

Abstract
AMPK is an endogenous energy sensor that regulates lipid and carbohydrate metabolism. Nonalcoholic fatty liver disease (NAFLD) is regarded as a hepatic manifestation of metabolic syndrome with impaired lipid and glucose metabolism and increased oxidative stress. Our recent study showed that folic acid supplementation attenuated hepatic oxidative stress and lipid accumulation in high-fat diet-fed mice. The aim of the present study was to investigate the effect of folic acid on hepatic AMPK during high-fat diet feeding and the mechanisms involved. Male C57BL/6J mice were fed a control diet (10% kcal fat), a high-fat diet (60% kcal fat), or a high-fat diet supplemented with folic acid (26 mg/kg diet) for 5 wk. Mice fed a high-fat diet exhibited hyperglycemia, hepatic cholesterol accumulation, and reduced hepatic AMPK phosphorylation. Folic acid supplementation restored AMPK phosphorylation (activation) and reduced blood glucose and hepatic cholesterol levels. Activation of AMPK by folic acid was mediated through an elevation of its allosteric activator AMP and activation of its upstream kinase, namely, liver kinase B1 (LKB1) in the liver. Consistent with in vivo findings, 5-methyltetrahydrofolate (bioactive form of folate) restored phosphorylation (activation) of both AMPK and LKB1 in palmitic acid-treated HepG2 cells. Activation of AMPK by folic acid might be responsible for AMPK-dependent phosphorylation of HMG-CoA reductase, leading to reduced hepatic cholesterol synthesis during high-fat diet feeding. These results suggest that folic acid supplementation may improve cholesterol and glucose metabolism by restoration of AMPK activation in the liver.
Keywords: AMP-activated protein kinase, liver kinase B1, AMP, nonalcoholic fatty liver disease, folic acid
ampk is a master regulator of whole body energy balance and metabolic homeostasis. It modulates anabolic and catabolic pathways involved in carbohydrate, lipid, and protein metabolism through phosphorylation of downstream enzymatic and transcriptional mediators (5, 18, 52). AMPK is a heterotrimeric complex that is composed of three subunits, namely a catalytic α subunit and regulatory subunits (β and γ). AMPK is activated by allosteric AMP interaction with its γ subunit (at adenine binding sites) and by phosphorylation (at Thr-172) of the α subunit via upstream kinases (17, 20, 47). Although several kinases have been identified, liver kinase B1 (LKB1) is thought to be the predominant upstream kinase that is responsible for phosphorylation of AMPK (activation) in the liver (58). Phosphorylation of LKB1 (Ser-428 in human or Ser-431 in mouse) is important for its activation (15, 22, 63). Upstream enzymes, including sirtuin 1 (SIRT1), PKA, and PKC-ζ have been implicated in LKB1 activation, which, in turn, leads to activation of AMPK (9, 22, 62). Since AMPK regulates metabolic pathways involved in glucose and lipid metabolism, it has been proposed as a potential therapeutic target in metabolic diseases, such as obesity, Type 2 diabetes mellitus, and nonalcoholic fatty liver disease (NAFLD) (48, 51).
NAFLD is regarded as a hepatic manifestation of metabolic syndrome and is characterized by impaired lipid and glucose metabolism, as well as increased oxidative stress in the liver. The spectrum of NAFLD ranges from steatosis (lipid accumulation in the liver) to nonalcoholic steatohepatitis and hepatic cirrhosis in its advanced stages (13, 50). Chronic consumption of high-fat diets is associated with obesity and NAFLD (1, 16, 19). The high-fat diet rodent model develops hepatic histopathological features in context of the metabolic syndrome, such as hyperglycemia and abnormal lipid metabolism, and, therefore, is commonly used to investigate the pathogenesis of NAFLD (21, 35, 43, 49). It is evident that hepatic regulation of glucose and cholesterol metabolism is also perturbed in high-fat diet-fed rodents (39, 60, 64), which is associated with a decrease in AMPK activation in the liver (39, 64). However, the regulation of hepatic AMPK during high-fat diet feeding is poorly understood. Understanding AMPK regulation in the liver during high-fat diet consumption might be important for improving the metabolism of glucose and cholesterol, which are often dysregulated in NAFLD (35, 36, 60).
Folate is a naturally occurring water-soluble B vitamin. It participates in intracellular methylation and one-carbon metabolism reactions and also contributes to nucleotide and amino acid biosynthesis in the body. Folic acid is the synthetic form of folate with a greater stability and is commonly fortified in foods and used for supplementation (24, 33). Folic acid fortification of the diet has been implemented to reduce the incidence of neural tube defects in newborns (28). The liver is the major organ responsible for folate storage and metabolism (33). Although folic acid supplied by a typical Western diet is sufficient to meet the requirements of generally healthy individuals, studies have revealed that serum folate levels are inversely correlated with obesity and are associated with the manifestation of liver disorders (30, 31, 34). Diets deficient in lipotropes such as methionine, choline, and/or folic acid have been shown to induce hepatic steatosis (7, 21). Supplementation with methyl donors (choline, methionine, vitamin B12, and folic acid) can attenuate hepatic lipid accumulation in the high-fat diet fed mice and may reduce the progression of NAFLD (11). In our previous study, we have observed that folic acid supplementation is hepatoprotective through reducing oxidative stress and lipid accumulation in the liver of high-fat diet-fed mice (43), as well as in hyperhomocysteinemic rats (56). The ability for folic acid to minimize lipid accumulation in the liver suggests that it may regulate hepatic lipid metabolism, but the underlying mechanisms remain to be defined. Because AMPK plays a crucial role in metabolic regulation, we hypothesize that folic acid supplementation may promote the activation of hepatic AMPK in high-fat diet-fed mice. In the present study, we aimed to investigate the mechanisms by which folic acid regulated AMPK in the liver during high-fat diet feeding.
MATERIALS AND METHODS
Animal model.
Male C57BL/6J mice aged 6 wk (Jackson Laboratory, Bar Harbor, ME) were fed either a control diet, high-fat diet, or high-fat diet supplemented with folic acid for 5 wk (23, 60). The formulation of the purified diets were as follows: control diet (D12450B) consisted of 10% kcal fat, 20% kcal protein, and 70% kcal carbohydrate with 2 mg of folic acid/kg of diet (3.85 kcal/g); the high-fat diet (D12492) contained 60% kcal fat, 20% kcal protein, and 20% kcal of carbohydrate with 2.6 mg of folic acid/kg of diet (5.24 kcal/g); and the high-fat diet supplemented with folic acid consisted of 26 mg of folic acid/kg of diet (Table 1). The sources of fat in both the control and high-fat diet are soybean oil and lard (Research Diets, Brunswick, NJ). The fat content in the high-fat diet is mainly derived from lard, while the main source of fat in the control diet is soybean oil (Table 1). In a pilot study, we conducted experiments in high-fat diet-fed mice that were supplemented with various doses of folic acid (5.6, 13, 26, 65, and 130 mg/kg of diet). We observed that increasing the folic acid contents to 26, 65, or 130 mg/kg of diet had glucose-lowering effects in high-fat diet-fed mice. In the present study, we used 26 mg/kg folic acid for supplementation, which was the lowest dose that displayed glucose-lowering effects in high-fat diet-fed mice. All the diets were prepared by Research Diets (23, 60). The mice were maintained on a 12:12-h light-dark cycle and had free access to water and food. Body weights were recorded prior to feeding and at the end of the experimental feeding period. At the end of the 5-wk period, mice were killed, and the blood was collected for serum preparation. The liver tissue was quickly removed and placed in liquid nitrogen, then frozen at −80°C until further analysis. Blood glucose was measured at the beginning and ending of the 5-wk feeding period. Mice were fasted for 5 h, and blood was sampled through the tail vein for glucose measurement. All procedures were performed in accordance with the Guide to the Care and Use of Experimental Animals published by the Canadian Council on Animal Care and approved by the University of Manitoba Protocol Management and Review Committee.
Table 1.
Diet composition
| Control (D12450B) | High-Fat Diet (D12492) | High-Fat Diet + 10x Folic Acid | |
|---|---|---|---|
| Energy content, % kcal | |||
| Protein | 20 | 20 | 20 |
| Carbohydrate | 70 | 20 | 20 |
| Fat | 10 | 60 | 60 |
| Fat content, g/kg | |||
| Soybean oil | 24 | 32 | 32 |
| Lard | 19 | 317 | 317 |
| Folic acid, mg/kg | 2 | 2.6 | 26 |
| Dietary energy, kcal/g | 3.85 | 5.24 | 5.24 |
Cell culture.
HepG2 cells (human hepatoblastoma cell line HB-8065; American Type Culture Collection, Manassas, VA) were cultured in high-glucose DMEM (Hyclone) supplemented with 10% FBS in a humidified incubator at 37°C with 5% CO2. HepG2 cells resemble many characteristics of hepatocytes and are widely used as a valuable model in metabolic and pharmacological studies (25, 55, 57, 60). HepG2 cells have also been used in studies of folate uptake and folate-dependent metabolism (41, 54). Treatments included palmitic acid (Sigma Aldrich), 5-methyltetrahydrofolate (5-MTHF; Sigma Aldrich), and folic acid (Sigma Aldrich). Palmitic acid was dissolved in 10% BSA and 5% ethanol with light shaking overnight at 37°C. Palmitic acid is the most abundant saturated fatty acid in the high-fat diet. It is also the major circulating saturated fatty acid in NAFLD patients (40) and in mice fed a high-fat diet (4). Palmitic acid (0.3 mM) has been shown to induce lipid accumulation in HepG2 cells, which was used as an in vitro model to study NAFLD pathogenesis (26, 60). 5-MTHF is the predominant form of folate detected in the circulation in humans and rodents. It is also the active form of folate that is taken up by the liver for storage and distribution to peripheral tissues (38, 65). It has been reported in humans that folic acid can also enter the hepatic portal circulation following intestinal absorption and is reduced to 5-MTHF in the liver (59). In a pilot study, we tested palmitic acid, 5-MTHF, and folic acid at various doses in HepG2 cells. We observed that palmitic acid (0.3 mM) reduced AMPK phosphorylation, while 5-MTHF (1 μg/ml) and folic acid (1 μg/ml) could restore AMPK activation. In the subsequent experiments, palmitic acid (0.3 mM), 5-MTHF (1 μg/ml), and folic acid (1 μg/ml) were used. In some experiments, nicotinamide (Sigma Aldrich), N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide dihydrochloride (H89; Sigma Aldrich), PKC-ζ pseudosubstrate (Sigma Aldrich), AMP sodium salt (Sigma Aldrich), and compound C (Calbiochem, Billerica, MA) were simultaneously incubated in cells treated with palmitic acid and/or 5-MTHF. The concentrations of the inhibitors or activators were tested to identify the lowest dose that could induce detectable changes in AMPK and LKB1 phosphorylation. Nicotinamide (10 mM) was used to inhibit SIRT1, an upstream deacetylase that activates LKB1 via deacetylation (22). H89 (10 μM) and PKC-ζ pseudosubstrate (1 μM) were used to suppress PKA and PKC-ζ activation, respectively (62). Compound C (10 μM) was used to inhibit activation of AMPK (60), and AMP (100 μM) was used to stimulate AMPK activation (17).
Western immunoblotting analysis.
Protein levels of phosphorylated and total AMPK, phosphorylated and total LKB1, and phosphorylated and total HMG-CoA reductase were determined by Western immunoblotting analysis. In brief, liver proteins (70 μg) were separated by electrophoresis in an 8% or 10% SDS polyacrylamide gel. Subsequently, proteins were transferred from the gel to a nitrocellulose membrane, which was then incubated with primary antibody overnight at 4°C. Primary antibodies include rabbit anti-AMPKα polyclonal antibody (New England Biolabs, Ipswich, MA), rabbit anti-phospho-AMPKα (Thr-172) polyclonal antibody (New England Biolabs), rabbit anti-LKB1 monoclonal antibody (New England Biolabs), rabbit anti-phospho-LKB1 (Ser-428/431) monoclonal antibody (New England Biolabs), and rabbit anti-phospho-HMG-CoA reductase (Ser-872) polyclonal antibody (Millipore). HRP-conjugated anti-mouse or anti-rabbit IgG antibodies (New England Biolabs) were used as secondary antibodies. Protein bands were detected in chemiluminescent HRP detection reagent (Luminata Crescendo; Millipore) for visualization. To ensure equal protein loading, the same membranes were reprobed with mouse anti-β-actin monoclonal antibody (New England Biolabs).
Determination of AMP levels.
The AMP in the liver tissue was measured by using the AMP-Glo assay kit (Promega, Madison, WI). The values were expressed in relative luminescence units (RLU), which were proportional to the AMP concentration. Data were plotted as a percentage of the control based on the changes in RLU (ΔRLU) calculated from the standard curve.
Measurement of cholesterol and HMG-CoA reductase activity.
Lipids in the liver tissue were extracted according to the Folch method (14). The levels of cholesterol in the liver and hepatocytes were determined by using enzymatic kits (Wako Chemicals) (60, 61). HMG-CoA reductase activity in the liver microsomes was measured by using [3-14C] HMG-CoA (PerkinElmer) as a substrate (60, 61).
Statistical analysis.
Results were analyzed using one-way ANOVA followed by Newman-Keuls post hoc test. P values less than 0.05 were considered statistically significant.
RESULTS
Body weight and biochemical parameters of mice.
Mice fed a high-fat diet for 5 wk exhibited a significant increase in body weight, similar to the results observed in our previous studies (23, 60). Folic acid supplementation had no effect on body weight gain in these animals (Fig. 1A). High-fat diet consumption for 5 wk was accompanied by elevated fasting blood glucose (Fig. 1B) and insulin levels (Fig. 1C). Folic acid supplementation attenuated an increase in fasting blood glucose levels (Fig. 1B) in mice fed a high-fat diet but did not significantly reduce fasting blood insulin levels (Fig. 1C). In addition, mice fed a high-fat diet exhibited higher levels of total cholesterol (Fig. 1D), free cholesterol (Fig. 1E), and triglyceride (Fig. 1F) in the liver. Folic acid supplementation during the high-fat diet feeding reduced total and free cholesterol levels (Fig. 1, D and E), but did not significantly change the triglyceride content in the liver (Fig. 1F).
Fig. 1.

Body weight, blood glucose, and liver lipids. Mice were fed a control diet, a high-fat diet (HFD), or a high-fat diet supplemented with folic acid (HFD+folic acid) for 5 wk. A: body weight was measured at the end of the 5-wk feeding period. B: fasting blood glucose was measured before and after the 5-wk feeding period. Open bars indicate the start of the feeding period, while solid bars indicate the end of the feeding period. C: fasting blood insulin was measured at the end of the 5-wk feeding period. Liver total cholesterol (D), free cholesterol (E), and triglycerides (F) were measured at the end of the 5-wk feeding period. Results are expressed as the means ± SE (n = 8). Different letters indicate statistical significance (P < 0.05).
Regulation of hepatic AMPK and LKB1 in mice.
In general, phosphorylation of AMPKα at Thr-172 leads to kinase activation, while dephosphorylation inactivates it. Western immunoblotting of phosphorylated AMPK is a recognized surrogate for AMPK activity (47). Relative to the control group, the protein levels of phosphorylated AMPK were markedly reduced (inactivation) in the liver of the high-fat diet-fed mice, while total AMPK protein levels remained constant in all of the groups (Fig. 2A). Under the same dietary condition, folic acid supplementation increased AMPK phosphorylation (activation) in the liver (Fig. 2A). Consistent with AMPK inactivation, the high-fat diet feeding caused a significant reduction in LKB1 phosphorylation (Fig. 2B). By contrast, folic acid supplementation markedly enhanced phosphorylation of LKB1 (activation), while total LKB1 levels were unchanged among the groups (Fig. 2B). These results suggested that folic acid may stimulate hepatic AMPK activation during high-fat diet consumption through inducing LKB1 activation.
Fig. 2.

AMPK and liver kinase B1 (LKB1) phosphorylation in mouse liver. Mice were fed a control diet, a HFD, or a HFD supplemented with folic acid (HFD+folic acid) for 5 wk. Western immunoblotting analysis was performed to measure the protein levels of phosphorylated AMPK (pAMPK), total AMPK, phosphorylated LKB1 (pLKB1), total LKB1, and β-actin in the liver. Results are depicted as pAMPK, total AMPK, and ratio of pAMPK to total AMPK (A), pLKB1, total LKB1, and ratio of pLKB1 to total LKB1 (B). Results are expressed as the means ± SE; n = 8. Different letters indicate statistical significance (P < 0.05).
Effect of fatty acid and folic acid on AMPK and LKB1 activation in hepatocytes.
To investigate the mechanisms by which folic acid supplementation regulated hepatic LKB1 and AMPK activation, experiments were performed in HepG2 cells that were incubated with palmitic acid. The protein level of phosphorylated AMPK was decreased in cells incubated with palmitic acid (Fig. 3A), indicating inactivation of the kinase. Incubation of cells with 5-MTHF effectively restored AMPK phosphorylation status in palmitic acid-treated cells, while total AMPK protein levels remained constant in all of the groups (Fig. 3A). In another set of experiments, incubation of cells with folic acid also restored AMPK phosphorylation status in palmitic acid-treated cells (Fig. 3B). As a control, experiments were conducted in HepG2 cells to examine the effect of 5-MTHF or folic acid without the influence of palmitic acid. Folic acid or 5-MTHF treatment did not alter AMPK phosphorylation status in control cells (Fig. 3C). As both 5-MTHF and folic acid could restore AMPK phosphorylation in palmitic acid-treated cells, 5-MTHF was used in the subsequent experiments. The level of phosphorylated LKB1 protein was significantly decreased in cells incubated with palmitic acid (Fig. 3D). Incubation of cells with 5-MTHF restored LKB1 phosphorylation status in palmitic acid-treated cells, while total LKB1 levels remained constant among the groups (Fig. 3D). Several inhibitors were employed to identify potential upstream targets that might be involved in LKB1 activation by 5-MTHF. Nicotinamide is an inhibitor of sirtuin 1 (SIRT 1) (2, 22), which is an upstream deacetylase that promotes LKB1 activation (22, 29). Although nicotinamide treatment slightly lowered 5-MTHF-induced phosphorylation of LKB1 in palmitic acid-treated cells, the effect was not significant (Fig. 4). PKA and PKC-ζ can also activate LKB1 via phosphorylation at its Ser-428 (9, 62). To determine whether 5-MTHF induced LKB1 phosphorylation was regulated by these kinases, hepatocytes were incubated with H89 (inhibitor of PKA) or PKC-ζ pseudosubstrate (inhibitor of PKC-ζ) (46, 62). Inhibition of PKA by H89 abolished 5-MTHF-induced LKB1 phosphorylation in hepatocytes, while incubation with the PKC-ζ inhibitor did not cause a significant change in LKB1 phosphorylation status (Fig. 4). Taken together, these results suggested that PKA might be involved in 5-MTHF-induced LKB1 activation in hepatocytes.
Fig. 3.

AMPK and LKB1 phosphorylation in HepG2 cells. HepG2 cells were incubated in the absence (control) or presence of palmitic acid (0.3 mM, PA) with or without 5-methyltetrahydrofolate (1 μg/ml, PA+5-MTHF) or folic acid (1 μg/ml, PA+folic acid) for 16 h. Control cells were also incubated in the absence or presence of 5-MTHF and folic acid treatment for 16 h. Western immunoblotting analysis was performed to measure the protein levels of phosphorylated AMPK (pAMPK), total AMPK, phosphorylated LKB1 (pLKB1), total LKB1 in HepG2 cells. β-actin was measured and used as a loading control. Results are depicted as pAMPK, total AMPK, ratio of pAMPK to total AMPK in palmitic acid-treated cells with or without 5-MTHF treatment (A), pAMPK, total AMPK, ratio of pAMPK to total AMPK in palmitic acid treated cells with or without folic acid treatment (B), pAMPK, total AMPK, ratio of pAMPK to total AMPK in cells incubated with 5-MTHF or folic acid in the absence of palmitic acid treatment (C), and pLKB1, total LKB1, ratio of pLKB1 to total LKB1 in palmitic acid treated cells (D). Results are expressed as the means ± SE; n = 4–6. Different letters indicate statistical significance (P < 0.05).
Fig. 4.

Regulation of LKB1 by folic acid in HepG2 cells. HepG2 cells were incubated with palmitic acid (0.3 mM, PA) in the absence or presence of 5-methyltetrahydrofolate (1 μg/ml, 5-MTHF) for 16 h. In some sets of experiments, nicotinamide (10 mM, NM), H89 (10 μM), or PKC-ζ pseudosubstrate (1 μM) were added to the culture medium. Western immunoblotting analysis was performed to measure the protein levels of phosphorylated LKB1 (pLKB1), total LKB1 and β-actin in cultured cells. Results are depicted as a ratio of pLKB1 to total LKB1 and expressed as the means ± SE; n = 4–6. Different letters indicate statistical significance (P < 0.05).
Role of AMP on AMPK activation in hepatocytes and liver tissue.
Aside from LKB1, AMPK is allosterically activated by AMP. Compound C is a selective inhibitor of AMPK that competes with adenine nucleotide binding on the kinase (66). Incubation of hepatocytes with compound C strongly suppressed 5-MTHF-induced AMPK phosphorylation (Fig. 5A). However, AMP effectively restored the phosphorylation of AMPK in palmitic acid-treated cells (Fig. 5A). In addition, AMP levels were measured in the liver tissue. While high-fat diet feeding caused a significant reduction in hepatic AMP levels, folic acid supplementation markedly increased AMP levels in the liver of mice fed a high-fat diet (Fig. 5B). These results indicated that under conditions of fatty acid overload or high-fat diet consumption, folic acid supplementation might also restore AMPK activation in the liver through the elevation of hepatic AMP levels.
Fig. 5.

AMPK phosphorylation in HepG2 cells and AMP levels in mouse liver. A: HepG2 cells were incubated with palmitic acid (0.3 mM, PA) in the absence or presence of 5-methyltetrahydrofolate (1 μg/ml; 5-MTHF) for 16 h. In one set of experiments, compound C (10 μM) or AMP (100 μM) was added to the culture medium. Western immunoblotting analysis was performed to measure the protein levels of phosphorylated AMPK (pAMPK), total AMPK, and β-actin in cultured cells. Results are depicted as a ratio of pAMPK to total AMPK and expressed as the means ± SE (n = 4–6). B: mice were fed a control diet, a high-fat diet (HFD) or a high-fat diet supplemented with folic acid (HFD+folic acid) for 5 wk. AMP levels in the liver were measured, and results were expressed as changes in relative luminescence units (ΔRLU). Results are expressed as the means ± SE (n = 6). Different letters indicate statistical significance (P < 0.05).
Effect of folic acid supplementation on hepatic cholesterol production.
HMG-CoA reductase regulates the rate-limiting step in cholesterol biosynthesis (60, 61). AMPK is identified as the major upstream kinase responsible for phosphorylation of HMG-CoA reductase (inactivation) (27, 37). High-fat diet feeding decreased the levels of phosphorylated HMG-CoA reductase in the liver (Fig. 6A), which was accompanied with a significant increase in HMG-CoA reductase activity (Fig. 6B). In contrast, folic acid supplementation increased HMG-CoA reductase phosphorylation (Fig. 6A) and reduced the activity of the reductase in high-fat diet-fed mice (Fig. 6B). Furthermore, incubation of HepG2 cells with palmitic acid resulted in a significant increase in cellular total cholesterol levels, while 5-MTHF treatment effectively reduced total cholesterol levels in palmitic acid-treated cells (Fig. 7). These results suggested that inhibition of HMG-CoA reductase activity by folic acid might be mediated through AMPK-dependent phosphorylation of the reductase.
Fig. 6.
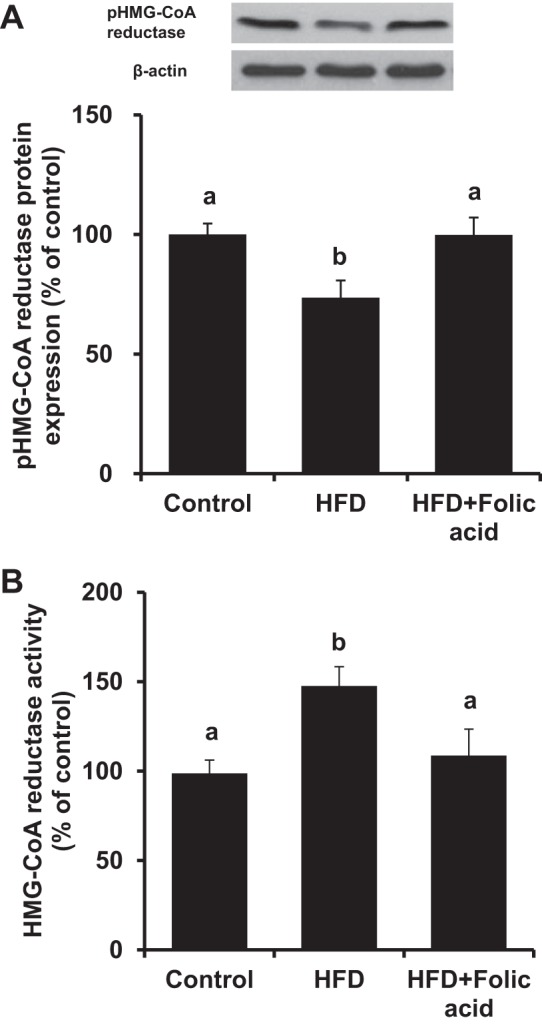
Determination of HMG-CoA reductase protein and enzyme activity in mouse liver. Mice were fed a control diet, a HFD, or a HFD+folic acid for 5 wk. A: phosphorylated HMG-CoA reductase (pHMG-CoA reductase) and β-actin in the liver were determined by Western immunoblotting analysis. B: HMG-CoA reductase enzyme activity was measured. Results are expressed as the means ± SE; n = 4. Different letters indicate statistical significance (P < 0.05).
Fig. 7.

Determination of cholesterol in HepG2 cells. HepG2 cells were incubated in the absence or presence of palmitic acid (0.3 mM; PA), with or without 5-methyltetrahydrofolate (5-MTHF; 1 μg/ml) for 24 h. Intracellular total cholesterol was measured. Results are expressed as means ± SE; n = 6. Different letters indicate statistical significance (P < 0.05).
DISCUSSION
AMPK plays a central role in sensing energy levels to favorably modulate hepatic metabolism. With widespread control over a variety of metabolic cascades, AMPK regulation might be important in NAFLD, in which both energy homeostasis and metabolic function are perturbed (42). The novel findings presented in this study are that folic acid supplementation effectively restores AMPK activity in the liver of high-fat diet-fed mice through 1) the elevation of AMP levels, and 2) the activation of its upstream kinase LKB1. Such effects of folic acid supplementation are associated with reduced blood glucose and hepatic cholesterol levels in high-fat diet-fed mice.
AMP, an allosteric activator of AMPK, mediates its effect via inducing a conformational change in AMPK and subsequently promotes its phosphorylation by upstream kinases (17, 18). Although studies have demonstrated that the high-fat diet feeding in rodents leads to AMPK inactivation in the liver (39, 63), hepatic AMP levels were not reported in these studies. For the first time, the present study demonstrated that hepatic AMP levels were significantly lower in mice fed a high-fat diet than those fed a control diet. Folic acid supplementation effectively increased hepatic AMP levels in high-fat diet-fed mice to the control level. Folic acid plays an important role in nucleotide biosynthesis (33). It is plausible that folic acid supplementation may contribute to increased formation of AMP during high-fat diet feeding and, therefore, stimulate AMPK activation in the liver. To investigate whether the restoration of hepatic AMP levels contributed to folic acid-mediated AMPK activation, experiments were carried out in cultured hepatocytes. While incubation of cells with palmitic acid significantly reduced AMPK phosphorylation (inactivation), its phosphorylation status was restored with 5-MTHF treatment. Furthermore, incubation of hepatocytes with AMP was able to reverse the inhibitory effect of palmitic acid on AMPK phosphorylation, indicating that increased AMP availability could stimulate AMPK activation. Taken together, these results suggested that AMPK activation by folic acid might be mediated, in part, through the elevation of hepatic AMP levels in high-fat diet-fed mice.
In addition to allosteric activation by AMP, the upstream kinase LKB1 also plays a crucial role in AMPK activation through direct phosphorylation at Thr-172 of AMPK (58). In the present study, high-fat diet feeding led to a significant decrease in LKB1 phosphorylation (inactivation) in the liver. By contrast, folic acid supplementation restored hepatic LKB1 phosphorylation status in these animals. Consistent with the in vivo findings, 5-MTHF and folic acid treatment restored AMPK phosphorylation (activation) in palmitic acid-treated cells. Treatment with 5-MTHF in HepG2 cells also stimulated LKB1 phosphorylation (activation). Upstream kinases, namely, PKA and PKC-ζ, have been implicated in the phosphorylation of LKB1 (9, 62). In the present study, incubation of cells with PKA inhibitor (H89) abolished 5-MTHF-induced LKB1 phosphorylation, while inhibition of PKC-ζ with PKC-ζ pseudosubstrate did not appear to affect LKB1 phosphorylation. Although deacetylation of LKB1 by SIRT1 is also regarded as another mechanism for LKB1 activation (22, 29), inhibition of SIRT1 by nicotinamide did not appear to significantly affect 5-MTHF-induced LKB1 phosphorylation in HepG2 cells. The effects of 5-MTHF observed in palmitic acid-treated HepG2 cells were in line with the findings obtained from the mice fed a high-fat diet supplemented with folic acid. HepG2 cells resemble many characteristics of hepatocytes and have been used as a valuable cell model in metabolic studies, including regulation of AMPK (22). Similar to primary human hepatocytes, HepG2 cells express the major enzymes that are involved in folate metabolism, such as dihydrofolate reductase (DHF) and methylene tetrahydrofolate reductase (MTHFR) (6). However, HepG2 cells are transformed cells in which the expression of certain genes may be different from that in primary human hepatocytes. For example, HepG2 cells express a lower level of some key enzymes involved in one-carbon or methyl group metabolism, such as phosphatidylethanolamine N-methyltransferase (PEMT). PEMT is an enzyme involved in the conversion of phosphatidylethanolamine to phosphatidylcholine in the liver. Reduced expression of this enzyme affects phosphatidylcholine synthesis, which, in turn, can impair hepatic assembly and secretion of very low-density lipoprotein (10, 32). Taken together, results from the present study suggested that PKA might be involved in folic acid-induced LKB1 phosphorylation, which, in turn, contributed to the restoration of hepatic AMPK activation in high-fat diet-fed mice supplemented with folic acid. It is intriguing that the insulin-sensitizing agent metformin, which is commonly used for the management of diabetes and other metabolic syndromes, also targets the LKB1-AMPK pathway (12, 44). The ability of folic acid to regulate AMPK activation through an AMP-LKB1-dependent mechanism suggests that it may also have a therapeutic application in metabolic disorders, such as high-fat diet-induced NAFLD.
AMPK plays a key role in regulating glucose and cholesterol metabolism (5, 52), both of which are dysregulated during NAFLD (35, 36). In the present study, folic acid supplementation effectively attenuated the increase in fasting blood glucose levels in mice fed a high-fat diet but did not significantly reduce blood insulin levels. It is plausible that the favorable effect of folic acid on blood glucose levels might be mediated, in part, by improving hepatic insulin sensitivity. On the other hand, increased cholesterol biosynthesis has been observed in patients with NAFLD (45). Our recent study has shown that high-fat diet feeding activates HMG-CoA reductase, which may contribute to hepatic cholesterol accumulation in NAFLD (60). The HMG-CoA reductase activity can be regulated by phosphorylation (inactivation) and dephosphorylation (activation) of the enzyme (8). AMPK is the major upstream kinase responsible for the phosphorylation of HMG-CoA reductase (27, 37). In the present study, folic acid supplementation reduced hepatic cholesterol levels in mice fed a high-fat diet. Such an effect was associated with increased phosphorylation of HMG-CoA reductase and decreased reductase enzyme activity. In addition, 5-MTHF reduced total cholesterol levels in palmitic acid-treated hepatocytes. Taken together, these results suggested that restoration of AMPK activation by folic acid might contribute to the improvement of glucose and cholesterol metabolism during high-fat diet feeding.
It is important to recognize the differences in folate metabolism between rodents and humans. Folate in the human diet consists of natural folates (generally present in polyglutamated form) and synthetic folic acid (monoglutamated form used for dietary fortification and supplements) while purified rodent diets are supplemented with synthetic folic acid. Folic acid is readily absorbed in the intestine via folate receptors. On the other hand, absorption of natural folates in the diet is less efficient since these folates must be hydrolyzed to a monoglutamate prior to intestinal absorption (38, 53). Upon absorption, folic acid undergoes biotransformation to become the biologically active form of folate, namely, tetrahydrofolate (THF), a precursor of 5-MTHF. Dihydrofolate reductase (DHFR) catalyzes the reduction of folic acid to dihydrofolate (DHF) and then to THF. The activity of DHFR in human liver is low compared with the rat liver (3). In the present study, mice fed a high-fat diet were supplemented with 26 mg/kg diet folic acid, a pharmacological dose, which was 10-fold higher than the folic acid content in the high-fat diet. Caution should be made when findings are translated from rodents to humans as rodents can efficiently metabolize folic acid and may be able to tolerate higher doses of folic acid compared with humans. A proper human clinical trial should be conducted to ascertain the optimal dose for the use of folic acid as a therapy for metabolic disorders. Another limitation of the present study was that we only investigated whether folic acid supplementation could restore hepatic AMPK activation that was altered by high-fat diet feeding. Future studies are warranted to investigate whether supplementation of folic acid at various doses affects hepatic metabolism under physiological conditions.
Perspectives and Significance
The present study demonstrates, for the first time, that folic acid supplementation during high-fat diet feeding can restore AMPK activation in the liver through the elevation of AMP levels and phosphorylation of LKB1. Restoration of AMPK function may contribute to the improvement of glucose and cholesterol metabolism that are impaired by high-fat diet consumption. Further studies are warranted to investigate whether the regulation of AMPK activation by folic acid is beneficial in metabolic disorders such as NAFLD.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: V.S., N.W., L.K.S., and K.O. performed experiments; V.S., N.W., and K.O. analyzed data; V.S., N.W., L.K.S., Y.L.S., and K.O. interpreted results of experiments; V.S. and N.W. prepared figures; V.S. and K.O. drafted manuscript; V.S., N.W., L.K.S., Y.L.S., J.D.H., and K.O. edited and revised manuscript; V.S., N.W., L.K.S., Y.L.S., J.D.H., and K.O. approved final version of manuscript; Y.L.S. and K.O. conception and design of research.
ACKNOWLEDGMENTS
This study was supported, in part, by grants from the Natural Sciences and Engineering Research Council of Canada and St. Boniface Hospital Research Centre.
REFERENCES
- 1.Angulo P. Obesity and nonalcoholic fatty liver disease. Nutr Rev 65: S57–S63, 2007. [DOI] [PubMed] [Google Scholar]
- 2.Avalos JL, Bever KM, Wolberger C. Mechanism of sirtuin inhibition by nicotinamide: altering the NAD+ cosubstrate specificity of a Sir2 enzyme. Mol Cell 17: 855–868, 2005. [DOI] [PubMed] [Google Scholar]
- 3.Bailey SW, Ayling JE. The extremely slow and variable activity of dihydrofolate reductase in human liver and its implications for high folic acid intake. Proc Natl Acad Sci USA 106: 15, 424–15, 429, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buettner R, Scholmerich J, Bollheimer LC. High-fat diets: modeling the metabolic disorders of human obesity in rodents. Obesity 15: 798–808, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Canto C, Auwerx J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci 67: 3407–3423, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chango A, Nour AA, Bousserouel S, Eveillard D, Anton PM, Gueant JL. Time course gene expression in the one-carbon metabolism network using HepG2 cell line grown in folate-deficient medium. J Nutr Biochem 20: 312–320, 2009. [DOI] [PubMed] [Google Scholar]
- 7.Christensen KE, Wu Q, Wang X, Deng L, Caudill MA, Rozen R. Steatosis in mice is associated with gender, folate intake, and expression of genes of one-carbon metabolism. J Nutr 140: 1736–1741, 2010. [DOI] [PubMed] [Google Scholar]
- 8.Clarke PR, Hardie DG. Regulation of HMG-CoA reductase: identification of the site phosphorylated by the AMP-activated protein kinase in vitro and in intact rat liver. EMBO J 9: 2439–2446, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins SP, Reoma JL, Gamm DM, Uhler MD. LKB1, a novel serine/threonine protein kinase and potential tumour suppressor, is phosphorylated by cAMP-dependent protein kinase (PKA) and prenylated in vivo. Biochem J 345: 673–680, 2000. [PMC free article] [PubMed] [Google Scholar]
- 10.Cui Z, Houweling M, Vance DE. Suppression of rat hepatoma cell growth by expression of phosphatidylethanolamine N-methyltransferase-2. J Biol Chem 269: 24, 531–24, 533, 1994. [PubMed] [Google Scholar]
- 11.Dahlhoff C, Worsch S, Sailer M, Hummel BA, Fiamoncini J, Uebel K, Obeid R, Scherling C, Geisel J, Bader BL, Daniel H. Methyl-donor supplementation in obese mice prevents the progression of NAFLD, activates AMPK, and decreases acyl-carnitine levels. Mol Metab 3: 565–580, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Despres JP. Potential contribution of metformin to the management of cardiovascular disease risk in patients with abdominal obesity, the metabolic syndrome and type 2 diabetes. Diabetes Metab 29: 6S53–6S61, 2003. [DOI] [PubMed] [Google Scholar]
- 13.Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology 43: S99–S112, 2006. [DOI] [PubMed] [Google Scholar]
- 14.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 226: 497–509, 1957. [PubMed] [Google Scholar]
- 15.Gan RY, Li HB. Recent progress on liver kinase B1 (LKB1): expression, regulation, downstream signaling and cancer suppressive function. Int J Mol Sci 15: 16, 698–16, 718, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Golay A, Bobbioni E. The role of dietary fat in obesity. Int J Obes Rel Metab Disord 21 Suppl 3: S2–S11, 1997. [PubMed] [Google Scholar]
- 17.Gowans GJ, Hawley SA, Ross FA, Hardie DG. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab 18: 556–566, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13: 251–262, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hariri N, Gougeon R, Thibault L. A highly saturated fat-rich diet is more obesogenic than diets with lower saturated fat content. Nutr Res 30: 632–643, 2010. [DOI] [PubMed] [Google Scholar]
- 20.Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem 271: 27,879–27,887, 1996. [DOI] [PubMed] [Google Scholar]
- 21.Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol 8: 35–44, 2011. [DOI] [PubMed] [Google Scholar]
- 22.Hou X, Xu S, Maitland-Toolan KA, Sato K, Jiang B, Ido Y, Lan F, Walsh K, Wierzbicki M, Verbeuren TJ, Cohen RA, Zang M. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J Biol Chem 283: 20,015–20,026, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hwang SY, Sarna LK, Siow YL, OK. High-fat diet stimulates hepatic cystathionine β-synthase and cystathionine γ-lyase expression. Can J Physiol Pharmacol 91: 913–919, 2013. [DOI] [PubMed] [Google Scholar]
- 24.Iyer R, Tomar SK. Folate: a functional food constituent. J Food Sci 74: R114–R122, 2009. [DOI] [PubMed] [Google Scholar]
- 25.Javitt NB. Hep G2 cells as a resource for metabolic studies: lipoprotein, cholesterol, and bile acids. FASEB J 4: 161–168, 1990. [DOI] [PubMed] [Google Scholar]
- 26.Joshi-Barve S, Barve SS, Amancherla K, Gobejishvili L, Hill D, Cave M, Hote P, McClain CJ. Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology 46: 823–830, 2007. [DOI] [PubMed] [Google Scholar]
- 27.Jurevics H, Hostettler J, Barrett C, Morell P, Toews AD. Diurnal and dietary-induced changes in cholesterol synthesis correlate with levels of mRNA for HMG-CoA reductase. J Lipid Res 41: 1048–1054, 2000. [PubMed] [Google Scholar]
- 28.Kim YI. Folic acid fortification and supplementation—good for some but not so good for others. Nutr Rev 65: 504–511, 2007. [DOI] [PubMed] [Google Scholar]
- 29.Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem 283: 27,628–27,635, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leevy CM, Baker H. Nutritional deficiencies in liver disease. Med Clin NA 54: 467–477, 1970. [PubMed] [Google Scholar]
- 31.Leevy CM, Thompson A, Baker H. Vitamins and liver injury. Am J Clin Nutr 23: 493–499, 1970. [DOI] [PubMed] [Google Scholar]
- 32.Ling J, Lewis J, Douglas D, Kneteman NM, Vance DE. Characterization of lipid and lipoprotein metabolism in primary human hepatocytes. Biochim Biophys Acta 1831: 387–397, 2013. [DOI] [PubMed] [Google Scholar]
- 33.Lucock M. Folic acid: nutritional biochemistry, molecular biology, and role in disease processes. Mol Genet Metab 71: 121–138, 2000. [DOI] [PubMed] [Google Scholar]
- 34.Mahabir S, Ettinger S, Johnson L, Baer DJ, Clevidence BA, Hartman TJ, Taylor PR. Measures of adiposity and body fat distribution in relation to serum folate levels in postmenopausal women in a feeding study. Eur J Clin Nutr 62: 644–650, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, Natale S, Vanni E, Villanova N, Melchionda N, Rizzetto M. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 37: 917–923, 2003. [DOI] [PubMed] [Google Scholar]
- 36.Min HK, Kapoor A, Fuchs M, Mirshahi F, Zhou H, Maher J, Kellum J, Warnick R, Contos MJ, Sanyal AJ. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab 15: 665–674, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Omkumar RV, Darnay BG, Rodwell VW. Modulation of Syrian hamster 3-hydroxy-3-methylglutaryl-CoA reductase activity by phosphorylation. Role of serine 871. J Biol Chem 269: 6810–6814, 1994. [PubMed] [Google Scholar]
- 38.Pietrzik K, Bailey L, Shane B. Folic acid and L-5-methyltetrahydrofolate: comparison of clinical pharmacokinetics and pharmacodynamics. Clin Pharmacokinet 49: 535–548, 2010. [DOI] [PubMed] [Google Scholar]
- 39.Pu P, Gao DM, Mohamed S, Chen J, Zhang J, Zhou XY, Zhou NJ, Xie J, Jiang H. Naringin ameliorates metabolic syndrome by activating AMP-activated protein kinase in mice fed a high-fat diet. Arch Biochem Biophys 518: 61–70, 2012. [DOI] [PubMed] [Google Scholar]
- 40.Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, Sargeant C, Contos MJ, Sanyal AJ. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 46: 1081–1090, 2007. [DOI] [PubMed] [Google Scholar]
- 41.Qiu A, Jansen M, Sakaris A, Min SH, Chattopadhyay S, Tsai E, Sandoval C, Zhao R, Akabas MH, Goldman ID. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 127: 917–928, 2006. [DOI] [PubMed] [Google Scholar]
- 42.Rui L. Energy metabolism in the liver. Compr Physiol 4: 177–197, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sarna LK, Wu N, Wang P, Hwang SY, Siow YL, OK. Folic acid supplementation attenuates high fat diet induced hepatic oxidative stress via regulation of NADPH oxidase. Can J Physiol Pharmacol 90: 155–165, 2012. [DOI] [PubMed] [Google Scholar]
- 44.Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310: 1642–1646, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simonen P, Kotronen A, Hallikainen M, Sevastianova K, Makkonen J, Hakkarainen A, Lundbom N, Miettinen TA, Gylling H, Yki-Jarvinen H. Cholesterol synthesis is increased and absorption decreased in non-alcoholic fatty liver disease independent of obesity. J Hepatol 54: 153–159, 2011. [DOI] [PubMed] [Google Scholar]
- 46.Song P, Xie Z, Wu Y, Xu J, Dong Y, Zou MH. Protein kinase Cζ-dependent LKB1 serine 428 phosphorylation increases LKB1 nucleus export and apoptosis in endothelial cells. J Biol Chem 283: 12,446–12,455, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Stein SC, Woods A, Jones NA, Davison MD, Carling D. The regulation of AMP-activated protein kinase by phosphorylation. Biochem J 345: 437–443, 2000. [PMC free article] [PubMed] [Google Scholar]
- 48.Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev 89: 1025–1078, 2009. [DOI] [PubMed] [Google Scholar]
- 49.Takahashi Y, Soejima Y, Fukusato T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol 18: 2300–2308, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tiniakos DG, Vos MB, Brunt EM. Nonalcoholic fatty liver disease: pathology and pathogenesis. Ann Rev Pathol 5: 145–171, 2010. [DOI] [PubMed] [Google Scholar]
- 51.Viollet B, Foretz M, Guigas B, Horman S, Dentin R, Bertrand L, Hue L, Andreelli F. Activation of AMP-activated protein kinase in the liver: a new strategy for the management of metabolic hepatic disorders. J Physiol 574: 41–53, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Viollet B, Guigas B, Leclerc J, Hebrard S, Lantier L, Mounier R, Andreelli F, Foretz M. AMP-activated protein kinase in the regulation of hepatic energy metabolism: from physiology to therapeutic perspectives. Acta Physiol (Oxf) 196: 81–98, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Visentin M, Diop-Bove N, Zhao R, Goldman ID. The intestinal absorption of folates. Annu Rev Physiol 76: 251–274, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang YC, Chen YM, Lin YJ, Liu SP, Chiang EP. GNMT expression increases hepatic folate contents and folate-dependent methionine synthase-mediated homocysteine remethylation. Mol Med 17: 486–494, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wilkening S, Stahl F, Bader A. Comparison of primary human hepatocytes and hepatoma cell line Hepg2 with regard to their biotransformation properties. Drug Metab Dispos 31: 1035–1042, 2003. [DOI] [PubMed] [Google Scholar]
- 56.Woo CW, Prathapasinghe GA, Siow YL, OK. Hyperhomocysteinemia induces liver injury in rat: Protective effect of folic acid supplementation. Biochim Biophys Acta 1762: 656–665, 2006. [DOI] [PubMed] [Google Scholar]
- 57.Woo CW, Siow YL, OK. Homocysteine activates cAMP-response element binding protein in HepG2 through cAMP/PKA signaling pathway. Arterioscler Thromb Vasc Biol 26: 1043–1050, 2006. [DOI] [PubMed] [Google Scholar]
- 58.Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 13: 2004–2008, 2003. [DOI] [PubMed] [Google Scholar]
- 59.Wright AJ, Dainty JR, Finglas PM. Folic acid metabolism in human subjects revisited: potential implications for proposed mandatory folic acid fortification in the UK. Br J Nutr 98: 667–675, 2007. [DOI] [PubMed] [Google Scholar]
- 60.Wu N, Sarna LK, Hwang SY, Zhu Q, Wang P, Siow YL, OK. Activation of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase during high fat diet feeding. Biochim Biophys Acta 1832: 1560–1568, 2013. [DOI] [PubMed] [Google Scholar]
- 61.Wu N, Sarna LK, Siow YL, OK. Regulation of hepatic cholesterol biosynthesis by berberine during hyperhomocysteinemia. Am J Physiol Regul Integr Comp Physiol 300: R635–R643, 2011. [DOI] [PubMed] [Google Scholar]
- 62.Xie Z, Dong Y, Scholz R, Neumann D, Zou MH. Phosphorylation of LKB1 at serine 428 by protein kinase C-ζ is required for metformin-enhanced activation of the AMP-activated protein kinase in endothelial cells. Circulation 117: 952–962, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yoneda M, Guo Y, Ono H, Nakatsu Y, Zhang J, Cui X, Iwashita M, Kumamoto S, Tsuchiya Y, Sakoda H, Fujishiro M, Kushiyama A, Koketsu Y, Kikuchi T, Kamata H, Nishimura F, Asano T. Decreased SIRT1 expression and LKB1 phosphorylation occur with long-term high-fat diet feeding, in addition to AMPK phosphorylation impairment in the early phase. Obes Res 4: e163–e246, 2010. [DOI] [PubMed] [Google Scholar]
- 64.Zhang S, Zheng L, Dong D, Xu L, Yin L, Qi Y, Han X, Lin Y, Liu K, Peng J. Effects of flavonoids from Rosa laevigata Michx fruit against high-fat diet-induced non-alcoholic fatty liver disease in rats. Food Chem 141: 2108–2116, 2013. [DOI] [PubMed] [Google Scholar]
- 65.Zhao R, Matherly LH, Goldman ID. Membrane transporters and folate homeostasis: intestinal absorption and transport into systemic compartments and tissues. Expert Rev Mol Med 11: e4, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108: 1167–1174, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]