

There is increasing interest in developmental programming of hypertension, yet little understanding of developmental misprogramming of smaller arteries where vascular resistance is determined. We show that behavioral stress accelerates postnatal programming of vasoconstrictor function suppressed by the α-adrenergic blocker terazosin, suggesting a mechanism and therapy for developmental programming of hypertension.
Keywords: smooth muscle, vasoconstriction, development, sympathetic, α-adrenergic
Abstract
We examined the effect of stress in the first 2 wk of life induced by brief periods of daily maternal separation on developmental programming of rat small resistance mesenteric arteries (MAs). In MAs of littermate controls, mRNAs encoding mediators of vasoconstriction, including the α1a-adrenergic receptor, smooth muscle myosin heavy chain, and CPI-17, the inhibitory subunit of myosin phosphatase, increased from after birth through sexual [postnatal day (PND) 35] and full maturity, up to ∼80-fold, as measured by quantitative PCR. This was commensurate with two- to fivefold increases in maximum force production to KCl depolarization, calcium, and the α-adrenergic agonist phenylephrine, and increasing systolic blood pressure. Rats exposed to maternal separation stress as neonates had markedly accelerated trajectories of maturation of arterial contractile gene expression and function measured at PND14 or PND21 (weaning), 1 wk after the end of the stress protocol. This was suppressed by the α-adrenergic receptor blocker terazosin (0.5 mg·kg ip−1·day−1), indicating dependence on stress activation of sympathetic signaling. Due to the continued maturation of MAs in control rats, by sexual maturity (PND35) and into adulthood, no differences were observed in arterial function or response to a second stressor in rats stressed as neonates. Thus early life stress misprograms resistance artery smooth muscle, increasing vasoconstrictor function and blood pressure. This effect wanes in later stages, suggesting plasticity during arterial maturation. Further studies are indicated to determine whether stress in different periods of arterial maturation may cause misprogramming persisting through maturity and the potential salutary effect of α-adrenergic blockade in suppression of this response.
NEW & NOTEWORTHY
There is increasing interest in developmental programming of hypertension, yet little understanding of developmental misprogramming of smaller arteries where vascular resistance is determined. We show that behavioral stress accelerates postnatal programming of vasoconstrictor function suppressed by the α-adrenergic blocker terazosin, suggesting a mechanism and therapy for developmental programming of hypertension.
observation of a strong geographical relationship between mortality from ischemic heart disease in the 1970s and infant mortality rates one-half a century earlier (5) formed the basis for the hypothesis that cardiovascular mortality in the adult is developmentally programmed (4). This relationship was confirmed in subsequent studies and extended to a variety of developmental stressors. Children raised in low socioeconomic status families under harsh and stressful conditions predicted increasing blood pressure (BP) over time and increased vascular reactivity to stress [CARDIA study (9, 34), reviewed in Ref. 56]. In the Bogalusa Heart (3) and other studies, those with the greatest trajectory of BP increase during childhood and adolescence had several-fold higher prevalence of hypertension (r ∼0.4, see Ref. 10 for meta-analysis) and increased cardiovascular mortality as adults (42). A natural human experiment was that of Finnish children voluntarily separated from their parents during World War II. As nonobese adults, children separated at ages 4–7 yr had systolic BP ∼9 mmHg higher than nonseparated controls (CON), with the effect stronger in men than women (2). Those separated at younger or older ages had less or no difference. Early intervention is thought to be important to prevent the long-term sequelae of elevated BP manifest in childhood, yet there is no consensus on the definition of childhood hypertension, its causes, when to treat, or with what medicines (26).
BP is largely determined and controlled by the contraction of resistance artery smooth muscle (RASM) determining systemic vascular resistance (SVR) to blood flow. Hypertension in adult humans is largely due to increased SVR (20). While there is now considerable interest in the role of early life stress (ELS) in programming of hypertension, little is known about the normal developmental programming or stress-induced misprogramming of RASM. In a previous study (53), we correlated the molecular and functional maturation of rat mesenteric RASM and demonstrated that it initiated after birth and was not complete until sexual maturity [postnatal day (PND) 35]. Expression of smooth muscle actin and myosin contractile proteins and maximal force increased concordantly. Additionally, there were specific changes in the expression of the myosin phosphatase (MP) subunits, key determinants of responses to vasoconstrictor and vasodilator signals that determine RASM tone (reviewed in Refs. 12, 13). CPI-17, the inhibitory subunit of MP, increased concordant with increased constrictor responses to the α-adrenergic agonist phenylephrine (PE) in intact and permeabilized/calcium-clamped arteries, reflecting increased inhibition of MP. The MP targeting subunit 1 (Mypt1) (regulatory) subunit switched from skipping of alternative exon 24 (E24), coding for a COOH-terminal leucine zipper (LZ) motif, to inclusion of E24 (LZ−), with reduced relaxant response to nitric oxide (NO)/cGMP in intact and permeabilized/calcium-clamped arteries, reflecting reduced activation of MP. We also provided evidence that RASM maturation was dependent on sympathetic innervation. These observations led to the hypothesis that stress-induced activation of sympathetic neural signaling at critical stages of development may cause misprogramming of RASM, increasing susceptibility to hypertension and cardiovascular mortality in adulthood.
A well-established model of ELS is that of maternal separation (MS) (35, 50) in which neonatal rats are separated from their mothers for several hours each day from PND2-14. Recent studies by Pollock and coworkers showed that rats exposed to this ELS had normal resting BP as adults, but exaggerated acute pressor responses to norepinephrine (37) and hypertensive responses to chronic angiotensin II infusion (39, 40). Their studies implicated changes in arterial and kidney function in the relationship between ELS and adult hypertension (38). Specific mechanisms for the presumed developmental misprogramming caused by ELS were not identified in this or other studies (reviewed in Ref. 27). Here we show that exposure of rat pups to MS stress accelerates the developmental programming and functional maturation of mesenteric resistance arteries. This developmental misprogramming was suppressed by coadministration of the α1-adrenergic receptor blocker terazosin, indicating dependency on stress-dependent sympathetic activation and suggesting a potential therapeutic strategy in this scenario.
MATERIALS AND METHODS
Animals.
All animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee at the University of Maryland and adhere to National Institutes of Health guidelines. Sprague-Dawley rats were purchased from Charles River Laboratories (Wilmington, MA) and maintained as a breeding colony at the University of Maryland-Baltimore. Offspring were weaned at PND21 and studied from as early as PND1 to 5–7 mo of age (n = 120 for investigation). All animals were maintained on a 12:12-hr light-dark cycle and were provided food and water ad libitum.
MS stress and drug treatments.
Neonatal rats were subject to the MS stress protocol, as previously described (35, 40), with litter sizes culled to 10 pups/litter. Neonates were separated from their mothers and placed in a heated cage (30°C) for 3 h/day from PND2-14. After the 3-h period, the neonates were returned to the mother's cage. Nonhandled littermates served as the control. MS rat pups were marked by tail snip and chemical cautery via silver nitrate. One MS pup and one littermate CON were used from each of five to six separate litters for each experiment to avoid litter and nested bias. MS and littermate CON were terminated at PND14, PND21, PND35, and as adults (5–7 mo). A separate set of MS and littermate CON pups (n = 5–6) received daily IP injections of the α-adrenergic receptor blocking drug terazosin (0.5 mg/kg in sterile saline, as described in Ref. 43) or vehicle (0.87% sterile saline) from PND2-14 and were terminated at PND21. A separate set of experiments was performed to test the effect of pharmacological blockade of the three different receptor families of sympathetic nervous signaling. CON rat pups received a daily intraperitoneal (IP) injection from PND1-35 of either: 1) terazosin (0.5 mg/kg; α-adrenergic antagonist); 2) BIBP3226 (100 μg/kg; Npy1r antagonist) (28); 3) NF449 [10 mg/kg; purinergic (P2X) receptor antagonist] (21); 4) all three drugs combined (denoted triple cocktail); or 5) vehicle (0.87% sterile saline). Rats were terminated after the last injection at PND35.
Adult restraint stress.
Adult male rats that had undergone MS stress as neonates and their littermate CON were subjected to 1-h restraint stress (RS) in a flat-bottom clear plastic restrainer (Kent Scientific) in their home cage, as previously described (57). After the restraint period, the rats were released in their home cage and terminated 3 h (n = 3–4) or 24 h (n = 3–4) later.
RNA analysis.
Mesenteric, femoral, and renal main and interlobar arteries were dissected and stored in RNALater for purification of total RNA. To isolate mesenteric arteries (MAs) from the small pups, the entire arterial arcade containing first- to fourth-order branches was stripped and harvested. Renal interlobar arteries were dissected by first bisecting the kidney along the longitudinal axis and removing the renal medulla. The small resistance interlobar arteries were exposed and carefully dissected distal to the arcuate artery. Total RNA was purified from tissue homogenates via column purification (Ambion), as per manufacturers' recommendations, and reverse transcribed using random hexamers and Superscript III enzyme (1,000 units, Invitrogen). Conventional PCR was used to quantify the ratios of Mypt1 alternative E24 splice variants using infrared-labeled oligonucleotide primers flanking the alternative exon, as previously described (53). PCR products were gel separated, and E24+ and E24− species quantified using a LiCor Odyssey imager and Image Studio 3.0 software, and reported as %Mypt1 E24−. mRNA levels were measured by quantitative real-time PCR (StepOnePlus) using Taqman-based assay probes and a Fast Advanced Taqman Master Mix (Applied Biosystems, Foster City, CA). Transcript abundance was calculated via the 2-ddCt method, normalized to cyclophilin A expression, which was invariant, and reported as fold-change. Due to the small size of these developing microvessels, assays of corresponding protein isoforms and abundance were not performed. In our previous study (53), we showed a good correlation in the expression of mRNAs and proteins that are the focus of this study.
Vascular function.
Rats of age PND21, PND35, and adult were used for these studies of arterial function; study of the microarteries of rats younger than PND21 was technically challenging. A section of the first-order MA (MA1) ∼2 mm in length was dissected free of connective tissue and placed in a physiological saline solution containing the following concentrations (mM): 112 NaCl, 25.7 NaHCO3, 4.9 KCl, 2.0 CaCl2, 1.2 MgSO4, 1.3 KH2PO4, 11.5 glucose, 10.0 HEPES, at pH 7.4, at 37°C. Arteries were mounted on a wire myograph (model 610M, Danish Myo Technology, Aarhus, Denmark) for measurements of force under isometric conditions. Starting tension was applied by stretching the vessel to IC90, as previously described (47). Arteries were primed with two doses of 10 μM PE. Force was continuously recorded over time in response to cumulative doses of PE (1 nM to 100 μM) or maximal depolarization with 100 mM KCl. Reductions in force to cumulative doses of the NO donor diethylamine (DEA)/NO (1 nM to 100 μM) were determined in arteries preconstricted with a submaximal concentration of PE. A separate set of MA1s was permeabilized with α-toxin (1,000 U/ml) or β-escin (400 μM) and fully relaxed in a high relaxing solution (pCa 9) (in mM): 60 potassium methane sulfonate, 5 EGTA, 0.02 CaCl2, 9.26 MgCl2, 5.2 Na2ATP, 25 creatine phosphate, and 25 N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid with intracellular pH 7.1 by 1 N KOH, as previously described (53). Force was continuously recorded over time in response to cumulative doses of calcium (pCa 9-4) and to the nonhydrolyzable analog of cGMP, 8-bromo-cGMP (8-Br-cGMP) (cumulative concentrations of 1 nM to 100 μM) at a submaximal concentration of calcium (pCa 6; 1 μM). Data are presented as force in milli-Newtons (mN) or percentage of maximum force. EC50 was calculated for dose-response curves using standard curve analysis. All reagents were purchased from Sigma.
BP analysis.
BP and heart rate were analyzed using a Millar MPVS-400 catheter, and data were collected using Chart software. Rats were lightly anesthetized with 2% isoflurane after which the catheter was placed into the left carotid artery for BP recordings. Systolic BP was recorded continuously for 30 min and presented as an average for the recording period.
Statistical analysis.
Sigma Plot software was used for data and graphical analysis with data presented as means ± SE (SYSTAT, Chicago, IL). mRNA expression data were analyzed with one-way ANOVA or Student's t-test, where applicable. Vascular function assays were analyzed with one-way ANOVA and a post hoc Bonferroni test. BP data were analyzed using one-way ANOVA. A Kruskal-Wallis ANOVA on ranks was used where applicable. Statistical significance was accepted at P < 0.05.
RESULTS
Postnatal maturation of arterial smooth muscle in regional circulations.
We first examined the expression of constituents of the smooth muscle contractile apparatus in the postnatal period, including 1) smooth muscle myosin heavy chain (smMHC), a major determinant of smooth muscle force production and marker specific for smooth muscle differentiation (45); 2) myosin light chain kinase (MLCK), the determinant of calcium activation of force (16); 3) MP regulatory (Mypt1) and inhibitory (CPI-17) subunits; MP mediates deactivation of force, while the expression of E24 splice variants of Mypt1 and the level of expression of CPI-17 determine sensitivity to NO/cGMP-mediated relaxation and α-adrenergic-mediated constriction, respectively (12, 13); and 4) the α1a-adrenergic receptor, a mediator of sympathetic vasoconstriction. We compared their expression in the mesenteric and renal circulations, major determinants of SVR and BP, and the muscular femoral artery as an additional reference vessel.
smMHC mRNA was detected in all of the arteries at PND1 and markedly induced in the MAs between PND1 and PND35 (Fig. 1A; P < 0.05; one-way ANOVA), consistent with the accretion of differentiated smooth muscle in the arteries through sexual maturity. The timing of the induction of smMHC mRNA varied in the other arteries examined: in the main renal artery it was complete by PND21 (weaning) (Fig. 1C); in the renal interlobar (Fig. 1D) and femoral arteries (Fig. 1B) it lagged and was complete at PND35 (sexual maturity); and in the MAs it continued through to adulthood (Fig. 1A). During this period of postnatal maturation, there were modest but significant increases in Mypt1 and MLCK mRNAs [P < 0.05; PND1 vs. PND35 (all vessels)], while CPI-17 mRNA was highly induced in all of the arteries and reached maximal levels by PND35 [P < 0.05; PND1 vs. PND35 (all vessels)]. The greatest induction of CPI-17 mRNA occurred in the MAs with much of the increase occurring from weaning (PND21) to sexual maturity (PND35), paralleling the increase in smMHC. The mRNA coding for the α1a-adrenergic receptor was also markedly induced in the arteries [P < 0.05; PND1 vs. PND35 (all vessels)] and followed a developmental time course similar to that of CPI-17 mRNA in all but the renal artery, in which it peaked at PND21 and markedly declined thereafter. With regards to the E24 splice variant isoforms of the MP regulatory subunit Mypt1, there was a near complete switch from skipping to inclusion of E24 in all of the arteries from PND1-35 [P < 0.05; PND1 vs. PND35 (all vessels)] (Fig. 2).
Fig. 1.
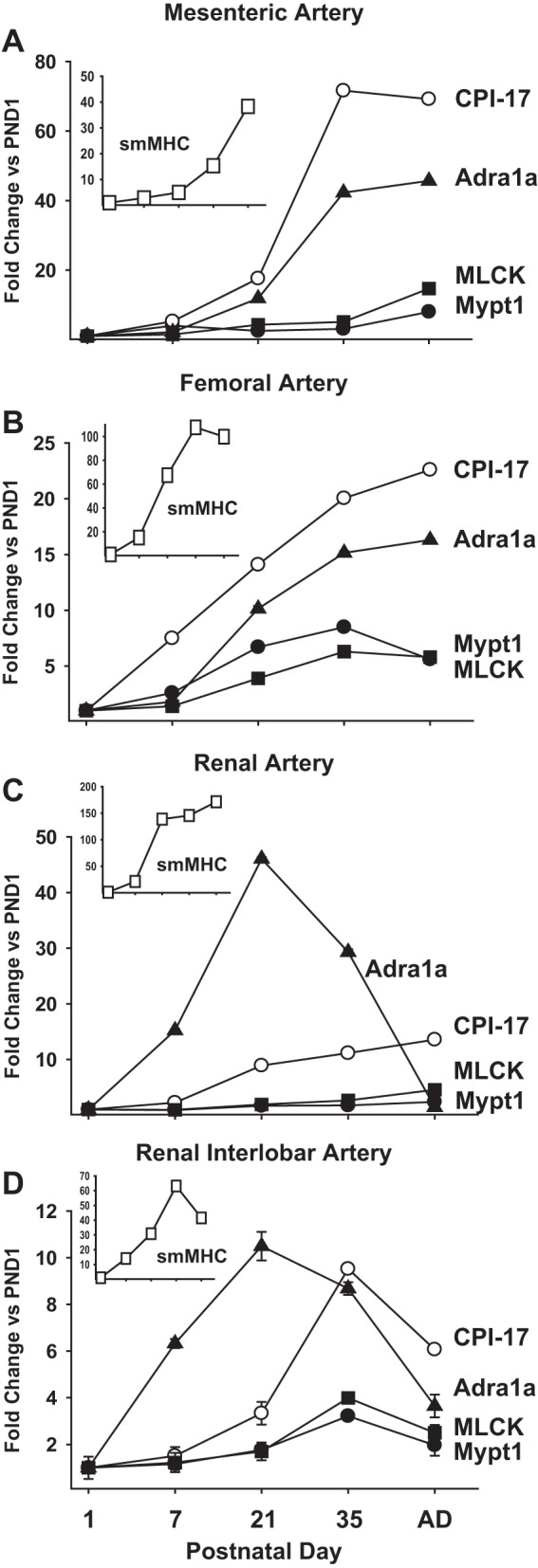
Postnatal maturation of rat arterial smooth muscle gene expression in regional circulations. RNA was isolated from the entire mesenteric arcade (A), femoral artery (B), main renal artery (C), and renal interlobar arteries (D) at the indicated postnatal day (PND). mRNAs were measured by quantitative PCR using Taqman probes, normalized to cyclophilin A, and reported as fold-change vs. PND1. Insets: smooth muscle myosin heavy chain (smMHC) mRNAs at different PNDs. Values are means ± SE with n = 5/PND. CPI-17, PKC-potentiated inhibitory protein of protein phosphatase-1; Mypt1, myosin phosphatase targeting subunit-1; MLCK, myosin light chain kinase; Adra1a, α1a-adrenergic receptor.
Fig. 2.
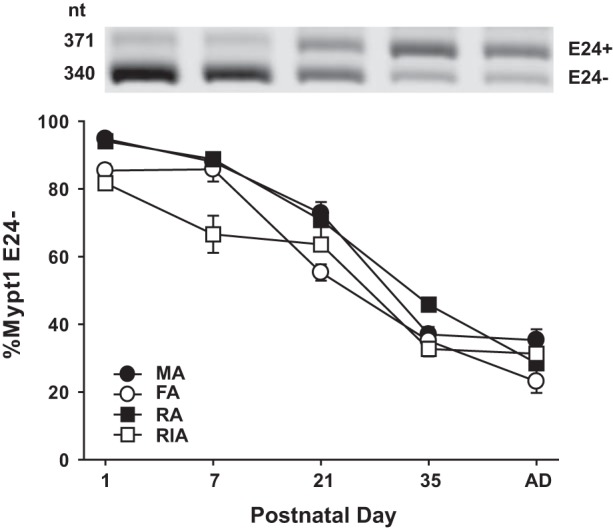
Postnatal switching of Mypt1 exon 24 (E24) splice variants in regional circulations. RNA was isolated from arteries as described in Fig. 1. Mypt1 E24 splice variants were amplified by conventional PCR with labeled primers that flank the alternative exon and PCR products separated by gel electrophoresis. %Mypt1 E24− was calculated as the signal from the E24− band divided by the combined signals of the E24+ and E24− bands. All arteries showed a similar postnatal maturational switch toward the Mypt1 E24+ splice variant, coding for the leucine zipper negative isoform. Thus only a single representative gel from the femoral artery (FA) is shown. Values are means ± SE, with n = 5/PND. MA, mesenteric artery; RA, renal artery; RIA, renal interlobar artery.
Behavioral stress accelerates arterial maturation.
We next examined how ELS in a critical developmental window affects arterial maturation. Neonatal rat pups were subject to MS stress for 3 h/day from PND2-14. MS caused a stress response in the MA, as indicated by 2.7-fold induction of heat shock protein 70 (HSP-70) mRNA (2.7 ± 0.4-fold; P < 0.05, n = 5) measured at the end of the stress protocol (PND14) and maintained at PND21 (2.9 ± 0.5-fold; P < 0.05, n = 5). MS stress accelerated the developmental maturation of the MA smooth muscle (Fig. 3, A–E). smMHC mRNA was approximately two- to threefold greater in MAs of MS stress rats measured at PND14 and PND21 compared with nonstressed littermate CON. In absolute terms, the smMHC mRNA was ∼5- to 10-fold greater in MS vs. CON MAs at PND14 and PND21. The other mRNAs measured followed a similar pattern. CPI-17 also showed a 2- to 3-fold relative induction by MS stress at PND14 and PND21, while the absolute increase of ∼20-fold at PND21 was the largest of any of the mRNAs measured. Mypt1, MLCK, and α1a-adrenergic receptor mRNAs followed similar trends in relative induction by MS stress at PND14 and PND21, while the absolute changes were much smaller, reflecting the lesser magnitude of their normal developmental induction. Similarly, the normal developmental switch of Mypt1 to the E24+ splice variant was accelerated by MS stress, as measured at PND14 and PND21 (Fig. 3F).
Fig. 3.

Maternal separation (MS) stress augments postnatal maturation of rat MA smooth muscle. Rats were subject to 3 h/day of MS stress on PND2-14. RNA was isolated from MAs of MS rats and their littermate controls (CON) at the indicated PND. A–E: mRNAs were measured by quantitative PCR with Taqman probes, normalized to cyclophilin A, and reported as fold-change vs. PND1 (A: smMHC; B: CPI-17; C: MLCK; D: Adra1a; E: Mypt1). F: Mypt1 E24 splice variants were measured as in Fig. 2 and plotted as %Mypt1 E24− at the indicated PND. Values are means ± SE with n = 5/PND. *P < 0.05 vs. CON.
The differences in mRNAs in the MAs of rats subject to MS stress had abated or significantly narrowed by 3 wk after the end of the MS stress protocol (PND35, sexual maturity) (Fig. 3). CPI-17 and α1a-adrenergic receptor mRNAs peaked in the MS stress MAs at PND21, while in the paired littermate CON they continued their normal developmental increase from PND21-35, such that there was no difference (CPI-17) or a much more modest difference (α1a) between the groups at PND35. smMHC and MLCK mRNAs continued to increase in MS stress group from PND21-35, but, due to the normal developmental increase in the paired littermate CON, the fold-difference between groups was diminished.
Programming and stress-induced misprogramming of MAs is α1-adrenergic receptor dependent.
To test the role of stress activation of sympathetic signaling in misprogramming of MAs, rat pups were treated with the α1-adrenergic receptor blocker terazosin (0.5 mg·kg IP−1·day−1) or vehicle during the stress protocol (PND2-14) and studied at PND21. In these experiments, the neonates were assigned to one of three groups: 1) CON pups injected with vehicle; 2) MS stress pups injected with vehicle; 3) MS stress pups injected with terazosin. There was no difference in mRNA levels between uninjected and vehicle-injected CON (data not shown). Rat pups subject to MS stress and injected with vehicle had induction of mRNAs at PND21 that was nearly identical to that of rats that had been subject to MS stress without IP injections (compare Fig. 4A and Fig. 3). Terazosin treatment during MS stress completely suppressed the stress-mediated induction of the contractile mRNAs and the switch to the Mypt1 E24+ splice variant in the MAs (Fig. 4). Terazosin treatment also suppressed stress induction of HSP-70 mRNA in the MAs (MS + vehicle: 2.9 ± 0.5; MS + terazosin: 1.5 ± 0.2; fold-change vs. CON; P = 0.12). Terazosin not only suppressed the stress-mediated induction of these mRNAs, but reduced their expression to levels less than that of the CON MAs (Fig. 4).
Fig. 4.

The α1-adrenergic receptor blocker terazosin suppresses MS stress augmentation of MA maturation. Neonatal rats were subject to MS stress from PND2-14, as described above, and at the same time treated with terazosin [T; 0.5 mg·kg intraperitoneal (IP)−1·day−1] or vehicle (VEH; 0.87% saline). Littermate CON were injected with VEH. A: mRNAs were measured by quantitative PCR, as described above, and reported as fold-change vs. PND1. B: Mypt1 E24 splice variants were measured as described above and plotted as %Mypt1 E24−. Values are means ± SE with n = 5/group. *P < 0.05 vs. PND1. †P < 0.05 vs. PND21 CON+VEH. ‡P < 0.05 vs. PND21 MS+VEH.
To test if α1-blockade suppresses the normal developmental maturation of MAs, an additional set of experiments was performed in which unstressed pups were treated with terazosin (0.5 mg/kg IP) or vehicle (0.87% sterile saline) daily from PND1-35 spanning postnatal maturation to sexual maturity. Terazosin markedly suppressed the normal developmental induction of smMHC, CPI-17, MLCK, Mypt1, and α1-adrenergic receptor mRNAs and the switch to Mypt1 E24 inclusion (Fig. 5). As the sympathetic nervous system may also provide trophic signals through activation of purinergic and neuropeptide Y (NPY) receptors (31), neonatal rats were treated in the same protocol with inhibitors of all three pathways (denoted triple cocktail). Addition of the purinergic and NPY receptors blockers NF449 and BIBP3226, respectively, to terazosin, had no additional effect on mRNA levels (Fig. 5). The NPY receptor blocker alone had a modest effect (data not shown). This indicates that α1-adrenergic receptor signaling is the main component of sympathetic developmental programming of MAs.
Fig. 5.

The α1-adrenergic receptor blocker terazosin suppresses normal developmental maturation of MA smooth muscle, and there is no additive effect of additional sympathetic receptor blockade. Neonatal rats were treated with terazosin (0.5 mg·kg IP−1·day−1), triple cocktail [0.5 mg/kg terazosin; 100 μg/kg BIBP3226 (neuropeptide Y receptor blocker); 10 mg/kg NF449 (purinergic receptor blocker; as a single IP injection/day], or VEH (0.87% saline) from PND1-35. mRNAs from MAs were assayed at PND35. A–E: mRNAs were measured by quantitiative PCR and normalized to cyclophilin A (A: smMHC; B: CPI-17; C: MLCK; D: Adra1a; E: Mypt1). Values are fold-change vs. PND1. F: Mypt1 E24 splice variants were measured by conventional PCR and plotted as %Mypt1 E24−. Values are means ± SE with n = 5/group. *P < 0.05 vs. PND1. †P < 0.05 vs. PND35 VEH. ‡P < 0.05 vs. PND35 VEH.
MS stress induces arterial hypercontractility.
To determine the functional significance of stress-induced developmental misprogramming of MAs, force, calcium sensitivity of force, and response to signaling pathways were measured in PND21 MA1 ex vivo. This was selected as the first time point for study, as maximal effects of stress on gene expression were evident, and measurement of contractile function of these small arteries was feasible. In these experiments, three groups were compared: 1) rat pups subject to MS stress from PND2-14; 2) littermate CON; and 3) rat pups subject to MS stress and treated with terazosin (0.5 mg·kg IP−1·day−1) from PND2-14. MA1s from rats subject to MS stress developed ∼50% more force to KCl depolarization (Fig. 6A) and ∼100% more force to calcium in permeabilized preparations (Fig. 6B), consistent with the stress induction of contractile mRNAs. Maximal force to the α1-adrenergic agonist PE was similarly increased by ∼100% (Fig. 6C), while the sensitivity was unchanged (EC50: CON: 1.2 ± 0.5 μM; MS: 1.7 ± 0.8 μM; MS + terazosin: 1.4 ± 0.5 μM). Relaxation to the NO donor DEA/NO (Fig. 6D), and its second messenger 8-Br-cGMP under calcium clamp (Fig. 6E), were reduced by MS stress, consistent with the accelerated switch to the E24+ splice variant of Mypt1 coding for the LZ− isoform. There were trends toward decreased sensitivity of the MAs from MS stress rats to DEA/NO (EC50: CON: 3.3 ± 1.4 μM; MS: 46 ± 24 μM; MS + terazosin: 6.3 ± 5.9 μM; P = 0.09) and 8-Br-cGMP (EC50: CON: 22 ± 2.4 nM; MS: 28 ± 3.0 nM; MS + terazosin: 29 ± 7 nM; P = 0.16) that did not reach statistical significance. The effects of MS stress on MA contractility were largely suppressed by coadministration of terazosin during the stress period (Fig. 6). Resting systolic BP measured in the PND21 rats under anesthesia were not different (CON: 77 ± 8 mmHg; MS: 79 ± 5 mmHg; MS + terazosin: 77 ± 10 mmHg, n = 5/group).
Fig. 6.

MS stress accelerates postnatal maturation of rat MA contractile function, which is suppressed by coadministration of terazosin. Neonatal rats were subject to MS stress from PND2-14 and treated with terazosin (T; 0.5 mg·kg IP−1·day−1), as described above. First-order MAs (MA1) were isolated from littermate CON, MS, and MS+T rats at PND21. Force generation was measured in intact or α-toxin permeabilized MA1s under isometric conditions in a wire myograph, as described in materials and methods. Data are plotted as developed force in milli-Newtons (mN) (A–C), or as percentage of force remaining in response to cumulative doses of vasodilator in arteries preconstricted at submaximal concentrations of phenylephrine (PE; 10 μM) (D–E). A: force generated to maximal KCl-induced depolarization (100 mM). B: dose response to calcium in α-toxin permeabilized MA1s. C: dose response to the α-adrenergic agonist PE. D: dose response to nitric oxide (NO) donor diethylamine (DEA)/NO in intact arteries. E: dose response to 8-bromo-cGMP (8-Br-cGMP) in α-toxin permeabilized MA1s activated at submaximal calcium (pCa6). Values are means ± SE with n = 5–6/group. *P < 0.05 vs. CON. †P < 0.05 vs. MS.
MS stress and arterial contractility at PND35.
Arterial function was again examined at PND35 (sexual maturity) in MA1s from MS stress rats and littermate CON. The enhanced contractility that was present in the MS stress MA1s at PND21 had dissipated by PND35. At PND35, MAs from rats subject to MS stress and CON developed identical force to KCl depolarization (Fig. 7A), to calcium in permeabilized preparations (Fig. 7B), and to PE (Fig. 7C). The dissipation of the differences in contractile function between groups by PND35 was due to a greater increment from PND21 to PND35 in force-generating capacity in the CON compared with rats that had been subject to MS stress. Thus, in the CON, force to KCl, calcium, and PE increased 2.5-, 1.9-, and 2.4-fold from PND21 to PND35, respectively (compare Figs. 6 and 7), while in the MS stress group these values were 1.5-, 0.9-, and 1.2-fold, respectively. Similarly, the response to DEA/NO and its second messenger 8-Br-cGMP decreased over this 2-wk time period (compare Figs. 6 and 7).
Fig. 7.

MS stress does not alter contractile function of MAs at PND35. Rats were stressed and MA1s studied as described above. Data are plotted as developed force in mN (A–C), or as percentage of force remaining in response to cumulative doses of vasodilator in arteries preconstricted at submaximal concentrations of PE (10 μM) (D–E). A: force generated to maximal KCl-induced depolarization (100 mM). B: dose response to calcium after α-toxin permeabilization. C: dose response to the α-adrenergic agonist PE. D: dose response to NO donor DEA/NO in intact arteries. E: dose response to 8-Br-cGMP in α-toxin permeabilized MA1s activated at submaximal calcium (pCa6). Values are means ± SE with n = 5–6/group.
The resting systolic BP measured in the PND35 rats under anesthesia had increased in both groups compared with PND21, but with a greater trajectory in the rats that had been subject to MS stress as neonates (CON: 91 ± 4 mmHg; MS: 110 ± 5 mmHg; P < 0.05; n = 5).
Effect of developmental stress on arterial stress responses of maturity.
We next examined if the ELS affected the arterial function of the mature rat. We had already observed that, in rats subject to MS stress, basal arterial function had normalized by PND35. We thus examined how MS stress may affect the adult arterial response to a second stressor, 1 h of restraint stress (RS), an established model of moderate stress (8) that induces a transcriptional response in the vascular smooth muscle (57). Consistent with the prior study, 1 h of RS induced HSP-70 mRNA in MAs measured at 3 h after RS (2.9 ± 0.3-fold; P < 0.05; n = 3), which returned to baseline by 24 h. Similarly, CPI-17 and smMHC mRNAs were induced at 3 h after RS (4.6 ± 0.1-fold, 4.2 ± 0.1-fold, respectively, P < 0.05, n = 3) and returned to baseline by 24 h.
We next examined if the ELS affected the arterial function of the adult rat MAs. Arterial function was studied in three groups of male rats: 1) CON not subject to any life stressors; 2) rats that had been subject to MS stress as neonates and 1 h RS as adults; and 3) littermate CON subject to 1 h RS as adults. MA1s studied at 3 h after RS developed similar force to KCl depolarization (Fig. 8A), to calcium in β-escin permeabilized preparations (Fig. 8B), and to PE (Fig. 8C), and showed similar relaxation response to the NO donor DEA/NO (Fig. 8D). Thus MS stress did not affect the contractile performance of the MA1s of the mature male rat under basal conditions, nor after a second heterotypic stress. Of note, the maximum force produced by the MAs to KCl depolarization and PE again increased from PND35 to full maturity, consistent with the gene expression data and indicating the continued functional maturation of these arteries.
Fig. 8.

MS stress does not affect MA function of adult rats subject to restraint stress (RS). MA1s were isolated from adult male rats that were either nonstressed CON, MS rats subjected to 1-h RS (MS+RS), and littermate CON subjected to 1-h RS (CON+RS). MA1s were harvested 3 h post-RS, and force was measured under isometric conditions in a wire myograph. Data are plotted as developed force in mN (A–C), or as percentage of force remaining in response to cumulative doses of vasodilator in arteries preconstricted at submaximal concentrations of PE (10 μM) (D). A: force generation to maximal depolarization with KCl (100 mM). B: dose response to calcium in β-escin permeabilized MA1s. Dose response to PE (C) and DEA/NO in intact arteries (D) is shown. Values are means ± SE with n = 3/group.
DISCUSSION
BP is largely determined by the state of contraction of the smooth muscle in the smaller arteries determining vascular resistance to blood flow. Vascular resistance and BP increase throughout postnatal maturation, along with dynamic changes in the regulation of blood flow in the regional circulations (7, 36, 48). It has been suggested, based mostly on epidemiological studies, that stressors in critical periods of developmental maturation may permanently reprogram arterial function and BP (reviewed in Ref. 27), yet there is surprisingly little understanding of normal or pathological developmental programming of the small arteries. In the present study we defined the molecular and functional maturation of the small arteries of several regional circulations extending from the postnatal period to maturity. Moderate behavioral stress applied during the early phase of postnatal arterial maturation caused molecular misprogramming, with the functional correlate of mesenteric arterial hypercontractility. This was suppressed by the α-adrenergic receptor blocker terazosin. However, this accelerated early trajectory of arterial maturation was not maintained throughout maturation such that, by sexual maturity (PND35) and into adulthood, there were no significant differences in arterial function. This suggests a substantial amount of plasticity in the vascular response to early life stressors, the long-term effect of which will likely depend on the timing, intensity, and chronicity (see Refs. 19, 23, 44).
Normal maturation.
Critical to the study of pathological developmental misprogramming of vascular function is an understanding of its normal developmental programming. In humans (10) and other mammals (61), there is a steady increase in SVR and BP from after birth to maturity. Most of this increase in rodents occurs from 3 wk (weaning) to 10 wk (maturity) (61). The basis for this functional maturation of the vascular system is not understood. Some of the increased vascular resistance is due to increasing overall size, as vascular resistance is proportional to vessel length, and adjustments of SVR and BP partially normalize developmental changes. However, there are also age-dependent functional changes in systemic regional circulations for which mechanisms have not been defined. In the present study, we demonstrate maturational changes in expression of contractile mRNAs correlated with predicted changes in arterial function in several regional circulations. The changes in MAs are consistent with our prior study (53), while changes in renal and femoral arteries indicate a more universal phenomenon of postnatal programming of the maturation of arteries in regional circulations. The timing of the changes in gene expression in the different circulations is variable. In our prior study, we provided evidence that maturation of MAs is under the influence of innervation, and whether this is also true of the other circulations requires further study.
In the study of the MAs, the increase in smMHC and maximal force throughout postnatal arterial maturation is indicative of accretion of differentiated smooth muscle and consistent with a histological study showing that, at birth, MA consists of a single layer of partially differentiated smooth muscle cells adjacent to the endothelium (59). In the present study, there were also specific changes in the regulatory enzymes that couple vasoactive signals to force development, with increased force to PE and reduced sensitivity to NO/cGMP-mediated relaxation concordant with changes in receptors and MP regulatory subunits mediating these responses. PE-induced maximal force was increased, but there was no change in efficacy, suggesting no change in signal transduction mechanisms.
These studies used wire myography to measure smooth muscle force production under isometric conditions as a direct indicator of arterial muscle function. It is possible that somewhat different results on sensitivity to agonists or antagonists would be obtained under isobaric conditions or in vivo. In this study, we selected isometric conditions so as to use force as the most direct indicator of muscle function, and the increasing force generated through maturation correlates well with increasing SVR and BP over the same period. The measured force was not normalized to wall thickness, and a significant portion of the increase in force is likely due to increase in muscle filaments (wall thickness). In summary, substantial changes in force production and expression of myosin and its regulatory enzymes occur throughout postnatal arterial maturation, concordant with increasing vascular resistance and BP.
Stress-dependent misprogramming.
The careful determination of the timing of molecular and functional mesenteric arterial maturation provided a basis for determining its susceptibility to behavioral stress in specific developmental windows. We tested the effect of separating the pups from their mothers for several hours each day, starting shortly after birth, targeting the initial period of MA maturation. This model of MS causes a moderate level of stress in the pups, as determined by measurement of serum corticosterone and catecholamines as indicators of activation of the hypothalamic-pituitary and sympathetic axes, respectively (37, 50). In the present study, we did not measure these parameters, but instead measured induction of HSP-70 in the target tissue as an indicator of a localized stress response. We demonstrated that MS augmented the normal developmental induction of smooth muscle contractile mRNAs and function, and this effect persisted for 1 wk after the end of MS, indicative of a persisting misprogramming effect. This was blocked by coadministration of the α-adrenergic blocker terazosin, indicating that the effect was mediated by signaling from the sympathetic nervous system. Terazosin is a hydrophilic α-blocker that does not cross the blood-brain barrier, suggesting that its suppression of MS misprogramming is due to blockade of α-adrenergic signaling at the arterial smooth muscle. These experiments cannot exclude the possibility that suppression by α-adrenergic blockade is secondary to an effect elsewhere in the animal. However, in vitro experiments have shown that α1-adrenergic signaling can directly program smooth muscle differentiation through autocrine transforming growth factor-β signaling (11), while in other cell types it couples to second messengers and transcriptional pathways, including calcium (nuclear factor of activated T cells), activator protein-1, cAMP (cAMP response element binding protein), and nuclear factor-κB (17, 46, 60). In the present study, α-adrenergic blockade suppressed the normal developmental maturation of MA smooth muscle and stress-induced augmentation of this process. This suggests that sympathetic signaling is required for both developmental programming and stress misprogramming of the arteries. Which transcriptional pathways mediate these programming effects requires further study. In contrast to the large blood vessels, there has been little study of the transcriptional or other controls of the gene program in the smaller arteries, while significant differences in the large vs. small artery gene programs are appreciated (51) (reviewed in Ref. 14). PND2-14 is a period when rat small MAs become innervated (22, 41), and further studies of this critical developmental period are indicated.
The effect of MS stress was maintained at 1 wk, but resolved by 3 wk after the end of the stress protocol. The studies by Pollock and coworkers showed that rats exposed to this ELS had normal resting BP as adults but exaggerated acute pressor (vasoconstrictor) responses to norepinephrine (37) and augmented hypertensive responses to chronic angiotensin II infusion (39, 40). Their studies implicated changes in arterial and kidney function in the relationship between ELS and adult hypertension (38). In the present study, we observed that PND21 MA1s from rats exposed to MS stress at PND2-14 had increased force to KCl depolarization, to PE, and to calcium in permeabilized preparations, all consistent with the acceleration of the functional maturation of these arteries. Similarly, there were reduced vasorelaxant responses to the NO donor DEA/NO and to its second messenger 8-Br-cGMP under calcium clamp in permeabilized preparations. The latter is the classic assay for activation of MP in situ (32), and the reduced response to cGMP in this assay is consistent with the shift in the Mypt1 E24/LZ isoforms required for cGMP-dependent protein kinase-1α activation of MP (25, 30, 52, 53), reviewed in Ref. 12. The magnitude of the change in vasorelaxation response to the NO donor DEA/NO was greater than that of cGMP under calcium clamp. This could reflect a combined effect on calcium cycling and myofilament calcium sensitivity, or even cGMP-independent signaling by NO, which in toto determine the response to NO. Further studies are required to investigate changes in constrictor and dilator signaling pathways that may occur during normal development and stress-induced misprogramming of these arteries.
In the present study, there was no effect of MS stress on arterial function at PND35 or adulthood, nor was there a conditioning effect of exposure to MS stress on response of male rats to RS as adults. Stress induction of HSP and contractile mRNAs, CPI-17 and smMHC, were maintained in adulthood, but not altered by the MS stress. It is possible that other indexes of arterial function or stress responses may be altered, and further study is required.
Variables that may determine the net effect of maturational stressors on adult vascular function include the timing (2, 23, 44), intensity, and genetic susceptibility (24). Regarding the timing, this stress was applied to the rats in the early postnatal period. As vascular maturation continues through maturity, it is possible that stressors applied throughout maturation or targeted to the postweaning juvenile to adolescent periods may have more lasting effects on vascular function, as has been observed in effects on behavior (23). PND15-25 were identified as the critical window of vulnerability to auditory stress-induced suppression of mouse cerebral microvascular angiogenesis, persistent to adulthood (58). Whether there is a similar window of vulnerability for developmental misprogramming of arterial function that will persist through adulthood requires further study. A final consideration is the extent to which genetic predisposition may influence the response to maturational stress. Hypertension in mature humans is contributed by genetic and environmental factors, but genetic variants that may underlie differing trajectories of BP increase during childhood and adolescence have not been identified (24). In rodent models, the genetically encoded spontaneously hypertensive rat shows increased sympathetic neural activity early in maturation, preceding the increase in BP (54). The development of hypertension in spontaneously hypertensive rats was suppressed by α1-adrenergic receptor blockade with terazosin, specifically from PND1-21 (43), or by sympathectomy with guanethidine plus antiserum to nerve growth factor in the first 4 wk after birth (33), but not by α1-blockade from 4.5 to 12 wk of age (29). This suggests that sympathetic nervous system hyperactivity in a critical phase of maturation may contribute to the development of hypertension in a genetically predisposed model.
Limitations of the present study.
A major limitation of the present study is that behavioral stress was applied in a narrow window of postnatal maturation, while very recent studies of humans suggest that multiple and recurrent stressors throughout one's lifetime are required to confer increased risk of hypertension and cardiovascular mortality (18, 55). In a related matter, studies are needed of children's small arteries to determine whether the same developmental programming and stress misprogramming occur as defined here in the rat. This study focused on the effects of stress on mesenteric arterial programming and function. The splanchnic circulation receives ∼25% of resting blood flow and thus makes a substantial contribution to resting SVR. Recent studies have suggested that increased sympathetic vasoconstrictor tone specifically in the splanchnic circulation may play a key role in hypertension (15, 49). However, this remains unsettled, and thus it will be important for future studies to determine whether behavioral stress causes developmental misprogramming in other circulations. Finally, in these studies of small arteries in small developing rodents, we collected the entire MA arcade (MA1-4) to have sufficient sample to measure mRNAs and used MA1 to measure force. Studies of other circulations have suggested heterogeneity in sympathetic vasoconstrictor control throughout the arterial arcade (1). We have not observed differences in gene expression between MA1 and the entire mesenteric arterial arcade, but further studies will be required to define potential variation in stress responses throughout the arcade. Similarly, MA1 was selected as a representative “resistance artery” for functional studies. This is a generalized term and consistent with the textbook definition of arteries less than or equal to ∼0.5-mm diameter, as well as classic studies indicating that a substantial fall in pressure occurs across the rat MA1 (6). However, it is possible that differing functional perturbations will occur, dependent on the arterial branch, requiring further study limited by this method of wire myography to arteries of ∼100 μm diameter.
Conclusion.
MS stress accelerated the trajectory of rat mesenteric arterial maturation during the first few weeks of life. This manifests as increased expression of smooth muscle contractile mRNAs and force production to depolarization and α-adrenergic stimulation. This stress-induced developmental misprogramming was suppressed by pharmacological α-adrenergic blockade. Rodents are born in a very immature state, and, while not specifically defined, this preweaning period of vascular maturation likely corresponds to the third trimester of in utero human development. In the rodents, arterial maturation continues from after birth through maturity, and, while an analogous arterial maturation has not been defined in humans, increasing BP and SVR similarly occur in both species through maturity. Humans raised under stressful conditions are more likely to have hypertension and vascular dysfunction as adults (56). While there is increasing interest in determining the basis for the presumed developmental programming of hypertension, there remains limited mechanistic understanding of this phenomenon. The present study in the rat model provides a strong rationale for defining the period of arterial functional maturation in humans and the effect of stress on misprogramming and its pharmacological suppression in critical developmental windows.
GRANTS
The authors acknowledge the funding for this investigation supported by National Institutes of Health Grants R01-HL-066171, T32-HL-072751 and T32-AR-007592.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.J.R. and S.A.F. conception and design of research; J.J.R. performed experiments; J.J.R. and S.A.F. analyzed data; J.J.R. and S.A.F. interpreted results of experiments; J.J.R. prepared figures; J.J.R. and S.A.F. drafted manuscript; J.J.R. and S.A.F. edited and revised manuscript; J.J.R. and S.A.F. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors are thankful for the technical expertise of Alex Lloyd and Shivani Kapoor.
REFERENCES
- 1.Al-Khazraji BK, Saleem A, Goldman D, Jackson DN. From one generation to the next: a comprehensive account of sympathetic receptor control in branching arteriolar trees. J Physiol 593: 3093–3108, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alastalo H, Raikkonen K, Pesonen AK, Osmond C, Barker DJP, Heinonen K, Kajantie E, Eriksson JG. Early life stress and blood pressure levels in late adulthood. J Hum Hypertens 27: 90–94, 2013. [DOI] [PubMed] [Google Scholar]
- 3.Bao W, Threefoot SA, Srinivasan SR, Berenson GS. Essential hypertension predicted by tracking of elevated blood pressure from childhood to adulthood: the Bogalusa Heart Study. Am J Hypertens 8: 657–665, 1995. [DOI] [PubMed] [Google Scholar]
- 4.Barker DJ, Bagby SP, Hanson MA. Mechanisms of disease: in utero programming in the pathogenesis of hypertension. Nat Clin Pract Nephrol 2: 700–707, 2006. [DOI] [PubMed] [Google Scholar]
- 5.Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 1: 1077–1081, 1986. [DOI] [PubMed] [Google Scholar]
- 6.Benoit JN, Granger DN. Intestinal microvascular adaptation to chronic portal hypertension in the rat. Gastroenterology 94: 471–476, 1988. [DOI] [PubMed] [Google Scholar]
- 7.Buckley NM. Maturation of circulatory system in three mammalian models of human development. Comp Biochem Physiol A Comp Physiol 83: 1–7, 1986. [DOI] [PubMed] [Google Scholar]
- 8.Buynitsky T, Mostofsky DI. Restraint stress in biobehavioral research: recent developments. Neurosci Biobehav Rev 33: 1089–1098, 2009. [DOI] [PubMed] [Google Scholar]
- 9.Carroll JE, Gruenewald TL, Taylor SE, Janicki-Deverts D, Matthews KA, Seeman TE. Childhood abuse, parental warmth, and adult multisystem biological risk in the Coronary Artery Risk Development in Young Adults study. Proc Natl Acad Sci U S A 110: 17149–17153, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen X, Wang Y. Tracking of blood pressure from childhood to adulthood: a systematic review and meta-regression analysis. Circulation 117: 3171–3180, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Damon DH. Sympathetic innervation promotes vascular smooth muscle differentiation. Am J Physiol Heart Circ Physiol 288: H2785–H2791, 2005. [DOI] [PubMed] [Google Scholar]
- 12.Dippold RP, Fisher SA. Myosin phosphatase isoforms as determinants of smooth muscle contractile function and calcium sensitivity of force production. Microcirculation 21: 239–248, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eto M. Regulation of cellular protein phosphatase-1 (PP1) by phosphorylation of the CPI-17 family, C-kinase-activated PP1 inhibitors. J Biol Chem 284: 35273–35277, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fisher SA. Vascular smooth muscle phenotypic diversity and function. Physiol Genomics 42A: 169–187, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foss JD, Fink GD, Osborn JW. Reversal of genetic salt-sensitive hypertension by targeted sympathetic ablation. Hypertension 61: 806–811, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao N, Huang J, He W, Zhu M, Kamm KE, Stull JT. Signaling through myosin light chain kinase in smooth muscles. J Biol Chem 288: 7596–7605, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonzalez Bosc LV, Wilkerson MK, Bradley KN, Eckman DM, Hill-Eubanks DC, Nelson MT. Intraluminal pressure is a stimulus for NFATc3 nuclear accumulation: role of calcium, endothelium-derived nitric oxide, and cGMP-dependent protein kinase. J Biol Chem 279: 10702–10709, 2004. [DOI] [PubMed] [Google Scholar]
- 18.Halonen JI, Stenholm S, Pentti J, Kawachi I, Subramanian SV, Kivimäki M, Vahtera J. Childhood psychosocial adversity and adult neighborhood disadvantage as predictors of cardiovascular disease: a cohort study. Circulation 132: 371–379, 2015. [DOI] [PubMed] [Google Scholar]
- 19.Hanson MA, Gluckman PD. Early developmental conditioning of later health and disease: physiology or pathophysiology? Physiol Rev 94: 1027–1076, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hart EC, Joyner MJ, Wallin BG, Charkoudian N. Sex, ageing and resting blood pressure: gaining insights from the integrated balance of neural and haemodynamic factors. J Physiol 590: 2069–2079, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hechler B, Magnenat S, Zighetti ML, Kassack MU, Ullmann H, Cazenave JP, Evans R, Cattaneo M, Gachet C. Inhibition of platelet functions and thrombosis through selective or nonselective inhibition of the platelet P2 receptors with increasing doses of NF449 [4,4′,4″,4″′-(carbonylbis(imino-5,1,3-benzenetriylbis-(carbonylimino)))tetrakis-benzene-1,3-disulfonic acid octasodium salt]. J Pharmacol Exp Ther 314: 232–243, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Hill CE. Selectivity in sympathetic innervation during development and regeneration in the rat. Experientia 41: 857–862, 1985. [DOI] [PubMed] [Google Scholar]
- 23.Horovitz O, Tsoory MM, Hall J, Jacobson-Pick S, Richter-Levin G. Post-weaning to pre-pubertal (“juvenile”) stress: a model of induced predisposition to stress-related disorders. Neuroendocrinology 95: 56–64, 2012. [DOI] [PubMed] [Google Scholar]
- 24.Howe LD, Parmar PG, Paternoster L, Warrington NM, Kemp JP, Briollais L, Newnham JP, Timpson NJ, Smith GD, Ring SM, Evans DM, Tilling K, Pennell CE, Beilin LJ, Palmer LJ, Lawlor DA. Genetic influences on trajectories of systolic blood pressure across childhood and adolescence. Circ Cardiovasc Genet 6: 608–614, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang QQ, Fisher SA, Brozovich FV. Unzipping the role of myosin light chain phosphatase in smooth muscle cell relaxation. J Biol Chem 279: 597–603, 2004. [DOI] [PubMed] [Google Scholar]
- 26.Ingelfinger JR. The child or adolescent with elevated blood pressure. N Engl J Med 370: 2316–2325, 2014. [DOI] [PubMed] [Google Scholar]
- 27.Intapad S, Ojeda NB, Dasinger JH, Alexander BT. Sex differences in the developmental origins of cardiovascular disease. Physiology 29: 122–132, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaaskelainen AE, Seppala S, Kakko T, Jaakkola U, Kallio J. Systemic treatment with neuropeptide Y receptor Y1-antagonist enhances atherosclerosis and stimulates IL-12 expression in ApoE deficient mice. Neuropeptides 47: 67–73, 2013. [DOI] [PubMed] [Google Scholar]
- 29.Jonsson JR, Head RJ, Frewin DB. Effect of alpha 1-adrenoceptor blockade on the development of hypertension in the spontaneously hypertensive rat. Eur J Pharmacol 211: 263–268, 1992. [DOI] [PubMed] [Google Scholar]
- 30.Khatri JJ, Joyce KM, Brozovich FV, Fisher SA. Role of myosin phosphatase isoforms in cGMP-mediated smooth muscle relaxation. J Biol Chem 276: 37250–37257, 2001. [DOI] [PubMed] [Google Scholar]
- 31.Kuo LE, Abe K, Zukowska Z. Stress, NPY and vascular remodeling: implications for stress-related diseases. Peptides 28: 435–440, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee MR, Li L, Kitazawa T. Cyclic GMP causes Ca2+ desnsitization in vascular smooth muscle by activating the myosin light chain phosphatase. J Biol Chem 272: 5063–5068, 1997. [DOI] [PubMed] [Google Scholar]
- 33.Lee RM, Triggle CR, Cheung DW, Coughlin MD. Structural and functional consequence of neonatal sympathectomy on the blood vessels of spontaneously hypertensive rats. Hypertension 10: 328–338, 1987. [DOI] [PubMed] [Google Scholar]
- 34.Lehman BJ, Taylor SE, Kiefe CI, Seeman TE. Relationship of early life stress and psychological functioning to blood pressure in the CARDIA study. Health Psychol 28: 338–346, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levine S. Infantile experience and resistance to physiological stress. Science 126: 405, 1957. [DOI] [PubMed] [Google Scholar]
- 36.Longo LD, Goyal R. Cerebral artery signal transduction mechanisms: developmental changes in dynamics and Ca2+ sensitivity. Curr Vasc Pharmacol 11: 655–711, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loria AS, Brands MW, Pollock DM, Pollock JS. Early life stress sensitizes the renal and systemic sympathetic system in rats. Am J Physiol Renal Physiol 305: F390–F395, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loria AS, Ho DH, Pollock JS. A mechanistic look at the effects of adversity early in life on cardiovascular disease risk during adulthood. Acta Physiol (Oxf) 210: 277–287, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loria AS, Kang KT, Pollock DM, Pollock JS. Early life stress enhances angiotensin II-mediated vasoconstriction by reduced endothelial nitric oxide buffering capacity. Hypertension 58: 619–626, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loria AS, Pollock DM, Pollock JS. Early life stress sensitizes rats to angiotensin II-induced hypertension and vascular inflammation in adult life. Hypertension 55: 494–499, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luff SE. Ultrastructure of sympathetic axons and their structural relationship with vascular smooth muscle. Anat Embryol (Berl) 193: 515–531, 1996. [DOI] [PubMed] [Google Scholar]
- 42.McCarron P, Smith GD, Okasha M, McEwen J. Blood pressure in young adulthood and mortality from cardiovascular disease. Lancet 355: 1430–1431, 2000. [DOI] [PubMed] [Google Scholar]
- 43.McCarty R, Lee JH. Preweanling administration of terazosin decreases blood pressure of hypertensive rats in adulthood. Hypertension 27: 1115–1120, 1996. [DOI] [PubMed] [Google Scholar]
- 44.McCormick CM, Green MR. From the stressed adolescent to the anxious and depressed adult: investigations in rodent models. Neuroscience 249: 242–257, 2013. [DOI] [PubMed] [Google Scholar]
- 45.Miano JM, Cserjesi P, Ligon KL, Periasamy M, Olson EN. Smooth muscle myosin heavy chain exclusively marks the smooth muscle lineage during mouse embryogenesis. Circ Res 75: 803–812, 1994. [DOI] [PubMed] [Google Scholar]
- 46.Minneman KP, Lee D, Zhong H, Berts A, Abbott KL, Murphy TJ. Transcriptional responses to growth factor and G protein-coupled receptors in PC12 cells: comparison of alpha(1)-adrenergic receptor subtypes. J Neurochem 74: 2392–2400, 2000. [DOI] [PubMed] [Google Scholar]
- 47.Mulvany MJ, Halpern W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ Res 41: 19–26, 1977. [DOI] [PubMed] [Google Scholar]
- 48.Nankervis CA, Reber KM, Nowicki PT. Age-dependent changes in the postnatal intestinal microcirculation. Microcirculation 8: 377–387, 2001. [DOI] [PubMed] [Google Scholar]
- 49.Osborn JW, Fink GD. Region-specific changes in sympathetic nerve activity in angiotensin II-salt hypertension in the rat. Exp Physiol 95: 61–68, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pryce CR, Feldon J. Long-term neurobehavioural impact of the postnatal environment in rats: manipulations, effects and mediating mechanisms. Neurosci Biobehav Rev 27: 57–71, 2003. [DOI] [PubMed] [Google Scholar]
- 51.Reho JJ, Shetty A, Dippold RP, Mahurkar A, Fisher SA. Unique gene program of rat small resistance mesenteric arteries as revealed by deep RNA sequencing. Physiol Rep 3: e12450, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reho JJ, Zheng X, Asico LD, Fisher SA. Redox signaling and splicing dependent change in myosin phosphatase underlie early versus late changes in NO vasodilator reserve in a mouse LPS model of sepsis. Am J Physiol Heart Circ Physiol 308: H1039–H1050, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Reho JJ, Zheng X, Benjamin JE, Fisher SA. Neural programming of mesenteric and renal arteries. Am J Physiol Heart Circ Physiol 307: H563–H573, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith PG, Poston CW, Mills E. Ontogeny of neural and non-neural contributions to arterial blood pressure in spontaneously hypertensive rats. Hypertension 6: 54–60, 1984. [DOI] [PubMed] [Google Scholar]
- 55.Su S, Wang X, Pollock JS, Treiber FA, Xu X, Snieder H, McCall WV, Stefanek M, Harshfield GA. Adverse childhood experiences and blood pressure trajectories from childhood to young adulthood: the Georgia Stress and Heart Study. Circulation 131: 1674–1681, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taylor SE. Mechanisms linking early life stress to adult health outcomes. Proc Natl Acad Sci U S A 107: 8507–8512, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Udelsman R, Blake MJ, Stagg CA, Li DG, Putney DJ, Holbrook NJ. Vascular heat shock protein expression in response to stress. Endocrine and autonomic regulation of this age-dependent response. J Clin Invest 91: 465–473, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Whiteus C, Freitas C, Grutzendler J. Perturbed neural activity disrupts cerebral angiogenesis during a postnatal critical period. Nature 505: 407–411, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Woolgar JR, Scott TM. The relationship between innervation and arterial structure in late prenatal and early postnatal development of the rat jejunal artery. J Anat 167: 57–70, 1989. [PMC free article] [PubMed] [Google Scholar]
- 60.Yaghi A, Sims SM. Constrictor-induced translocation of NFAT3 in human and rat pulmonary artery smooth muscle. Am J Physiol Lung Cell Mol Physiol 289: L1061–L1074, 2005. [DOI] [PubMed] [Google Scholar]
- 61.Zicha J, Kuneš J. Ontogenetic aspects of hypertension development: analysis in the rat. Physiol Rev 79: 1227–1282, 1999. [DOI] [PubMed] [Google Scholar]