

Abstract
Myelinating cells synthesize large amounts of membrane protein through the secretory pathway, which makes these cells particularly sensitive to perturbations of the endoplasmic reticulum (ER). Ig binding protein (BiP), also known as glucose-regulated protein 78 (GRP78), is a critical ER chaperone that also plays a pivotal role in controlling the cellular response to ER stress. To examine the potential importance of BiP to myelinating cells, we used a conditional knock-out approach to BiP gene inactivation in oligodendrocytes during development, in adulthood, and in response to experimental autoimmune encephalomyelitis (EAE), an animal model of the inflammatory demyelinating disorder multiple sclerosis (MS). During development, mice lacking functional BiP gene expression in oligodendrocytes developed tremors and ataxia and died before reaching maturity. When BiP gene inactivation in oligodendrocytes was initiated in adulthood, the mice displayed severe neurological symptoms including tremors and hind-limb paralysis. The inactivation of BiP in oligodendrocytes during development or in adulthood resulted in oligodendrocyte loss and corresponding severe myelin abnormalities. Mice heterozygous for the oligodendrocyte-specific inactivation of BiP, which were phenotypically normal without evidence of neuropathology, displayed an exacerbated response to EAE that correlated with an increased loss of oligodendrocytes. Furthermore, mice in which the BiP gene was specifically inactivated in developing Schwann cells displayed tremor that progressed to hindlimb paralysis, which correlated with diminished numbers of myelinating Schwann cells and severe PNS hypomyelination. These studies demonstrate that BiP is critical for myelinating cell survival and contributes to the protective response of oligodendrocyte against inflammatory demyelination.
SIGNIFICANCE STATEMENT The myelinating cells, oligodendrocytes in the CNS and Schwann cells in the PNS, are responsible for synthesizing an enormous amount of cellular membrane during the active phase of myelination. Therefore, these cells are particularly sensitive to insults that disrupt the function of the secretory pathway. Here, we show that the endoplasmic reticulum (ER) resident chaperone protein Ig binding protein (BiP) plays an essential role in the survival and function of myelinating cells both during the myelination process and in adult animals. Moreover, we demonstrate that BiP participates in the protective response of oligodendrocytes to inflammatory demyelinating insults. The work described here suggests that a compromised response to perturbations to the ER could contribute to myelin disorders of the CNS and PNS.
Keywords: ER stress, mouse models, protein homeostasis, unfolded protein response
Introduction
To form the myelin sheath, myelinating cells synthesize large amounts of lipids and proteins in the endoplasmic reticulum (ER) (Pfeiffer et al., 1993), which increases the sensitivity of these cells to perturbations of the secretory pathway (Lin and Popko, 2009). This increased sensitivity to secretory pathway abnormalities likely contributes to genetic and acquired myelin abnormalities. Notably, ER stress markers are expressed in the demyelinated lesions found in multiple sclerosis (MS), as well as in experimental autoimmune encephalomyelitis (EAE), an animal model of MS (Mháille et al., 2008; Cunnea et al., 2011; McMahon et al., 2012; Ni Fhlathartaigh et al., 2013).
Ig binding protein (BiP), also known as glucose-regulated protein 78 kDa (Grp78), is an ER chaperone protein that belongs to the heat shock protein 70 family (Lee, 1992; Jolly and Morimoto, 2000; Ni and Lee, 2007). BiP is one of several chaperone proteins localized to the ER that play a critical role in facilitating the proper folding of membrane and secreted proteins (King et al., 2001; Kleizen and Braakman, 2004). In addition, BiP plays a key role in the cellular response to the accumulation of misfolded proteins in the ER (ER stress), including acting as a regulator of the unfolded protein response (UPR) (Hendershot, 2004; Lee, 2005; Li and Lee, 2006), which allows cells to adapt to ER stress (Malhotra and Kaufman, 2007). BiP modulates the UPR by binding and inhibiting the activation of the ER stress sensors pancreatic ER kinase (PERK), inositol-requiring enzyme-1 (IRE1), and activating transcription factor-6 (ATF6) (Bertolotti et al., 2000; Shen et al., 2002). In ER stress situations, BiP is released from the sensors, leading to UPR activation (Bertolotti et al., 2000). The UPR provides protection to cells against ER stress, although prolonged UPR activation leads to the apoptotic death of the stressed cells (Faitova et al., 2006; Szegezdi et al., 2006; Wang and Kaufmann, 2012).
The importance of the secretory pathway in the myelination process inspired our interest in exploring BiP function in myelinating cells. Mice homozygous for a null mutation in the BiP gene die embryonically (Luo et al., 2006), so here we used a conditional gene inactivation approach to eliminate BiP specifically from mouse myelinating cells to study the effects of BiP deletion during development, adulthood, and in response to EAE. The inactivation of BiP in oligodendrocytes during development and adulthood results in a phenotype consistent with a myelination deficit associated with the loss of oligodendrocytes, indicating that BiP function is critical to oligodendrocyte viability. We also found that, although mice heterozygous for the inactivate allele of BiP in oligodendrocytes displayed a normal phenotype without myelin abnormalities, these animals experienced an exacerbated EAE disease course associated with an increased loss of oligodendrocytes, which suggests that BiP plays a protective role in these cells in response to inflammation. Moreover, we demonstrate that BiP is essential to the myelinating function of Schwann cells. In total, these results demonstrate the critical role that BiP plays in the survival and function of myelinating cells.
Materials and Methods
Generation of developmental oligodendrocyte BiP knock-out (OL-BiPko/ko) mice.
Because the conventional BiP/Grp78 knock-out mice showed embryonic lethality (Luo et al., 2006), we used the 2′, 3′ cyclic nucleotide 3′ phosphodiesterase (Cnp) promoter-driven Cre-recombinase to inactivate BiP specifically in oligodendrocytes (Lappe-Siefke et al., 2003). C57BL/6 mice carrying a conditional allele of the BiP gene, in which exons 5, 6, and 7 encoding the ATPase and peptide-binding domains essential for BiP function and in which LoxP recombination showed no truncated protein, were used (Luo et al., 2006). C57BL/6 mice expressing the Cre recombinase under the transcriptional control of the (Cnp) gene were kindly provided by Dr. Klaus Nave's group (Lappe-Siefke et al., 2003). Mice heterozygous for the floxed BiP allele and heterozygous for the Cnp/Cre allele were bred and then mated to mice heterozygous for the floxed BiP allele to generate litters containing mice homozygous for the floxed BiP allele and heterozygous for the Cnp/Cre allele (OL-BiPko/ko) and control littermates that were homozygous for the floxed BiP allele but lacked the Cnp/Cre allele (BiPflox/flox). These mice were used to study the effect of the BiP deletion in oligodendrocytes during development and both male and female mice were studied. Mouse breeding also generated mice heterozygous for the floxed BiP allele and heterozygous for the Cnp/Cre allele (OL-BiPwt/ko) and control littermates that were heterozygous for the floxed BiP allele but lacked the Cnp/Cre allele (BiPwt/flox); female mice were used for the EAE study. LoxP recombination was examined as described previously (Luo et al., 2006; Fu et al., 2008). Briefly, genomic DNA isolated from the indicated tissues was used for genotyping with the following PCR primers, PF3: 5′-GATTTGAACTCAGGACCTTCGGAAGAGCAG-3′, PR3: 5′-GCAATAGCAGCTGCTGTACTGTGAGGATGA-3′ and PTR: 5′-TTGTTAGGGG TCGTTCACCTAGA-3′. All animals were housed under pathogen-free conditions and all animal procedures were approved by the Institutional Animal Care and Use Committees of the University of Chicago.
Generation of adult oligodendrocyte BiP knock-out (BiPko/ko) mice.
To delete the BiP gene in adult oligodendrocytes, we used an inducible conditional gene inactivation approach. Mice homozygous for the floxed BiP allele (Luo et al., 2006) and hemizygous for a transgene that expresses the Cre recombinase under the transcriptional control of the myelin proteolipid protein transcriptional control region (PLP/CreERT) (Doerflinger et al., 2003) were generated and then mated to mice homozygous for the floxed BiP allele to produce mice homozygous for the floxed BiP allele and hemizygous for the Plp/CreERT transgene. Eight-week-old mice were treated with 4-hydroxytamoxifen (4OHT; Sigma-Aldrich), as described previously (Traka et al., 2010). Briefly, 4OHT was dissolved in 90% sunflower seed oil and 10% ethanol to 10 mg/ml. Mice were given a daily intraperitoneal injection of 1 mg (100 μl) for 5 consecutive days. The first day of injections was designated as day 0. Littermates were given a daily intraperitoneal injection of 100 μl of sunflower seed oil and were used as a control.
Generation of Schwann cell-specific BiP knock-out (Sch-BiPko/ko) mice.
To study the effect of BiP inactivation in Schwann cells, mice carrying the floxed BiP allele were crossed with a mouse strain carrying a transgene in which the Cre recombinase was placed under the transcriptional control of the myelin protein zero (P0) gene (Feltri et al., 1999). Mice heterozygous for the floxed BiP allele and hemizygous for the P0/Cre transgene were bred to mice homozygous for the floxed BiP allele to generate litters containing mice homozygous for the floxed BiP allele and hemizygous for the P0/Cre transgene (Sch-BiPko/ko) and control littermates that were homozygous for the floxed BiP allele but lacked the P0/Cre transgene (BiPflox/flox). Both male and female mice were studied.
EAE immunization.
EAE was induced as described previously (Iglesias et al., 2001; Lin et al., 2006). Briefly, 6-week-old females received subcutaneous injections of 200 μg of myelin oligodendrocyte glycoprotein (MOG) 35–55 peptide (Genemed Synthesis), emulsified in incomplete Freund's adjuvant (BD Biosciences), and supplemented with 600 μg of Mycobacterium tuberculosis (strain H37Ra; BD Biosciences) in the flank and tail base. The mice also received two 400 ng of intraperitoneal injections of pertussis toxin (List Biological Laboratories) in 100 μl of PBS immediately and 48 h later. Clinical severity scores were recorded daily using a 0–5 point scale (0 = healthy, 1 = flaccid tail, 2 = ataxia and/or paresis of hind limbs, 3 = paralysis of hind limbs and/or paresis of fore limbs, 4 = tetraparalysis, and 5 = moribund or dead). To eliminate diagnostic bias, mice were scored blindly.
Immunohistochemistry and TUNEL staining.
Anesthetized mice were perfused through the left cardiac ventricle with 4% paraformaldehyde in PBS. The tissues were removed, postfixed with paraformaldehyde, cryopreserved in 30% sucrose, embedded in optimal cutting temperature compound, and frozen on dry ice. Frozen sections were cut in a cryostat at a thickness of 10 μm and stored at −80°C until further use. For immunohistochemistry, the sections were treated with −20°C acetone, blocked with PBS containing 5% goat serum and 0.1% Triton X-100, and incubated overnight with the primary antibody diluted in blocking solution. Antigen retrieval was used for BiP and CC1 double staining, as described previously (Hussien et al., 2014). Briefly, the sections were boiled in 0.01 m trisodium citrate buffer, pH 6, for 10 min and then cooled at room temperature for 20 min. The sections were incubated with 0.3% hydrogen peroxide for 30 min to block endogenous peroxidases, preincubated with 5% BSA with 0.1% Triton X-100 for 2 h at room temperature to block nonspecific binding, and then incubated overnight with the primary antibody diluted in blocking solution. The appropriate fluorescein- or enzyme-labeled secondary antibodies (Vector Laboratories) were used for detection. The immunohistochemical detections of BiP (1:50; Santa Cruz Biotechnology), the N terminus of adenomatous polyposis coli (CC1; 1:50; Calbiochem), Egr2 (Krox-20; 1:250; Covance), platelet-derived growth factor α (PDGFRα; 1:50; clone APA5; Millipore), C/EBP homologous protein (CHOP; 1:50; Santa Cruz Biotechnology), myelin basic protein (Madhavarao et al., 2004) (MBP; 1:1000; Covance), and aspartoacylase (1:1000; kindly provided by Dr. M.A. Namboodiri) were performed and analyzed as described previously (Madhavarao et al., 2004; Traka et al., 2010; Lin et al., 2013; Hussien et al., 2014). TUNEL staining was performed using the DeadEnd Fluorometric TUNEL system (Promega) according to the manufacturer's instructions.
Western blot analysis.
Brains were collected from mice, rinsed in ice-cold PBS, and then homogenized in cold lysis buffer with a motorized homogenizer, as described previously (Lin et al., 2005; Lin et al., 2008). After incubation on ice for 15 min, the extracts were cleared twice by centrifugation at 14,000 rpm for 15 min each. The protein concentrations were determined by the DC Protein Assay (Bio-Rad Laboratories) and the protein samples (40–80 μg) were separated by SDS-PAGE and transferred onto nitrocellulose (Bio-Rad Laboratories). The blots were incubated with the following primary antibodies: anti-MBP (1:1000; Covance), anti-PLP and anti-MAG (1:50; Cell Signaling Technology), anti-phosphorylated PERK (p-PERK; 1:200; Santa Cruz Biotechnology), anti-ATF6 (for both total and cleaved ATF6; 1:500; Cosmo Bio), and anti-β-actin (1:1000; Sigma-Aldrich). The blots were then incubated with an HRP-conjugated secondary antibody (1:5000; GE Healthcare) and the signals were detected by chemiluminescence.
Real-time PCR and X box-binding protein (Xbp-1) mRNA splicing assay.
Deeply anesthetized OL-BiPko/ko mice or BiPflox/flox mice were perfused with ice-cold PBS. Total RNA from brain was extracted by using an Aurum Total RNA Mini Kit (Bio-Rad Laboratories). The first-strand cDNA was synthesized by using an iScript cDNA Synthesis Kit (Bio-Rad Laboratories). TaqMan real-time PCR was performed with IQ TaqMan Supermix (Bio-Rad Laboratories) and a Bio-Rad CFX96 Real-Time PCR detection system, as described previously (Lin et al., 2006; Lin et al., 2007). The Xbp-1 splicing assay was performed with total mRNA isolated from mouse brain to amplify the PCR products of the unspliced and spliced Xbp-1 mRNAs, with sizes of 171 and 145 base pairs, respectively.
Toluidine blue staining and electron microscopy analysis.
Mice were anesthetized and perfused with 0.1 m sodium cacodylate buffer containing 4% paraformaldehyde and 2.5% glutaraldehyde. The brain, spinal cord, and sciatic nerve tissues were processed, embedded, sectioned, and analyzed as described previously (Traka et al., 2010; Hussien et al., 2014). Toluidine-blue-stained, 1-μm-thick sections of the tissues were assessed for hypomyelination and demyelination. Images were collected using the Tecnai electron microscope at the University of Chicago Electron Microscope Facility.
Ex vivo recall and flow cytometry during EAE.
For T-cell analysis from MOG35-55/CFA primed mice, spleens were dissociated into single-cell suspensions and red blood cells lysed. Total splenocytes (0.5 × 106 cells/well) were assessed for ex vivo responses in the presence of anti-CD3 (1 μg/ml), OVA323-339, or MOG35-55 (20 μg/ml). Twenty-four hours after culture initiation, the wells for cellular proliferation analysis were pulsed with 1 μCi of tritiated thymidine (3H-TdR) and the cultures harvested at day +3 and 3H-TdR incorporation detected using a Topcount Microplate Scintillation Counter. Results are expressed as the mean counts per minute of triplicate cultures. For cytokine analysis, replicate wells were harvested on day +3 of culture and the level of cytokine secreted determined via multiplex Luminex LiquiChip (Millipore).
For the CNS leukocytes, single-cell suspensions were prepared as described previously (McMahon et al., 2005) from spinal cords of individual mice perfused with 20 ml of PBS. For flow cytometric analysis, splenocytes and CNS cells were cultured for 2 h in the presence of phorbol ester (50 ng/ml) and ionomycin (500 ng/ml), followed by the addition of brefeldin A (10 μg/ml; Sigma-Aldrich) for an additional 2 h. The cells were washed in PBS, stained with LIVE/DEAD Fixable Aqua Dead Cell Stain (Life Technologies), blocked with anti-CD16/32 (eBioscience), and then specifically stained with the indicated antibodies. Cells were stained with anti-CD45 (clone 30-F11), anti-CD3 (clone 145-2C11), anti-CD4 (clone RM4-5), anti-CD8 (clone 53-6.7), anti-CD25 (clone PC61), anti-IL-17 (clone eBio17B7), anti-IFN-γ (clone XMG1.2), and anti-Ki67 (clone SolA15) (all from eBioscience).
To assess the oligodendrocyte populations, total CNS cells were stained with LIVE/DEAD Fixable Aqua Dead Cell Stain (Life Technologies), blocked with anti-CD16/32 (eBioscience), and then specifically stained with the following antibodies: anti-A2B5 (clone 105; R&D Systems); anti-NG2 (unspecified clone), anti-O4 (clone 81), and anti-GALC (unspecified clone) (Millipore); and anti-CHOP (9C8; Thermo Scientific). 1 × 106 viable cells were analyzed per individual sample using a Fortessa cytometer (BD Bioscience) and the data analyzed using FloJo Version 9.5.2 software (TreeStar).
Statistics.
Student's t test was used for two-group comparisons. For multiple comparisons, a series of repeated-measures ANOVA with a Greenhous–Geisser adjustment was used, followed by a Bonferroni test, using Stata 12.0 software (StataCorp). p < 0.05 was considered statistically significant. For the EAE studies, a series of nonparametric Wilcoxon rank-sum tests were also performed for each time point.
Results
Generation of OL-BiPko/ko mice
To inactive BIP expression in oligodendrocytes during development, we used BiPflox/flox mice that carry a conditional allele of the BiP gene that functions normally in the absence of the cre recombinase (Luo et al., 2006). We mated these mice with animals in which the gene encoding the cre recombinase was placed under the transcriptional control of the gene that encodes the myelin enzyme 2′, 3′-cyclic nucleotide-3′-phosphodiesterase (CNP) (Lappe-Siefke et al., 2003) to generate OL-BiPko/ko mice in which the BiP gene was inactivated in oligodendrocytes during development. BiP gene recombination was detected in DNA isolated from the brains, sciatic nerves, and tails of the OL-BiPko/ko mice (Fig. 1A), whereas no recombination was detected in DNA from the hearts, lungs, spleens, or kidneys of these mice or in BiPflox/flox control mice (Fig. 1A and data not shown). Both male and female OL-BiPko/ko mice exhibited tremors and ataxia around postnatal day 10 (P10) and died ∼P13. In addition, these mice showed smaller body sizes and lower body weights compared with the BiPflox/flox mice (Fig. 1B,C). The double-immunolabeling analysis of 12-d-old mice with CC1 antibodies, a marker for mature oligodendrocytes, and BiP antibodies revealed that only 10% of the oligodendrocytes in the OL-BiPko/ko mice expressed BiP compared with 95% of the oligodendrocytes in BiPflox/flox mice (Fig. 1D,E). These data indicate that BiP was inactivated in the oligodendrocytes of the OL-BiPko/ko mice.
Figure 1.

Generation of OL-BiPko/ko mice. A, Genotyping PCR using genomic DNA of different tissues from OL-BiPko/ko mice, BiPflox/flox mice, and wild-type mice showing LoxP recombination (650 bp; knock-out BiP allele) in the brains, sciatic nerves, and tails, whereas no recombination was detected in the heart tissue of OL-BiPko/ko mice. Both OL-BiPko/ko and BiPflox/flox mice, but not WT mice, had the floxed BiP allele (550 bp) in all tissues examined. Only WT mice had the WT BiP allele (515 bp) in all tissues examined. n = 3 animals. B, C, Twelve-day-old OL-BiPko/ko mice showing smaller body sizes and loss of body weight compared with BiPflox/flox mice. D, E, Immunohistochemical analysis of tissues from 12-d-old mice using antibodies against BiP and CC1, a marker for mature oligodendrocytes, showing lower numbers of BiP and CC1 double-positive cells in OL-BiPko/ko mice compared with BiPflox/flox mice. Scale bars, 20 μm. n = 3 animals; graph indicates mean ± SD; *p < 0.004; **p < 0.001. SC-WM, Spinal cord white matter.
Homozygous deletion of BiP impacts the survival of mature oligodendrocytes during development
Immunohistochemical staining using CC1 antibodies confirmed the loss of oligodendrocytes at P12 in the OL-BiPko/ko mice (Fig. 2A,B). In contrast, using PDGFRα antibodies, a marker for oligodendrocyte precursor cells (OPCs), we found a higher proportion of these cells in OL-BiPko/ko mice compared with BiPflox/flox mice (Fig. 2C,D). TUNEL staining was detected in ∼35% of the oligodendrocytes in 12-d-old OL-BiPko/ko mice, but was not detected in BiPflox/flox mice (Fig. 2E,F). Collectively, these results establish that BiP is required for the survival of developing oligodendrocytes and that the pool of OPCs increases to compensate for the loss of mature oligodendrocytes.
Figure 2.

Deletion of BiP affects oligodendrocyte survival during development. A, B, Immunohistochemical analysis of tissues from 12-d-old mice using antibodies against CC1 revealing lower numbers of oligodendrocytes in different regions of the CNS in OL-BiPko/ko mice compared with BiPflox/flox mice. Scale bars, 20 μm. n = 3 animals; graph indicates mean ± SD; *p < 0.04; ****p < 0.01; # #p < 0.003; # # #p < 0.001. C, D, Immunohistochemical analysis of tissues from 12-d-old mice using antibodies against PDGFRα, a marker for OPCs, showing higher OPC numbers in different regions of the CNS in OL-BiPko/ko mice compared with BiPflox/flox mice. Scale bars, 20 μm. n = 3 animals; graph indicates mean ± SD; ***p < 0.02; ****p < 0.01; #p < 0.004. E, F, Immunohistochemical staining of 12-d-old mice for TUNEL and CC1 demonstrating higher percentages of cells expressing TUNEL and CC1 in the spinal cord white matter of OL-BiPko/ko mice compared with BiPflox/flox mice. Scale bars, 20 μm. n = 3 animals; graph indicates mean ± SD; **p < 0.03. CC, Corpus callosum; Cer, cerebellum; BS, brainstem.
Hypomyelination in OL-BiPko/ko mice during development
To assess the myelin status in OL-BiPko/ko mice, MBP expression was examined by immunohistochemistry, which revealed that the expression of MBP was dramatically lower in OL-BiPko/ko mice compared with BiPflox/flox mice (Fig. 3A). Furthermore, Western blot analysis confirmed the loss of MBP, MAG, and PLP protein expression in OL-BiPko/ko mice compared with BiPflox/flox mice (Fig. 3B), again suggesting dramatically reduced myelin levels. We used quantitative PCR to assess whether the mRNAs showed the same kinetics as the proteins and found lower expression of the Mbp, Mag, and Plp mRNAs in OL-BiPko/ko mice compared with BiPflox/flox mice (Fig. 3C). In addition, an electron microscopy analysis of various regions of the brain and spinal cord in 12-d-old OL-BiPko/ko mice revealed hypomyelination compared with BiPflox/flox mice (Fig. 3D). Unexpectedly, we found no difference in optic nerve myelination in OL-BiPko/ko mice compared with BiPflox/flox mice (Fig. 3E). Assessments of the number of oligodendrocytes in the optic nerve also revealed no difference between OL-BiPko/ko and BiPflox/flox mice (Fig. 3F,G). Nevertheless, we found that a higher percentage of the oligodendrocytes in the optic nerve of OL-BiPko/ko mice exhibited TUNEL expression compared with those of BiPflox/flox mice (Fig. 3H), suggesting that these cells are on the path of apoptosis. Collectively, these data confirm that the deletion of BiP from oligodendrocytes during development affects myelin sheath synthesis in different regions of the brain, but not in the optic nerve.
Figure 3.
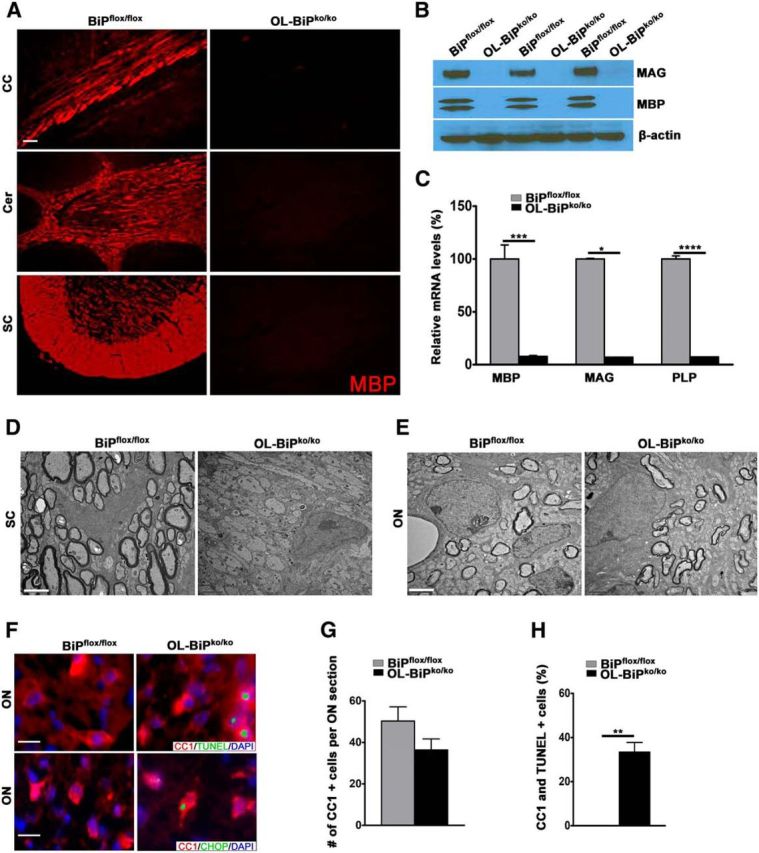
BiP is required for myelin synthesis during development. A, Immunohistochemical staining of tissues from 12-d-old mice demonstrating the lack of MBP expression in different CNS areas in OL-BiPko/ko mice compared with BiPflox/flox mice. n = 3 animals. Scale bars represent 20 μm. B, Western blot analysis of the total brain protein extracts from 12-d-old mice demonstrating the lack of MBP and MAG protein expression in OL-BiPko/ko mice compared with BiPflox/flox mice. n = 3 animals. C, qPCR results showing that the deletion of BiP specifically in oligodendrocytes suppressed the Mbp, Mag, and Plp mRNA levels in the brains of OL-BiPko/ko mice compared with BiPflox/flox mice. All data presented are mean ± SD; n = 3 animals; *p < 0.01, **p < 0.004, ***p < 0.002. D, E, EM analysis of various regions of the brain and spinal cord white matter of 12-d-old mice showing no myelin abnormalities in the optic nerve in OL-BiPko/ko mice compared with BiPflox/flox mice. However, there is no myelination in the spinal cord white matter in OL-BiPko/ko mice compared with BiPflox/flox mice. n = 3 animals. Scale bars, 1 μm. F, G, Double immunostaining for TUNEL or CHOP and CC1 in the ON demonstrating no difference in the numbers of oligodendrocytes in OL-BiPko/ko mice compared with BiPflox/flox mice. Scale bars, 20 μm. n = 3 animals; graph indicates mean ± SD. H, Quantitative analysis of the immunostaining data showing a higher percentage of double-positive cells for TUNEL and CC1 in the ON of OL-BiPwt/ko mice compared with BiPwt/flox mice. n = 3 animals; graph indicates mean ± SD; **p < 0.01.
Modulation of ER stress sensors and expression of ER apoptotic factor CHOP in OL-BiPko/ko mice during development
In response to ER stress, BiP is released from the ER stress sensors PERK, ATF6, and IRE1, resulting in UPR activation (Bertolotti et al., 2000). Although the UPR provides protection against ER stress, persistent UPR activation leads to apoptosis (Walter and Ron, 2011). To examine the effect of BiP gene deletion on the ER stress sensors, we performed analyses for p-PERK, cleaved ATF6 and Xbp-1 splicing, to assess the activated state of these pathways. Using Western blots and Xbp-1 mRNA assays, we found that the deletion of BiP in oligodendrocytes increased the expression of p-PERK and cleaved ATF6, but had no effect on the splicing of Xbp-1 mRNA, in the brains of OL-BiPko/ko mice at P12 compared with BiPflox/flox mice (Fig. 4A–F). BiP suppresses the expression of CHOP (Wang et al., 2010), a transcription factor induced downstream of PERK and ATF6, which orchestrates cellular apoptosis in response to persistent UPR activation (Zinszner et al., 1998; Oyadomari and Mori, 2004). Therefore, we assessed the expression of CHOP in the OL-BiPko/ko mice. We found that the deletion of BiP in oligodendrocytes during development caused a signification increase in the expression of CHOP in oligodendrocytes in P12 OL-BiPko/ko mice compared with BiPflox/flox mice (Fig. 4G,H). Interestingly, we also detected increased expression of CHOP in mature oligodendrocytes of the optic nerve (Fig. 4H), which correlates with the increased percentage of TUNEL-positive cells detected in this tissue of the OL-BiPko/ko mice (Fig. 3H). In addition, the deletion of BiP in oligodendrocytes increased the expression of CHOP mRNA in P12 OL-BiPko/ko mice compared with BiPflox/flox mice (Fig. 4I). Collectively, these results establish that the deletion of BiP in oligodendrocytes leads to the activation of the PERK and ATF6 signaling pathways, resulting in the expression of the pro-apoptotic factor CHOP.
Figure 4.
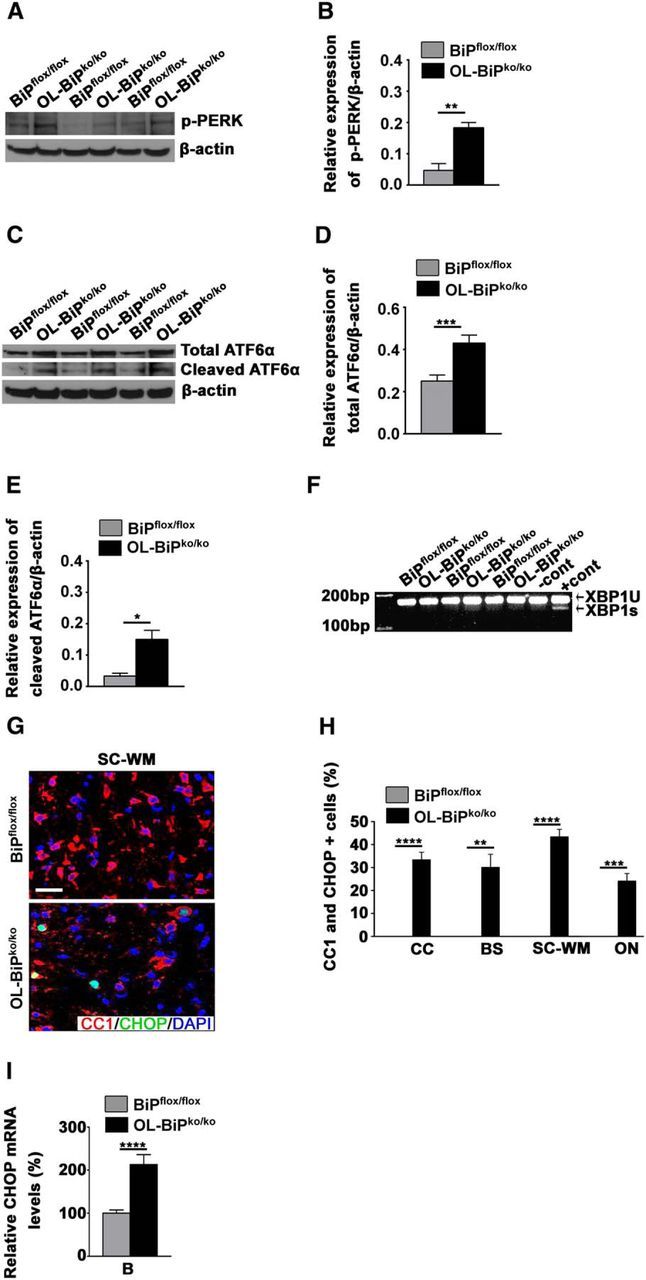
Homozygous deletion of BiP in oligodendrocytes during development activates the PERK and IRE1 signaling pathways and increases CHOP expression. A, B, Western blot analysis of total protein extracts from mouse brains demonstrating the higher expression of p-PERK in OL-BiPko/ko mice compared with BiPflox/flox mice. n = 3 animals; graph indicates mean ± SD; **p < 0.03. C, Western blot analysis of total brain protein extracts from mice demonstrating the higher expression of total and cleaved ATF6α in OL-BiPko/ko mice compared with BiPflox/flox mice, and confirmed by quantitative analysis (D, E). n = 3 animals; graph indicates mean ± SD; *p < 0.04; ***p < 0.02. F, Xbp-1 splicing assay. Total mRNAs isolated from mouse brain were used to amplify the products of spliced and unspliced Xbp-1 mRNA, which showed no splicing product for Xbp-1 in OL-BiPko/ko and BiPflox/flox mice. mRNA isolated from 3T3 cells treated with thapsigargin was used as a positive control (+cont), and mRNA isolated from untreated 3T3 cells was used as a negative control (−cont). G, H, Immunohistochemical staining of different CNS areas of 12-d-old mice using antibodies against CHOP and CC1 demonstrating a higher percentage of double-positive cells for CHOP and CC1 in OL-BiPko/ko mice compared with BiPflox/flox mice. Scale bars, 20 μm. n = 3 animals; graph indicates mean ± SD; **p < 0.03; ***p < 0.02., ****p < 0.01. I, qPCR showing elevated CHOP mRNA levels in total mRNAs isolated from the brains of OL-BiPko/ko mice compared with BiPflox/flox mice. n = 4 animals; graph indicates mean ± SD; ****p < 0.01. CC, Corpus callosum; Cer, cerebellum; BS, brainstem; B, brain.
BiP is required for oligodendrocyte survival and myelin maintenance during adulthood
To address the roles that BiP plays in adult oligodendrocytes and myelin maintenance, we allowed animals to reach adulthood before inactivating BiP expression. To achieve BiP inactivation in adult mice, we used transgenic animals that express a tamoxifen-inducible Cre recombinase under the transcriptional control of the gene that encodes the myelin protein PLP. Eight-week-old mice homozygous for the floxed BiP allele and hemizygous for the PLP/CreERT transgene received tamoxifen (BiPko/ko) or sunflower seed oil (control) by IP injection for 5 consecutive days. These mice displayed tremors, which initiated ∼2 weeks after the initial tamoxifen dose and progressed to hind-limb paralysis 1–2 weeks later. Because this phenotype closely resembled that displayed by our DTA mouse model in which oligodendrocytes are ablated in adulthood (Traka et al., 2010), we examined oligodendrocyte viability in the absence of BiP expression. Using CC1 antibodies, we detected the loss of oligodendrocytes in different regions of the CNS in mice on day 12 (BiPko/ko D12), day 19 (BiPko/ko D19), and day 26 (BiPko/ko D26) after tamoxifen injections compared with control mice (Fig. 5A,B). In addition, we found that a higher percentage of the oligodendrocytes of BiPko/ko mice at D12, D19, and D26 expressed TUNEL compared with control mice (Fig. 5A,C). Moreover, we also found that a higher percentage of the oligodendrocytes of BiPko/ko mice at D12, D19, and D26 expressed CHOP compared with control mice (Fig. 5D,E). These data indicated that deletion of BiP in oligodendrocytes resulted in their death by apoptosis. Analyses of toluidine blue-stained sections of various regions of the brain and spinal cord at day 59 after tamoxifen injections revealed extensive demyelination of the BiPko/ko mice, but not control mice (Fig. 5F). Collectively, these results establish that BiP is required for oligodendrocyte survival and myelin maintenance during adulthood.
Figure 5.
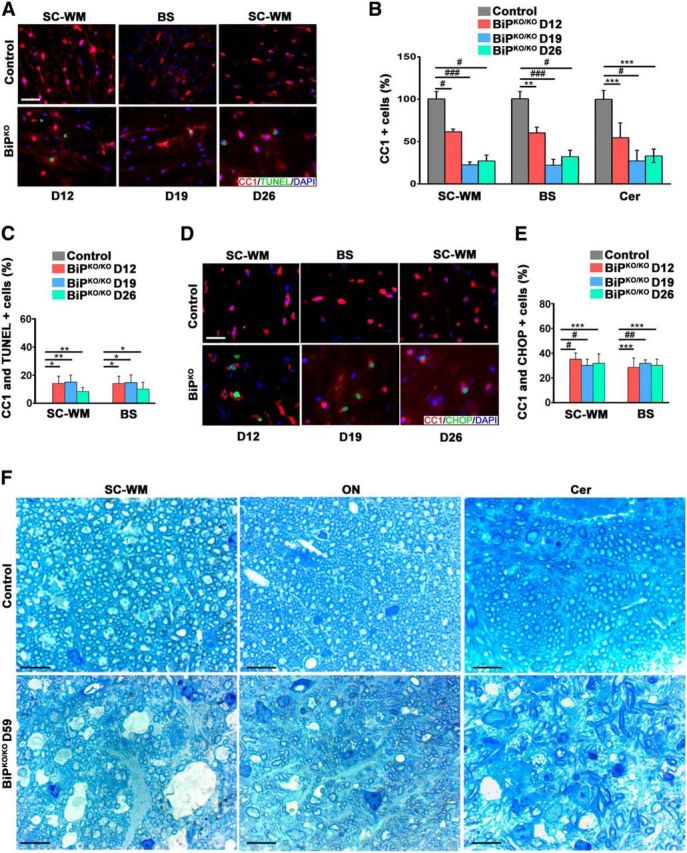
Deletion of BiP affects oligodendrocyte survival and myelination in adulthood. A, Immunohistochemical analysis of mouse tissues at 12 d (BiPko/ko D12), D19, and D26 after tamoxifen injections using antibodies against CC1 and TUNEL showing lower numbers of oligodendrocytes (B) and higher numbers of double-positive cells for CC1 and TUNEL (C) in mice that received tamoxifen at different time points after the tamoxifen injections compared with control mice (control). Scale bar, 20 μm. n = 3 animals; graph indicates mean ± SD; *p < 0.05; **p < 0.04; ***p < 0.02, #p < 0.01; ###p < 0.001. D, E, Immunohistochemical analysis of different CNS areas of mouse tissues at D12, D19, and D26 after tamoxifen injections using antibodies against CC1 and CHOP demonstrating higher numbers of double-positive oligodendrocytes for CC1 and CHOP at different time points after tamoxifen injections in BiPko/ko mice compared with control mice. Scale bar, 20 μm. n = 3 animals; graph indicates mean ± SD; ***p < 0.02; #p < 0.01; ##p < 0.002. F, Toluidine blue staining of tissues from different regions of the brain and spinal cord white matter showing myelin damage in these regions in mice that received tamoxifen injections at D59 compared with the control mice. Scale bar, 20 μm. n = 3 animals.
Heterozygous deletion of BiP in oligodendrocytes does not affect survival during development, but worsens the EAE disease course
Because mice with homozygous deletion of BiP in oligodendrocytes die at ∼P13, we studied the functional effect of BiP in oligodendrocytes in the pathogenesis of EAE by the heterozygous deletion of the BiP gene specifically in oligodendrocytes, again using the CNP/Cre driver line. Mice with the heterozygous deletion of BiP in oligodendrocytes (OL-BiPwt/ko mice) displayed a normal clinical phenotype, normal numbers of mature oligodendrocytes, and myelin in different regions of the CNS (Fig. 6A–C). Six-week-old female OL-BiPwt/ko mice were immunized with the MOG35-55 peptide to induce EAE. CNP/Cre, BiPwt/flox and WT mice served as controls for the EAE studies. All immunized mice developed a typical EAE clinical phenotype. Nevertheless, the onset was earlier in OL-BiPwt/ko mice starting at approximately postimmunization day (PID) 9, whereas the other groups showed disease symptoms beginning ∼PID14. In addition, whereas the control mice followed a typical EAE disease course, reaching a mean maximal clinical score of 2.9, OL-BiPwt/ko mice displayed a higher clinical score that reached a mean maximum of 4 (Fig. 6D). These data indicate that the heterozygous deletion of BiP in oligodendrocytes has no effect on their survival during normal development, but exacerbates the EAE disease course.
Figure 6.

Heterozygous deletion of BiP in oligodendrocytes exacerbates the course of EAE. A, B, CC1 immunostaining of 8-week-old mice showing no difference in the numbers of mature oligodendrocytes in different regions of the CNS in mice with one allele of BiP (OL-BiPwt/ko mice) compared with the control (OL-BiPwt/flox mice). Scale bars, 20 μm. n = 3 animals. C, MBP immunostaining of 8-week-old mice showing normal expression of MBP in different regions of the CNS of OL-BiPwt/ko mice compared with OL-BiPwt/flox mice. Scale bars, 20 μm. n = 3 animals. D, OL-BiPwt/flox mice showed exacerbated EAE disease severity compared with OL-BiPwt/flox mice. Error bars indicate SED; *p < 0.05; #p < 0.001. In addition, a series of nonparametric Wilcoxon rank-sum tests were performed for each EAE time point and the ANOVA results were confirmed.
OL-BiPwt/ko mice experiencing EAE display an increased loss of oligodendrocytes
To explore the underlying reason for the exacerbated clinical EAE disease course in the OL-BiPwt/ko mice, we examined oligodendrocyte viability in these animals. We found lower numbers of oligodendrocytes within the lumbar spinal cord white matter during EAE at PID17 in the OL-BiPwt/ko mice compared with either BiPwt/flox mice or unimmunized mice of both genotypes (Fig. 7A,B). In addition, we found that OL-BiPwt/ko mice had a higher percentage of TUNEL-positive oligodendrocytes in the lumbar spinal cord white matter during EAE at PID17 compared with either BiPwt/flox mice or unimmunized mice of both genotypes (Fig. 7C). Flow cytometry analysis of oligodendrocyte lineage cells in the brain and spinal cord of the OL-BiPwt/ko and littermate control animals did not detect a statistically significant difference in mature oligodendrocytes, although the OL-BiPwt/ko mice tended to contain a reduced number of these cells (Fig. 7D), which is consistent with the immunohistochemistry data described above (Fig. 7B). Moreover, a significantly higher percentage of mature oligodendrocytes were positive for the CHOP protein in the OL-BiPwt/ko EAE mice compared with control EAE animals (Fig. 7E), suggesting that there is an enhanced response to ER stress in the BiP-haploinsufficient mice. These results indicate that BiP has a protective effect in oligodendrocytes and the heterozygous deletion of BiP has a significant impact on the onset and severity of EAE.
Figure 7.
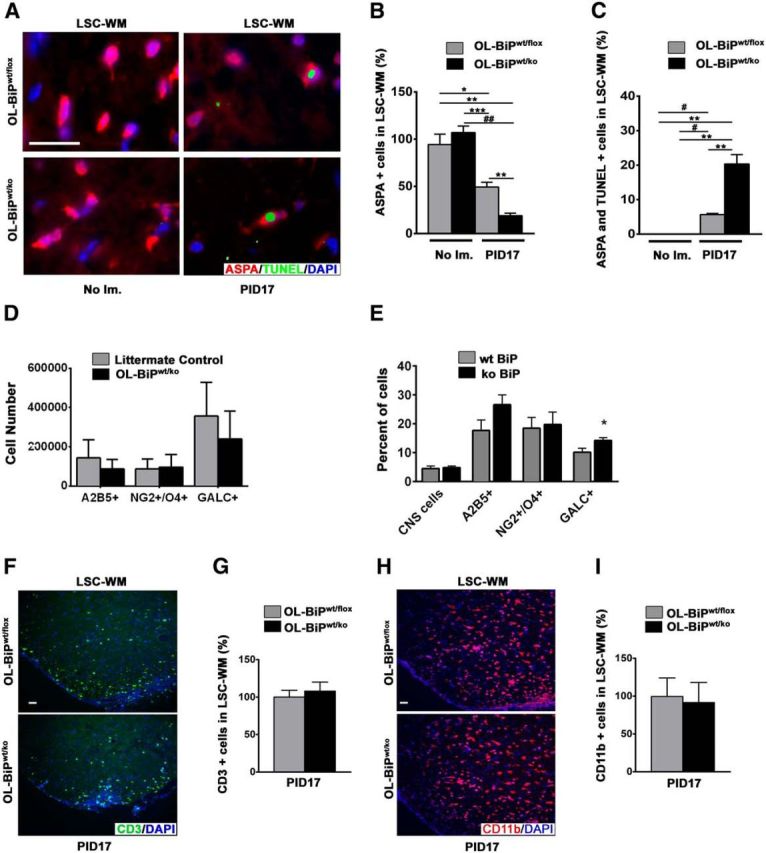
Heterozygous deletion of BiP exacerbates oligodendrocyte loss during EAE without altering T-cell infiltration and macrophage/microglial activation. A, Double immunostaining for TUNEL and ASPA, a marker for mature oligodendrocytes, showing that the deletion of one allele of BiP in oligodendrocytes resulted in lower numbers of oligodendrocytes in lumbar spinal cord white matter during EAE at PID17 in OL-BiPwt/ko mice compared with OL-BiPwt/flox mice or nonimmunized mice of both genotypes. Quantitative analysis of the immunostaining data showing lower numbers of oligodendrocytes (B) and higher percentages of double-positive cells for TUNEL and ASPA (C) in the lumbar spinal cord white matter of OL-BiPwt/ko mice during EAE at PID17 compared with OL-BiPwt/flox mice or nonimmunized mice of both genotypes. n = 3 animals. Scale bars, 20 μm. *p < 0.02; **p < 0.01; ***p < 0.004; #p < 0.003; ##p < 0.002. D, Number of A2B5+, NG2+/O4+, and GALC+ oligodendrocyte lineage cells was assessed by flow cytometry. E, Percentage of total CNS cells and A2B5+, NG2+/O4+, and GALC+ oligodendrocyte lineage cells that express the CHOP protein was assessed by flow cytometry. *p < 0.05. F, G, CD3 immunostaining showed no difference in the numbers of T cells in the lumbar spinal cord white matter from the OL-BiPwt/ko mice compared with the OL-BiPwt/flox mice. n = 3 animals. Scale bars, 20 μm. H, I, CD11b immunostaining showing no difference in the numbers of activated macrophages/microglia in the lumbar spinal cord white matter of the OL-BiPwt/ko mice compared with the OL-BiPwt/flox mice. n = 3 animals. Scale bars, 20 μm. LSC-WM, Lumbar spinal cord white matter; No Im., no immunization.
OL-BiPwt/ko mice experiencing EAE display a normal inflammatory response
To determine whether the altered response to EAE induction by the OL-BiPwt/ko mice was the result of an altered inflammatory response after the induction of EAE, we characterized the immune response in these animals at the peak of acute disease. Using an immunohistochemical approach, we found no differences in T-cell infiltration in the CNS between the OL-BiPwt/ko and BiPwt/flox mice (Fig. 7F,G). Macrophage/microglia activation in situ also appeared normal (Fig. 7H,I). In addition, splenocytes were isolated from OL-BiPwt/ko and littermate control mice at the peak of EAE disease and reactivated ex vivo in the presence of a negative control antigen (OVA323-339), a pan T cell activator (anti-CD3), or the immunizing antigen (MOG35-55) for 3 d. The levels of IFN-γ, IL-17, and GM-CSF secreted by splenocytes from the OL-BiPwt/ko and littermate control mice were not significantly different (Fig. 8A–C). Spleen and CNS immune cell populations were also characterized by flow cytometry at the peak of acute disease. No significant differences in T-cell populations were detected in either tissue, suggesting that the OL-BiPwt/ko mice developed a normal immune response to the immunizing myelin antigen (Fig. 8D–I) and presented with an earlier onset of disease compared with littermate control mice.
Figure 8.

The absence of BiP does not modulate inflammatory CD4+ T-cell function. Littermate control mice and OL-Bipwt/ko mice (n = 9–10/group) were primed with MOG35-55/CFA and followed for disease until the peak of acute phase of disease. On PID17, spleens and CNS were harvested for analysis. Splenocytes (0.5 × 106 cells/well in a 96-well plate) from the littermate control mice (n = 10) and OL-Bipwt/ko mice (n = 9) were reactivated ex vivo in the presence of OVA323-339 (10 μg/ml), anti-CD3 (1 μg/ml), or MOG35-55 (10 μg/ml) for 3 d. The level of the secreted cytokines IFN-γ (A), IL-17 (B), and GM-CSF (C) was assessed after activation. The numbers of total live CD45hi cells (D, G), total CD4+ and CD8+ T cells (CD3+/CD4+/CD8− vs CD3+/CD4−/CD8+; E, H), and CD4+ T cells expressing IFN-γ, IL-17, Ki67, and CD25 (F, I) was determined by flow cytometry in the spleen and CNS (J).
Peripheral nerve abnormalities in Sch-BiPko/ko mice
We also examined the importance of BiP expression by Schwann cells, the myelinating cells of the PNS. To inactive BIP expression in Schwann cells during development, BiPflox/flox mice were mated to mice that express the cre recombinase under the transcriptional control of the gene that encodes the abundant peripheral myelin protein P0 (Feltri et al., 1999) to generate the Sch-BiPko/ko mice. At P9, the Sch-BiPko/ko mice displayed hindlimb tremor and weakness that gradually progressed to complete paralysis by P25. Ambulatory capability was lost completely by P35, when the mutant mice had to be killed. Morphologically, the sciatic nerves displayed a significant reduction of myelinating Schwann cells and a paucity of myelin (Fig. 9). PNS myelination therefore appears to be dependent on BiP expression in Schwann cells.
Figure 9.

Deletion of BiP affects Schwann cell survival during development. A, B, Immunohistochemical analysis of sciatic nerves from 25-d-old mice using Krox-20 antibodies showing lower numbers of Schwann cells in Sch-BiPko/ko mice compared with BiPflox/flox mice. n = 3 animals; graph indicates mean ± SD; *p < 0.01. Scale bars, 20 μm. C, Immunohistochemical analysis of sciatic nerve from 25-d-old mice using antibodies against MBP demonstrating lower MBP expression in Sch-BiPko/ko mice compared with BiPflox/flox mice. n = 3. Scale bars, 20 μm. D, Toluidine blue staining of sciatic nerve from 25-d-old mice showing myelin damage in Sch-BiPko/ko mice compared with BiPflox/flox mice. n = 3. Scale bars, 20 μm. E, Electron microscopy analysis of sciatic nerves of 25-d-old mice showing unmyelinated axons in Sch-BiPko/ko mice compared with BiPflox/flox mice (arrows). n = 3 animals. Scale bars, 2 μm.
Discussion
The ER resident chaperone protein BiP, which is a member of the heat shock protein 70 family, plays a number of distinct roles in ER function, including facilitating nascent protein folding, modulating calcium homeostasis, and regulation of the UPR. Although homozygous knock-out of the BiP gene in prostate epithelial cells has little effect on mouse prostate development and function (Fu et al., 2008), most cells and organs in which BiP function has been analyzed have shown a critical need for basal BiP expression (Wang et al., 2010; Zhu et al., 2013). In the current study, we examined the role that BiP plays in oligodendrocytes using a cell-specific gene inactivation approach. Myelinating cells synthesize an enormous amount of membrane protein so their secretory pathway plays a critical role in the function of these specialized glial cells. Our results demonstrate that the elimination of BiP expression in oligodendrocytes and Schwann cells during development results in a dramatic loss of myelinating cells and severe hypomyelination of the CNS and PNS, respectively. Moreover, BiP gene inactivation in adult oligodendrocytes also results in the rapid loss of these cells, which is accompanied by extensive CNS demyelination. We also show that mice that are heterozygous for the oligodendrocyte-specific inactivated BiP allele display increased sensitivity to EAE, which is correlated with an increased loss of oligodendrocytes, suggesting that BiP normally provides protection to oligodendrocytes against inflammatory insults.
The profound effect of BiP elimination on actively myelinating oligodendrocytes and Schwann cells is not surprising because the secretory pathway of these cells is responsible for generating a considerable amount of membrane and membrane protein during a relatively short developmental window during which myelination is most active. More unexpected was the rapid demise of mature oligodendrocytes in adult animals after the elimination of BiP expression in these cells. Although mature oligodendrocytes remain associated with the myelin sheath, their metabolic demands are dramatically reduced because the myelin components have exceptionally long half-lives (Toyama et al., 2013). Despite the reduced load on their secretory pathway, mature oligodendrocytes were eliminated within days of BiP inactivation.
One possible explanation for the rapid demise of both myelinating and mature oligodendrocytes when BiP is eliminated is related to the role that the BiP protein plays in the regulation of the UPR. BiP associates with and inhibits the activation of the ER stress sensors PERK, ATF6, and IRE1. The elimination of the BiP protein should allow these sensors to become activated, which is consistent with our observations in developing oligodendrocytes from the OL-BiPko/ko mice. Both PERK and ATF6 display evidence of being activated in the BiP-deficient animals, which likely triggers an UPR response in oligodendrocytes. The activation of the UPR results in the upregulation of a number of cytoprotective pathways that restore cellular homeostasis by dampening new protein synthesis and increasing the cell's protein-folding capacity (Walter and Ron, 2011). Nevertheless, persistent UPR activation leads to cellular apoptosis, which is associated with the induction of expression of the pro-apoptotic transcription factor CHOP, the expression of which we have observed in the BiP deficient animals. Therefore, it seems likely that the loss of oligodendrocytes in the BiP-deficient animals is associated at least in part with the release of UPR sensor inhibition. Nevertheless, BiP also plays critical roles in protein folding and ER calcium homeostasis, the disruption of which might also contribute to the demise of oligodendrocytes in these animals. Interestingly, in contrast to BiP deletion leading to upregulation of all three branches of the UPR in hematopoietic stem cells (Wey et al., 2012), BiP elimination does not appear to result in the activation of the IRE1 arm of the UPR in oligodendrocytes. We did not detect evidence of increased XBP1 splicing, which occurs as a result of IRE1 activation, in the mutant animals. This is consistent with our previous failures to detect XBP1 splicing in oligodendrocytes undergoing ER stress (unpublished observations), suggesting that the IRE1 arm of the UPR is not a significant participant in the ER stress response of oligodendrocytes.
The role that CHOP plays in the increased apoptotic cell death of the BiP-deficient oligodendrocytes remains unresolved. As mentioned above, we have detected increased CHOP expression in oligodendrocytes in which the BiP gene has been specifically inactivated, which correlates with the increased apoptotic death of these cells. Although in most situations examined, CHOP expression is believed to be pro-apoptotic (Tabas and Ron, 2011), it has been reported that CHOP inactivation might actually be harmful to oligodendrocytes experiencing severe ER stress. Southwood et al. (2002) demonstrate that, in the rumpshaker mouse, CHOP expression appears protective to oligodendrocytes and CNS myelin. Rumpshaker mice represent a mouse model of the human leukodystrophy Pelizaeus–Merzbacher disease, which is caused by a point mutation in the gene that encodes the abundantly expressed myelin proteolipid protein. Nevertheless, in preliminary studies in our laboratory, we have found that, in response to the stress activated by inflammation, CHOP induction is detrimental to oligodendrocyte survival (Y. Dzhashiashvili, H. Bommiasamy, R.B. Kunjamma, J.R. Podojil, S.D. Miller, B. Popko, unpublished observations.) It thus appears that the effect of CHOP induction is likely cell and stress specific (Gow and Wrabetz, 2009).
Considering that GRP78 is believed to play a protective role under physiologic and pathologic stress (Pfaffenbach and Lee, 2011), we also used mice with an oligodendrocyte-specific heterozygous BiP-null mutation to study the role that BiP plays in the pathogenesis of EAE. The OL-BiPWT/KO mice were phenotypically normal and fertile, in agreement with previous studies showing that BiP heterozygosity does not affect mouse development (Luo et al., 2006; Dong et al., 2008). In addition, we found that deletion of one allele of BiP in oligodendrocytes during development had no effect on oligodendrocytes numbers or CNS myelination, which allowed us to use these animals to study the effect of reduced BiP expression on the response of oligodendrocytes to CNS inflammation. The OL-BiPwt/ko mice displayed an exacerbated EAE disease course that correlated with a greater loss of oligodendrocytes, which expressed elevated levels of the CHOP protein. Importantly, the inflammatory response was unaltered in the OL-BiPwt/ko mice relative to wild-type control mice that had been immunized to induce EAE. BiP is normally upregulated in MS and EAE lesions (Mháille et al., 2008; Cunnea et al., 2011; McMahon et al., 2012; Ni Fhlathartaigh et al., 2013) and our current results demonstrate that the UPR likely plays a protective role in the normal response of oligodendrocytes to CNS inflammation.
These results support our previous observation that mice with a compromised ability to respond to ER stress are more susceptible to inflammatory demyelination (Lin et al., 2005; Lin et al., 2007; Hussien et al., 2014). Moreover, these findings bolster our efforts to enhance the UPR either genetically (Lin et al., 2008; Lin et al., 2013) or pharmacologically (Way et al., 2015) in an attempt to provide increased protection to oligodendrocytes against inflammatory demyelinating insults. The continued detailed analysis of the response of oligodendrocytes to ER stress is essential to efforts to enhance the natural cytoprotective response of these cells for therapeutic benefit. Oligodendrocyte protection would likely contribute to the restorative (myelin repair) and anti-inflammatory approaches to MS therapy.
Although BiP inactivation resulted in widespread oligodendrocyte loss and severe hypomyelination throughout much of the CNS in the OL-BiPko/ko mice, the optic nerve appeared spared: oligodendrocyte numbers and myelin appeared similar to what is observed in control animals. Nevertheless, at the time that these animals had reached the end of their dramatically shortened lifespan, we did detect a higher number of CHOP-positive and TUNEL-positive oligodendrocytes in the optic nerve of the OL-BiPko/ko mice, suggesting that these cells might be on the pathway to apoptosis. Perhaps the effect of BiP inactivation is somehow delayed in the optic nerve. The myelination of the optic nerve occurs relatively late during development (Foran and Peterson, 1992), which might explain the persistence of oligodendrocytes in this region of the OL-BiPko/ko mice.
In summary, our studies establish that BiP is required for oligodendrocyte survival during development and in adulthood. The elimination of BiP specifically in oligodendrocytes resulted in the activation of the UPR, which likely contributed to the death of these cells. Moreover, our data revealed that the heterozygous deletion of BiP in oligodendrocytes results in worsened EAE symptoms, which correlated with an increased loss of oligodendrocytes that was correlated with increased CHOP expression. These results strongly suggest that BiP functions to protect oligodendrocytes against apoptosis during development, in adulthood, and in response to CNS inflammation. In addition, we show that BiP expression by Schwann cells is required for PNS myelination. Collectively, our data suggest that a compromised response to ER stress could contribute to myelin disorders of the CNS and PNS.
Footnotes
This work was supported by the National Institutes of Health (Grant NS34939 to B.P. and Grant CA027607 to A.S.L.), and the Myelin Repair Foundation (B.P. and S.D.M.). We thank Klaus Nave for providing the CNP/Cre mice, Lawrence Wrabetz and Laura Feltri for providing the P0/Cre mice, Aryan Namboodiri for providing the ASPA antisera, Benjamin Clayton for providing the 3T3 cells treated with thapsigargin, and Gloria Wright for help preparing the figures. The University of Chicago Biostatistics Laboratory provided assistance with data analysis.
The authors declare no competing financial interests.
References
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol. 2000;2:326–332. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- Cunnea P, Mháille AN, McQuaid S, Farrell M, McMahon J, FitzGerald U. Expression profiles of endoplasmic reticulum stress-related molecules in demyelinating lesions and multiple sclerosis. Mult Scler. 2011;17:808–818. doi: 10.1177/1352458511399114. [DOI] [PubMed] [Google Scholar]
- Doerflinger NH, Macklin WB, Popko B. Inducible site-specific recombination in myelinating cells. Genesis. 2003;35:63–72. doi: 10.1002/gene.10154. [DOI] [PubMed] [Google Scholar]
- Dong D, Ni M, Li J, Xiong S, Ye W, Virrey JJ, Mao C, Ye R, Wang M, Pen L, Dubeau L, Groshen S, Hofman FM, Lee AS. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008;68:498–505. doi: 10.1158/0008-5472.CAN-07-2950. [DOI] [PubMed] [Google Scholar]
- Faitova J, Krekac D, Hrstka R, Vojtesek B. Endoplasmic reticulum stress and apoptosis. Cell Mol Biol Lett. 2006;11:488–505. doi: 10.2478/s11658-006-0040-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltri ML, D'Antonio M, Previtali S, Fasolini M, Messing A, Wrabetz L. P0-Cre transgenic mice for inactivation of adhesion molecules in Schwann cells. Ann N Y Acad Sci. 1999;883:116–123. doi: 10.1111/j.1749-6632.1999.tb08574.x. [DOI] [PubMed] [Google Scholar]
- Foran DR, Peterson AC. Myelin acquisition in the central nervous system of the mouse revealed by an MBP-Lac Z transgene. J Neurosci. 1992;12:4890–4897. doi: 10.1523/JNEUROSCI.12-12-04890.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Wey S, Wang M, Ye R, Liao CP, Roy-Burman P, Lee AS. Pten null prostate tumorigenesis and AKT activation are blocked by targeted knockout of ER chaperone GRP78/BiP in prostate epithelium. Proc Natl Acad Sci U S A. 2008;105:19444–19449. doi: 10.1073/pnas.0807691105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow A, Wrabetz L. CHOP and the endoplasmic reticulum stress response in myelinating glia. Curr Opin Neurobiol. 2009;19:505–510. doi: 10.1016/j.conb.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendershot LM. The ER function BiP is a master regulator of ER function. Mt Sinai J Med. 2004;71:289–297. [PubMed] [Google Scholar]
- Hussien Y, Cavener DR, Popko B. Genetic inactivation of PERK signaling in mouse oligodendrocytes: normal developmental myelination with increased susceptibility to inflammatory demyelination. Glia. 2014;62:680–691. doi: 10.1002/glia.22634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias A, Bauer J, Litzenburger T, Schubart A, Linington C. T- and B-cell responses to myelin oligodendrocyte glycoprotein in experimental autoimmune encephalomyelitis and multiple sclerosis. Glia. 2001;36:220–234. doi: 10.1002/glia.1111. [DOI] [PubMed] [Google Scholar]
- Jolly C, Morimoto RI. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J Natl Cancer Inst. 2000;92:1564–1572. doi: 10.1093/jnci/92.19.1564. [DOI] [PubMed] [Google Scholar]
- King LS, Berg M, Chevalier M, Carey A, Elguindi EC, Blond SY. Isolation, expression, and characterization of fully functional nontoxic BiP/GRP78 mutants. Protein Expr Purif. 2001;22:148–158. doi: 10.1006/prep.2001.1424. [DOI] [PubMed] [Google Scholar]
- Kleizen B, Braakman I. Protein folding and quality control in the endoplasmic reticulum. Curr Opin Cell Biol. 2004;16:343–349. doi: 10.1016/j.ceb.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Lee AS. Mammalian stress response: induction of the glucose-regulated protein family. Curr Opin Cell Biol. 1992;4:267–273. doi: 10.1016/0955-0674(92)90042-B. [DOI] [PubMed] [Google Scholar]
- Lee AS. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods. 2005;35:373–381. doi: 10.1016/j.ymeth.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Li J, Lee AS. Stress induction of GRP78/BiP and its role in cancer. Curr Mol Med. 2006;6:45–54. doi: 10.2174/156652406775574523. [DOI] [PubMed] [Google Scholar]
- Lin W, Popko B. Endoplasmic reticulum stress in disorders of myelinating cells. Nat Neurosci. 2009;12:379–385. doi: 10.1038/nn.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Harding HP, Ron D, Popko B. Endoplasmic reticulum stress modulates the response of myelinating oligodendrocytes to the immune cytokine interferon-γ. J Cell Biol. 2005;169:603–612. doi: 10.1083/jcb.200502086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Kemper A, Dupree JL, Harding HP, Ron D, Popko B. Interferon-γ inhibits central nervous system remyelination through a process modulated by endoplasmic reticulum stress. Brain. 2006;129:1306–1318. doi: 10.1093/brain/awl044. [DOI] [PubMed] [Google Scholar]
- Lin W, Bailey SL, Ho H, Harding HP, Ron D, Miller SD, Popko B. The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J Clin Invest. 2007;117:448–456. doi: 10.1172/JCI29571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Kunkler PE, Harding HP, Ron D, Kraig RP, Popko B. Enhanced integrated stress response promotes myelinating oligodendrocyte survival in response to interferon-gamma. Am J Pathol. 2008;173:1508–1517. doi: 10.2353/ajpath.2008.080449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Lin Y, Li J, Fenstermaker AG, Way SW, Clayton B, Jamison S, Harding HP, Ron D, Popko B. Oligodendrocyte-specific activation of PERK signaling protects mice against experimental autoimmune encephalomyelitis. J Neurosci. 2013;33:5980–5991. doi: 10.1523/JNEUROSCI.1636-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S, Mao C, Lee B, Lee AS. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol Cell Biol. 2006;26:5688–5697. doi: 10.1128/MCB.00779-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavarao CN, Moffett JR, Moore RA, Viola RE, Namboodiri MA, Jacobowitz DM. Immunohistochemical localization of aspartoacylase in the rat central nervous system. J Comp Neurol. 2004;472:318–329. doi: 10.1002/cne.20080. [DOI] [PubMed] [Google Scholar]
- Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18:716–731. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med. 2005;11:335–339. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- McMahon JM, McQuaid S, Reynolds R, FitzGerald UF. Increased expression of ER stress- and hypoxia-associated molecules in grey matter lesions in multiple sclerosis. Mult Scler. 2012;18:1437–1447. doi: 10.1177/1352458512438455. [DOI] [PubMed] [Google Scholar]
- Mháille AN, McQuaid S, Windebank A, Cunnea P, McMahon J, Samali A, FitzGerald U. Increased expression of endoplasmic reticulum stress-related signaling pathway molecules in multiple sclerosis lesions. J Neuropathol Exp Neurol. 2008;67:200–211. doi: 10.1097/NEN.0b013e318165b239. [DOI] [PubMed] [Google Scholar]
- Ni M, Lee AS. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007;581:3641–3651. doi: 10.1016/j.febslet.2007.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ní Fhlathartaigh M, McMahon J, Reynolds R, Connolly D, Higgins E, Counihan T, Fitzgerald U. Calreticulin and other components of endoplasmic reticulum stress in rat and human inflammatory demyelination. Acta Neuropathol Commun. 2013;1:37. doi: 10.1186/2051-5960-1-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- Pfaffenbach KT, Lee AS. The critical role of GRP78 in physiologic and pathologic stress. Curr Opin Cell Biol. 2011;23:150–156. doi: 10.1016/j.ceb.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer SE, Warrington AE, Bansal R. The oligodendrocyte and its many cellular processes. Trends Cell Biol. 1993;3:191–197. doi: 10.1016/0962-8924(93)90213-K. [DOI] [PubMed] [Google Scholar]
- Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3:99–111. doi: 10.1016/S1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- Southwood CM, Garbern J, Jiang W, Gow A. The unfolded protein response modulates disease severity in Pelizaeus–Merzbacher disease. Neuron. 2002;36:585–596. doi: 10.1016/S0896-6273(02)01045-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama BH, Savas JN, Park SK, Harris MS, Ingolia NT, Yates JR, 3rd, Hetzer MW. Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell. 2013;154:971–982. doi: 10.1016/j.cell.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traka M, Arasi K, Avila RL, Podojil JR, Christakos A, Miller SD, Soliven B, Popko B. A genetic mouse model of adult-onset, pervasive central nervous system demyelination with robust remyelination. Brain. 2010;133:3017–3029. doi: 10.1093/brain/awq247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang M, Ye R, Barron E, Baumeister P, Mao C, Luo S, Fu Y, Luo B, Dubeau L, Hinton DR, Lee AS. Essential role of the unfolded protein response regulator GRP78/BiP in protection from neuronal apoptosis. Cell Death Differ. 2010;17:488–498. doi: 10.1038/cdd.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way SW, Podojil JR, Clayton BL, Zaremba A, Collins TL, Kunjamma RB, Robinson AP, Brugarolas P, Miller RH, Miller SD, Popko B. Pharmaceutical integrated stress response enhancement protects oligodendrocytes and provides a potential multiple sclerosis therapeutic. Nat Commun. 2015;6:6532. doi: 10.1038/ncomms7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wey S, Luo B, Lee AS. Acute inducible ablation of GRP78 reveals its role in hematopoietic stem cell survival, lymphogenesis and regulation of stress signaling. PLoS One. 2012;7:e39047. doi: 10.1371/journal.pone.0039047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Ye R, Jung DY, Barron E, Friedline RH, Benoit VM, Hinton DR, Kim JK, Lee AS. GRP78 plays an essential role in adipogenesis and postnatal growth in mice. FASEB J. 2013;27:955–964. doi: 10.1096/fj.12-213330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]