

Mitochondrial flash activity is tightly coupled with the electron flow along mitochondrial electron transport chain in intact cardiac myocytes and intact heart. The positive correlation between flash frequency and oxygen consumption rate highly suggests that flash frequency could be used as a novel biomarker for mitochondrial respiration in vivo.
Keywords: mitochondrial respiration, mitochondrial flash, electron transport chain, real-time confocal imaging, biomarker
Abstract
Mitochondrial respiration through electron transport chain (ETC) activity generates ATP and reactive oxygen species in eukaryotic cells. The modulation of mitochondrial respiration in vivo or under physiological conditions remains elusive largely due to the lack of appropriate approach to monitor ETC activity in a real-time manner. Here, we show that ETC-coupled mitochondrial flash is a novel biomarker for monitoring mitochondrial respiration under pathophysiological conditions in cultured adult cardiac myocyte and perfused beating heart. Through real-time confocal imaging, we follow the frequency of a transient bursting fluorescent signal, named mitochondrial flash, from individual mitochondria within intact cells expressing a mitochondrial matrix-targeted probe, mt-cpYFP (mitochondrial-circularly permuted yellow fluorescent protein). This mt-cpYFP recorded mitochondrial flash has been shown to be composed of a major superoxide signal with a minor alkalization signal within the mitochondrial matrix. Through manipulating physiological substrates for mitochondrial respiration, we find a close coupling between flash frequency and the ETC electron flow, as measured by oxygen consumption rate in cardiac myocyte. Stimulating electron flow under physiological conditions increases flash frequency. On the other hand, partially block or slowdown electron flow by inhibiting the F0F1 ATPase, which represents a pathological condition, transiently increases then decreases flash frequency. Limiting electron entrance at complex I by knocking out Ndufs4, an assembling subunit of complex I, suppresses mitochondrial flash activity. These results suggest that mitochondrial electron flow can be monitored by real-time imaging of mitochondrial flash. The mitochondrial flash frequency could be used as a novel biomarker for mitochondrial respiration under physiological and pathological conditions.
NEW & NOTEWORTHY
Mitochondrial flash activity is tightly coupled with the electron flow along mitochondrial electron transport chain in intact cardiac myocytes and intact heart. The positive correlation between flash frequency and oxygen consumption rate highly suggests that flash frequency could be used as a novel biomarker for mitochondrial respiration in vivo.
mitochondrial respiration is at the center stage of bioenergetics in eukaryotic cells, especially the cells in energy craving tissues, such as the nervous system and the heart. It is estimated that the human heart generates and utilizes ∼6 kg of ATP per day (54), and this high rate of ATP and energy flow mainly relies on mitochondrial metabolism (24). Central to mitochondrial metabolism is the oxidative phosphorylation, also known as mitochondrial respiration, which is the sequential process of electron flow along the electron transport chain (ETC) and ATP generation by F0F1 ATPase (42). Mitochondrial respiration is also the driving force for establishing the negative inner membrane potential, ion transportation (4), and generation of reactive oxygen species (ROS), which are physiologically indispensable signaling molecules (17, 38), as well as culprit of a variety of diseases and aging (6, 24, 46). Meanwhile, mitochondrial dysfunction, which is originated from impaired mitochondrial respiration, has been linked to numerous human diseases and is the target for therapy (12, 49). However, evaluating mitochondrial respiration mainly relies on in vitro measurement of oxygen consumption rate (OCR), which may not reflect mitochondrial respiration status in vivo or under physiologically relevant conditions (9, 32). The lack of a real-time, in vivo, and tissue/cell-specific approach for monitoring mitochondrial respiration is a major challenge for the study of mitochondrial function and dysfunction in health and disease.
We have discovered that individual mitochondrion under physiological condition and in vivo display a transient and bursting signal, named superoxide flash, which is detected by a mitochondrial targeted superoxide indicator, mt-cpYFP (mitochondrial-circularly permuted yellow fluorescent protein) (51). Since then, other groups have also used this probe and other probes and detected similar bursting single mitochondrial events, which are associated with mitochondrial ROS production and redox signaling and play critical roles in cell physiology and pathology (10, 40, 45, 55, 57). However, concerns over the pH and superoxide sensitivity of the cpYFP probe have also been raised, which suggested that the bursting flash events detected by cpYFP could be interpreted as transient alkalization signals in mitochondrial matrix, named pH flashes (41, 43, 44). This controversy could arise from the differences in in vitro calibration of cpYFP and will need to be resolved most likely with the elucidation of crystal structure of cpYFP (14, 44). Nevertheless, we simultaneously monitored the cpYFP signal and a pH indicator loaded in mitochondrial matrix and found a major contribution of superoxide with a minor component of alkalization in each of the flash events (52). Considering the mixed signal in the flash event and the ongoing debate, superoxide flash has been renamed to mitoflash (45) or mitochondrial flash. Meanwhile, regardless of the interpretation of its nature, accumulated evidence has suggested that flash event is closely associated with mitochondrial energy metabolism and depends on mitochondrial ETC activity (10, 20, 41, 51). Indeed, both superoxide production and proton pumping are originated from and can be integrated by ETC electron flow. Moreover, a simultaneous loss of mitochondrial NADH signal was observed during each of the single mitochondrial flashes (52). Taken together, it is highly probable that mitochondrial flash is a composite event arising from mitochondrial respiration in vivo or in intact cells. Although mitochondrial flash has been monitored under several disease conditions, no study has systematically investigated the electron flow dependence of flash and whether flash is coupled with mitochondrial respiration (e.g., oxygen consumption). The aim of this study is to answer these important questions and provide new evidence to justify the use of flash as a novel biomarker for mitochondrial respiration in vivo or under pathophysiological conditions.
To achieve this aim, we manipulated mitochondrial substrate availability in Langendorff perfused heart and in intact adult cardiac myocytes. We found that respiration substrates initiated electron flow to stimulate oxygen consumption and induce mitochondrial flashes. In permeabilized myocytes, initiation of forward or reverse electron flows increased flash frequency. Interestingly, inhibition of F0F1 ATP synthase, which slows down electron flow, augmented electron accumulation and transiently increased mitochondrial flash frequency. Cardiac-specific deletion of an assembling subunit of complex I, Ndufs4, which resulted in 75% decrease in complex I content and activity (30), inhibited mitochondrial flashes in cardiac myocytes. These results provide convincing evidence to show the tight coupling between ETC electron flow and mitochondrial flash under physiological conditions and as such justify the use of flash as a biomarker of mitochondrial respiration.
METHODS
Recombinant adenovirus vectors and transgenic mice.
All of the animal procedures used in this study were approved by Internal Review Board of the Institutional Animal Care and Use Committee at the University of Washington. Animals were maintained on rodent diet with water available ad libitum and in a vivarium with a 12:12-h light-dark cycle at 22°C. Generation of pan-tissue mt-cpYFP transgenic (TG) mice used pUC-CAGGS-mt-cpYFP vector and pronuclear microinjection was conducted by transgenic core facility at the University of Washington. Linearized expression vector was injected into pronucleus of fertilized C57BL/6 mouse (Charles River) oocytes. Genotyping used primers for cpYFP (upstream: 5′-GCGAGGAGCTGTTCACCG-3′; downstream: 5′-AGCCACCACTTCTGATAGGCA-3′). GADPH was used as internal control (upstream: 5′-TGCCTGCTTCACCACCTTCT-3′; downstream: 5′-AGGCGGGTGCTGAGTATGTC-3′). The mt-cpYFP TG mice were housed and bred at the animal facility of the South Lake Union Campus of the University of Washington. Generation of cardiac-specific Ndufs4 knockout (cKO) mice have been reported previously (30). Genotyping of cKO mice used primers for floxed Ndufs4 (upstream: 5′-AGCCTGTTCTCATACCTCGG-3′; downstream: 5′-GCTCTCTATGAGGGTACAGAG-3′) and primers for αMHC-Cre (upstream: 5′-CGATGCAACGAGTGATGAGG-3′; downstream: 5′-CGAATAACCAGTGAAACAGC-3′). cKO mice were further bred with mt-cpYFP TG mice to obtain cKO + mt-cpYFP mice. cKO + mt-cpYFP mice at 3–4 mo were used. At this age the Ndufs4 protein level is significantly decreased, while cardiac function is normal.
Construction of recombinant adenovirus vectors containing mt-cpYFP (Ad-mt-cpYFP) has been described previously (51). Ad-mt-cpYFP was amplified in human embryonic kidney-293 cells and purified by standard CsCl gradient centrifugation followed by overnight dialysis. The titer of virus was determined to be ∼1 × 1011 viral particles/ml. Ad-SOD1 and Ad-SOD2 viruses were kind gifts from Dr. John F. Engelhardt (University of Iowa). All adenoviruses were divided into aliquots and stored at −80°C.
Confocal imaging of perfused heart.
To imaging mitochondrial flashes in the perfused heart, mt-cpYFP TG mouse was heparinized (intraperitoneal heparin, 50 U/mouse) and euthanized (intraperitoneal pentobarbital, 150 mg/kg). The heart was quickly removed and perfused in Langendorff mode using a custom-designed perfusion system that allows mounting of the heart onto a chamber on the confocal microscope stage (23). The heart was perfused with physiological solutions containing 118 mM NaCl, 25 mM NaHCO3, 5.3 mM KCl, 2 mM CaCl2, 1.2 mM MgSO4, 0.5 mM EDTA, and equilibrated with 95% O2 and 5% CO2 (pH 7.4) at 37°C. In addition, substrates were added to generate glucose-only solution (10 mM glucose and 0.5 mM pyruvate) or mixed substrate solution [5.5 mM glucose, 0.4 mM mixed long-chain fatty acids (palmitic acid 56.7%, palmitoleic acid 11.7%, stearic acid 1.9%, oleic acid 17.1%, linoleic acid 10.8%, and linolenic acid 1.7%) bound with 1.2% albumin, 1.2 mM lactate, 1.3 mM ketone, and 50 μU/ml insulin] (30, 36). Mitochondrial membrane potential indicator, tetramethylrhodamine, methyl ester (TMRM) (100 nM, Invitrogen) was included in the perfusion solution. For substrate removal and restoration, the heart was perfused with oxygenated and substrate-free solution for 30–40 min before changing back to substrate-containing solution. Blebbistatin (10 μM) was added in the perfusion solution to suppress (but not completely stop) the heartbeat, and gentle pressure was applied to further prevent motion artifact when taking the images. Confocal imaging followed the procedure developed in our laboratory's previous report (23).
Adult cardiac myocyte culture and gene transfer.
Adult rat cardiac myocytes were isolated from female Sprague-Dawley rats (200 ∼ 250 g, Harlan) following the protocol reported previously (50). Briefly, rats were anesthetized by intraperitoneal injection of 100 mg/kg pentobarbital. The heart was quickly removed and cannulated via ascending aorta and mounted on a modified Langendorff perfusion system. The heart was perfused with oxygenated Krebs-Henseleit Buffer supplemented with collagenase II (80 U/ml, Worthington) and hyaluronidase (0.15 mg/ml, Sigma) at 37°C for 30 min. The heart was cut into small pieces for further digestion under gentle agitation in enzyme solution. Rod-shaped adult rat cardiac myocytes were collected by brief centrifugation.
Adult mouse cardiac myocytes were isolated from mt-cpYFP TG mice (20 ∼ 25 g) following a protocol reported previously (29). Briefly, mouse was anesthetized by intraperitoneal injection of 150 mg/kg pentobarbital. The heart was quickly removed and cannulated via aorta and mounted on a perfusion system. The heart was perfused with oxygenated myocyte isolation solution (113 mM NaCl, 4.7 mM KCl, 0.6 mM KH2PO4, 0.6 mM NaH2PO4, 1.2 mM MgSO4, 11 mM HEPES and 20 mM glucose) supplemented with collagenase II (300 U/ml, Worthington), hyaluronidase (0.5 mg/ml, Sigma), 50 μM CaCl2, and 10 μM blebbistatin at 37°C for 10 min. The heart was taken down, disaggregated with forceps, and gently triturated five to six times and incubated in enzyme solution for 5 min at room temperature to allow further digestion. Rod-shaped adult mouse cardiac myocytes were collected by brief centrifugation.
The isolated myocytes from rat or mouse were plated on coverslips (20 mm, round) precoated with laminin (40 μg/ml for 1 h, Invitrogen) at a density of 2 ∼ 3 × 104 cells per coverslip and in M199 medium (Sigma) supplemented with 10 mM glutathione, 26.2 mM sodium bicarbonate, 5 mM creatine, 2 mM l-carnitine, 5 mM taurine, 0.1% insulin-transferrin-selenium-X, 0.02% bovine serum albumin, 50 U/ml penicillin-streptomycin, and 5% fetal bovine serum. Two hours after the plating, the medium was changed to serum-free M199. Freshly isolated mouse myocytes right after 2-h plating were used for confocal imaging. Adenovirus-mediated gene transfer was done at a multiplicity of infection of 50–100 on rat cardiac myocytes. The rat myocytes were kept in culture for 48 ∼ 72 h to allow adequate gene expression before imaging.
Determination of gene expression.
Protein levels of SOD1 and SOD2 after adenovirus-mediated gene expression in cultured myocytes were determined by Western blot. Rat cardiac myocytes were harvested 48–72 h after gene transfer. Protein samples were collected in 1× lysis buffer (Cell Signaling), quantified by a BCA kit (Pierce), and loaded (20 μg) onto SDS-PAGE gel. Separated proteins were transferred to nitrocellulose membrane and probed with antibodies specific for SOD1 (1:1,000, Calbiochem), SOD2 (1:1,000, Calbiochem), or actin (1:2,000, Sigma). Secondary antibodies were conjugated to IRDye 800 (Rockland) or Alexa Fluor 680 (Invitrogen), and signals were visualized and quantified using Odyssey system (Licor).
Confocal imaging of cultured adult cardiac myocyte.
Confocal imaging used a Zeiss LSM 510 Meta confocal microscope equipped with a 40× 1.3 numerical aperture oil immersion objective and followed a procedure developed previously (51). Intact myocytes were incubated in physiological solutions containing 138 mM NaCl, 3.7 mM KCl, 1.2 mM KH2PO4, 5 mM glucose, 20 mM HEPES and 1 mM CaCl2 at room temperature and in a custom-designed perfusion chamber mounted on the confocal microscope stage. Dual-excitation images of mt-cpYFP were taken on randomly selected cells by alternating excitation at 405 and 488 nm and collecting emissions at >505 nm. Time-lapse x,y images were acquired at 1,024 resolution for 100 frames and at a sampling rate of 1 s/frame. For substrate stimulation, myocytes were first incubated in glucose-free solution for 30–40 min before changing to the solution with various substrates.
To detect mitochondrial flashes in permeabilized rat myocytes, the cells were first incubated in Ca2+-free solution for 3 min, changed to internal solution containing 120 mM potassium aspartate, 3 mM MgATP (free [Mg2+] ∼ 1 mM), 0.1 mM EGTA, 10 mM phosphocreatine, 5 U/ml creatine phosphokinase, 8% dextran (40,000), and 50 μg/ml saponin (pH 7.2) for 30 s and then maintained in saponin-free internal solution (35). In a subset of experiments, permeabilization protocol was verified by adding rhod-2 salt (5 μM, Invitrogen), a membrane-impermeable indicator, right after permeabilization, and visualizing the intracellular rhod-2 signals. To test substrate-induced respiration and flash activity, permeabilized cells were incubated in mitochondrial respiration solution containing 0.5 mM EGTA, 3 mM MgCl2, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 60 mM potassium-lactobionate, 110 mM mannitol, 0.3 mM dithiothreitol, and 1 g/l BSA (32). After the baseline recording was obtained, various substrates were added [10 mM pyruvate, 5 mM malate, and 1 mM ADP; 10 mM succinate and 1 mM ADP; or 0.5 mM N,N,N′,N′ tetramethyl p-phenylenediamine (TMPD), 2 mM ascorbate, and 1 mM ADP], and inhibitors were added subsequently (0.5 μM rotenone, 5 μM antimycin A, 1 mM NaCN, or 1 μM FCCP).
To monitor mitochondrial superoxide, intact cardiac myocytes were loaded with MitoSOX Red (5 μM, Invitrogen) at 37°C for 10 min, followed by washing two times. Dual-wavelength imaging was done by tandem excitation at 405 and 514 nm, and emission collected at >560 nm (52). Since MitoSOX signal increases irreversibly and reaches saturation quickly, the experiments were completed within 30 min after MitoSOX loading.
To monitor mitochondrial membrane potential, TMRM (20 nM, Invitrogen) was loaded into intact myocytes at room temperature for 20 min. Tri-wavelength excitation imaging of mt-cpYFP and TMRM was done by tandem excitation at 405, 488 and 543 nm, and emission collected at 505–545, 505–545, and 560–657 nm, respectively (52).
Polarography assays for OCR.
Measurement of OCR in permeabilized rat cardiac myocytes used a Clark electrode and the same solution and substrates as for the confocal imaging of permeabilized cells and followed the protocol reported previously (30, 32).
Statistics.
Data are presented as means ± SE. One-way ANOVA was used to compare the differences among three or more groups, followed by unpaired Student's t-test to compare between any two groups. In some experiments (see Fig. 3, B and C), when the two groups were from the same set of samples (before and after a treatment), paired Student's t-test was used. Nonparametric correlation was used to determine the relationship between OCR and mitochondrial flash frequency or OCR and MitoSOX signal. A P value < 0.05 was deemed significant.
Fig. 3.

Manipulation of ETC electron flow in permeabilized adult rat cardiac myocyte. A: experimental protocol for manipulating substrate in permeabilized rat cardiac myocytes. B: oxygen consumption rate (OCR) measured in permeabilized rat myocytes with substrates for complex I (Pyr/Mal/ADP: 10 mM Pyr, 5 mM malate, and 2 mM ADP), complex II (Succ/ADP: 10 mM succinate and 2 mM ADP), or complex IV (TMPD/Asc/ADP: 0.5 mM N,N,N′,N′ tetramethyl p-phenylenediamine, 2 mM ascorbate, and 2 mM ADP) and subsequent addition of ETC inhibitors [0.5 μM Rot, 5 μM antimycin A (AA)]. N = 5–10 experiments from 3–4 rats. C: summarized results showing increased MitoSOX Red signals (slope of 405-nm or 514-nm excitation) in permeabilized rat myocytes during substrate stimulation. N = 9–25 cells from 3–4 rats. D: correlation between OCR (B) and MitoSOX signal (C) in permeabilized rat myocytes. ○, 405-nm excitation of MitoSOX vs. OCR; ▲: 514-nm excitation of MitoSOX vs. OCR. Values are means ± SE. †P < 0.001 vs. No sub. #P < 0.01 vs. with substrate.
RESULTS
Respiration substrates modulated mitochondrial flash in the perfused heart.
To determine mitochondrial flash activity under physiologically relevant condition, we manipulated the substrate availability in Langendorff perfused beating hearts from mt-cpYFP TG mice (Fig. 1). The respiration manipulation protocol was carried out by removing substrate (perfuse with oxygenated and no substrate solution for 30–40 min) and then restoring substrate (Fig. 1A). TMRM (100 nM) was used to evaluate mitochondrial membrane potential simultaneously. We tested mixed substrate (glucose plus mixed fatty acids) and glucose-only substrate, both of which have been used for functional and metabolic evaluation of the perfused heart (30, 56). We found that, in the intact myocardium, mitochondrial flash frequency decreased after substrate removal and rebounded after substrate restoration (Fig. 1, B–D). Each flash is accompanied with loss of membrane potential (Fig. 1, B and C). These results suggest that flash frequency is modulated by respiration substrates in the perfused beating heart.
Fig. 1.

Physiological substrates modulated mitochondrial flash in the perfused heart. A: experimental protocol for manipulating physiological substrates in the perfused mouse heart. B: representative images of the myocardium of mt-cpYFP (mitochondrial-circularly permuted yellow fluorescent protein) hearts perfused with mixed substrate (left) or glucose-only substrate (right) showing a representative mitochondrial flash at its peak (boxed areas and the inset enlarged images). C: traces showing fluorescence changes of the two flashes highlighted in B. D: summarized data showing mitochondrial flash frequency during substrate manipulations in the perfused mt-cpYFP transgenic (TG) mouse hearts. TMRM, tetramethylrhodamine, methyl ester; ΔF/F0, amplitude, where F0 refers to basal fluorescence intensity. Values are means ± SE; N = 30–70 cells from 3–4 mice for each group. #P < 0.01 and †P < 0.001 vs. control (with substrate). *P < 0.05 vs. no substrate.
ETC dependence of substrate-induced mitochondrial flash in intact myocytes.
To further determine whether the substrate-induced mitochondrial flash depends on mitochondrial respiration via ETC activity, we used adult rat cardiac myocytes, which can be cultured for 3–4 days to facilitate adenovirus-mediated gene manipulation. The physiological substrates, glucose, palmitate, or a mixture of both, acutely increased the frequency of mitochondrial flash in cultured rat cardiac myocytes (Fig. 2, A and B). These effects were abolished by rotenone, an inhibitor of complex I of the ETC (Fig. 2B). Importantly, galactose, which is not a respiratory or glycolytic substrate for the heart (39), was unable to induce flashes in the cultured rat myocytes, while pyruvate, a substrate that mainly generates NADH and provides electrons entering ETC at complex I, significantly increased flash activity (Fig. 2C). The glucose-induced flash activity was sensitive to antioxidants (Tiron or SOD2 overexpression) and transient mitochondrial permeability transition (tMPT) inhibition by cyclosporine A (Fig. 2D), consistent with previous reports (51). Unitary properties of flashes, including amplitude (ΔF/F0), time to peak, and time to 50% decay were mildly altered by these physiological substrates (Fig. 2E). These results suggest that substrate-induced mitochondrial flash is dependent on ETC activity.
Fig. 2.

Electron transport chain (ETC) activity underlay substrate-induced mitochondrial flashes in intact adult cardiac myocyte. A: representative traces of typical mitochondrial flashes in cultured adult rat cardiac myocytes supplemented with various substrates [5 mM glucose (Glu), 0.5 mM palmitate (Pal), or 5 mM Glu plus 0.5 mM Pal]. B: stimulation of mitochondrial flash frequency by physiological substrates and the subsequent inhibition by rotenone (+Rot, 0.5 μM). C: galactose (Gal; 10 mM) had no effect on mitochondrial flash frequency, while pyruvate (Pyr; 10 mM) increased flash frequency. N = 11–22 cells from 3–4 rats. †P < 0.001 and #P < 0.01 vs. no substrate (No sub). D: manipulation of Glu-supported mitochondrial flash frequency by cyclosporine A (CsA, 1 μM), Tiron (1 mM), or adenovirus-mediated SOD2 overexpression (SOD2). N = 17–26 cells from 3–4 rats. #P < 0.01 vs. No sub. †P < 0.001 vs. Glu. E: unitary properties of mitochondrial flash in cultured rat myocytes. Tpk, time to peak; T50, time from peak to 50% decay. N = 143–323 flashes from 11–46 cells in 3–7 rats. #P < 0.01 and †P < 0.001 vs. No sub. Values are means ± SE.
ETC electron flow underlay mitochondrial flash generation.
To further determine whether substrate-induced mitochondrial flash generation requires the electron flow along ETC, we monitored flash in permeabilized adult rat cardiac myocytes, which largely preserved the intracellular environment/structure, while allowing the assessment of electron flow associated with specific substrates of the ETC complexes (Fig. 3A) (32). Successful permeabilization of plasma membrane was confirmed by visualization of the fluorescence of rhod-2 salt, a membrane-impermeable dye, in the cytosol (data not shown). We then supplemented the permeabilized rat myocytes with ETC substrates to start electron flow from complex I [pyruvate, malate, and ADP (Pyr/Mal/ADP)], complex II (succinate and ADP), or complex IV (TMPD, ascorbate, and ADP) (Fig. 3, B–D). We first determined the OCR, which confirmed the stimulation of electron flow by these substrates and subsequent blockade by ETC inhibition (8, 31, 32) (Fig. 3B). Next, we monitored the fluorescence of MitoSOX, a pH-insensitive and mitochondrial targeted superoxide indicator, in permeabilized rat myocytes. We found increased MitoSOX signal when complex I (22), complex II, or complex IV substrate was added (Fig. 3C), consistent with increased superoxide production upon the initiation of ETC electron flow. There is a positive correlation between OCR and MitoSOX fluorescence during the above substrate manipulations (Fig. 3D). These results suggest that, in permeabilized myocytes, the specific substrates for ETC complexes can stimulate electron flow along the ETC.
Next, we monitored mitochondrial flash in permeabilized rat myocytes (Fig. 4). Addition of Pyr/Mal/ADP significantly induced flash activity (Fig. 4A). This effect was abolished by blockers of ETC electron flow or uncoupler (Fig. 4B). Although uncoupler maximizes ETC electron flow and OCR, it completely dissipates membrane potential and diminishes mitochondrial flashes (51). A similar increase in flash frequency was found when using complex II or IV substrate to initiate electron flow in permeabilized rat myocytes (Fig. 4B). In addition, flash amplitude was increased and kinetics was largely unchanged by these substrates (Fig. 4C). Interestingly, the OCR in permeabilized rat myocytes was positively correlated with mitochondrial flash frequency during respiration stimulation by these substrates (Fig. 4D). Taken together, ETC electron flow underlies mitochondrial flash generation. Regarding the direction of electron flow, complex I substrates mainly induce forward electron flow, complex II substrates can induce both forward and reverse flows (8, 31, 47), and complex IV substrates has been shown to induce reverse flow to quinone pool or NAD+ (13, 37). However, we speculate that forward electron flow may be the major direction that supports mitochondrial flash in intact cells, in the perfused hearts, and in vivo (20, 23, 51).
Fig. 4.

ETC electron flow supported mitochondrial flash generation in permeabilized adult rat cardiac myocyte. A, left: representative images of a permeabilized rat cardiac myocyte showing flash events (highlighted in white boxes) during the 100-s scan in the absence (No sub) or presence of complex I substrates (Pyr/Mal/ADP). Scale bar = 10 μm. Right: representative traces showing a typical flash before or after the complex I substrate. B: flash frequency supported by Pyr/Mal/ADP, Succ/ADP, or TMPD/Asc/ADP and subsequent inhibition by ETC inhibitors: Rot (0.5 μM), AA (5 μM), FCCP (1 μM) or NaCN (1 mM). N = 11–58 cells from 3–8 rats. C: unitary properties of mitochondrial flash in permeabilized rat myocytes with or without substrates. N = 194–524 flashes in 16–57 cells from 6–8 rats. D: correlation between OCR and mitochondrial flash frequency in permeabilized rat myocytes. The data points are from Figs. 3B and 4B. Values are means ± SE. †P < 0.001 vs. No sub.
Slowdown ETC electron flow transiently increased mitochondrial flashes.
ETC electron flow is controlled by downstream ATP generation and utilization (4, 5). Our laboratory has shown that long-term blockade of F0F1 ATP synthase (complex V) by oligomycin A, which decreases ETC electron flow, abolished flashes (51). This is in line with the above results showing electron flow along ETC underlies flash activity. To further explore how ETC electron flow supports mitochondrial flashes, we followed the time-dependent effect of oligomycin A on flash frequency in intact myocytes supplemented with glucose as substrate. Surprisingly, we found a biphasic change of flash frequency: an initial increase peaked at 20 min, followed by a decline and eventually a decrease to a level lower than control at 40 min (Fig. 5A). The transient increase in flash frequency was abolished by addition of uncoupler or ETC inhibitors, likely due to the fact that they either dissipate membrane potential or totally block electron flow (Fig. 5B). In intact myocytes without substrate, oligomycin A only moderately induced flashes. Subsequent addition of pyruvate further increased flash frequency (Fig. 5C). The additive effect suggests that providing more electrons to a slowed ETC further augments mitochondrial flash generation. When oligomycin A was added after pyruvate in intact (Fig. 5C) or permeabilized myocytes (Fig. 5D), no further increase in flash frequency was observed, probably due to the already saturated electron flow by pyruvate. Oligomycin A-stimulated mitochondrial flashes were attenuated by tMPT inhibitor, cyclosporine A (Fig. 5E), or mitochondrial overexpression of the superoxide scavenger, SOD2, but not overexpression of the cytosolic SOD1 (Fig. 5F). Finally, oligomycin A induced a significant increase in MitoSOX signal (Fig. 5G), supporting that oligomycin A increased ROS production. Oligomycin A has been shown to slow down electron flow (1), block transition from state 2 to state 3 respiration, and induce mitochondrial ROS production (48). Previously, our laboratory has shown that excessive electrons in ETC, such as during state 4 respiration in isolated mitochondria, supported the maximal mitochondrial flash activity (52). Therefore, in the presence of substrates, slowdown electron flow transiently induces mitochondrial flash, probably through promoting electron accumulation and/or reverse electron flow to enhance proton motive force and ROS production. Taken together, these results further support that flash generation relies on electron flow.
Fig. 5.

Slowdown electron flow transiently increased mitochondrial flashes in intact rat cardiac myocyte. A: time-dependent change of mitochondrial flash frequency in intact rat myocytes after oligomycin A (OA; 5 μM) treatment. N = 12–38 cells from 3–8 rats. B: OA (5 μM)-induced flashes were sensitive to ETC blockers. The inhibitors used were Rot (0.5 μM), AA (5 μM), and FCCP (1 μM). Top shows representative traces of flashes before and after OA treatment. C: OA (5 μM) or Pyr (10 mM) added separately or sequentially increased flash frequency in intact rat myocytes. N = 10–17 cells from 2–3 rats. #P < 0.01 and †P < 0.001 vs. No sub. D: OA (5 μM) had no effect on Pyr/Mal/ADP-induced flashes in permeabilized rat myocytes. N = 12–38 cells from 3–9 rats. †P < 0.001 vs. No sub. E: OA-induced flashes in intact rat myocytes were blocked by CsA (1 μM). N = 15–41 cells from 3–8 rats. †P < 0.001 vs. control. F: adenovirus-mediated overexpression of human SOD2 but not SOD1 suppressed basal and OA (5 μM)-induced flashes. N = 15–32 cells from 3–5 rats. #P < 0.01 and †P < 0.001 vs. −OA. G: representative traces showing OA (5 μM) accelerated the increase rate of MitoSOX Red signal in intact rat myocytes. Similar effects were observed in 12–25 cells from 3 rats. Values are means ± SE.
Complex I deficiency suppressed mitochondrial flash.
The above results suggest that electron flow through ETC is responsible for mitochondrial flash generation in the heart and adult cardiac myocytes. Next, we sought to determine the molecular mechanism of ETC complexes in controlling mitochondrial flash. Complexes I and III have been shown repeatedly as the major complexes for electron transport, proton pumping, and ROS production within the ETC, and complex I likely plays a more important role in vivo, since it is the major acceptor of electrons transported from reducing equivalents (8, 31, 33, 38, 47). Therefore, we focused on complex I in mitochondrial flash generation. We crossed mt-cpYFP TG mice with cardiac-specific complex I-deficient mice (Ndufs4 knockout, cKO), which had lost 75% of complex I content and activity in the heart with unchanged activity of other complexes (30). Freshly isolated adult cardiac myocytes from wild-type or cKO mice exhibited mitochondrial flash activity accompanied by loss of membrane potential (Fig. 6, A and B). Flash frequency was significantly decreased in cKO myocytes under basal conditions, while unitary properties of flash remained unchanged (Fig. 6, C and E). Furthermore, pyruvate-induced flash activity was diminished in cKO myocytes compared with wild-type myocytes (Fig. 6D). In addition, we have shown that basal mitochondrial superoxide and H2O2 levels are decreased in the cKO hearts (30). Taken together, these results suggest that complex I, which accepts electrons mainly from pyruvate-generated NADH, plays a critical role in mitochondrial flash generation in the heart.
Fig. 6.

Complex I deficiency suppressed mitochondrial flashes in mouse cardiac myocyte. A: representative images of a freshly isolated and intact cardiac myocyte from heart-specific Ndufs4 knockout and mt-cpYFP TG mouse (cKO). White boxes indicate the onset of a mitochondrial flash. B: representative traces showing the flash highlighted in A. C: frequency of mitochondrial flash in freshly isolated myocytes from wild-type (WT) or cKO mice in physiological solutions containing 5 mM Glu as substrate. N = 32–40 cells from 5 mice. D: complex I substrate (Pyr 10 mM)-induced flashes in WT and cKO myocytes. N = 17–31 cells from 3–5 mice. E: unitary properties of flashes in freshly isolated myocytes from WT or cKO mice. N = 66–159 flashes in 32–40 cells from 5 mice. Values are means ± SE. *P < 0.05 vs. WT. †P < 0.001 vs. No sub.
DISCUSSION
In this study, we sought to explore the mechanistic coupling between mitochondrial respiration and mitochondrial flash generation in intact cardiac myocyte and the beating heart under physiological relevant conditions. The major findings are that metabolic substrates initiated electron flow along ETC to support mitochondrial flash generation. Slowdown of the electron flow transiently enhanced mitochondrial flash activity, while restricting electron entering complex I attenuated flashes. These results provide new evidence to show, for the first time, the tight coupling between ETC electron flow and mitochondrial flash generation (Fig. 7). These findings also justify the use of mitochondrial flash as a novel biomarker for monitoring mitochondrial respiration in situ or in vivo, at single mitochondrion resolution, and under physiologically relevant conditions.
Fig. 7.
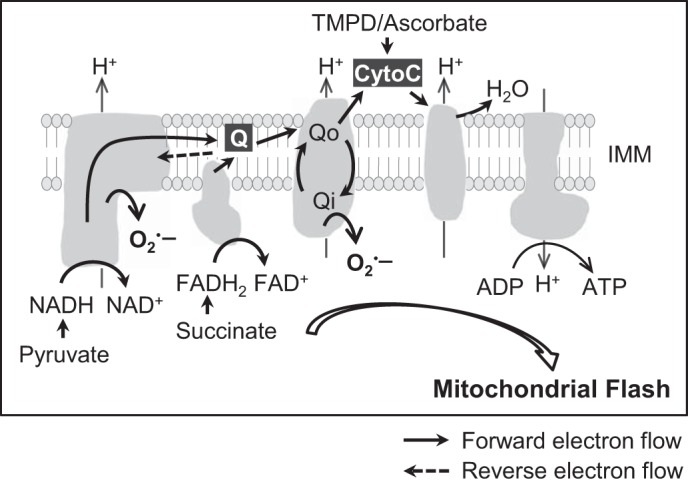
Model of the coupling between ETC electron flow and mitochondrial flash. Respiration substrate initiates electron flow to support mitochondrial respiration and flash generation. The amount of electrons transported along ETC is a determining factor for the respiration-flash coupling. The electron flow leads to superoxide production and matrix alkalization, which are components of mitochondrial flash. IMM, inner mitochondrial membrane; Qo, quinol-oxidizing center; Qi, quinone-reducing center; CytoC, cytochrome C.
The gold standard for studying mitochondrial respiration is the OCR, which is measured by using a Clark electrode on isolated mitochondria or permeabilized cells (9). The Seahorse XF Analyzer and Oroboros Oxygraph system are newly developed techniques that can be used for intact cell OCR measurement. However, these methods require sophisticated systems, have limited ability for real-time manipulation of respiration status, and are not suitable for in vivo measurement. Here, we show that the frequency of a recently discovered transient and single mitochondrial event, mitochondrial flash, is tightly coupled with the electron flow along ETC and has the potential to be used as a novel biomarker for mitochondrial respiration evaluation. Previously, we and others have shown that mitochondrial flash requires intact ETC activity (51), and its frequency can be modulated by metabolic perturbations in intact cells and in vivo (20, 22, 52). In this study, we systematically evaluated the coupling between mitochondrial flash and ETC electron flow and found a positive correlation between OCR of the population of mitochondria and the flash frequency of single mitochondria. It should be noted that flash frequency is usually low, and the flash events are confined within single mitochondria. However, the positive correlation between flash frequency and OCR and the fact that under various conditions the electron flow along ETC determines flash generation highly suggest that flash frequency is an appropriate readout for real-time evaluation of mitochondrial respiration status in intact cells, intact heart, and in vivo.
We and others have shown that mitochondrial flashes are triggered by tMPT pore openings (28, 51), which are mainly regulated by ROS and Ca2+ (11). The tMPT opening under physiological condition has been shown to bear important roles in mitochondrial Ca2+ regulation and preventing matrix Ca2+ overload (11, 18, 19, 26). Results from this study further suggest that tMPT may play a role in mitochondrial respiration regulation under physiological conditions, as reflected by the triggering of single mitochondrial flashes. Besides ROS and Ca2+, a number of factors can also modulate tMPT activity, including proton motive force, phosphate, Mg2+, and ADP/ATP (11, 16, 25). Importantly, all of these factors are integrated by mitochondrial respiration and ETC electron flow. In this regard, a positive feedback mechanism may exist, in which mitochondrial respiration through ETC electron flow may provide the triggering signal, such as ROS and Ca2+ (22, 27), to stimulate the stochastic opening of tMPT in individual mitochondrion and accelerate respiration in single mitochondria. The accelerated electron flow in the flashing mitochondria may further support the ROS and pH changes for local signaling. Recent studies have shed light on a close association between tMPT and ETC. For instance, the tMPT regulator, cyclophilin D, can bind ATP synthase, modulate local phosphate level, and regulate permeability transition (15). Furthermore, it has been proposed that ATP synthase may be part of the permeability transition pore or its subunit, or dimerization may form the pore (2, 7, 21). These data indicate that a physical coupling between ETC and tMPT may exist. Finally, the ETC-coupled tMPT and flash activity is compatible but not identical to ROS-induced ROS release phenomenon (58) or whole cell ROS oscillations (3), which are laser-induced phenomena in stressed cells. Moreover, the ROS-induced ROS release or whole cell ROS oscillations are propagating processes initiated by ROS released from individual mitochondria that subsequently trigger ROS release from the adjacent mitochondria. Here, the tMPT regulation of respiration and ROS production is stochastic and locally confined within the same mitochondrion, with no signs of propagation.
The application of mitochondrial flash in monitoring mitochondrial respiration in intact cells, tissues, and in vivo has clear advantage over the current in vitro methods. However, interpretation of the changes in flash frequency under various situations should take into consideration the context and other regulatory factors. In most cases, when ETC electron flow is not blocked, mitochondrial flash frequency reliably reflects substrate availability (this study), electron flow rate (measured by OCR in this study), intracellular energy utilization/demand (22), and mitochondrial metabolic status (20, 52, 53). In other cases, however, slowdown electron flow, such as by oligomycin A (this study) or under state 4 respiration (52), will further stimulate flashes. This could be explained by the increased electron accumulation in ETC, which may facilitate reverse electron flow and lead to increased flash activity. Moreover, it should be noted that uncoupling ATP production with ETC electron flow (e.g., by FCCP) abolishes flashes, largely due to the dissipation of mitochondrial membrane potential. In addition and as we discussed above, since flash is a composite event arising from serial processes, including tMPT and ETC activity within single mitochondria, flash frequency can be modulated by factors/regulators within ETC or targeting tMPT, such as Ca2+ and basal levels of ROS. Therefore, simultaneous or parallel monitoring of other key parameters, including OCR, membrane potential, ATP, pH, and ROS, may be needed to provide a comprehensive picture of mitochondrial respiration status under certain conditions. Although this study focuses on the flash-respiration coupling under physiological condition, the same mechanism may apply to pathological conditions, such as heart failure, ischemia reperfusion, neurotoxicity, and muscle disorders (22, 34, 45, 51, 53). We speculate that the establishment of flash-respiration coupling will stimulate its application in diseases and, together with other indicators, advance our understanding of the role of mitochondria in human disease.
In summary, through monitoring mitochondrial flashes and OCR in parallel, we found a close coupling between mitochondrial respiration and flash frequency in permeabilized cells, intact cells, and the perfused beating heart. The frequency of mitochondrial flash is positively correlated with increased ETC electron flow under physiological conditions. Under pathological conditions, when electron flow is blocked or slowed down, the accumulated electrons in ETC can also transiently support increased flash activity. Complex I is a critical site in ETC for the respiration-coupled mitochondrial flash activity. Therefore, we propose that flash frequency could be a useful biomarker for mitochondrial respiration evaluation in vivo and under pathophysiological conditions.
GRANTS
This work was supported by the National Heart, Lung, and Blood Institute (W. Wang; HL-114760, HL-110349) and the American Heart Association (W. Wang; 10SDG3450009).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: G.G., X.L., H.Z., and W.W. performed experiments; G.G., X.L., H.Z., and W.W. analyzed data; G.G., X.L., H.Z., S.-S.S., and W.W. interpreted results of experiments; G.G., H.Z., and W.W. prepared figures; G.G. and W.W. drafted manuscript; G.G., S.-S.S., and W.W. edited and revised manuscript; S.-S.S. and W.W. conception and design of research; S.-S.S. and W.W. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Drs. Stephen Kolwicz Jr., Zhen Zhang, and Chi Fung Lee for technical support and helpful discussions. We thank Dr. John F. Engelhardt (University of Iowa) for providing Ad-SOD1 and Ad-SOD2 viruses.
REFERENCES
- 1.Abe Y, Sakairi T, Kajiyama H, Shrivastav S, Beeson C, Kopp JB. Bioenergetic characterization of mouse podocytes. Am J Physiol Cell Physiol 299: C464–C476, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M, Porter GA Jr, Jonas EA. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci U S A 111: 10580–10585, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aon MA, Cortassa S, Marban E, O'Rourke B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J Biol Chem 278: 44735–44744, 2003. [DOI] [PubMed] [Google Scholar]
- 4.Balaban RS. Cardiac energy metabolism homeostasis: role of cytosolic calcium. J Mol Cell Cardiol 34: 1259–1271, 2002. [DOI] [PubMed] [Google Scholar]
- 5.Balaban RS. Perspectives on SGP symposium on mitochondrial physiology and medicine: metabolic homeostasis of the heart. J Gen Physiol 139: 407–414, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev 78: 547–581, 1998. [DOI] [PubMed] [Google Scholar]
- 7.Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, Patergnani S, Rimessi A, Suski JM, Wojtala A, Wieckowski MR, Kroemer G, Galluzzi L, Pinton P. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 12: 674–683, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J 134: 707–716, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J 435: 297–312, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Breckwoldt MO, Pfister FM, Bradley PM, Marinkovic P, Williams PR, Brill MS, Plomer B, Schmalz A, St Clair DK, Naumann R, Griesbeck O, Schwarzlander M, Godinho L, Bareyre FM, Dick TP, Kerschensteiner M, Misgeld T. Multiparametric optical analysis of mitochondrial redox signals during neuronal physiology and pathology in vivo. Nat Med 20: 555–560, 2014. [DOI] [PubMed] [Google Scholar]
- 11.Brenner C, Moulin M. Physiological roles of the permeability transition pore. Circ Res 111: 1237–1247, 2012. [DOI] [PubMed] [Google Scholar]
- 12.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol 287: C817–C833, 2004. [DOI] [PubMed] [Google Scholar]
- 13.Chance B, Hollunger G. Energy-linked reduction of mitochondrial pyridine nucleotide. Nature 185: 666–672, 1960. [DOI] [PubMed] [Google Scholar]
- 14.Cheng H, Wang W, Wang X, Sheu SS, Dirksen RT, Dong MQ. Cheng et al. reply. Nature 514: E14–E15, 2014. [DOI] [PubMed] [Google Scholar]
- 15.Chinopoulos C, Adam-Vizi V. Modulation of the mitochondrial permeability transition by cyclophilin D: moving closer to F(0)-F(1) ATP synthase? Mitochondrion 12: 41–45, 2012. [DOI] [PubMed] [Google Scholar]
- 16.Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J 341: 233–249, 1999. [PMC free article] [PubMed] [Google Scholar]
- 17.Droge W. Free radicals in the physiological control of cell function. Physiol Rev 82: 47–95, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Elrod JW, Molkentin JD. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ J 77: 1111–1122, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA, Karch J, Gabel S, Farber J, Force T, Brown JH, Murphy E, Molkentin JD. Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest 120: 3680–3687, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fang H, Chen M, Ding Y, Shang W, Xu J, Zhang X, Zhang W, Li K, Xiao Y, Gao F, Shang S, Li JC, Tian XL, Wang SQ, Zhou J, Weisleder N, Ma J, Ouyang K, Chen J, Wang X, Zheng M, Wang W, Cheng H. Imaging superoxide flash and metabolism-coupled mitochondrial permeability transition in living animals. Cell Res 21: 1295–1304, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabo I, Lippe G, Bernardi P. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A 110: 5887–5892, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gong G, Liu X, Wang W. Regulation of metabolism in individual mitochondria during excitation-contraction coupling. J Mol Cell Cardiol 76C: 235–246, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gong G, Wang W. Confocal imaging of single mitochondrial superoxide flashes in intact heart or in vivo. J Vis Exp 81: e50818, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Griffiths EJ. Mitochondria and heart disease. Adv Exp Med Biol 942: 249–267, 2012. [DOI] [PubMed] [Google Scholar]
- 25.Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol 46: 821–831, 2009. [DOI] [PubMed] [Google Scholar]
- 26.Hom JR, Quintanilla RA, Hoffman DL, de Mesy Bentley KL, Molkentin JD, Sheu SS, Porter GA Jr. The permeability transition pore controls cardiac mitochondrial maturation and myocyte differentiation. Dev Cell 21: 469–478, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hou T, Zhang X, Xu J, Jian C, Huang Z, Ye T, Hu K, Zheng M, Gao F, Wang X, Cheng H. Synergistic triggering of superoxide flashes by mitochondrial Ca2+ uniport and basal reactive oxygen species elevation. J Biol Chem 288: 4602–4612, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hou Y, Mattson MP, Cheng A. Permeability transition pore-mediated mitochondrial superoxide flashes regulate cortical neural progenitor differentiation. PLoS One 8: e76721, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kabaeva Z, Zhao M, Michele DE. Blebbistatin extends culture life of adult mouse cardiac myocytes and allows efficient and stable transgene expression. Am J Physiol Heart Circ Physiol 294: H1667–H1674, 2008. [DOI] [PubMed] [Google Scholar]
- 30.Karamanlidis G, Lee Chi F, Garcia-Menendez L, Kolwicz Stephen C, Suthammarak W, Gong G, Sedensky Margaret M, Morgan Philip G, Wang W, Tian R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab 18: 239–250, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett 416: 15–18, 1997. [DOI] [PubMed] [Google Scholar]
- 32.Kuznetsov AV, Veksler V, Gellerich FN, Saks V, Margreiter R, Kunz WS. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat Protoc 3: 965–976, 2008. [DOI] [PubMed] [Google Scholar]
- 33.Lambert AJ, Brand MD. Reactive oxygen species production by mitochondria. Methods Mol Biol 554: 165–181, 2009. [DOI] [PubMed] [Google Scholar]
- 34.Liu X, Xu S, Wang P, Wang W. Transient mitochondrial permeability transition mediates excitotoxicity in glutamate-sensitive NSC34 motor neuron-like cells. Exp Neurol 271: 122–130, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lukyanenko V, Gyorke S. Ca2+ sparks and Ca2+ waves in saponin-permeabilized rat ventricular myocytes. J Physiol 521: 575–585, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luptak I, Yan J, Cui L, Jain M, Liao R, Tian R. Long-term effects of increased glucose entry on mouse hearts during normal aging and ischemic stress. Circulation 116: 901–909, 2007. [DOI] [PubMed] [Google Scholar]
- 37.Miki T, Miki M, Orii Y. Membrane potential-linked reversed electron transfer in the beef heart cytochrome bc1 complex reconstituted into potassium-loaded phospholipid vesicles. J Biol Chem 269: 1827–1833, 1994. [PubMed] [Google Scholar]
- 38.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 417: 1–13, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Musick WD, Wells WW. Studies on galactose metabolism in heart and brain: the identification of d-galactose 6-phosphate in brains of galactose-intoxicated chicks and rat hearts perfused with galactose. Arch Biochem Biophys 165: 217–228, 1974. [DOI] [PubMed] [Google Scholar]
- 40.Pouvreau S. Superoxide flashes in mouse skeletal muscle are produced by discrete arrays of active mitochondria operating coherently. PLoS One 5: e13035, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santo-Domingo J, Giacomello M, Poburko D, Scorrano L, Demaurex N. OPA1 promotes pH flashes that spread between contiguous mitochondria without matrix protein exchange. EMBO J 32: 1927–1940, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scheffler IE. Mitochondria. New York: Wiley; 2008, p. 484. [Google Scholar]
- 43.Schwarzlander M, Logan DC, Fricker MD, Sweetlove LJ. The circularly permuted yellow fluorescent protein cpYFP that has been used as a superoxide probe is highly responsive to pH but not superoxide in mitochondria: implications for the existence of superoxide “flashes”. Biochem J 437: 381–387, 2011. [DOI] [PubMed] [Google Scholar]
- 44.Schwarzlander M, Wagner S, Ermakova YG, Belousov VV, Radi R, Beckman JS, Buettner GR, Demaurex N, Duchen MR, Forman HJ, Fricker MD, Gems D, Halestrap AP, Halliwell B, Jakob U, Johnston IG, Jones NS, Logan DC, Morgan B, Muller FL, Nicholls DG, Remington SJ, Schumacker PT, Winterbourn CC, Sweetlove LJ, Meyer AJ, Dick TP, Murphy MP. The “mitoflash” probe cpYFP does not respond to superoxide. Nature 514: E12–E14, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shen EZ, Song CQ, Lin Y, Zhang WH, Su PF, Liu WY, Zhang P, Xu J, Lin N, Zhan C, Wang X, Shyr Y, Cheng H, Dong MQ. Mitoflash frequency in early adulthood predicts lifespan in Caenorhabditis elegans. Nature 508: 128–132, 2014. [DOI] [PubMed] [Google Scholar]
- 46.Sugamura K, Keaney JF Jr. Reactive oxygen species in cardiovascular disease. Free Radic Biol Med 51: 978–992, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tahara EB, Navarete FD, Kowaltowski AJ. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic Biol Med 46: 1283–1297, 2009. [DOI] [PubMed] [Google Scholar]
- 48.Tocchetti CG, Caceres V, Stanley BA, Xie C, Shi S, Watson WH, O'Rourke B, Spadari-Bratfisch RC, Cortassa S, Akar FG, Paolocci N, Aon MA. GSH or palmitate preserves mitochondrial energetic/redox balance, preventing mechanical dysfunction in metabolically challenged myocytes/hearts from type 2 diabetic mice. Diabetes 61: 3094–3105, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walters AM, Porter GA Jr, Brookes PS. Mitochondria as a drug target in ischemic heart disease and cardiomyopathy. Circ Res 111: 1222–1236, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang W, Barnabei MS, Asp ML, Heinis FI, Arden E, Davis J, Braunlin E, Li Q, Davis JP, Potter JD, Metzger JM. Noncanonical EF-hand motif strategically delays Ca(2+) buffering to enhance cardiac performance. Nat Med 19: 305–312, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, Wang X, Li K, Han P, Zheng M, Yin J, Mattson MP, Kao JP, Lakatta EG, Sheu SS, Ouyang K, Chen J, Dirksen RT, Cheng H. Superoxide flashes in single mitochondria. Cell 134: 279–290, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wei-LaPierre L, Gong G, Gerstner BJ, Ducreux S, Yule DI, Pouvreau S, Wang X, Sheu SS, Cheng H, Dirksen RT, Wang W. Respective contribution of mitochondrial superoxide and pH to mitochondria-targeted circularly permuted yellow fluorescent protein (mt-cpYFP) flash activity. J Biol Chem 288: 10567–10577, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wei L, Salahura G, Boncompagni S, Kasischke KA, Protasi F, Sheu SS, Dirksen RT. Mitochondrial superoxide flashes: metabolic biomarkers of skeletal muscle activity and disease. FASEB J 25: 3068–3078, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weiss RG, Gerstenblith G, Bottomley PA. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc Natl Acad Sci U S A 102: 808–813, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu S, Chisholm AD. C. elegans epidermal wounding induces a mitochondrial ROS burst that promotes wound repair. Dev Cell 31: 48–60, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yan J, Young ME, Cui L, Lopaschuk GD, Liao R, Tian R. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet-induced obesity. Circulation 119: 2818–2828, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang X, Huang Z, Hou T, Xu J, Wang Y, Shang W, Ye T, Cheng H, Gao F, Wang X. Superoxide constitutes a major signal of mitochondrial superoxide flash. Life Sci 93: 178–186, 2013. [DOI] [PubMed] [Google Scholar]
- 58.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta 1757: 509–517, 2006. [DOI] [PubMed] [Google Scholar]