

With the use of commercial and primary preparations along with pharmacology and adenovirus-mediated overexpression, this study highlights a potential new transforming growth factor-β/Smad3-dependent mechanism for AMP kinase in reducing vascular smooth muscle growth. These findings offer insights into AMP kinase signaling and provide support for a novel therapeutic target to combat vascular proliferative disorders.
Keywords: adenosine monophosphate-activated protein kinase, proliferation
Abstract
Dysfunctional vascular growth is a major contributor to cardiovascular disease, the leading cause of morbidity and mortality worldwide. Growth factor-induced activation of vascular smooth muscle cells (VSMCs) results in a phenotypic switch from a quiescent, contractile state to a proliferative state foundational to vessel pathology. Transforming growth factor-β (TGF-β) is a multifunctional signaling protein capable of growth stimulation via Smad signaling. Although Smad signaling is well characterized in many tissues, its role in VSM growth disorders remains controversial. Recent data from our lab and others implicate the metabolic regulator AMP-activated protein kinase (AMPK) in VSM growth inhibition. We hypothesized that AMPK inhibits VSMC proliferation by reducing TGF-β-mediated growth in a Smad-dependent fashion. Treatment of rat VSMCs with the AMPK agonist AICAR significantly decreased TGF-β-mediated activation of synthetic Smad2 and Smad3 and increased inhibitory Smad7. Flow cytometry and automated cell counting revealed that AICAR reversed TGF-β-mediated cell cycle progression at 24 h and elevated cell numbers at 48 h. TGF-β/Smad signaling increased the G0/G1 inducers cyclin D1/cyclin-dependent kinase (CDK) 4 and cyclin E/CDK2; however, AICAR reversed these events while increasing cytostatic p21. The specific role of Smad3 in AMPK-mediated reversal of TGF-β-induced growth was then explored using adenovirus-mediated Smad3 overexpression (Ad-Smad3). Ad-Smad3 cells increased cell cycle progression and cell numbers compared with Ad-GFP control cells, and these were restored to basal levels with concomitant AICAR treatment. These findings support a novel AMPK target in TGF-β/Smad3 for VSMC growth control and support continued investigation of AMPK as a possible therapeutic target for reducing vascular growth disorders.
NEW & NOTEWORTHY
With the use of commercial and primary preparations along with pharmacology and adenovirus-mediated overexpression, this study highlights a potential new transforming growth factor-β/Smad3-dependent mechanism for AMP kinase in reducing vascular smooth muscle growth. These findings offer insights into AMP kinase signaling and provide support for a novel therapeutic target to combat vascular proliferative disorders.
cardiovascular disease (CVD) is the number one cause of fatality in the United States and worldwide, and according to the American Heart Association abnormal vascular smooth muscle (VSM) growth is a key underpinning for many of these pathologies (6). Despite ample research the molecular events leading to mitogenic activation of vascular smooth muscle cells (VSMCs) and their ensuing involvement in CVD pathogenesis are not fully understood.
Adenosine monophosphate-activated protein kinase (AMPK) is a constitutively expressed serine/threonine kinase that responds to a rise in AMP/ATP under metabolic challenge (8, 17, 20). Activation of AMPK requires AMP binding and catalytic phosphorylation (17, 20) and leads to energy-producing pathways being upregulated while energy-consuming pathways are shut off or rendered minimally active (7, 8, 17, 22–24). AMPK has many tissue-dependent functional targets leading to phosphorylation of key metabolic enzymes and, specifically in the vasculature, has been implicated in increased endothelial nitric oxide synthase activity and nitric oxide bioavailability, the promotion of angiogenesis, and cytostasis of commercial and primary VSMCs via inhibition of cell cycle progression (10, 13, 22–24). Moreover, AMPK activation has been shown to inhibit neointima formation in rat arteries following injury (13, 23). Together, these studies suggest growth-regulatory capacity by AMPK; however, specific mechanisms of AMPK in VSMCs, particularly related to proliferation and migration, remain elusive. Of these, the role that AMPK may play in the inhibition of cell proliferation under cytokine stimulation during disease is especially intriguing and is critical for a better understanding of vascular and associated proliferative pathologies.
Transforming growth factor-β1 (TGF-β1) is a multifunctional cytokine acting canonically through Smad signaling to exert effects in a wide range of cell types. In VSMCs, TGF-β has historically been considered antiproliferative (2, 14–16); however, other findings suggest that TGF-β and its downstream Smads stimulate growth in primary VSMCs (9, 18, 19, 25–27). Adding to this uncertainty, in cultured cells TGF-β1 is suggested to switch between growth stimulation and growth suppression depending on concentration (3, 18) and cell density (9). This pleiotropic and often discordant nature of TGF-β/Smad signaling and its control of tissue growth demand clarity.
TGF-β elicits effects by binding to cell surface receptors, whereupon it stimulates phosphorylation of cytoplasmic receptor-activated Smads (R-Smads), Smad2 and Smad3, that combine with common Smad4, and the oligomer translocates to the nucleus where it activates growth regulatory genes (18, 21, 26, 28). Another component of this signal transduction pathway is inhibitory Smad7 that suppresses TGF-β signaling by interfering with activation of the R-Smads (18, 21, 28). The growth-stimulating capacity of TGF-β in VSMCs has recently been suggested to involve Smad3-mediated phosphorylation and nuclear export of p27, a cyclin-dependent kinase inhibitor (27). The many uncertain aspects of TGF-β/Smad signaling, particularly in VSM, justify continued study and present an attractive target for future therapeutic interests.
To date, no studies have examined the potential role of AMPK in modulating TGF-β-induced VSMC growth. Based on our previous observations showing growth inhibition by AMPK in VSM (23, 24), and considering the potential growth-promoting nature of TGF-β/Smad3 in VSM, an interesting yet unexplored relationship may exist between these two signaling molecules. The purpose of this investigation was to characterize the capacity of AMPK to modulate growth in response to TGF-β in rat VSMCs. We hypothesized that AMPK has the capacity to reduce TGF-β-stimulated VSMC growth via reduction in Smad signaling and inhibition of cell cycle regulatory proteins. Novel results shed light on the relationship between AMPK and TGF-β/Smads in VSM and provide support for continued investigation of AMPK as a valuable target for therapies aimed at reducing vascular growth disorders.
MATERIALS AND METHODS
This investigation was approved by the East Carolina University Animal Care and Use Committee (U.S. National Institutes of Health, Publication No. 85-23, revised 1996).
Materials
AICAR was purchased from Toronto Research Chemicals (North York, Ontario) and Invitrogen (Carlsbad, CA). Recombinant TGF-β1 was purchased from R&D Systems (Minneapolis, MN). All primary antibodies were purchased from Abcam (Cambridge, MA) or Cell Signaling (Danvers, MA), were diluted 1:1,000 for In-Cell Western or 1:500 for immunofluorescence detected by flow cytometry and iCys laser-scanning cytometric (LSC) analysis, and were targeted against the following: Smad2, pSmad2Thr8, Smad3, pSmad3Ser423/425, Smad7; cyclin D, cyclin E, cyclin-dependent kinase (CDK) 2, CDK4, CDK6, p21, p27; α-tubulin, and β-actin. Antibodies were diluted in IRDye blocking buffer (Rockland, Gilbertsville, PA) or 1% BSA in PBS (Gemini, Stoneham, MA). IRDye secondary antibodies (1:1,000; Rockland) and FITC- or Texas red-conjugated secondary antibodies (1:10,000; Rockland) were used for protein detection while Draq 5 (1:10,000; Cell Signaling) and Sapphire 700 (1:1,000; Li-Cor, Lincoln, NE) were used for DNA staining and protein normalization, respectively. Propidium iodide and RNase or DRAQ5 for cell cycle analysis with flow cytometry and DAPI for cell cycle analysis with iCys LSC were purchased from Invitrogen. Adenoviruses (type 5) encoding GFP (Ad-GFP) or Smad3 (Ad-Smad3) were purchased from Vector Biolabs (Philadelphia, PA).
Methods
Cell culture.
Rat thoracic aorta VSMCs (A7r5) were purchased from ATCC (Manassas, VA). Primary VSMCs were obtained from thoracic aorta of male Sprague Dawley rats (∼125 g; Charles River Labs) by collagenase and elastase digestion and characterized morphologically as described (5). VSMCs were cultured in Dulbecco's modified Eagles medium (DMEM) supplemented with fetal bovine serum (FBS, 0.5–10%), 2 mM l-glutamine, and 1:500 dilution of 50 μg/ml Primocin at 37°C in 95% air-5% CO2, and were serially split and used through passage 10 (commercial) or passage 6 (primary) to avoid onset of phenotypic switching (1, 11, 23, 24) unless otherwise stated.
Protein detection.
IN-CELL WESTERN.
VSMCs were seeded in 96-well plates and, once confluent, were treated with specified agents. Cells were then formalin fixed, and protein expression was determined by In-Cell Western analysis as described (1, 11, 24). Briefly, fixed cells were permeabilized with 0.1% Triton X, blocked with IRDye blocking buffer (Invitrogen), and treated with rabbit antirat primary antibodies (1 h; room temperature) in 96-well plates. Primary antibodies targeting specific proteins were detected using antirabbit secondary antibodies conjugated with an IR800 fluorophore, and DNA was stained using DRAQ5 and Sapphire700 and used for normalization. Fluorescence was detected and quantified at 800 nm and DNA at 700 nm using a Li-Cor Odyssey Infrared Imaging System and analysis software.
IMMUNOFLUORESCENCE WITH FLOW CYTOMETRY.
Confluent VSMCs were treated with select pharmacological agents for 1–24 h and then trypsinized, fixed with 4% formalin, washed with PBS, permeabilized with 0.1% Triton X in PBS, washed, and blocked with 1% BSA in PBS. Select proteins were detected by primary/secondary antibody conjugation in 1% BSA and analyzed by flow cytometry (Accuri C6 Flow Cytometer) using CFlow Plus software (Accuri).
Cell cycle analysis.
Cells were plated in 12-well plates at 80,000 cells/well in complete media until ∼50% confluent. Cells were quiesced in 0.5% FBS for 24 h followed by treatment in complete growth media (DMEM, 10% FBS; Primocin) containing select pharmacological agents for 24 h. Cells were trypsinized, fixed, and stained with propidium iodide or Draq5 for flow cytometry or DAPI for iCys per the manufacturer's recommendations (11, 24). The fraction of cells in each phase of the cell cycle was assessed by flow cytometry or iCys LSC.
Cell proliferation and viability analyses.
VSMCs were plated in six-well plates at 180,000 cells/well, 12-well plates at 50,000 cell/well, or Ibidi 6.4 slides at 25,000 cells/channel in complete media until ∼50% confluent. Cells were quiesced in 0.5% FBS for 24 h followed by treatment in complete growth media for 48 h. For LSC analysis, cells were fixed and stained with DAPI, and cell cycle progression was determined through iCys LSC software. Proliferation and viability of suspended cells were accomplished through automated cell counting using trypan blue exclusion staining (ViCell; Beckman Coulter).
Adenovirus overexpression of Smad3.
Adenoviral titers incorporating GFP or Smad3 (4 × 1010 and 1 × 1010 plaque formation units/ml, respectively) were determined using the end-point dilution assay previously described (4). Sufficient protein expression in the absence of cytotoxicity was observed after infecting VSMCs for 6 h using Ad-GFP at a 1:10,000 dilution followed by washout and 24 h incubation in complete media. Cell viability was determined using trypan blue exclusion 42 h after virus washout.
Statistical Analyses
Data were analyzed using Excel 2011 (Microsoft) and Sigma Plot 11.2 (SPSS). All datasets were tested for normal distribution and met the homogeneity prerequisites for analysis of variance (ANOVA). One-way ANOVA and Tukey's post hoc multiple-comparison tests were used to detect changes between individual groups. Two-way ANOVA with multiple comparisons and Tukey's post hoc tests were used for cell cycle analysis to detect significance between groups. Data are expressed as means ± SE with P < 0.05 considered statistically significant.
RESULTS
TGF-β Promotes VSMC Growth
Our initial series of experiments aimed to verify capacity of TGF-β1 to induce Smad signaling and to regulate growth in rat VSMCs. To determine the lowest bioactive concentration of recombinant TGF-β1 (rTGF-β1) in VSMCs, serial dosing experiments were performed on two different preparations: commercial A7R5 cells (Fig. 1A) and rat primary cells (Fig. 1B). In both cases, 10 ng/ml rTGF-β1 was the lowest concentration that yielded the maximal effect on Smad3 phosphorylation and thus was the rTGF-β1 concentration used in subsequent experiments. Also, given these comparable responses in commercial vs. primary cells in rTGF-β1-induced Smad3 phosphorylation as well as parallel growth responses to rTGF-β1 with/without AICAR (discussed below), commercial VSMCs were used in all experiments unless otherwise specified. Cells were then treated with rTGF-β1 (60 min), and Smad2, Smad3, and Smad7 were analyzed. Results showed significantly elevated levels of phosphorylated Smad2 (at Thr8) and Smad3 (at Ser423/425) as well as moderately (≈20%; P = 0.35) decreased expression of Smad7 in rTGF-β1-treated cells compared with vehicle controls (Fig. 2). Treatment with rTGF-β1 significantly increased cell numbers in the G2/M phase of the cell cycle in both commercial (Fig. 3A) and primary (Fig. 3C) cells after 24 h, and significantly elevated viable cell numbers in commercial (Fig. 3B) and primary (Fig. 3D) cells after 48 h compared with respective vehicle controls. Treatment with rTGF-β1 significantly increased growth-promoting cyclins D1 and E (Fig. 4, A and B) and the catalytic CDK2 and CDK4 (Fig. 4, C and D) compared with vehicle controls, yet only marginally reduced expression of the CDK inhibitors (CDK-I) p21 and p27 compared with vehicle controls (Fig. 4, F and G).
Fig. 1.
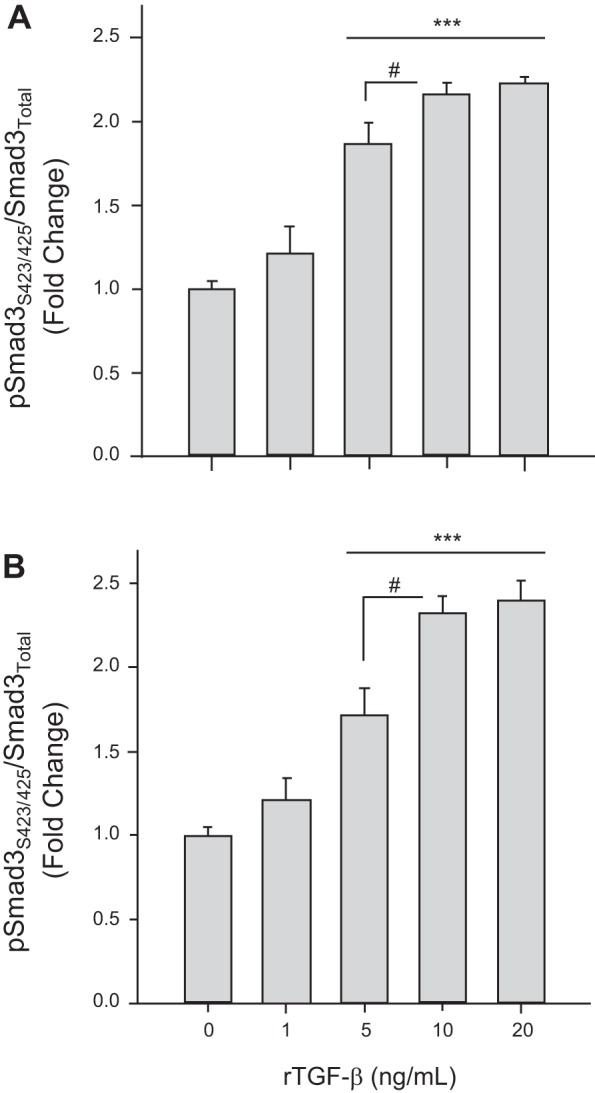
Recombinant transforming growth factor-β (TGF-β1) stimulates Smad3 signaling in rat vascular smooth muscle cells (VSMCs). Rat VSMCs were treated with vehicle or recombinant TGF-β1 (rTGF-β1; 1–20 ng/ml) for 60 min, and Smad3 phosphorylation (at Ser423/425) was analyzed by immunofluorescence in rat commercial A7R5 VSMCs (A) and rat primary VSMCs (B). Data are presented as pSmad3/total Smad3 with P values <0.05 considered statistically significant for 3 independent experiments, each run in triplicate. ***P < 0.001 compared with vehicle controls, and #P < 0.05 within groups as indicated.
Fig. 2.
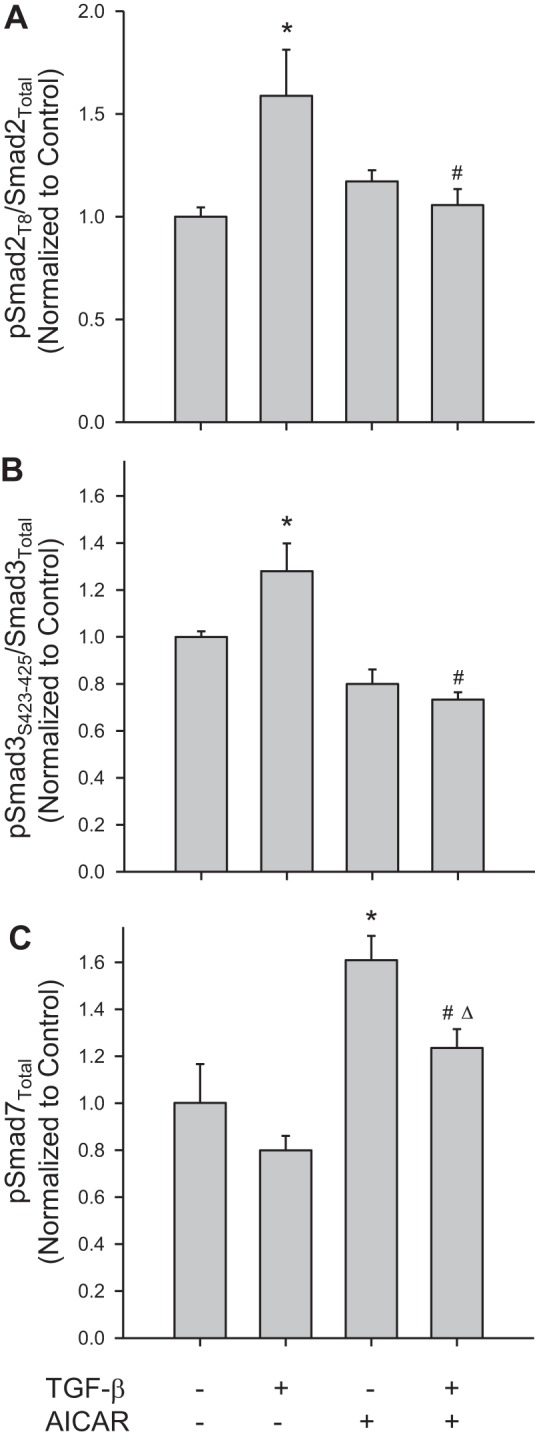
The AMP-activated protein kinase (AMPK) stimulator AICAR reverses TGF-β1-induced Smad signaling. Rat A7R5 VSMCs were treated with vehicle or rTGF-β1 (10 ng/ml, 60 min) with/without the AMPK stimulator AICAR (1 mM, 60 min), and Smad proteins were analyzed by immunofluorescence with In-Cell Western and flow cytometry for phosphorylation of Smad2 at Thr8 (A), phosphorylation of Smad3 at Ser423/425 (B), and Smad7 (C). For phosphorylation of Smad2 and Smad3, data were normalized to total Smad2 or Smad3, respectively, and presented as pSmad/total Smad. Five independent experiments were performed, each in triplicate. *P < 0.05 compared with vehicle controls, #P < 0.05 compared with recombinant TGF-β1 treatment alone, and ΔP < 0.05 compared with AICAR treatment alone.
Fig. 3.

AICAR reverses TGF-β1-induced cell cycle progression. Rat VSMCs were treated with vehicle or rTGF-β1 (10 ng/ml) with/without AICAR (1 mM) following overnight quiescence. After 24 h cell cycle progression was analyzed by flow cytometry (A), and after 48 h cell numbers were assessed via automated cell counting with trypan blue exclusion staining to estimate cytotoxicity in rat A7R5 VSMCs (B). Parallel experiments were performed in rat primary VSMCs for cell cycle progression at 24 h by flow cytometry (C) and cell numbers at 48 h (D). E: representative flow cytometry data are presented for cells treated with vehicle (control), AICAR, rTGF-β1, or rTGF-β1 + AICAR, respectively, for 24 h. Three independent experiments were performed, each in triplicate. *P < 0.05, **P < 0.01, and ***P < 0.001 compared with vehicle controls and #P < 0.05 compared with TGF-β1 treatment alone.
Fig. 4.

AICAR controls TGF-β1-induced cell cycle regulatory proteins. Rat A7R5 VSMCs were treated with vehicle or rTGF-β1 (10 ng/ml) with/without AICAR (1 mM) for 24 h following overnight quiescence, and cell cycle regulatory proteins cyclin D1 and cyclin E, the cyclin-dependent kinases (CDK) 2, CDK4, and CDK6, and the cyclin-dependent kinase inhibitors (CDK-I) p21 and p27 were analyzed by immunofluorescence with flow cytometry. Treatment with rTGF-β1 alone significantly increased expression of cyclin D1 (A) and cyclin E (B), and these were reversed in the presence of AICAR. AICAR alone did not significantly alter expression of cyclins D1 or E compared with vehicle controls. Treatment with rTGF-β1 alone induced expression of the cyclin E/cyclin A agonist CDK2 (C) and the cyclin D agonist CDK4 (D) compared with vehicle controls, and both were significantly reversed in the presence of AICAR. AICAR alone modestly (nonsignificantly) reduced CDK2 and significantly reduced CDK4 expression compared with controls, yet no changes were observed in CDK6 expression with any TGF-β1/AICAR regimen (E). Treatment with TGF-β1 alone failed to significantly alter p21 (F) or p27 (G) expression compared with vehicle controls, yet sole AICAR treatment induced significant elevations in both p21 and p27 compared with controls. Concomitant TGF-β1 and AICAR reversed p27 induction observed with TGF-β1 treatment alone but did not markedly alter TGF-β1-induced p21 induction. Data are presented as protein of interest normalized to total protein, with n = 3–5/treatment group. P values < 0.05 were considered statistically significant. *P < 0.05, **P < 0.005, and ***P < 0.001 compared with vehicle controls, #P < 0.05 compared with the TGF-β1 treatment group, and ΔP < 0.05 compared with AICAR treatment alone.
AICAR Inhibits TGF-β1-Induced VSMC Growth Via Inhibition of Smad Signaling
We recently demonstrated that activation of AMPK via the AMP mimetic AICAR (1 mM) effectively reduces proliferation and migration of rat VSMCs (23, 24). Therefore, in an effort to examine the influence of AMPK on the observed TGF-β1/Smad-induced VSMC growth, VSMCs were treated with AICAR (1 mM in all experiments) in the presence or absence of rTGF-β1 and Smad signals, and indexes of vascular growth were evaluated. As shown in Fig. 2, AICAR alone had modest effects on pSmad2 and pSmad3 (normalized to total Smad2 or Smad3, respectively), yet significantly increased Smad7 levels compared with vehicle controls. Interestingly, in the presence of rTGF-β1, AICAR significantly and completely reversed the increases in pSmad2 and pSmad3 observed with rTGF-β1 alone and sustained significant increases in Smad7 (Fig. 2).
In line with our previous observations (23, 24), sole AICAR treatment induced significant increases in S phase cell numbers, significantly reduced G2/M phase cell numbers after 24 h (Fig. 3A), and significantly reduced viable cell numbers after 48 h (Fig. 3B) in commercial VSMCs compared with vehicle controls, and analogous findings were observed in primary cells (Fig. 3, C and D). Moreover, AICAR completely reversed the increases in G2/M cell numbers observed with rTGF-β1 alone after 24 h (Fig. 3, A and C) and completely abrogated the rTGF-β1-induced increases in viable cell numbers at 48 h (Fig. 3, B and D) in both VSMC preparations. No significant changes were observed in G0/G1 at 24 h or in cell viability at 48 h (P = 0.08; data not shown) following rTGF-β1 with/without AICAR.
AICAR Reverses TGF-β1-Mediated Alterations in Cell Cycle Regulatory Proteins
To determine possible mechanisms by which AMPK inhibits TGF-β1-induced cell growth, we investigated expression of the key cell cycle regulators observed to be altered by rTGF-β1 in the presence of AICAR. AICAR alone had no effect on cyclin expression (Fig. 4, A and B); however, a nonsignificant trend for reduction of CDK2 (P = 0.07; Fig. 4C) and a significant reduction in CDK4 (Fig. 4D) were observed in nonstimulated cells. Intriguingly, in stimulated cells, AICAR reversed rTGF-β1-induced increases in cyclins D1 and E and CDK2 and CDK4 (Fig. 4, A–D, respectively). Additionally, AICAR significantly increased p21 expression under both nonstimulated and rTGF-β1-stimulated conditions (Fig. 4F), while only under nonstimulated conditions did AICAR significantly increase p27 content (Fig. 4G).
Smad3 Specifically Increases VSMC Growth
In an effort to determine if TGF-β1 acts through canonical Smad3 to elevate VSMC growth, we used an adenovirus to overexpress Smad3 as we previously described (4). Figure 5 shows significantly increased expression of GFP (Ad-GFP; Fig. 5A; photomicrograph shows positive GFP staining in Ad-GFP-infected cells) or Smad3 (Ad-Smad3; Fig. 5B) compared with vehicle controls after 24 h. As before, we analyzed cell cycle progression and viable cell numbers of Smad3-infected VSMCs with/without AICAR. Smad3 overexpression induced cell cycle progression by reducing the number of cells in G0/G1 and significantly increasing the number of cells in G2/M compared with Ad-GFP controls (Fig. 5C); however, concomitant incubation of cells with AICAR fully reversed these observations (Fig. 5C). Moreover, Ad-Smad3 increased viable cell numbers after 48 h compared with Ad-GFP controls, and this too was completely reversed with concomitant AICAR (Fig. 5D).
Fig. 5.

AICAR reverses Smad3-induced cell cycle progression and cell proliferation in Smad3-overexpressing VSMCs. Rat commercial A7R5 VSMCs were infected with empty vehicle, adenovirus (type 5) encoding green fluorescent protein (Ad-GFP), or adenovirus (type 5) encoding Smad3 (Ad-Smad3) [at optimal multiplicity of infection (MOI)] for 6 h, quiesced overnight, and grown in complete media for 24 h after which expression of GFP (A) and Smad 3 (B) was analyzed by immunofluorescent flow cytometry and In-Cell Westerns. Inset: GFP expression in Ad-GFP-infected VSMCs after 24 h treatment. Smad3- and GFP-overexpressing cells were treated with/without AICAR (1 mM), after which cell cycle progression was analyzed after 24 h (C), and viable cell numbers were quantified by automated cell counting with trypan blue exclusion staining to estimate cytotoxicity after 48 h (D). Three independent experiments were performed, each in triplicate. ***P < 0.001 compared with vehicle control cells and #P < 0.001 compared with Ad-Smad3-treated cells.
To verify that transient Smad3 overexpression was causative for the reversal of rTGF-β1-mediated VSMC growth by AICAR, we serially passaged Ad-GFP and Ad-Smad3 cells (to between passages 3 and 6 following infection) and performed immunofluorescence for Smad3 content and cell cycle analysis. Expression of GFP and total Smad3 as well as Ad-Smad3-mediated stimulation of cell cycle progression returned to control levels after repeated passaging of VSMCs (data not shown).
DISCUSSION
Data presented in this study support the hypothesis that AMPK inhibits VSM growth through a mechanism at least partly dependent on TGF-β/Smad signals. Novel findings show that the metabolic regulator AMPK, stimulated to biologically active levels by AICAR as demonstrated recently (23, 24), serves to reduce TGF-β/Smad signaling and its growth-stimulating effects in rat VSMCs. These findings are among the first to suggest that AMPK inhibits VSMC growth associated with the proliferative and synthetic TGF-β signaling network. Data presented here in conjunction with our recently published findings (23, 24) provide unique insights into AMPK signaling as a biologically capable system to offer remediation of vascular growth, including that induced by TGF-β/Smad.
In light of previously reported controversial findings, results from this research support the capacity of TGF-β to increase VSMC growth evidenced through cell cycle progression and cell proliferation analyses. Recombinant TGF-β alone significantly increased cell numbers entering the G2/M phase after 24 h (Fig. 3, A and C) and induced an ∼50% increase in viable cell numbers after 48 h (Fig. 3, B and D). Moreover, Smad3 overexpression (Fig. 5B) produced similar and significant increases in cell numbers in G2/M as well as viable cell numbers (Fig. 5, C and D). Importantly, in both rTGF-β-stimulated cells and Smad3-overexpressing cells AICAR was able to significantly reduce cell cycle progression revealed by reduced cell numbers in G2/M after 24 h (Figs. 3A and 5C) and reduced cell numbers after 48 h (Figs. 3B and 5D) compared with rTGF-β and Ad-Smad3 stimulation alone. Intriguingly, this reversal of TGF-β1-induced proliferation by AICAR, although not significant compared with control conditions, was significant compared with solitary AICAR treatment, suggesting that the effects of AICAR are targeted to TGF/Smad signaling. Together these data support recent reports that suggest TGF-β not only promotes collagen synthesis and secretion but also acts to promote proliferation and adhesion of VSMCs (18, 21, 25–27). Moreover, our results provide insights suggesting that TGF-β acts, at least in part, in a Smad3-dependent fashion and that AMPK has the ability to reduce TGF-β-induced VSM growth in a Smad3-dependent manner. This does not rule out the possibility, though, that AMPK via AICAR may have non-TGF-β/Smad actions in VSM. Nonetheless, because VSMC growth and migration are foundational in the progression of vascular growth disorders, implication of AMPK as an inhibitor of TGF-β signaling has great biological impact.
To determine possible mechanisms by which AMPK inhibits TGF-β/Smad3 signaling and cellular growth, we investigated the influence of AICAR on the TGF-β/Smad network. Figure 2 demonstrates that, while AICAR alone had no effect on expression of pSmad2 or pSmad3, in rTGF-β-stimulated cells AICAR completely reversed phosphorylation of both Smad2 and Smad3. Additionally, both stimulated and nonstimulated VSMCs showed significant increases in the expression of inhibitory Smad7 when treated with AICAR. These data suggest that, while under basal conditions AMPK may play little or no role in regulating TGF-β signaling, under cytokine-induced growth-provoking conditions, AMPK plays a key inhibitory role on TGF-β at least in part through the inhibitory actions of Smad7. An increase in Smad7 inhibits oligomerization of pSmad2/pSmad3 with cytoplasmic Smad4. In addition, a recent report by Zhao et al. (29) showed that AMPK can inhibit nuclear translocation of Smad4 via reduction of Smad4 expression. Taken together, these data strongly suggest that AMPK can inhibit translocation of the Smad oligomer into the nucleus thus inhibiting activation of TGF-β-dependent progrowth gene transcription (18, 21, 29). These findings, in light of our cytostatic data, suggest AMPK-mediated inhibition of TGF-β signal transduction further occurs by increasing cytoplasmic Smad7 thereby indirectly reducing cell cycle progression and cell proliferation.
In this study we also investigated the cytostatic role of AMPK on TGF-β-stimulated VSMCs by examining cell cycle regulatory proteins. Early G0/G1-dependent cyclin D and late G0/G1-dependent cyclin E were both significantly elevated by rTGF-β; however, concomitant treatment with AICAR significantly reduced both cyclin D1 and cyclin E compared with respective control and rTGF-β-stimulated conditions (Fig. 4, A and B). Both cyclin E-associated CDK2 and cyclin D1-associated CDK4 were significantly elevated by rTGF-β but were completely reversed to below basal levels with concomitant AICAR (Fig. 4, C and D). Intriguingly, no change was observed in cyclin D-associated CDK6, possibly suggesting that rTGF-β promotes cell cycle progression in a sequential cyclin D/CDK4- and cyclin E/CDK2-specific fashion. Furthermore, AICAR elevated both CDK-I p21 and p27, yet only p21 remained significantly elevated following cotreatment with rTGF-β (Fig. 4, F and G). Together, these data suggest that AMPK has the ability to inhibit rTGF-β-induced cell cycle progression via reduction in G0/G1 cyclin D/CDK4 and cyclin E/CDK2 complexes possibly through CDK inhibition via increased p21.
Regarding potential concerns with commercial or repeatedly passaged cells compared with primary preparations, it should be noted that initial experiments incorporated commercial A7R5 rat aortic VSMCs as well as primary rat aortic VSMCs, and their responsiveness to exogenous rTGF-β and AICAR stimulation were compared. Results show remarkable similarities between commercial and primary VSMCs in the ability of rTGF-β to induce downstream Smad3 phosphorylation (Fig. 1). Additionally, comparable findings were observed between commercial and primary cells in the ability for rTGF-β to induce cell cycle progression and to increase cell numbers and the ability for AICAR to reverse these effects (Fig. 3). Also, neither baseline nor AICAR-stimulated activation of AMPK nor phosphorylation of its downstream targets, acetyl-CoA carboxylase and vasodilator-stimulated phosphoprotein, were markedly different in commercial cells compared with primary cells as previously reported (23, 24). Thus, data presented in this study using commercial cells mimic those observed in primary preparations and are considered sound, translatable, and biologically relevant.
In summary, findings in this study provide support for a discrete signaling network by which AMPK inhibits growth-promoting actions of TGF-β/Smad3, and this concept is presented as a theoretical schematic in Fig. 6. We propose that AMPK inhibits pSmad2/3 by promoting Smad7. Resulting inhibition of TGF-β/Smad signaling leads to reversal of G0/G1 cell cycle progression via inhibition of cyclins/CDKs D/4 and E/2 that we suggest is mediated by cytostatic p21. Cumulatively, these data highlight a novel and biologically important signaling cascade by which a metabolically activated protein such as AMPK has capability to inhibit cytokine-induced progrowth signaling events. This has clear biological importance as a therapeutically desirable approach to reverse vascular cell growth associated with disease.
Fig. 6.

Schematic depicting the proposed inhibitory actions of AICAR-stimulated AMPK on TGF-β1-induced VSMC growth. Data presented in this study suggest that AICAR-stimulated AMPK has the ability to inhibit TGF-β1-mediated Smad signaling via promoting inhibitory Smad7 and by preventing growth-promoting Smad2/Smad3. Prevention of Smad2/3 phosphorylation and oligomerization with Smad4 prevents their nuclear translocation and transcriptome activation of growth regulatory genes. We propose a possible prosynthetic mechanism of TGF-β1/Smad signaling in VSMCs in the promotion of cell cycle progression and cell proliferation through cyclin D/CDK4 and cyclin E/CDK2. Additionally, AMPK possesses the ability to enhance the CDK inhibitor p21 that acts to further inhibit TGF-β1-mediated G0/G1 cell cycle progression. Solid lines, signaling events previously characterized in the literature; broken lines, signaling events supported by new findings from this study.
GRANTS
This project was supported by Award No. 12PRE12060400 from the American Heart Association (J. D. Stone), Award No. r83783 from the Bahner Summer Scholars Program (J. D. Stone), Award No. R01-HL-81720 from the National Heart, Lung, and Blood Institute (D. A. Tulis), an East Carolina University Brody School of Medicine Seed/Bridge Grant (D. A. Tulis), and a Brody Brothers Endowment Fund Award (D. A. Tulis).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors. The content is solely the responsibility of the authors and does not necessarily represent the official views of the American Heart Association, the Bahner Scholars Program, the National Heart, Lung, and Blood Institute, the National Institutes of Health, or the Brody Brothers Endowment Fund.
AUTHOR CONTRIBUTIONS
Author contributions: J.D.S. and D.A.T. conception and design of research; J.D.S., A.W.H., J.R.V., and J.J.B. performed experiments; J.D.S., A.W.H., J.R.V., J.J.B., and D.A.T. analyzed data; J.D.S., A.W.H., J.J.B., and D.A.T. interpreted results of experiments; J.D.S. and A.W.H. prepared figures; J.D.S. drafted manuscript; J.D.S., A.W.H., J.J.B., and D.A.T. edited and revised manuscript; J.D.S., A.W.H., J.R.V., J.J.B., and D.A.T. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the academic and support staff of the East Carolina University Department of Physiology for assistance. We also thank Dr. Robert M. Lust for support and guidance as Chair, Department of Physiology at East Carolina University. We thank Hannah Kiser, Andrew Prince, and Frankie Terrell, undergraduate student researchers from Carson-Newman University, for efforts in the completion of this project.
REFERENCES
- 1.Adderley SP, Joshi CN, Martin DN, Tulis DA. Phosphodiesterases regulate BAY 41-2272-induced VASP phosphorylation in vascular smooth muscle cells. Frontiers Pharmacol 3: 10, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akhurst RJ, Hata A. Targeting the TGFbeta signalling pathway in disease. Nat Rev 11: 790–811, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Battegay EJ, Raines EW, Seifert RA, Bowen-Pope DF, Ross R. TGF-beta induces bimodal proliferation of connective tissue cells via complex control of an autocrine PDGF loop. Cell 63: 515–524, 1990. [DOI] [PubMed] [Google Scholar]
- 4.Bollinger LM, Witczak CA, Houmard JA, Brault JJ. SMAD3 augments FoxO3-induced MuRF-1 promoter activity in a DNA-binding-dependent manner. Am J Physiol Cell Physiol 307: C278–C287, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Durante W, Schini VB, Catovsky S, Kroll MH, Vanhoutte PM, Schafer AI. Plasmin potentiates induction of nitric oxide synthesis by interleukin-1β in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 264: H617–H624, 1993. [DOI] [PubMed] [Google Scholar]
- 6.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation 127: e6–e245, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev 25: 1895–1908, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev 8: 774–785, 2007. [DOI] [PubMed] [Google Scholar]
- 9.Hneino M, Bouazza L, Bricca G, Li JY, Langlois D. Density-dependent shift of transforming growth factor-beta-1 from inhibition to stimulation of vascular smooth muscle cell growth is based on unconventional regulation of proliferation, apoptosis and contact inhibition. J Vasc Res 46: 85–97, 2009. [DOI] [PubMed] [Google Scholar]
- 10.Igata M, Motoshima H, Tsuruzoe K, Kojima K, Matsumura T, Kondo T, Taguchi T, Nakamaru K, Yano M, Kukidome D, Matsumoto K, Toyonaga T, Asano T, Nishikawa T, Araki E. Adenosine monophosphate-activated protein kinase suppresses vascular smooth muscle cell proliferation through the inhibition of cell cycle progression. Circ Res 97: 837–844, 2005. [DOI] [PubMed] [Google Scholar]
- 11.Joshi CN, Martin DN, Fox JC, Mendelev NN, Brown TA, Tulis DA. The soluble guanylate cyclase stimulator BAY 41–2272 inhibits vascular smooth muscle growth through the cAMP-dependent protein kinase and cGMP-dependent protein kinase pathways. J Pharmacol Exp Ther 339: 394–402, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagata D, Takeda R, Sata M, Satonaka H, Suzuki E, Nagano T, Hirata Y. AMP-activated protein kinase inhibits angiotensin II-stimulated vascular smooth muscle cell proliferation. Circulation 110: 444–451, 2004. [DOI] [PubMed] [Google Scholar]
- 14.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84: 767–801, 2004. [DOI] [PubMed] [Google Scholar]
- 15.Polyak K, Kato JY, Solomon MJ, Sherr CJ, Massague J, Roberts JM, Koff A. p27Kip1, a cyclin-Cdk inhibitor, links transforming growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev 8: 9–22, 1994. [DOI] [PubMed] [Google Scholar]
- 16.Reardon C, McKay DM. TGF-beta suppresses IFN-gamma-STAT1-dependent gene transcription by enhancing STAT1-PIAS1 interactions in epithelia but not monocytes/macrophages. J Immunol 178: 4284–4295, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Rubin LJ, Magliola L, Feng X, Jones AW, Hale CC. Metabolic activation of AMP kinase in vascular smooth muscle. J Appl Physiol 98: 296–306, 2005. [DOI] [PubMed] [Google Scholar]
- 18.Ruiz-Ortega M, Rodriguez-Vita J, Sanchez-Lopez E, Carvajal G, Egido J. TGF-beta signaling in vascular fibrosis. Cardiovasc Res 74: 196–206, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Samarakoon R, Higgins SP, Higgins CE, Higgins PJ. TGF-beta1-induced plasminogen activator inhibitor-1 expression in vascular smooth muscle cells requires pp60(c-src)/EGFR(Y845) and Rho/ROCK signaling. J Mol Cell Cardiol 44: 527–538, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J 403: 139–148, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santibanez JF, Quintanilla M, Bernabeu C. TGF-beta/TGF-beta receptor system and its role in physiological and pathological conditions. Clin Sci (Lond) 121: 233–251, 2011. [DOI] [PubMed] [Google Scholar]
- 22.Shirwany NA, Zou MH. AMPK in cardiovascular health and disease. Acta Pharmacol 31: 1075–1084, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stone JD, Narine A, Shaver PR, Fox JC, Vuncannon JR, Tulis DA. AMP-activated protein kinase inhibits vascular smooth muscle cell proliferation and migration and vascular remodeling following injury. Am J Physiol Heart Circ Physiol 304: H369–H381, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stone JD, Narine A, Tulis DA. Inhibition of vascular smooth muscle growth via signaling crosstalk between AMP-activated protein kinase and cAMP-dependent protein kinase. Frontiers Physiol 3: 409, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suwanabol PA, Seedial SM, Shi X, Zhang F, Yamanouchi D, Roenneburg D, Liu B, Kent KC. Transforming growth factor-beta increases vascular smooth muscle cell proliferation through the Smad3 and extracellular signal-regulated kinase mitogen-activated protein kinases pathways. J Vasc Surg 56: 446–454, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suwanabol PA, Seedial SM, Zhang F, Shi X, Si Y, Liu B, Kent KC. TGF-β and Smad3 modulate PI3K/Akt signaling pathway in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 302: H2211–H2219, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsai S, Hollenbeck ST, Ryer EJ, Edlin R, Yamanouchi D, Kundi R, Wang C, Liu B, Kent KC. TGF-β through Smad3 signaling stimulates vascular smooth muscle cell proliferation and neointimal formation. Am J Physiol Heart Circ Physiol 297: H540–H549, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yokote K, Kobayashi K, Saito Y. The role of Smad3-dependent TGF-beta signal in vascular response to injury. Trends Cardiovasc Med 16: 240–245, 2006. [DOI] [PubMed] [Google Scholar]
- 29.Zhao J, Miyamoto S, You YH, Sharma K. AMP-activated protein kinase (AMPK) activation inhibits nuclear translocation of Smad4 in mesangial cells and diabetic kidneys. Am J Physiol Renal Physiol 308: F1167–F1177, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]