olig1 and CBP increased in the nucleus of differentiated cells compared with that in OPCs (Fig. 7B). Presumably, Olig1 acetylation must be regulated both by acetyltransferases and by deacetylases. To identify the Olig1 deacetylase, we coexpressed Olig1 with HDAC1 through HDAC10, Sirt1, or Sirt2 and immunoprecipitated samples to assess potential interactions. Olig1 interacted with HDAC1, HDAC2, HDAC3, HDAC4, HDAC10, and Sirt2 (data not shown). When Olig1 was coexpressed with these candidate HDACs plus excess CBP, HDAC1, HDAC3, and HDAC10 decreased Olig1 acetylation (Fig. 7C), but HDAC2, HDAC4, and Sirt2 did not (data not shown). HDAC1, HDAC3, and HDAC10 deacetylated Olig1 K150, the validated CBP acetylation site (Fig. 7C). Because HDAC1, HDAC3, and HDAC10 are also highly expressed in oligodendrocytes in the corpus callosum in vivo (Shen et al., 2005), these results suggested that HDAC1, HDAC3, and/or HDAC10 are present in oligodendrocytes and could potentially deacetylate Olig1 specifically at lysine 150 in vivo.
Figure 7.

Olig1 acetylation is regulated by preferential interaction with CBP and HDAC1. A, Olig1 was acetylated in cultured rat oligodendrocytes. Cultured rat OPCs or oligodendrocyte cell lysates were immunoprecipitated with an Olig1 antibody and detected with an acetyl-lysine antibody (top) or Olig1 antibody (middle). Merged Western bands are shown in the bottom panel. B, Olig1 interacted with CBP in nuclei of cultured rat oligodendrocytes and this interaction increased as oligodendrocytes differentiated. Nuclear lysates from rat OPCs or differentiated oligodendrocytes were immunoprecipitated with CBP antibody and detected with Olig1 antibody. Quantification of the ratio of immunoprecipitated Olig1 to total Olig1 indicated below in red, normalized to that from OPCs. C, HDAC1, HDAC3, and HDAC10 were potential HDACs responsible for Olig1 acetyl-K150 deacetylation in vivo. The indicated constructs were cotransfected into HEK293T cells. Six hours after transfection, cells were treated with 500 nm TSA overnight and fresh medium was then added for another 10 h. Cell homogenates were immunoprecipitated with Olig1 antibody and detected with acetyl-Olig1-K150 antibody. Quantification of the ratio of acetylated Olig1 to total Olig1 is indicated below in red, normalized to that of empty vector. D, Olig1 preferentially interacted with HDAC1 or CBP in developing mouse brain. Endogenous HDAC1 was immunoprecipitated from subcortical white matter homogenates from different postnatal developmental ages and bound Olig1 was detected with Olig1 antibody as indicated (top row). Endogenous Olig1 was immunoprecipitated from the same samples and bound CBP was detected with CBP antibody (middle row). Quantification of the ratio of immunoprecipitated Olig1 to total Olig1 (normalized to that from P3 tissue) and immunoprecipitated CBP to total CBP indicated below in red, normalized to that from OPCs. Note the high association of Olig1 and HDAC1 in P3 tissue and high association of Olig1 and CBP in P10 tissue.
Interestingly, in vivo the Olig1 interaction was higher with HDAC1 in P3 brain (Fig. 7D) when deacetylated Olig1 was greater than acetylated Olig1 (Fig. 7D). Conversely, the increased interaction of Olig1 and CBP in P10 brain correlated with elevated Olig1 acetylation (Fig. 7E). Collectively, these data suggest that interaction between Olig1 and CBP or HDAC1 is developmentally regulated and this interaction could be responsible for the increased Olig1 acetylation as oligodendrocytes mature in vivo.
Olig1 is phosphorylated at serine 149 in vitro independent of its acetylation
Phosphorylaton of Olig1 at serine 149 has been shown to regulate the nuclear export of Olig1 in primary cultured OPCs (Niu et al., 2012). Because the lysine 150 site that we identified is adjacent to this potential phosphorylation site, it is possible that phosphorylation of serine 149 can disrupt the acetylation at lysine 150. To test directly the impact of Ser149 phosphorylation on Olig1 nuclear export, S149 was mutated to aspartate (D) to mimic phosphorylation or to alanine to mimic the nonphosphorylated form. WT-Olig1-GFP, Olig1-S149A-GFP, and Olig1-S149D-GFP were transfected into primary cultured rat OPCs (Fig. 8A–C). Surprisingly, in contrast to the dramatic influence of Olig1 acetylation on its nuclear export, neither the phosphorylated nor the nonphosphorylated Olig1 mimic showed different subcellular distribution from wild-type Olig1 (Fig. 8A–D). These data suggest that serine 149 phosphorylation is not a major player in regulating Olig1 nuclear export in cultured oligodendrocytes. To examine directly the S149 phosphorylation of Olig1 in vivo, we raised an antibody against the phosphorylated S149 site of Olig1. Protein kinase A (PKA) was proposed to be the potential kinase responsible for Olig1 S149 phosphorylation (Niu et al., 2012). Indeed, overexpression of the PKA catalytic subunit (PKA-cat) in HEK293T cells increased WT Olig1 phosphorylation at S149, but not that of the S149 de-phosphorylated mimic Olig1-S149A (Fig. 8E). Therefore, Olig1 was phosphorylated by PKA at S149 in vitro and this phosphorylated S149 could be recognized specifically by the phospho-Olig1 antibody. Unfortunately, this antibody only detected the phosphorylation of endogenous Olig1 in primary cultured oligodendrocytes treated with PKA agonist forskolin, but not the basal level of Olig1 phosphorylation either in cultured oligodendrocytes or in developing mouse brain (Fig. 8F,G). After forskolin treatment, S149 phosphorylated Olig1 was predominantly found in the nuclear fraction of differentiated oligodendrocytes (Fig. 8F, top). This nuclear enrichment of S149 phosphorylated Olig1 contrasted with the cytoplasmic enrichment of acetylated Olig1 as oligodendrocytes differentiated (Fig. 6E, middle). It indicated that Olig1 S149 phosphorylation and K150 acetylation may be independent. To examine directly the impact of K150 acetylation on Olig1 S149 phosphorylation, Olig1 K150 acetylation and deacetylation mimic mutants were transfected with PKA-Cat into HEK293T cells. Phosphorylation of both the Olig1 acetylation and deacetylation mimics, as detected by the phosphorylation-specific antibody, was decreased >3-fold compared with wild-type Olig1 (Fig. 8H). These data suggest that neither K150 acetylation nor deacetylation has a specific impact on Olig1 S149 phosphorylation because either loss or gain of K150 acetylation of Olig1 had the same impact. It is possible that the reduction of apparent phosphorylation of S149 after K150 mutation is caused by reduced phospho-Olig1 antibody affinity because of a conformational change caused by amino acid substitution at K150 site. Collectively, our results suggest that the S149 site of Olig1 can be phosphorylated independently of K150 acetylation. However, the basal level of Olig1 S149 phosphorylation in vivo is too low to be detected and this phosphorylation does not regulate Olig1 nuclear export.
Figure 8.

Olig1 is phosphorylated at serine 149 in vitro independent of its acetylation. A–C, Phosphorylation of serine (S)149 on Olig1 did not alter its subcellular localization in cultured rat OPCs. Olig1-GFP (A), Olig1-S149A-GFP (B), or Olig1-S149D-GFP (C) mutants were transfected into the rat OPCs and Olig1 subcellular location was determined 48 h later in transfected OLs, using Olig2 (red) and DAPI (blue) to localize nuclei. Scale bar, 10 μm. D, Quantitative analysis of the subcellular distribution of Olig1-GFP or its S149 mutants shown in A–C. Data were quantified as N < C (black), N = C (red), or N > C (blue). Data represent the mean ± SEM from at least three independent experiments and, for each different construct in each experiment, at least 300 tranfected cells were counted. The percentage of nuclear or cytoplasmic localization of indicated mutants was compared with each other. No significant different were found between different mutants (one-way ANOVA). E, Olig1 was phosphorylated at serine 149 by PKA in HEK293T cells detected by phospho-Olig1-S149 specific antibody. Olig1-myc, Olig1-S149A-myc, or Olig1-S149D-myc was cotransfected with catalytic subunit of PKA (PKA-cat) into HEK293T cells. Twenty-four hours later, cells were collected and lysate was immunoprecipitated with myc tag antibody and detected with phospho-Olig1-S149 antibody. F, Olig1 was phosphorylated at serine 149 in rat oligodendrocytes only after forskolin treatment. Primary cultured OPCs or differentiated oligodendrocytes were treated with DMSO or forskolin (20 μm) for 16 h, fractionated and the subcellular fractions were immunoprecipitated with Olig1 antibody and detected with phospho-Olig1-S149 antibody. G, Olig1 phosphoryaltion at serine 149 site was not detected in developing mouse brain. Endogenous Olig1 was immunoprecipitated from subcortical white matter homogenates of different postnatal ages and Olig1 phosphorylation was detected with phospho-Olig1-S149 antibody. H, Acetylation at K150 had little specific impact on phosphorylation of serine 149 site of Olig1. Olig1-myc, Olig1-S149A-myc, Olig1-S149D-myc, Olig1-K50R-myc, or Olig1-K150Q-myc was cotransfected with catalytic subunit of PKA (PKA-cat) into HEK293T cells. Twenty-four hours later, cells were collected and lysate was immunoprecipitated with myc tag antibody and detected with phospho-Olig1-S149 antibody. All data represent the results from three independent experiments. Quantification of the ratio of phosphorylated Olig1 to total Olig1 indicated below in red, normalized to control.
Increased acetylation of Olig1 decreased its chromatin binding and preceded its active nuclear export
For many transcription factors or chromatin-binding complexes, dissociation from chromatin precedes their inactivation or nuclear export (Niida et al., 2007; Nakamura et al., 2012). In the developing mouse brain, complete nuclear extrusion of Olig1 is seen in actively myelinating oligodendrocytes (Arnett et al., 2004; Dai et al., 2015). However, we saw only partial cytoplasmic translocation of Olig1 in fully differentiated oligodendrocytes in vitro (Fig. 9A–C). We hypothesized that Olig1 nuclear export is a three successive step process: (1) Olig1 dissociation from chromatin, (2) reduced interaction of Olig1 with potential nuclear retention proteins, and (3) active export of Olig1 from the nucleus. To test whether the chromatin dissociation of Olig1 and its subsequent nuclear export was partially uncoupled in cultured oligodendrocytes, endogenous Olig1 was immunoprecipitated from the nuclear fraction of OPCs and mature oligodendrocytes and analyzed for its histone association. Histone H3 was strongly associated with Olig1 in OPCs, but not in mature oligodendrocytes, which had elevated Olig1 acetylation (Fig. 9E). Therefore, although some Olig1 was present in the nucleus in mature oligodendrocytes in vitro, most nuclear Olig1 in these differentiating cells was not bound to the chromatin. Olig1 chromatin association was examined directly by separating the chromatin-bound or chromatin-free proteins in the nuclear fraction. More Olig1 was found in the chromatin-free nuclear fraction in mature oligodendrocytes than in OPCs (Fig. 9D).
Interestingly, in adult samples in vivo, there was increased Olig1 acetylation at K150 in the nuclear fraction (Fig. 6E, bottom). Because nuclear Olig1 is exclusively found in OPCs, this suggests that, in adult OPCs, Olig1 may dissociate from chromatin, but the additional mechanism required for subsequent nuclear export is lacking, which is different from the coupled chromatin dissociation and nuclear export of Olig1 in OPCs at P10. To examine directly the impact of Olig1 acetylation on its association with chromatin in vivo, endogenous Olig1 was immunoprecipitated from the nuclear fraction of subcortical white matter obtained from P3, P10, or 3 month brains. More histone H3 interacted with Olig1 in P3 brain nuclei than in P10 nuclei, and much less association was seen in 3 month brain nuclei (Fig. 9F). Conversely, despite reduced binding to chromatin in adult brain, Olig1 from all ages tested had comparable binding to DNA itself (Fig. 9G). Therefore, changes in the direct DNA binding of Olig1 cannot account for the dissociation of Olig1 from chromatin in differentiating oligodendrocytes, suggesting that additional mechanisms must be involved. Collectively, these data suggest that, as oligodendrocytes mature, Olig1 acetylation facilitates its dissociation from chromatin, which precedes its subsequent nuclear export.
Acetylation of Lysine 150 on Olig1 increased its binding affinity for Id2
We hypothesize that Olig1 acetylation reduces its binding with potential nuclear retention proteins and facilitates its nuclear export. Olig1 can dimerize with other bHLH transcription factors such as Olig2, Olig1, Id2, Id4, and E47 or the non-bHLH transcription factor Sox10 (Samanta and Kessler, 2004; Li et al., 2007). We therefore investigated whether Olig1 acetylation changes its binding affinity for these known binding partners. We analyzed the interactions of WT Olig1, Olig1-K150R, or Olig1-K150Q with Olig1, Olig2, E12/E47, Id4, or Id2 upon overexpression in HEK293T cells. All proteins interacted with WT Olig1 or the acetylation mimic Olig1-K150Q and Olig1, Olig2, and E12/E47 bound comparably to WT and the deacetylation mimic Olig1-K150R (data not shown). Sox10 had slightly increased binding to Olig1-K150R and reduced binding to Olig1-K150Q (Fig. 10A), suggesting that it binds preferentially to deacetylated Olig1. However, Olig1-K150R had reduced binding affinity for Id2 (Fig. 10B), but not Id4 (Fig. 10C), suggesting that Id2 might be a specific partner for acetylated Olig1.
Figure 10.
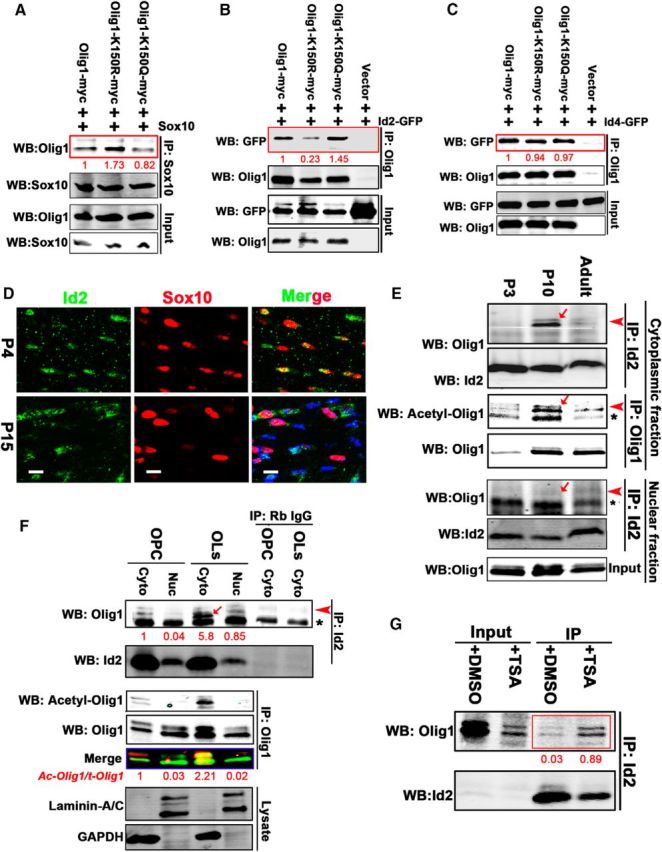
Acetylation of Lysine 150 on Olig1 increased its binding affinity for Inhibitor of DNA binding 2 (Id2). A–C, Lysine 150 acetylation increased Olig1/Id2 interaction. Olig1-myc, deacetylation mimic Olig1-K150R-myc or acetylation mimic Olig1-K150Q-myc were cotransfected with pCMV5-Sox10 (A), Id2-GFP (B), or Id4-GFP (C) into HEK293T cells. Protein interactions were detected by coimmunoprecipitation with the indicated antibodies. Note increased association of deacetylated Olig1 (Olig1-K150R-myc) with Sox10 (A), strong increase in association of acetylated Olig1 (Olig1-K150Q-myc) with Id2 (B), and no impact of acetylation on association of Olig1 with Id4 (C). Quantification of the ratio of coimmunoprecipitated protein to total protein input indicated below in red, normalized to control. D, Oligodendrocytes in P4 and P15 mouse corpus callosum were costained with Id2 (green) and Sox10 (red). Note cytoplasmic localization of Id2 at P15. Scale bar, 10 μm. E, Id2 interacted with Olig1 at P10 in the cytoplasmic fraction, but not in the nuclear fraction. Mouse subcortical white matter from different postnatal developmental time points was fractionated. Endogenous Id2 was immunoprecipitated from the cytoplasmic or nuclear fraction with Id2 antibody and Olig1 association was determined by Western blot for Olig1. Arrowheads, Olig1; arrow, coimmunoprecipitated Olig1; asterisks, rabbit IgG. F, The Olig1/Id2 interaction increased as oligodendrocytes differentiated in vitro. Primary cultured OPCs or differentiated oligodendrocytes were treated with TSA (500 nm) for 10 h, fractionated, and the subcellular fractions were immunoprecipitated with Id2 antibody and detected with Olig1 antibody (top row) or immunoprecipitated with Olig1 antibody and detected with acetyl-Olig1-K150 antibody (middle row). Laminin A/C and GAPDH were used as markers for nuclear and cytoplasmic fractions respectively (bottom row). Arrowheads, Olig1; arrow, coimmunoprecipitated Olig1; asterisks, rabbit IgG. Quantification of the ratio of coimmunoprecipitated Olig1 to total Olig1 (top row) and ratio of acetylated Olig1 to total Olig1 (middle row) is indicated below in red, normalized to that from cytoplasmic of OPCs. G, Excess acetylation increased the interaction of Olig1 and Id2. Mouse OPCs were induced to differentiate for 4 d and treated with DMSO or TSA (100 nm) for 1 d. Cell lysates were immunoprecipitated with anti-Id2 antibody and detected with Olig1 antibody. Quantification of the ratio of coimmunoprecipitated Olig1 to total Olig1 indicated below in red. All data represent the results from three independent experiments.
Id2 is a negative regulator of oligodendrocyte differentiation and undergoes nuclear to cytoplasmic translocation upon oligodendrocyte differentiation in vitro (Wang et al., 2001). The subcellular localization of Id2 in the developing mouse brain was predominantly in the nucleus of OPCs at P4 and it either translocated to the cytoplasm or was downregulated in mature oligodendrocytes at P15 (Fig. 10D). The nuclear to cytoplasmic translocation of Id2 and Olig1 occurred over the same time period in vivo. To assess possible interactions of these proteins in vivo, we analyzed cytoplasmic and nuclear fractions from P3, P10 or 3 month mouse subcortical white matter for Id2 localization and interactions. Endogenous Id2 was immunoprecipitated from the cytoplasmic or nuclear fractions and immunoblotted with Olig1 antibody. Olig1 was found to interact strongly with Id2 in the cytoplasmic fraction at P10, but not at 3 months (Fig. 10E). The Olig1/Id2 interaction was not found in the nuclear fraction (Fig. 10E, bottom). The strong Olig1/Id2 interaction in the P10 mouse cerebrum cytoplasm correlated with high acetylation of Olig1 at K150 (Fig. 10E). Because Id2 is also expressed in neurons and astrocytes in CNS, we used primary cultured oligodendrocytes to test the role of Olig1 K150 acetylation in regulating the Olig1/Id2 interaction. Consistent with earlier studies, the acetylated level of Olig1 K150 increased as oligodendrocytes differentiated and Olig1 acetylation increased in the cytoplasm of differentiated oligodendrocytes in vitro (Fig. 10G). The interaction between Olig1 and Id2 was strongest in the cytoplasm of differentiated oligodendrocytes, which correlated with the higher acetylation of Olig1 in these cells (Fig. 10G). Furthermore, increased Olig1/Id2 interaction was observed when differentiating oligodendrocytes were treated with the general HDAC inhibitor trichostatin A (Fig. 10G), which increased general protein acetylation. These studies demonstrated that Olig1 acetylation at K150 increased the interaction of Olig1 and Id2. The interactions of Olig1 and Id2 would likely facilitate Id2 retention in the cytoplasm, which would repress its inhibitory impact on oligodendrocyte differentiation.
Discussion
Mechanism of nuclear export of Olig1
In this study, we first investigated protein sequences that could explain the differences in subcellular localization of Olig1 and Olig2 by identifying two additional NES in Olig1 that are not found in Olig2. NES1 was exactly conserved between Olig1 and Olig2, but Olig1 contained two extra NES, which were stronger than NES1. Intriguingly, ectopic insertion of NES3 of Olig1 into Olig2 was sufficient to drive Olig2 translocation exclusively to the cytoplasm. Therefore, the export signal of NES1 and NES3 together is strong enough to extrude Olig2 from the nucleus. In contrast, despite the presence of both NES1 and NES3, the Olig1-dNES2 mutant localized more in the nucleus. Furthermore, even with the three nuclear export sequences, in OPCs, Olig1 is predominantly found in the nucleus, suggesting dynamic regulation of the nuclear export sequences.
Our results demonstrate that Olig1 nuclear export is a finely regulated, multistep process (Fig. 11). The interplay of nuclear retention and nuclear export signals likely determine the subcellular localization of Olig1 developmentally, regulated by the acetylation state of lysine150 in NES1 (lysine139 in mouse). In proliferating OPCs, deacetylated Olig1 is tightly bound to chromatin and its genomic targets. As noted above, the presence of the three NES is insufficient to export Olig1 from the OPC nucleus, suggesting that deacetylated Olig1 may have unique nuclear binding partners that retain it in OPC nuclei. Acetylation of lysine 150 would neutralize the positive charge on this lysine, which could change the protein-binding partner preference of Olig1 or increase the strength of NES1 as a nuclear export signal (Güttler et al., 2010; Regot et al., 2014). As OPCs differentiate and Olig1 is acetylated, it dissociates from the chromatin. Figure 9 shows that this chromatin dissociation preceded, but was not sufficient for its nuclear export, but as OPCs differentiate, Olig1 nuclear-binding proteins may be downregulated or their interaction with acetylated Olig1 may be disrupted, allowing export. In cultured oligodendrocytes and in adult OPCs in vivo, Olig1 chromatin dissociation and nuclear export appear to be uncoupled, with non-chromatin-associated acetylated Olig1 remaining in the nucleus. We conclude that, in OPCs, deacetylated Olig1, along with its potential partners, bind chromatin, which serves as a positive regulator of OPC cell cycle exit. As OPCs differentiate, Olig1 is acetylated and dissociates from chromatin, likely reducing its interaction with potential nuclear retention signals and facilitating its subsequent nuclear export, which is important for morphological differentiation of newly formed oligodendrocytes (Fig. 11).
Figure 11.

Proposed model of the mechanism by which Olig1 acetylation regulates its nuclear to cytoplasmic translocation and function. Deacetylated Olig1 localized in the nucleus is associated with chromatin and functions as a positive regulator for cell cycle exit of OPCs: (1) acetylation of Olig1 facilitates its dissociation from chromatin and (2) its active nuclear export also requires decreased interaction with potential nuclear retention proteins. (3) The last step of its nuclear export depends on its consensus NES. Olig1 acetylation and its nuclear export is required for morphogenesis of newly formed oligodendrocytes. Acetylated Olig1 and Id2 interact in mature oligodendrocytes and the retention of Id2/acetylated Olig1 in the oligodendrocyte cytoplasm could reduce the Id2 inhibition of oligodendrocyte differentiation.
Potential role of Olig1 acetylation in temporospatial control of myelin initiation
Oligodendrocyte lineage progression and myelin initiation are tightly controlled both temporally and spatially in the developing mouse brain (Trapp et al., 1997; Baumann and Pham-Dinh, 2001). Both extrinsic and intrinsic cues play critical roles in oligodendrocyte differentiation and myelination (for reviews, see Emery, 2010a,b; Wood et al., 2013). However, the mechanism by which oligodendrocytes integrate extracellular signaling with their intrinsic transcriptional program to determine when and where to start myelination is largely unknown. Although Olig1 is clearly important for oligodendrocyte specification and OPC differentiation (Lu et al., 2002; Zhou and Anderson, 2002; Silbereis et al., 2014; Dai et al., 2015), tight temporal control of Olig1 expression and localization as oligodendrocytes initiate myelination implies a potentially negative function of Olig1 when present in the nucleus of myelinating cells. Acetylation of nonhistone proteins is emerging as an important regulator of protein function (Choudhary et al., 2009; Zhao et al., 2010). How the acetylation of Olig1 is temporally regulated during myelinogenesis is still unclear. Preferentially, interaction with HDAC1 and CBP in the developing mouse brain provides one mechanism for regulating Olig1 acetylation at distinct stages. The interaction between Olig1 and CBP or HDAC1 may be regulated by extracellular cues, including signaling from axons. For example, neuronal-activity-induced phosphorylation of MeCP2 has been shown to serve as a molecular switch to integrate extrinsic signals with intrinsic transcriptional programs in developing brain (Zhou et al., 2006; Tao et al., 2009; Cohen et al., 2011). Differential regulation of Olig1 acetylation in OPCs versus myelinating oligodendrocytes regulates its chromatin association and subcellular translocation. This fine-tuned regulation of Olig1 function at the initiation of myelinating may serve as an important molecular switch to ensure proper temporal–spatial myelination onset.
Nuclear versus cytoplasmic function of Olig1
In a variety of developing systems, transcription factors important for early cell development are downregulated at later developmental stages (Jessell, 2000; Isshiki et al., 2001; Braun and Gautel, 2011). Therefore, an important question is why Olig1 remains in the cytoplasm as the oligodendrocyte matures. It is possible that Olig1 is important for initial differentiation, but it becomes a negative regulator of later oligodendrocyte differentiation and must be downregulated or translocated. As OPCs begin to differentiate in vivo, they become premyelinating oligodendrocytes before starting to wrap axons (Trapp et al., 1997). Interestingly, Olig1 is greatly reduced in premyelinating oligodendrocytes (Dai et al., 2015), which suggests that sustained nuclear Olig1 may be inhibitory to late oligodendrocyte differentiation and may need to be eliminated in this stage. Consistent with this hypothesis, deacetylated Olig1 in OPC nuclei was associated with chromatin and this was important for its nuclear function. However, sustained deacetylated Olig1 nuclear retention inhibited subsequent morphological differentiation (Fig. 5J). These data support an inhibitory effect of sustained nuclear Olig1 on late oligodendrocyte differentiation during development.
Intriguingly, whereas Olig1 is downregulated in the premyelinating cell, it reappears and is retained in the cytoplasm of myelinating oligodendrocytes. Olig1 expression in the oligodendrocyte cytoplasm long after active myelination is finished suggests its importance in mature oligodendrocytes, but not for myelinogenesis per se (Dai et al., 2015). Nucleocytoplasmic shuttling of transcription factor has long been appreciated as a mechanism that mediates the transcriptional response to the intracellular or extracellular stimuli (Cartwright and Helin, 2000; Gama-Carvalho and Carmo-Fonseca, 2001; Meyer and Vinkemeier, 2004). Dynamic nucleocytoplasmic shuttling provides a mechanism by which transcription factors can quickly turn on or off target gene expression in response to the changing microenvironment. For example, cytoplasmic-localized NF-κB, NF-AT, Smad, and STAT family transcription factors instantly translocate to the nucleus to activate their target genes upon cytokine treatment. After removal of the stimuli, they quickly translocate back to the cytoplasm to quench the signaling cascade (Zhu and McKeon, 2000; Herrmann et al., 2007; Hill, 2009; Oeckinghaus and Ghosh, 2009). Nucleocytoplasmic shuttling of transcription factors such as the DNA-damaging sensor P53 (Sharpless and DePinho, 2002), the oxidative stress sensor AP-1 (Yan et al., 1998), and others (Xu and Massagué, 2004) also serves as a mechanism to sense cellular stress, transducing the signal to the nucleus to maintain cellular homeostasis. It is possible that cytoplasmic Olig1 might serve as a sensor for injury to myelinating oligodendrocytes. To further investigate the role of cytoplasmic Olig1 at the molecular level, identification of Olig1-interacting proteins in the cytoplasm will be necessary.
Olig1 translocation and remyelination
Recent studies indicate extensive myelin remodeling in the adult mouse brain, mainly attributed to newly generated oligodendrocytes (Rivers et al., 2008; Young et al., 2013). However, retrospective birth-dating of oligodendrocytes with 14C in human brain indicates that oligodendrocyte number does not change with age and, in fact, very little turnover of oligodendrocytes occurs through life in human white matter. In contrast, comparable analysis of the myelin itself in these postmortem samples indicated that myelin was generated and remodeled throughout life (Yeung et al., 2014). This raises the question of how mature oligodendrocytes remodel myelin. Whether mature oligodendrocytes with cytoplasmic Olig1 are involved in myelin remodeling in the mouse has not been investigated. Newly generated myelin is laid down by existing mature oligodendrocytes in response to learning (Bengtsson et al., 2005; Scholz et al., 2009) and it is intriguing to consider the possibility that movement of cytoplasmic Olig1 back to the nucleus might reactivate myelin production in these cells.
Although Olig1 is required for oligodendrocyte development only in mouse brain (Dai et al., 2015), Olig1 function is essential for remyelination in both spinal cord and brain in several different demyelination models (Arnett et al., 2004; Whitman et al., 2012). The differentiation block in Olig1 null mice during remyelination resembles the finding in MS lesions in which abundant OPCs fail to differentiate (Chang et al., 2000; Chang et al., 2002; Kuhlmann et al., 2008). Considering the role of Olig1 acetylation in regulating its function in oligodendrocyte differentiation, it is possible that Olig1 acetylation is disrupted in OPCs in MS lesions and affects its function in oligodendrocytes. This disruption could result from the altered acetylation patterns seen in MS lesions (Pedre et al., 2011). Therefore, whereas Olig1 is still present in the nuclei in MS lesions (Arnett et al., 2004), its nuclear function may be disrupted by dysregulation of acetylation. Promoting Olig1 function in chronic MS lesions or modulating the Olig1 activity by its acetylation might be meaningful approaches to promote oligodendrocyte differentiation and subsequent myelination in demyelinated lesions.
Footnotes
This work was supported by the NIH (Grant NS08223 to W.B.M.) and by the National Natural Science Foundation of China (Grant 31171033 to W.J.). We thank Katherine E. Saul and Hilary Sachs for technical assistance, and Dr. Danielle Harlow and Dr. Marnie Preston for helpful discussion.
The authors declare no competing financial interests.
References
- Arnett HA, Fancy SP, Alberta JA, Zhao C, Plant SR, Kaing S, Raine CS, Rowitch DH, Franklin RJ, Stiles CD. bHLH transcription factor Olig1 is required to repair demyelinated lesions in the CNS. Science. 2004;306:2111–2115. doi: 10.1126/science.1103709. [DOI] [PubMed] [Google Scholar]
- Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- Bengtsson SL, Nagy Z, Skare S, Forsman L, Forssberg H, Ullén F. Extensive piano practicing has regionally specific effects on white matter development. Nat Neurosci. 2005;8:1148–1150. doi: 10.1038/nn1516. [DOI] [PubMed] [Google Scholar]
- Braun T, Gautel M. Transcriptional mechanisms regulating skeletal muscle differentiation, growth and homeostasis. Nat Rev Mol Cell Biol. 2011;12:349–361. doi: 10.1038/nrm3118. [DOI] [PubMed] [Google Scholar]
- Bujalka H, Koenning M, Jackson S, Perreau VM, Pope B, Hay CM, Mitew S, Hill AF, Lu QR, Wegner M, Srinivasan R, Svaren J, Willingham M, Barres BA, Emery B. MYRF is a membrane-associated transcription factor that autoproteolytically cleaves to directly activate myelin genes. PLoS Biol. 2013;11:e1001625. doi: 10.1371/journal.pbio.1001625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock TH, Moore JK, Fields RD. Evolution of myelin sheaths: both lamprey and hagfish lack myelin. Neurosci Lett. 1984;48:145–148. doi: 10.1016/0304-3940(84)90010-7. [DOI] [PubMed] [Google Scholar]
- Cartwright P, Helin K. Nucleocytoplasmic shuttling of transcription factors. Cell Mol Life Sci. 2000;57:1193–1206. doi: 10.1007/PL00000759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A, Nishiyama A, Peterson J, Prineas J, Trapp BD. NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J Neurosci. 2000;20:6404–6412. doi: 10.1523/JNEUROSCI.20-17-06404.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A, Tourtellotte WW, Rudick R, Trapp BD. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med. 2002;346:165–173. doi: 10.1056/NEJMoa010994. [DOI] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Cohen S, Gabel HW, Hemberg M, Hutchinson AN, Sadacca LA, Ebert DH, Harmin DA, Greenberg RS, Verdine VK, Zhou Z, Wetsel WC, West AE, Greenberg ME. Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron. 2011;72:72–85. doi: 10.1016/j.neuron.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Bercury KK, Macklin WB. Interaction of mTOR and Erk1/2 signaling to regulate oligodendrocyte differentiation. Glia. 2014;62:2096–2109. doi: 10.1002/glia.22729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Bercury KK, Ahrendsen JT, Macklin WB. Olig1 function is required for oligodendrocyte differentiation in the mouse brain. J Neurosci. 2015;35:4386–4402. doi: 10.1523/JNEUROSCI.4962-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- di Bari MG, Ciuffini L, Mingardi M, Testi R, Soddu S, Barilà D. c-Abl acetylation by histone acetyltransferases regulates its nuclear-cytoplasmic localization. EMBO Rep. 2006;7:727–733. doi: 10.1038/sj.embor.7400700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery B. Transcriptional and post-transcriptional control of CNS myelination. Curr Opin Neurobiol. 2010a;20:601–607. doi: 10.1016/j.conb.2010.05.005. [DOI] [PubMed] [Google Scholar]
- Emery B. Regulation of oligodendrocyte differentiation and myelination. Science. 2010b;330:779–782. doi: 10.1126/science.1190927. [DOI] [PubMed] [Google Scholar]
- Franklin RJ, Ffrench-Constant C. Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci. 2008;9:839–855. doi: 10.1038/nrn2480. [DOI] [PubMed] [Google Scholar]
- Gama-Carvalho M, Carmo-Fonseca M. The rules and roles of nucleocytoplasmic shuttling proteins. FEBS Lett. 2001;498:157–163. doi: 10.1016/S0014-5793(01)02487-5. [DOI] [PubMed] [Google Scholar]
- Güttler T, Madl T, Neumann P, Deichsel D, Corsini L, Monecke T, Ficner R, Sattler M, Görlich D. NES consensus redefined by structures of PKI-type and Rev-type nuclear export signals bound to CRM1. Nat Struct Mol Biol. 2010;17:1367–1376. doi: 10.1038/nsmb.1931. [DOI] [PubMed] [Google Scholar]
- Herrmann A, Vogt M, Mönnigmann M, Clahsen T, Sommer U, Haan S, Poli V, Heinrich PC, Müller-Newen G. Nucleocytoplasmic shuttling of persistently activated STAT3. J Cell Sci. 2007;120:3249–3261. doi: 10.1242/jcs.03482. [DOI] [PubMed] [Google Scholar]
- Hill CS. Nucleocytoplasmic shuttling of Smad proteins. Cell Res. 2009;19:36–46. doi: 10.1038/cr.2008.325. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, McConnell KW, Bullok K, Davis CG, Chang KC, Schwulst SJ, Dunne JC, Dietz GP, Bähr M, McDunn JE, Karl IE, Wagner TH, Cobb JP, Coopersmith CM, Piwnica-Worms D. TAT-BH4 and TAT-Bcl-xL peptides protect against sepsis-induced lymphocyte apoptosis in vivo. J Immunol. 2006;176:5471–5477. doi: 10.4049/jimmunol.176.9.5471. [DOI] [PubMed] [Google Scholar]
- Isshiki T, Pearson B, Holbrook S, Doe CQ. Drosophila neuroblasts sequentially express transcription factors which specify the temporal identity of their neuronal progeny. Cell. 2001;106:511–521. doi: 10.1016/S0092-8674(01)00465-2. [DOI] [PubMed] [Google Scholar]
- Jakovcevski I, Zecevic N. Olig transcription factors are expressed in oligodendrocyte and neuronal cells in human fetal CNS. J Neurosci. 2005;25:10064–10073. doi: 10.1523/JNEUROSCI.2324-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessell TM. Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat Rev Genet. 2000;1:20–29. doi: 10.1038/35049541. [DOI] [PubMed] [Google Scholar]
- Kitada M, Rowitch DH. Transcription factor co-expression patterns indicate heterogeneity of oligodendroglial subpopulations in adult spinal cord. Glia. 2006;54:35–46. doi: 10.1002/glia.20354. [DOI] [PubMed] [Google Scholar]
- Krämer OH, Knauer SK, Greiner G, Jandt E, Reichardt S, Gührs KH, Stauber RH, Böhmer FD, Heinzel T. A phosphorylation-acetylation switch regulates STAT1 signaling. Genes Dev. 2009;23:223–235. doi: 10.1101/gad.479209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlmann T, Miron V, Cuo Q, Wegner C, Antel J, Brück W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain. 2008;131:1749–1758. doi: 10.1093/brain/awn096. [DOI] [PubMed] [Google Scholar]
- Kurooka H, Yokota Y. Nucleo-cytoplasmic shuttling of Id2, a negative regulator of basic helix-loop-helix transcription factors. J Biol Chem. 2005;280:4313–4320. doi: 10.1074/jbc.M412614200. [DOI] [PubMed] [Google Scholar]
- Li A, Xue Y, Jin C, Wang M, Yao X. Prediction of Nepsilon-acetylation on internal lysines implemented in Bayesian discriminant method. Biochem Biophys Res Comm. 2006;350:818–824. doi: 10.1016/j.bbrc.2006.08.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Lu Y, Smith HK, Richardson WD. Olig1 and Sox10 interact synergistically to drive myelin basic protein transcription in oligodendrocytes. J Neurosci. 2007;27:14375–14382. doi: 10.1523/JNEUROSCI.4456-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, Rowitch DH. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell. 2002;109:75–86. doi: 10.1016/S0092-8674(02)00678-5. [DOI] [PubMed] [Google Scholar]
- MacDonald JI, Kubu CJ, Meakin SO. Nesca, a novel adapter, translocates to the nuclear envelope and regulates neurotrophin-induced neurite outgrowth. J Cell Biol. 2004;164:851–862. doi: 10.1083/jcb.200309081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meek DW, Anderson CW. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol. 2009;1:a000950. doi: 10.1101/cshperspect.a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei F, Wang H, Liu S, Niu J, Wang L, He Y, Etxeberria A, Chan JR, Xiao L. Stage-specific deletion of Olig2 conveys opposing functions on differentiation and maturation of oligodendrocytes. J Neurosci. 2013;33:8454–8462. doi: 10.1523/JNEUROSCI.2453-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer T, Vinkemeier U. Nucleocytoplasmic shuttling of STAT transcription factors. Eur J Biochem. 2004;271:4606–4612. doi: 10.1111/j.1432-1033.2004.04423.x. [DOI] [PubMed] [Google Scholar]
- Mi YJ, Hou B, Liao QM, Ma Y, Luo Q, Dai YK, Ju G, Jin WL. Amino-Nogo-A antagonizes reactive oxygen species generation and protects immature primary cortical neurons from oxidative toxicity. Cell Death Differ. 2012;19:1175–1186. doi: 10.1038/cdd.2011.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Liu YJ, Nakashima H, Umehara H, Inoue K, Matoba S, Tachibana M, Ogura A, Shinkai Y, Nakano T. PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature. 2012;486:415–419. doi: 10.1038/nature11093. [DOI] [PubMed] [Google Scholar]
- Niida H, Katsuno Y, Banerjee B, Hande MP, Nakanishi M. Specific role of Chk1 phosphorylations in cell survival and checkpoint activation. Mol Cell Biol. 2007;27:2572–2581. doi: 10.1128/MCB.01611-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu J, Mei F, Wang L, Liu S, Tian Y, Mo W, Li H, Lu QR, Xiao L. Phosphorylated olig1 localizes to the cytosol of oligodendrocytes and promotes membrane expansion and maturation. Glia. 2012;60:1427–1436. doi: 10.1002/glia.22364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1:a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Meara RW, Michalski JP, Anderson C, Bhanot K, Rippstein P, Kothary R. Integrin-linked kinase regulates process extension in oligodendrocytes via control of actin cytoskeletal dynamics. J Neurosci. 2013;33:9781–9793. doi: 10.1523/JNEUROSCI.5582-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paes de Faria J, Kessaris N, Andrew P, Richardson WD, Li H. New Olig1 null mice confirm a non-essential role for Olig1 in oligodendrocyte development. BMC Neurosci. 2014;15:12. doi: 10.1186/1471-2202-15-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedraza CE, Monk R, Lei J, Hao Q, Macklin WB. Production, characterization, and efficient transfection of highly pure oligodendrocyte precursor cultures from mouse embryonic neural progenitors. Glia. 2008;56:1339–1352. doi: 10.1002/glia.20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedre X, Mastronardi F, Brück W, López-Rodas G, Kuhlmann T, Casaccia P. Changed histone acetylation patterns in normal-appearing white matter and early multiple sclerosis lesions. J Neurosci. 2011;31:3435–3445. doi: 10.1523/JNEUROSCI.4507-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regot S, Hughey JJ, Bajar BT, Carrasco S, Covert MW. High-sensitivity measurements of multiple kinase activities in live single cells. Cell. 2014;157:1724–1734. doi: 10.1016/j.cell.2014.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivers LE, Young KM, Rizzi M, Jamen F, Psachoulia K, Wade A, Kessaris N, Richardson WD. PDGFRA/NG2 glia generate myelinating oligodendrocytes and piriform projection neurons in adult mice. Nat Neurosci. 2008;11:1392–1401. doi: 10.1038/nn.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta J, Kessler JA. Interactions between ID and OLIG proteins mediate the inhibitory effects of BMP4 on oligodendroglial differentiation. Development. 2004;131:4131–4142. doi: 10.1242/dev.01273. [DOI] [PubMed] [Google Scholar]
- Scholz J, Klein MC, Behrens TE, Johansen-Berg H. Training induces changes in white-matter architecture. Nat Neurosci. 2009;12:1370–1371. doi: 10.1038/nn.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpless NE, DePinho RA. p53: good cop/bad cop. Cell. 2002;110:9–12. doi: 10.1016/S0092-8674(02)00818-8. [DOI] [PubMed] [Google Scholar]
- Shen S, Li J, Casaccia-Bonnefil P. Histone modifications affect timing of oligodendrocyte progenitor differentiation in the developing rat brain. J Cell Biol. 2005;169:577–589. doi: 10.1083/jcb.200412101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silbereis JC, Nobuta H, Tsai HH, Heine VM, McKinsey GL, Meijer DH, Howard MA, Petryniak MA, Potter GB, Alberta JA, Baraban SC, Stiles CD, Rubenstein JL, Rowitch DH. Olig1 function is required to repress dlx1/2 and interneuron production in mammalian brain. Neuron. 2014;81:574–587. doi: 10.1016/j.neuron.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soane L, Fiskum G. TAT-mediated endocytotic delivery of the loop deletion Bcl-2 protein protects neurons against cell death. J Neurochem. 2005;95:230–243. doi: 10.1111/j.1471-4159.2005.03359.x. [DOI] [PubMed] [Google Scholar]
- Tao J, Hu K, Chang Q, Wu H, Sherman NE, Martinowich K, Klose RJ, Schanen C, Jaenisch R, Wang W, Sun YE. Phosphorylation of MeCP2 at Serine 80 regulates its chromatin association and neurological function. Proc Natl Acad Sci U S A. 2009;106:4882–4887. doi: 10.1073/pnas.0811648106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci. 2008;31:247–269. doi: 10.1146/annurev.neuro.30.051606.094313. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Nishiyama A, Cheng D, Macklin W. Differentiation and death of premyelinating oligodendrocytes in developing rodent brain. J Cell Biol. 1997;137:459–468. doi: 10.1083/jcb.137.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Sdrulla A, Johnson JE, Yokota Y, Barres BA. A role for the helix-loop-helix protein Id2 in the control of oligodendrocyte development. Neuron. 2001;29:603–614. doi: 10.1016/S0896-6273(01)00237-9. [DOI] [PubMed] [Google Scholar]
- Whitman LM, Blanc CA, Schaumburg CS, Rowitch DH, Lane TE. Olig1 function is required for remyelination potential of transplanted neural progenitor cells in a model of viral-induced demyelination. Exp Neurol. 2012;235:380–387. doi: 10.1016/j.expneurol.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolswijk G. Oligodendrocyte regeneration in the adult rodent CNS and the failure of this process in multiple sclerosis. Prog Brain Res. 1998;117:233–247. doi: 10.1016/S0079-6123(08)64019-4. [DOI] [PubMed] [Google Scholar]
- Wolswijk G. Oligodendrocyte survival, loss and birth in lesions of chronic-stage multiple sclerosis. Brain. 2000;123:105–115. doi: 10.1093/brain/123.1.105. [DOI] [PubMed] [Google Scholar]
- Wood TL, Bercury KK, Cifelli SE, Mursch LE, Min J, Dai J, Macklin WB. mTOR: a link from the extracellular milieu to transcriptional regulation of oligodendrocyte development. ASN Neurol. 2013;5:e00108. doi: 10.1042/AN20120092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin M, Yue T, Ma Z, Wu FF, Gow A, Lu QR. Myelinogenesis and axonal recognition by oligodendrocytes in brain are uncoupled in Olig1-null mice. J Neurosci. 2005;25:1354–1365. doi: 10.1523/JNEUROSCI.3034-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Massagué J. Nucleocytoplasmic shuttling of signal transducers. Nat Rev Mol Cell Biol. 2004;5:209–219. doi: 10.1038/nrm1331. [DOI] [PubMed] [Google Scholar]
- Yan C, Lee LH, Davis LI. Crm1p mediates regulated nuclear export of a yeast AP-1-like transcription factor. EMBO J. 1998;17:7416–7429. doi: 10.1093/emboj/17.24.7416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung MS, Zdunek S, Bergmann O, Bernard S, Salehpour M, Alkass K, Perl S, Tisdale J, Possnert G, Brundin L, Druid H, Frisén J. Dynamics of oligodendrocyte generation and myelination in the human brain. Cell. 2014;159:766–774. doi: 10.1016/j.cell.2014.10.011. [DOI] [PubMed] [Google Scholar]
- Young KM, Psachoulia K, Tripathi RB, Dunn SJ, Cossell L, Attwell D, Tohyama K, Richardson WD. Oligodendrocyte dynamics in the healthy adult CNS: evidence for myelin remodeling. Neuron. 2013;77:873–885. doi: 10.1016/j.neuron.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Anderson DJ. The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell. 2002;109:61–73. doi: 10.1016/S0092-8674(02)00677-3. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, McKeon F. Nucleocytoplasmic shuttling and the control of NF-AT signaling. Cell Mol Life Sci. 2000;57:411–420. doi: 10.1007/PL00000703. [DOI] [PMC free article] [PubMed] [Google Scholar]