

Despite successful resuscitation, cardiac contractile function following ischemia and ventricular fibrillation is markedly impaired, contributing to the high mortality rate after cardiac arrest. Our findings on the beneficial effects of enhancing pyruvate dehydrogenase activation shed light on the novel therapeutic strategy of modulating cardiac metabolism to mitigate this post-ventricular fibrillation contractile dysfunction.
Keywords: cardiac metabolism, myocardial infarction, myocardial contractile dysfunction, ventricular fibrillation
Abstract
Ventricular fibrillation (VF) is an important cause of sudden cardiac arrest following myocardial infarction. Following resuscitation from VF, decreased cardiac contractile function is a common problem. During and following myocardial ischemia, decreased glucose oxidation, increased anaerobic glycolysis for cardiac energy production are harmful and energetically expensive. The objective of the present study is to determine the effects of dichloroacetate (DCA), a glucose oxidation stimulator, on cardiac contractile dysfunction following ischemia-induced VF. Male Sprague-Dawley rat hearts were Langendorff perfused in Tyrode's buffer. Once stabilized, hearts were subjected to 15 min of global ischemia and 5 min of aerobic reperfusion in the presence or absence of DCA. At the 6th min of reperfusion, VF was induced electrically, and terminated. Left ventricular (LV) pressure was measured using a balloon. Pretreatment with DCA significantly improved post-VF left ventricular developed pressure (LVDP) and dp/dtmax. In DCA-pretreated hearts, post-VF lactate production and pyruvate dehydrogenase (PDH) phosphorylation were significantly reduced, indicative of stimulated glucose oxidation, and inhibited anaerobic glycolysis by activation of PDH. Epicardial NADH fluorescence was increased during global ischemia above preischemic levels, but decreased below preischemia levels following VF, with no differences between nontreated controls and DCA-pretreated hearts, whereas DCA pretreatment increased NADH production in nonischemic hearts. With exogenous fatty acids (FA) added to the perfusion solution, DCA pretreatment also resulted in improvements in post-VF LVDP and dp/dtmax, indicating that the presence of exogenous FA did not affect the beneficial actions of DCA. In conclusion, enhancement of PDH activation by DCA mitigates cardiac contractile dysfunction following ischemia-induced VF.
NEW & NOTEWORTHY
Despite successful resuscitation, cardiac contractile function following ischemia and ventricular fibrillation is markedly impaired, contributing to the high mortality rate after cardiac arrest. Our findings on the beneficial effects of enhancing pyruvate dehydrogenase activation shed light on the novel therapeutic strategy of modulating cardiac metabolism to mitigate this post-ventricular fibrillation contractile dysfunction.
ventricular fibrillation (VF) is the most common cause of sudden cardiac death (SCD) following myocardial infarction. SCD is an important health problem for patients with cardiovascular diseases. In coronary heart diseases, a significant proportion of total mortality is due to SCD (2, 46). Despite effective cardiopulmonary resuscitation attempts and successful resuscitation, the survival to discharge has been poor among these patients (2). Decreased cardiac contractile function following resuscitation from VF is the most common problem contributing to poor outcomes (12, 19).
In a healthy heart, most of the energy needed for cardiac contractile function is produced from the oxidation of fatty acid (FA) while the oxidation of glucose or lactate primarily provides the rest of the energy (24, 26, 32, 37, 39, 41). Decreased glucose oxidation, increased anaerobic glycolysis, and a dominant role of FA oxidation for cardiac energy production during and following myocardial ischemia are harmful and energetically expensive (23, 25, 44). Increased anaerobic glycolysis and decreased mitochondrial pyruvate oxidation are associated with increased lactate, increased proton production, and decreased ATP production, which is responsible for decreased cardiac contractile function (21, 22). While regional ischemia may lead to VF, VF itself causes a massive global ischemic insult to the heart and therefore would be expected to result in metabolic alterations detailed above and contractile dysfunction.
Mitochondrial modulators that promote glucose oxidation and inhibit FA oxidation have been shown to improve the recovery of cardiac mechanical function following ischemia (13, 21, 22, 27, 44). Dichloroacetate (DCA) is a mitochondrial metabolic modulator that enhances glucose oxidation and decreases FA oxidation (18, 29). Although metabolic modulators have been studied in ischemia-reperfusion, the impact of altering cardiac fuel utilization following massive global ischemia and recovery of cardiac contractile function following VF has not been studied in detail. The purpose of this study was to determine the effects of DCA on cardiac contractile function, redox state of the myocardial mitochondria, levels of metabolic intermediates in the ischemic heart following VF, and incidence of arrhythmias. We hypothesized that, in globally ischemic hearts, DCA pretreatment will mitigate cardiac contractile dysfunction following VF and reduce the incidence of cardiac arrhythmias.
MATERIALS AND METHODS
Materials.
DCA, palmitate, and glucose assay kits were bought from Sigma-Aldrich. Lactate assay kits were bought from BioVision. Phospho Detect anti-pyruvate dehydrogenase (PDH)-E1α (pSer232) rabbit antibody was purchased from Calbiochem, and PDH antibody was bought from Cell Signaling.
Rat heart Langendorff model.
The use of animals was approved by the Animal Care Committee of the University Health Network based on National Institutes of Health guidelines. Male Sprague-Dawley rats (450–550 g) were deeply anesthetized with 4–5% isoflurane in oxygen. The heart was rapidly excised through a midline thoracotomy and immersed in +4°C modified Tyrode's solution containing (in mM) 118 NaCl, 4.7 KCl, 1.25 CaCl2, 0.6 MgSO4, 1.2 KH2PO4, 25 NaHCO3, and 6 glucose, gassed with 95% O2 and 5% CO2 (28, 31). The aorta was cannulated, and the heart was mounted to a Langendorff setup. After equilibration, the perfused hearts were subjected to 15 min of no-flow global ischemia to simulate pathological conditions similar to cardiac arrest. Hearts were then reperfused with or without DCA (1 mM) in the perfusion solution for 5 min. The concentration of DCA was chosen based on a previous study showing stimulation of glucose oxidation by DCA at this dose (30). At the 6th min of reperfusion, VF was induced electrically and terminated after 1 min. Left ventricular pressure (LVP) and pseudo-ECG were recorded throughout the protocol using a CYQ pressure transducer and our custom-made data acquisition systems. To ensure that the observed reduced left ventricular developed pressure (LVDP) following ischemia-induced VF is at least in part due to VF and not just due to ischemia, we performed additional experiments where we compared the LVDP between ischemia alone and ischemia with a subsequent VF.
VF induction, resuscitation, and determination of arrhythmogenic threshold.
Langendorff-perfused rat hearts were rested on a plastic chair with four epicardial leads attached to the chair (11). Two electrodes were used for recording the pseudosurface ECG and two for electrical stimulation; electrodes were in direct contact with the lateral wall of the left ventricle (LV) as described previously (11). Short-duration burst pacing at a cycle length of 20 ms (50 pulses/s), a pulse width of 4 ms, and an output level of 12 V was applied up to five times to induce VF. An episode of induced VF with a duration of 60 s was considered to be sustained VF. This technique has been used in our laboratory for a variety of Langendorff-perfused heart models (11, 17). After 60 s, VF was terminated with a defibrillation shock (2 J) using a defibrillator pad custom-made for small animal hearts. The total number of burst-pacing attempts needed to induce sustained VF and the total number of defibrillation attempts needed to terminate VF were documented. The incidence of VF was expressed as a percentage of sustained VF episodes in each group. The arrhythmogenic threshold was expressed as the total number of burst-pacing attempts and defibrillation attempts needed for induction of sustained VF and successful defibrillation, respectively (5).
LVDP measurement.
LV contractile function was determined by measuring pressure with a balloon inserted in the LV through an incision in the left atrium. LV end systolic pressure (LVESP) and LV end diastolic pressure (LVEDP) were continuously recorded using a CYQ pressure transducer connected to the balloon and our custom-made data acquisition system. Data were analyzed using custom programming developed in MATLAB. LVDP was calculated as LVESP − LVEDP (4).
Myocardial glucose uptake measurement.
Samples of coronary effluent leaving the Langendorff-perfused heart were collected at the end of the experimental protocol in an Eppendorf tube and stored at −20°C for later analysis. Glucose concentration was measured using a glucose hexokinase assay kit. Myocardial glucose uptake was calculated using the formula: (inflow glucose concentration − outflow glucose concentration) × coronary flow rate (43).
Myocardial lactate production measurement.
Lactate concentration in the coronary effluent was measured using a lactate assay kit. Lactate production was calculated using the formula: (outflow lactate concentration − inflow lactate concentration) × coronary flow rate (45).
Myocardial PDH expression and activation assay.
At the end of the experimental protocol, LV tissue was excised and snap-frozen in liquid nitrogen. Protein extracts were prepared from frozen LV tissue specimens by homogenizing small tissue chunks in a lysis buffer containing 50 mM Tris, 500 mM NaCl, 10 mM MgCl2, 1% Triton X-100, 0.5% deoxycholic acid sodium salt, and 0.1% SDS in the presence of Roche protease and phosphatase inhibitors, using a mechanical tissue homogenizer. Protein concentrations were measured by the BCA method using the QuantiPro BCA Assay Kit (Sigma). Extracts containing 16 μg of proteins were loaded in a 4–15% gradient gel and resolved by SDS-PAGE. Separated proteins were transferred onto polyvinylidene difluoride membrane. After being blocked for nonspecific protein binding using skim milk, the membrane was incubated with primary antibodies against PDH, or phospho-PDH at Ser232, followed by the incubation with a specific secondary antibody conjugated with horseradish peroxidase, which allowed specific membrane-bound protein to be visualized on X-ray film using an enhanced chemiluminescence kit. Developed films were scanned and digitalized using a GS 800 Calibrated Densitometer (Bio-Rad), and the Quantity One software (Bio-Rad) was used to measure specific protein band density in the scanned images. Band density for specific proteins was normalized to that of GAPDH and total protein band in each sample, and the quantitative data of normalized amounts of the specific protein were expressed as a multiple over corresponding values in nontreated controls. For comparative purposes, the level of each protein of interest in nontreated controls was expressed as 1.00.
Malonyl-CoA, triglyceride, and acetyl-CoA determination.
Malonyl-CoA regulates FA oxidation by inhibiting carnitine palmitoyltransferase I (CPT-I), an enzyme required in the translocation of long-chain FA into the mitochondrial matrix, the subcellular site where FA oxidation takes place. Inhibition of CPT-I by malonyl-CoA decreases mitochondrial FA uptake and thus prevents FA from oxidation. To explore the effects of DCA on FA oxidation, we measured the LV tissue content of malonyl-CoA, triglyceride (TG), and acetyl-CoA. LV tissue samples frozen in liquid nitrogen were subjected to biochemical analysis to measure malonyl-CoA, TG, and acetyl-CoA contents as previously described (35). Briefly, malonyl-CoA was extracted from 40 mg of tissue using 6% perchloric acid, and the supernatant was subjected to high-performance liquid chromatographic (HPLC) analysis to determine malonyl-CoA levels. Acetyl-CoA was extracted from 20 mg of tissue using 6% perchloric acid, and the supernatant was subjected to HPLC analysis to determine acetyl-CoA levels. TG was measured in LV tissue that was homogenized in 2:1 chloroform-methanol, followed by centrifugation and collection of supernatant. The supernatant was applied to a silicic acid column, and TG was eluted in chloroform, dried at 60°C, and quantitated colorimetrically using enzymatic assay kits (Wako Pure Chemical Industries).
NADH fluorescence imaging.
NADH is an important coenzyme in mitochondrial respiration, redox state, and ATP synthesis. Oxidation of NADH drives the outward transport of protons and builds an electrochemical potential across the inner mitochondrial membrane, which then drives F1Fo-ATP synthase to produce ATP. Changes in NADH abundance, which can be quantified by measuring the fluorescence emitted by NADH under ultraviolet (UV) light, provide a window into the myocardial redox state before and after VF, allowing for the study of intervention. Epicardial NADH fluorescence was recorded from Langendorff-perfused rat hearts under UV illumination from a 365-nm light-emitting diode spotlight (PLS-0365-030-07-S; Mightex Systems, Toronto, Canada). Emitted fluorescence was band-pass filtered (460 ± 25 nm) and imaged using an EMCCD camera (AndoriXon 860BV; Andor PLC, Belfast, Ireland), as previously described (3, 14). Because NADH binding to complex I of the electron transport chain (ETC) results in amplification of the NADH signal, the captured fluorescence was assumed to originate from mitochondria (6, 7). To understand how DCA may alter redox status and metabolic activity, the effects of DCA on NADH fluorescence were also studied in both nonischemic hearts and in hearts subjected to the VF resuscitation protocol.
Effects of DCA on NADH fluorescence in nonischemic hearts.
For nonischemic studies, rat hearts were perfused as described above, and pseudo-ECG and LVP were continuously measured. After each heart was allowed to equilibrate for 15 min under normal sinus rhythm, epicardial NADH fluorescence was imaged for 20 min (2 frames/s). DCA was then added to the perfusate (final concentration at 1 mM), and epicardial NADH fluorescence was imaged for an additional 20 min. Changes in NADH fluorescence (ΔNADH) were computed as:
where meanpost-DCA is the average NADH fluorescence counts after DCA addition, and meanpre-DCA is the average NADH fluorescence counts before DCA was added. Average fluorescence was assessed for 1–5 min, based on signal stability and noise.
Effects of DCA on NADH fluorescence in hearts subjected to global no-flow ischemia.
In the VF resuscitation studies, Langendorff-perfused rat hearts were allowed to equilibrate for 15 min and were then subjected to 15 min of global no-flow ischemia, followed by reperfusion with or without DCA (1 mM) in the perfusion solution. Short-duration burst pacing (50 Hz, 12 V) was used to electrically induce VF at the 6th min of reperfusion. ECG, LVP, and epicardial NADH fluorescence were continuously monitored and recorded. Average values of NADH fluorescence were computed at periodic intervals throughout the experimental protocol. NADH fluorescence was normalized to the range of fluorescence from baseline preischemia to ischemia such that normalized NADH at a given time (t) was computed as
where meant is the average NADH fluorescence counts at a given time, and meanbaseline and meanischemia are the average NADH fluorescence counts during baseline preischemia and ischemia, respectively.
Measurement of cardiac hemodynamics in the presence of exogenous FA.
All experimentation described above was performed in the absence of exogenous FA. As our data demonstrated, in the absence of exogenous FA in the perfusion buffer, pretreatment of DCA is associated with improvements in cardiac hemodynamics. To explore the effects of DCA pretreatment on cardiac hemodynamics and incidence of VF in the presence of exogenous FA, we conducted a separate set of experiments using perfusion solution containing 1.2 mM of exogenous palmitate, following the same protocol as described in Rat heart Langendorff model. The fatty acid palmitate was dissolved in perfusion buffer bound with 3% BSA. High FA was chosen based on earlier studies showing high levels of FA in the blood following a myocardial infarction (1, 16, 33). In this set of experiments, only cardiac hemodynamics and incidences of VF were assessed.
Statistical analysis.
Values are presented as means ± SE. The incidences of VF were compared using the Fisher's exact test. Continuous variables (such as LVDP, dp/dtmax, and heart rate) were analyzed using analysis of variance where the variable of interest was treated as the outcome variable while treatment groups (control vs. treatment) and time were treated as response variables. If significant, a post hoc Bonferroni adjustment for multiple comparisons was performed. For comparing baseline and post-VF LVDP, HR, and dp/dt, paired t-test was performed. For other measurements with nonparametric parameters, the Mann-Whitney test was performed. A P value of <0.05 is considered as statistically significant. Data were analyzed using GraphPad Prism 5.
RESULTS
Effects of ischemia and VF on cardiac hemodynamics.
There were no significant differences in baseline body weight, heart rate (HR), LVDP, or LV dp/dtmax between the groups. Following ischemia and VF, there were significant reductions in LVDP from 96.99 ± 4.36 mmHg at baseline to 57.02 ± 5.83 mmHg (P < 0.001; Fig. 1A), dp/dtmax from 3,695 ± 247 mmHg/s at baseline to 1,936 ± 222 mmHg/s (P < 0.001; Fig. 1B), and HR from 271.40 ± 6.73 beats/min at baseline to 200 ± 14.31 beats/min (P < 0.001; Fig. 1C). To determine the relative contribution of VF on the observed reduction in LDVP, we performed additional experiments where we compared the LVDP following ischemia alone vs. LVDP following ischemia and a subsequent VF; we found that ischemia alone reduced LVDP by 15.26 ± 5.73%, whereas the induction of VF following ischemia resulted in a reduction of LVDP by 41.89 ± 5.42% (P = 0.02 compared with baseline LVDP; Fig. 1D).
Fig. 1.
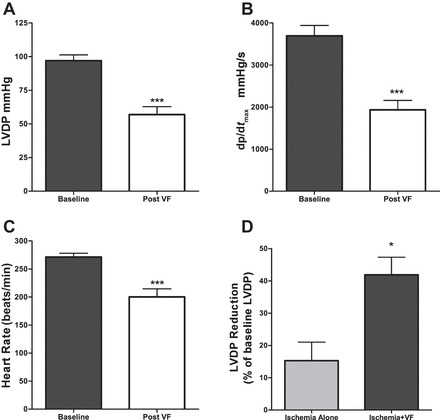
Post-ventricular fibrillation (VF) cardiac hemodynamics compared with baseline in nontreated controls. A: left ventricular developed pressure (LVDP) following 15 min ischemia and 1 min VF. B: left ventricle (LV) dp/dtmax following 15 min ischemia and 1 min VF. C: heart rate (HR) following 15 min ischemia and 1 min VF; n = 12 in each group. D: %reduction of LVDP compared with baseline following 15 min ischemia alone (n = 4) vs. following 15 min ischemia and a subsequent 1-min VF (n = 12). *P < 0.05 and ***P < 0.001.
Effects of DCA on cardiac contractile function following VF.
Pretreatment with DCA significantly improved post-VF LVDP compared with nontreated controls (P < 0.0001; Fig. 2A). LV dp/dtmax following VF was significantly improved in DCA-pretreated hearts compared with nontreated control hearts (P < 0.0001; Fig. 2B). Pretreatment with DCA had no effects on HR following VF (P = 0.8; Fig. 2C).
Fig. 2.
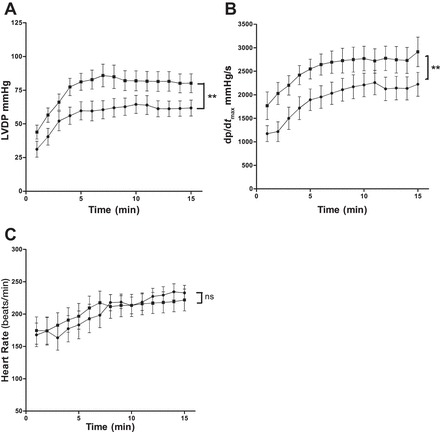
Effects of dichloroacetate (DCA) on cardiac contractile function following VF. LVDP (A), LV dp/dtmax (B), and HR (C) in nontreated controls and DCA-pretreated hearts; n = 12 in each group. **P < 0.01. ns, Nonsignificant compared with nontreated control. ●, Nontreated controls; ■, DCA pretreated.
Effects of DCA on glucose metabolism following VF.
Myocardial lactate production rate following VF was significantly reduced in DCA-pretreated hearts (0.97 ± 0.30 vs. 1.92 ± 0.25 μM·g−1·min−1 in nontreated controls, P = 0.04; Fig. 3A). Following VF, there was no significant difference in glucose uptake in the DCA-pretreated hearts (0.47 ± 0.12 vs. 0.28 ± 0.07 µg·mg−1·min−1 in controls, P = 0.21; Fig. 3B). These data suggest that DCA pretreatment decreases anaerobic glycolysis and enhances glucose oxidation. To determine the mechanism by which DCA increases glucose oxidation, we assessed the abundance and the phosphorylation state of PDH following VF in LV tissues. We found a significant reduction in PDH phosphorylation at Ser232 in DCA-pretreated hearts (0.40 ± 0.02 vs. 1.0 ± 0.14 from control hearts in LV tissue samples, P = 0.0023; Fig. 3, C and E), whereas there was no significant difference in total PDH protein expression between the nontreated controls and DCA-pretreated hearts (1 ± 0.04 vs. 0.90 ± 0.04, P = 0.1; Fig. 3, D and E). These findings suggest that DCA stimulates glucose oxidation by activating PDH following VF.
Fig. 3.
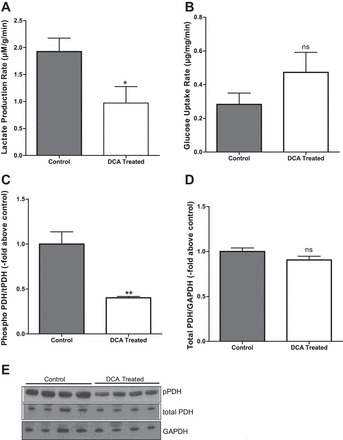
Effects of DCA on glucose metabolism following VF. A: myocardial lactate production rate. B: myocardial glucose uptake rate; n = 11 in each group. C: pyruvate dehydrogenase (PDH) phosphorylation. D: PDH expression. Phospho-PDH (pPDH) at Ser232 and total PDH were detected by Western blotting; n = 6 in each group. *P < 0.05 and **P < 0.01. E: representative Western blot.
Effects of DCA on cardiac mitochondrial redox state.
Administering DCA to a nonischemic heart that experienced no VF caused NADH fluorescence to increase 7.5 ± 0.9% (P = 0.002; Fig. 4, A and B). This increase was preceded by a short bout of premature ventricular contractions, which caused compensatory LVP spikes. LVP and HR returned to baseline levels within two to three beats while the increase in NADH fluorescence was maintained.
Fig. 4.
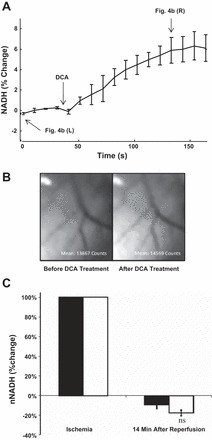
Effects of DCA on cardiac mitochondrial redox state. A: NADH fluorescence in normoxic hearts performed before and after DCA treatment; n = 4. P = 0.002 compared with before DCA treatment. B: representative NADH fluorescence images. Left, epicardial NADH fluorescence before DCA treatment, at time 0 in A. Right, epicardial NADH fluorescence after DCA addition, at the time indicated in A. C: %change in NADH fluorescence in the presence (n = 8) and absence (n = 5) of DCA. nNADH, normalized NADH. Open bars, Nontreated control; filled bars, DCA pretreated.
Global ischemia resulted in a large increase in epicardial NADH fluorescence by 83.2 ± 12.4% above preischemic levels (P < 0.01) (data not shown). Upon reperfusion, NADH fluorescence rapidly decreased to sub-baseline levels in both controls and DCA-pretreated groups (−14 ± 3 and −23 ± 13%, respectively, P > 0.05). Following VF resuscitation, NADH remained lower than baseline in both groups (Fig. 4C). NADH fluorescence did not significantly differ between DCA-pretreated and nontreated controls at any time point during reperfusion.
Effects of DCA on endogenous FA metabolism following VF.
No significant difference in LV malonyl-CoA content was observed between DCA-pretreated hearts and nontreated control hearts (0.85 ± 0.11 vs. 0.98 ± 0.09 nmol/g, respectively, P = 0.4; Fig. 5A). Myocardial TG is an important source of FA. We therefore measured TG content in the LV tissue samples, but we found no significant difference in myocardial TG content between the DCA-pretreated hearts and nontreated controls (1.40 ± 0.11 vs. 1.38 ± 0.10 μmol/g, respectively, P = 0.8; Fig. 5B), indicating that DCA has no effects on myocardial TG turnover. There was also no significant difference in acetyl-CoA content between the DCA-pretreated hearts and nontreated controls (19.43 ± 3.44 and 19.34 ± 1.86 nmol/g, respectively, P = 0.64; Fig. 5C). All of these data together suggested that DCA has no effects on myocardial endogenous FA oxidation following VF.
Fig. 5.
Effects of DCA on endogenous FA metabolism. Malonyl-CoA (A), triglyceride (B), and acetyl-CoA (C) content in LV tissue; n = 11 in each group.
Effects of DCA on the incidence of VF.
To explore the effects of DCA on the inducibility of VF, the incidence of VF was documented in isolated perfused hearts. We found no significant difference in incidence of sustained VF in DCA- pretreated hearts (66%) compared with nontreated controls (60%, P = 1.0; Fig. 6A).
Fig. 6.
Effects of DCA on VF inducibility and defibrillation. A: incidence of VF; n = 20 in each group. B: no. of burst pacing attempts needed to induce sustained VF in nontreated controls (n = 12) and the DCA-pretreated group (n = 13). C: total no. of defibrillation attempts needed to terminate VF in nontreated controls (n = 12) and the DCA-pretreated group (n = 13).
Effects of DCA on the arrhythmogenic threshold.
As a surrogate of arrhythmogenic threshold, we documented the total number of burst-pacing attempts required to induce sustained VF in the perfused hearts and found no difference in the number of burst-pacing attempts in DCA-pretreated hearts (2.53 ± 0.43 bursts) compared with nontreated controls (1.83 ± 0.36 bursts, P = 0.2; Fig. 6B). In terms of successful defibrillation, there was no significant difference in the total number of defibrillation attempts between the nontreated controls and DCA-pretreated hearts (1.08 ± 0.08 and 1.15 ± 0.10, respectively, P = 0.6; Fig. 6C).
Effects of DCA on cardiac hemodynamics in the presence of exogenous FA.
Thus far, our data demonstrated that DCA has no effects on myocardial endogenous FA oxidation, and earlier studies (1, 16, 33) suggest high levels of FA in the blood following a myocardial infarction. In a separate set of experiments, the effects of DCA pretreatment on post-VF cardiac hemodynamics were explored in hearts perfused with solution supplemented with 1.2 mM of sodium palmitate. In the presence of exogenous palmitate, post-VF LVDP was significantly improved in DCA-pretreated hearts compared with nontreated controls (P < 0.0001; Fig. 7A). LV dp/dtmax following VF was also significantly improved in DCA-pretreated hearts compared with nontreated controls (P < 0.0001; Fig. 7B). DCA pretreatment showed no effects on HR following VF (P = 0.4) in the presence of exogenous FA (Fig. 7C). In the presence of exogenous FA in the perfusion buffer, there was no significant difference in the incidence of VF (81.82 vs. 85.71%) in DCA-pretreated and nontreated hearts (Fig. 7D). These data suggested that presence of exogenous FA did not affect the beneficial actions of DCA.
Fig. 7.
Effects of DCA on cardiac hemodynamics in the presence of exogenous FA. LVDP (A), LV dp/dtmax (B), and HR (C) in nontreated controls and in DCA-pretreated hearts; n = 6 in each group. ●, Nontreated controls; ■, DCA-pretreated hearts. D: incidence of VF in the presence of 1.2 mM palmitate. **P < 0.01.
DISCUSSION
In this study, we demonstrated that DCA pretreatment improves cardiac contractile function following VF in ischemic hearts and is associated with a concomitant decrease in phosphorylation of PDH. This improvement in contractile function persists irrespective of the absence or presence of FA in our experimental preparation. NADH optical mapping suggests that the beneficial effects of DCA in the post-VF period are due to increased NADH production and increased efficiency of cardiac mitochondria in utilization of NADH.
DCA, cardiac fuel utilization, and contractility following VF.
Our data showed decreases in lactate production and in PDH phosphorylation without changes in glucose uptake in hearts pretreated with DCA. These results indicate decreased anaerobic glycolysis and increased glucose oxidation in DCA-pretreated hearts by enhancement of PDH activation. Our findings are consistent with previous studies that have demonstrated increased glucose uptake during reperfusion, although not necessarily an increase in pyruvate oxidation by the mitochondria. As a result, glucose undergoes anaerobic glycolysis, producing lactate and hydrogen ions, which lower ATP production and are responsible for reduced cardiac contractile function and increased risk for arrhythmias (21, 22, 39, 41). During ischemia and reperfusion, a large portion of produced ATP is used to clear intracellular lactate and hydrogen ions and to maintain ion channel function, while sacrificing the energy needed for cardiac contractile function (38). The enhancement of PDH activation by DCA treatment would diminish anaerobic glycolysis and hence diminish the production of lactate and hydrogen ions. The resultant amelioration in ATP turnover would then improve cardiac contractility following VF, as demonstrated in our experiments.
An earlier report showed that DCA also inhibits FA oxidation (18). We explored this inhibition as a potential mechanism of drug action in our experimental model, but we could not find any difference in malonyl-CoA content in myocardial tissue after DCA treatment, suggesting that DCA did not affect endogenous FA oxidation. The concentration of malonyl-CoA depends on its synthesis by acetyl-CoA carboxylase and degradation by malonyl-CoA decarboxylase. A previous study showed that DCA increased malonyl-CoA concentration in normal, perfused hearts in the working Langendorff heart model with no ischemia and VF (35). Our experimental protocol included 15 min of no-flow global ischemia, followed by 1 min of VF. It has been previously reported that ischemia results in a significant decrease in cardiac malonyl-CoA levels, due to a decrease in acetyl-CoA carboxylase activity with no changes in malonyl-CoA decarboxylase activity (15). This could explain why we did not observe a DCA-induced increase in malonyl-CoA levels in our experimental model. We were also unable to detect the changes of acetyl-CoA in our experimental model, and the supply of acetyl-CoA is an important regulator of malonyl-CoA production (35). The inconsistency of the effect of DCA on malonyl-CoA content could be due to differences in the experimental model. There were also no differences in endogenous left ventricular TG levels following DCA pretreatment. Thus, our data suggested that endogenous FA oxidation was not affected by DCA following VF. Although previous studies have reported that DCA stimulates glucose oxidation and inhibits FA oxidation (18, 29), we could only detect stimulated PDH activation in DCA-pretreated hearts following VF resuscitation, with no effects on endogenous FA oxidation. An earlier report suggests that myocardial TG turnover is inhibited during ischemia (10). Other studies also demonstrated that, depending on the presence and absence of exogenous FA, accelerated TG oxidation can be the major source of energy for the heart (9, 36). Because FA activation to long-chain acyl-CoA (a precursor of TG) requires two high-energy phosphates following the incorporation of the fatty acyl group into TG, the subsequent hydrolysis of TG results in the production of FAs, which need to be reesterified to long-chain acyl-CoA before further metabolism. Hence, an accelerated TG turnover is energetically expensive and inefficient in the ischemic heart. We could not detect increases in TG turnover in the absence of exogenous FA, and we therefore conclude that the observed increased cardiac contractile function following VF in DCA-pretreated hearts is due to the stimulation of PDH activation.
DCA and mitochondrial redox state following VF.
One of the intriguing and unique findings of this study is that the effect of DCA on mitochondrial NADH depended upon whether or not hearts experienced ischemia. This is highlighted by the data showing an increase in NADH with DCA in hearts that did not fibrillate. This increase was preceded by a short bout of premature ventricular contractions. At normal levels of oxygen and ATP, the addition of DCA resulted in increased mitochondrial NADH production without changes in LV pressure because baseline NADH and ATP levels are sufficient to support cardiac contractile function. An earlier study showed that higher NADH is correlated with increased production of reactive oxygen species (42). Specifically, DCA increased NADH in normal, nonischemic hearts but did not result in higher NADH fluorescence after global ischemia and VF in DCA-pretreated hearts. This could be explained by the ATP deficit caused by ischemia. In brief, NADH is produced during glycolysis and complete glucose oxidation, as well as during FA oxidation. NADH oxidation at the ETC and subsequent electron flow down the ETC to complex IV drives the outward transport of hydrogen ions and builds an electrochemical potential. The resultant gradient of hydrogen ions drives ATP synthesis. During ischemia, oxygen supply is limited; because O2 is the ultimate electron acceptor at complex IV, ETC flux slows down. This results in an accumulation of NADH, a depletion of ATP, and a decrease in cardiac contractile function. Hearts subjected to global ischemia in the present study displayed a large increase in NADH fluorescence and reduced pressures during ischemia. Upon reperfusion, both nontreated controls and DCA-pretreated hearts had lower NADH fluorescence compared with baseline. Upon restoration of flow, oxygen is again present, and mitochondrial respiration rapidly increases ATP production. This results in a rapid decrease in NADH, since NADH is rapidly oxidized to NAD+. Because DCA increases the abundance of the active form of PDH, which produces acetyl-CoA from pyruvate for entry into the tricarboxylic acid cycle, and PDH is an NADH-producing enzyme itself, we expected that DCA would increase NADH fluorescence under any conditions. However in our experiments NADH fluorescence did not differ between controls and DCA-pretreated hearts, we therefore conclude that either 1) DCA did not result in an increase in NADH production or 2) the increase in NADH production caused by DCA was immediately matched by an increase in the rate of its oxidation. Our investigation demonstrated decreased phosphorylation of PDH and decreased lactate accumulation, as well as higher LV pressures in the DCA-pretreated hearts following VF and increased NADH production in nonischemic heart. These data predict a greater NADH production, and an increase in ATP consumption for contractile function, suggesting the increase in NADH production was matched by an increase in NADH utilization in the DCA-pretreated hearts. This suggests the improved efficiency of the cardiac mitochondrial function in the DCA-pretreated hearts. Previous reports (8, 20, 34) showed that, during reperfusion, the mitochondrial ETC is damaged and activities are decreased, which also support our notion that DCA improves mitochondrial ETC efficiency following resuscitation from VF.
DCA and cardiac arrhythmia.
Our results showed that there was no difference in the incidence of VF and arrhythmogenic threshold between DCA-pretreated and nontreated controls. In our study, DCA may not have inhibited endogenous FA oxidation as expected. This may explain why we did not detect a DCA-dependent decrease in arrhythmias in our experimental model. Our data demonstrated that, in the absence of exogenous FA, pretreatment with DCA had no effect on endogenous FA oxidation following VF. However, previous reports demonstrated that enhancement of glucose oxidation by using DCA is associated with inhibition of FA oxidation (35, 40), and circulatory FA levels are elevated following myocardial infarction (1, 16, 33); therefore, we wanted to confirm that, with FA present in the perfusion solution, DCA would be an effective therapeutic strategy under such realistic pathophysiological conditions. In the presence of exogenous FA, improvement of cardiac contractile function following VF persisted in DCA-pretreated hearts; however, there were no differences in incidence to VF in the presence of exogenous FA between DCA-pretreated hearts and nontreated controls, demonstrating that DCA can exert its beneficial effects in the presence of exogenous FA following VF.
In conclusion, enhancement of pyruvate dehydrogenase activation by DCA mitigates cardiac contractile dysfunction following ischemia-induced VF. We have identified that enhancement of PDH activation is a potential new target for developing therapeutic strategies. To the best of our knowledge, this study is the first to propose a novel strategy of enhancing cardiac PDH activation for the management of cardiac contractile function following ischemia-induced VF.
GRANTS
M. A. Azam is a recipient of the Heart and Stroke Foundation of Canada Junior Personnel Award. This research work was supported by grants from the Heart and Stroke Foundation of Ontario (NA 7152), Canadian Institutes of Health Research (MOP 130501), and the National Heart, Lung, and Blood Institute (HL-095828).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: M.A.A., M.W.K., G.D.L., and K.N. conception and design of research; M.A.A., C.S.W., S.M., P.F.L., M.K., J.A., R.J., and S.K.-G. performed experiments; M.A.A., C.S.W., S.M., T.F., M.K., R.J., and S.K.-G. analyzed data; M.A.A., T.F., P.F.L., M.K., J.A., R.J., S.K.-G., M.W.K., G.D.L., and K.N. interpreted results of experiments; M.A.A., S.M., R.J., and S.K.-G. prepared figures; M.A.A. drafted manuscript; M.A.A., P.F.L., M.K., R.J., S.K.-G., M.W.K., G.D.L., and K.N. edited and revised manuscript; M.W.K., G.D.L., and K.N. approved final version of manuscript.
REFERENCES
- 1.Allison SP, Chamberlain MJ, Hinton P. Intravenous glucose tolerance, insulin, glucose, and free fatty acid levels after myocardial infarction. Br Med J 4: 776–778, 1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arking DE, Sotoodehnia N. The genetics of sudden cardiac death. Annu Rev Genomics Hum Genet 13: 223–239, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asfour H, Wengrowski AM, Jaimes R 3rd, Swift LM, Kay MW. Nadh fluorescence imaging of isolated biventricular working rabbit hearts. J Vis Exp 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ban K, Noyan-Ashraf MH, Hoefer J, Bolz SS, Drucker DJ, Husain M. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation 117: 2340–2350, 2008. [DOI] [PubMed] [Google Scholar]
- 5.Bauer A, Becker R, Dreyhaupt J, Voss F, Kraft P, Kelemen K, Senges-Becker JC, Katus HA, Schoels W. Role of katp channels in repetitive induction of ventricular fibrillation. Europace 9: 154–161, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Blinova K, Levine RL, Boja ES, Griffiths GL, Shi ZD, Ruddy B, Balaban RS. Mitochondrial nadh fluorescence is enhanced by complex i binding. Biochemistry 47: 9636–9645, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chapman JB. Fluorometric studies of oxidative metabolism in isolated papillary muscle of the rabbit. J Gen Physiol 59: 135–154, 1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol 294: C460–C466, 2008. [DOI] [PubMed] [Google Scholar]
- 9.Crass MF., 3rd Exogenous substrate effects on endogenous lipid metabolism in the working rat heart. Biochim Biophys Acta 280: 71–81, 1972. [DOI] [PubMed] [Google Scholar]
- 10.Crass MF, Pieper GM 3rd. Lipid and glycogen metabolism in the hypoxic heart: Effects of epinephrine. Am J Physiol 229: 885–889, 1975. [DOI] [PubMed] [Google Scholar]
- 11.Farid TA, Nair K, Masse S, Azam MA, Maguy A, Lai PF, Umapathy K, Dorian P, Chauhan V, Varro A, Al-Hesayen A, Waxman M, Nattel S, Nanthakumar K. Role of katp channels in the maintenance of ventricular fibrillation in cardiomyopathic human hearts. Circ Res 109: 1309–1318, 2011. [DOI] [PubMed] [Google Scholar]
- 12.Gazmuri RJ, Weil MH, Bisera J, Tang W, Fukui M, McKee D. Myocardial dysfunction after successful resuscitation from cardiac arrest. Crit Care Med 24: 992–1000, 1996. [DOI] [PubMed] [Google Scholar]
- 13.Griffin JL, White LT, Lewandowski ED. Substrate-dependent proton load and recovery of stunned hearts during pyruvate dehydrogenase stimulation. Am J Physiol Heart Circ Physiol 279: H361–H367, 2000. [DOI] [PubMed] [Google Scholar]
- 14.Kay M, Swift L, Martell B, Arutunyan A, Sarvazyan N. Locations of ectopic beats coincide with spatial gradients of nadh in a regional model of low-flow reperfusion. Am J Physiol Heart Circ Physiol 294: H2400–H2405, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kudo N, Barr AJ, Barr RL, Desai S, Lopaschuk GD. High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-coa levels due to an increase in 5′-amp-activated protein kinase inhibition of acetyl-coa carboxylase. J Biol Chem 270: 17513–17520, 1995. [DOI] [PubMed] [Google Scholar]
- 16.Kurien VA, Oliver MF. Serum-free-fatty-acids after acute myocardial infarction and cerebral vascular occlusion. Lancet 2: 122–127, 1966. [DOI] [PubMed] [Google Scholar]
- 17.Lai PF, Panama BK, Masse S, Li G, Zhang Y, Kusha M, Farid TA, Asta J, Backx PH, Yau TM, Nanthakumar K. Mesenchymal stem cell transplantation mitigates electrophysiological remodeling in a rat model of myocardial infarction. J Cardiovasc Electrophysiol 24: 813–821, 2013. [DOI] [PubMed] [Google Scholar]
- 18.Latipaa PM, Hiltunen JK, Peuhkurinen KJ, Hassinen IE. Regulation of fatty acid oxidation in heart muscle. Effects of pyruvate and dichloroacetate. Biochim Biophys Acta 752: 162–171, 1983. [DOI] [PubMed] [Google Scholar]
- 19.Laurent I, Monchi M, Chiche JD, Joly LM, Spaulding C, Bourgeois B, Cariou A, Rozenberg A, Carli P, Weber S, Dhainaut JF. Reversible myocardial dysfunction in survivors of out-of-hospital cardiac arrest. J Am Coll Cardiol 40: 2110–2116, 2002. [DOI] [PubMed] [Google Scholar]
- 20.Lee HL, Chen CL, Yeh ST, Zweier JL, Chen YR. Biphasic modulation of the mitochondrial electron transport chain in myocardial ischemia and reperfusion. Am J Physiol Heart Circ Physiol 302: H1410–H1422, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu B, Clanachan AS, Schulz R, Lopaschuk GD. Cardiac efficiency is improved after ischemia by altering both the source and fate of protons. Circ Res 79: 940–948, 1996. [DOI] [PubMed] [Google Scholar]
- 22.Liu B, el Alaoui-Talibi Z, Clanachan AS, Schulz R, Lopaschuk GD. Uncoupling of contractile function from mitochondrial tca cycle activity and mvo2 during reperfusion of ischemic hearts. Am J Physiol Heart Circ Physiol 270: H72–H80, 1996. [DOI] [PubMed] [Google Scholar]
- 23.Liu Q, Docherty JC, Rendell JC, Clanachan AS, Lopaschuk GD. High levels of fatty acids delay the recovery of intracellular ph and cardiac efficiency in post-ischemic hearts by inhibiting glucose oxidation. J Am Coll Cardiol 39: 718–725, 2002. [DOI] [PubMed] [Google Scholar]
- 24.Lloyd S, Brocks C, Chatham JC. Differential modulation of glucose, lactate, and pyruvate oxidation by insulin and dichloroacetate in the rat heart. Am J Physiol Heart Circ Physiol 285: H163–H172, 2003. [DOI] [PubMed] [Google Scholar]
- 25.Lopaschuk GD, Spafford MA, Davies NJ, Wall SR. Glucose and palmitate oxidation in isolated working rat hearts reperfused after a period of transient global ischemia. Circ Res 66: 546–553, 1990. [DOI] [PubMed] [Google Scholar]
- 26.Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev 90: 207–258, 2010. [DOI] [PubMed] [Google Scholar]
- 27.Lopaschuk GD, Wambolt RB, Barr RL. An imbalance between glycolysis and glucose oxidation is a possible explanation for the detrimental effects of high levels of fatty acids during aerobic reperfusion of ischemic hearts. J Pharmacol Exp Ther 264: 135–144, 1993. [PubMed] [Google Scholar]
- 28.Masse S, Farid T, Dorian P, Umapathy K, Nair K, Asta J, Ross H, Rao V, Sevaptsidis E, Nanthakumar K. Effect of global ischemia and reperfusion during ventricular fibrillation in myopathic human hearts. Am J Physiol Heart Circ Physiol 297: H1984–H1991, 2009. [DOI] [PubMed] [Google Scholar]
- 29.McAllister A, Allison SP, Randle PJ. Effects of dichloroacetate on the metabolism of glucose, pyruvate, acetate, 3-hydroxybutyrate and palmitate in rat diaphragm and heart muscle in vitro and on extraction of glucose, lactate, pyruvate and free fatty acids by dog heart in vivo. Biochem J 134: 1067–1081, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McVeigh JJ, Lopaschuk GD. Dichloroacetate stimulation of glucose oxidation improves recovery of ischemic rat hearts. Am J Physiol Heart Circ Physiol 259: H1079–H1085, 1990. [DOI] [PubMed] [Google Scholar]
- 31.Nanthakumar K, Jalife J, Masse S, Downar E, Pop M, Asta J, Ross H, Rao V, Mironov S, Sevaptsidis E, Rogers J, Wright G, Dhopeshwarkar R. Optical mapping of langendorff-perfused human hearts: Establishing a model for the study of ventricular fibrillation in humans. Am J Physiol Heart Circ Physiol 293: H875–H880, 2007. [DOI] [PubMed] [Google Scholar]
- 32.Neely JR, Morgan HE. Relationship between carbohydrate and lipid metabolism and the energy balance of heart muscle. Annu Rev Physiol 36: 413–459, 1974. [DOI] [PubMed] [Google Scholar]
- 33.Opie LH. Metabolism of free fatty acids, glucose and catecholamines in acute myocardial infarction. Relation to myocardial ischemia and infarct size. Am J Cardiol 36: 938–953, 1975. [DOI] [PubMed] [Google Scholar]
- 34.Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Federici A, Ruggiero FM. Decrease in mitochondrial complex i activity in ischemic/reperfused rat heart: Involvement of reactive oxygen species and cardiolipin. Circ Res 94: 53–59, 2004. [DOI] [PubMed] [Google Scholar]
- 35.Saddik M, Gamble J, Witters LA, Lopaschuk GD. Acetyl-coa carboxylase regulation of fatty acid oxidation in the heart. J Biol Chem 268: 25836–25845, 1993. [PubMed] [Google Scholar]
- 36.Saddik M, Lopaschuk GD. Myocardial triglyceride turnover and contribution to energy substrate utilization in isolated working rat hearts. J Biol Chem 266: 8162–8170, 1991. [PubMed] [Google Scholar]
- 37.Shipp JC, Opie LH, Challeoner D. Fatty acid and glucose metabolism in the perfused heart. Nature 189: 1018–1019, 1961. [Google Scholar]
- 38.Skierczynska A, Beresewicz A. Demand-induced ischemia in volume expanded isolated rat heart the effect of dichloroacetate and trimetazidine. J Physiol Pharmacol 61: 153–162, 2010. [PubMed] [Google Scholar]
- 39.Stanley WC. Cardiac energetics during ischaemia and the rationale for metabolic interventions. Coron Artery Dis 12, Suppl 1: S3–S7, 2001. [PubMed] [Google Scholar]
- 40.Stanley WC, Hernandez LA, Spires D, Bringas J, Wallace S, McCormack JG. Pyruvate dehydrogenase activity and malonyl coa levels in normal and ischemic swine myocardium: effects of dichloroacetate. J Mol Cell Cardiol 28: 905–914, 1996. [DOI] [PubMed] [Google Scholar]
- 41.Stanley WC, Lopaschuk GD, Hall JL, McCormack JG. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc Res 33: 243–257, 1997. [DOI] [PubMed] [Google Scholar]
- 42.Starkov AA, Fiskum G. Regulation of brain mitochondrial h2o2 production by membrane potential and nad(p)h redox state. J Neurochem 86: 1101–1107, 2003. [DOI] [PubMed] [Google Scholar]
- 43.Tada H, Thompson CI, Recchia FA, Loke KE, Ochoa M, Smith CJ, Shesely EG, Kaley G, Hintze TH. Myocardial glucose uptake is regulated by nitric oxide via endothelial nitric oxide synthase in langendorff mouse heart. Circ Res 86: 270–274, 2000. [DOI] [PubMed] [Google Scholar]
- 44.Ussher JR, Wang W, Gandhi M, Keung W, Samokhvalov V, Oka T, Wagg CS, Jaswal JS, Harris RA, Clanachan AS, Dyck JR, Lopaschuk GD. Stimulation of glucose oxidation protects against acute myocardial infarction and reperfusion injury. Cardiovasc Res 94: 359–369, 2012. [DOI] [PubMed] [Google Scholar]
- 45.Zhao T, Parikh P, Bhashyam S, Bolukoglu H, Poornima I, Shen YT, Shannon RP. Direct effects of glucagon-like peptide-1 on myocardial contractility and glucose uptake in normal and postischemic isolated rat hearts. J Pharmacol Exp Ther 317: 1106–1113, 2006. [DOI] [PubMed] [Google Scholar]
- 46.Zheng ZJ, Croft JB, Giles WH, Mensah GA. Sudden cardiac death in the united states, 1989 to 1998. Circulation 104: 2158–2163, 2001. [DOI] [PubMed] [Google Scholar]