

Neuropeptide Y1–36 (NPY1–36) and peptide YY1–36 (PYY1–36) (PP-fold peptides), via the Y1 receptor/receptor for activated C kinase 1/Gi/phospholipase C/PKC signaling pathway, stimulate proliferation of and collagen production by cardiac fibroblasts (more potent than ANG II). Dipeptidyl peptidase 4 inhibitors and hypertension augment the effects of PP-fold peptides. PP-fold peptides may contribute to heart failure, and this may be exacerbated by hypertension/dipeptidyl peptidase 4 inhibitors.
Keywords: dipeptidyl peptidase 4, peptide YY1–36, neuropeptide Y1–36, cardiac fibroblasts, spontaneously hypertensive rats, cell proliferation, receptor for activated C kinase 1
Abstract
Cardiac sympathetic nerves release neuropeptide Y (NPY)1–36, and peptide YY (PYY)1–36 is a circulating peptide; therefore, these PP-fold peptides could affect cardiac fibroblasts (CFs). We examined the effects of NPY1–36 and PYY1–36 on the proliferation of and collagen production ([3H]proline incorporation) by CFs isolated from Wistar-Kyoto (WKY) normotensive rats and spontaneously hypertensive rats (SHRs). Experiments were performed with and without sitagliptin, an inhibitor of dipeptidyl peptidase 4 [DPP4; an ectoenzyme that metabolizes NPY1–36 and PYY1–36 (Y1 receptor agonists) to NPY3–36 and PYY3–36 (inactive at Y1 receptors), respectively]. NPY1–36 and PYY1–36, but not NPY3–36 or PYY3–36, stimulated proliferation of CFs, and these effects were more potent than ANG II, enhanced by sitagliptin, blocked by BIBP3226 (Y1 receptor antagonist), and greater in SHR CFs. SHR CF membranes expressed more receptor for activated C kinase (RACK)1 [which scaffolds the Gi/phospholipase C (PLC)/PKC pathway] compared with WKY CF membranes. RACK1 knockdown (short hairpin RNA) and inhibition of Gi (pertussis toxin), PLC (U73122), and PKC (GF109203X) blocked the proliferative effects of NPY1–36. NPY1–36 and PYY1–36 stimulated collagen production more potently than did ANG II, and this was enhanced by sitagliptin and greater in SHR CFs. In conclusion, 1) NPY1–36 and PYY1–36, via the Y1 receptor/Gi/PLC/PKC pathway, activate CFs, and this pathway is enhanced in SHR CFs due to increased localization of RACK1 in membranes; and 2) DPP4 inhibition enhances the effects of NPY1–36 and PYY1–36 on CFs, likely by inhibiting the metabolism of NPY1–36 and PYY1–36. The implications are that endogenous NPY1–36 and PYY1–36 could adversely affect cardiac structure/function by activating CFs, and this may be exacerbated in genetic hypertension and by DPP4 inhibitors.
NEW & NOTEWORTHY
Neuropeptide Y1–36 (NPY1–36) and peptide YY1–36 (PYY1–36) (PP-fold peptides), via the Y1 receptor/receptor for activated C kinase 1/Gi/phospholipase C/PKC signaling pathway, stimulate proliferation of and collagen production by cardiac fibroblasts (more potent than ANG II). Dipeptidyl peptidase 4 inhibitors and hypertension augment the effects of PP-fold peptides. PP-fold peptides may contribute to heart failure, and this may be exacerbated by hypertension/dipeptidyl peptidase 4 inhibitors.
neuropeptide y (NPY)1–36 and peptide YY (PYY)1–36 are members of the pancreatic polypeptide-fold (PP-fold) family and have potent biological activities (5). In this regard, our recent study (24) has shown that both NPY1–36 or PYY1–36 stimulate proliferation of and collagen production by preglomerular vascular smooth muscle cells (VSMCs) and glomerular mesangial cells (GMCs). The fact that NPY1–36 and PYY1–36 stimulate proliferation of and collagen production by preglomerular VSMCs and GMCs (24) suggests that these peptides could similarly affect cardiac fibroblasts; and if biologically active concentrations of PP-fold peptides exist in the cardiac fibroblast biophase, this could be of pathophysiological significance.
There is now strong evidence that PP-fold peptides can achieve biologically active concentrations in the cardiac fibroblast biophase. For example, cardiac sympathetic nerves release NPY1–36 directly into the myocardium (35, 52, 55). Moreover, as discussed by Sosunov et al. (46), the heart releases NPY1–36 from intrinsic intracardiac neurons and granular-containing cells. Endocrine L cells in the intestines release PYY peptides directly into the plasma (1, 17). As discussed by Manning and Batterham (29) and Ballantyne (2), although PYY peptides were viewed for 20 yr after their discovery as primarily locally acting gut hormones, since 2002 the paradigm has undergone a major shift, and there is now overwhelming evidence that PYY peptides are circulating hormones that can have large effects in remote organ systems, including the entire gastrointestinal tract, pancreas, brain, kidneys, and cardiovascular system. Therefore, just as cardiac fibroblasts are under the influence of circulating peptides such as ANG II (44), likely circulating PYY1–36 also reaches receptors in cardiac fibroblasts. It is probable, therefore, that pharmacologically significant levels of PP-fold peptides exist in the biophase of cardiac fibroblasts.
In some disease states, the exposure of cardiac fibroblasts to PP-fold peptides may be exacerbated. For example, the release of NPY1–36 by cardiac sympathetic nerves is accelerated in heart failure (27), and type 2 diabetics with metabolic syndrome often ingest fatty meals that can promote release of PYY1–36 into the circulation (16). Thus, type 2 diabetics, particularly those with metabolic syndrome or heart failure, likely would have high cardiac levels of NPY1–36 or PYY1–36. As reviewed by Shanks and Herring (41), levels of PP-fold peptides correlate with the severity of heart failure and 1-yr mortality. Hulting et al. (23) reported raised levels of NPY in heart failure patients, and Gouya et al. (20) reported significantly elevated PYY levels in heart failure patients with advanced disease. These findings suggest that PP-fold peptides may influence cardiac fibroblasts because activation of cardiac fibroblasts is critically involved in the pathogenesis of heart failure (7). Although cardiac fibroblasts may be exposed to pharmacologically active levels of NPY1–36 and PYY1–36 and even though such exposure may contribute to heart failure via activation of cardiac fibroblasts, the concept that PP-fold peptides affect cardiac fibroblasts has never been directly examined, and currently nothing is known regarding the effects of NPY1–36 and PYY1–36 on fibroblasts in general or cardiac fibroblasts specifically.
Because cardiac fibroblasts contribute to cardiac fibrosis in heart failure, hypertension, and myocardial infarction (7) and because NPY1–36 and PYY1–36 likely are elevated in these conditions, it is important to determine whether NPY1–36 and PYY1–36 have progrowth effects on cardiac fibroblasts. Therefore, the main objective of the present investigation was to determine whether PP-fold peptides stimulate proliferation of and collagen production by cardiac fibroblasts. Recent studies have shown that in vivo right atrial levels of NPY are twofold elevated in the hearts of spontaneously hypertensive rats (SHR) versus Wistar-Kyoto (WKY) rats (42) and that preglomerular VSMCs and GMCs from SHRs are more susceptible to the progrowth effects of NPY1–36 and PYY1–36 (24). Therefore, we examined the effects of PP-fold peptides in cardiac fibroblasts isolated from both SHR and WKY rats.
Dipeptidyl peptidase (DPP)4 inhibitors represent a novel class of antidiabetic drugs that are widely used for the treatment of type 2 diabetes and afford sustained reductions in hemoglobin A1c with a low risk of hypoglycemia and little effect on body weight (3). DPP4 is anchored to the cell surface and efficiently converts PYY1–36 to PYY3–36 and NPY1–36 to NPY3–36 by cleaving two amino acids from the NH2-termini of PYY1–36 and NPY1–36 (31, 32). DPP4 should be considered a “NPY-converting enzyme” because the kcat/Km of DPP4 for NPY1–36 is ∼36-fold and 73-fold greater for NPY1–36 compared with glucagon-like peptide-1 and gastric inhibitory polypeptide, respectively, i.e., incretins that are substrates for DPP4 (32). Whereas PYY1–36 and NPY1–36 are potent Y1 receptor agonists, PYY3–36 and NPY3–36 are inactive at Y1 receptors but are selective Y2 receptor agonists (33). Because the progrowth effects of NPY1–36 and PYY1–36 on preglomerular VSMCs and GMCs are mediated by Y1 receptors, DPP4 inhibitors likely would augment any progrowth effects of either NPY1–36 or PYY1–36 on cardiac fibroblasts by preventing their metabolism to NPY3–36 and PYY3–36 (which are inactive at Y1 receptors). Therefore, a second objective of the present investigation was to determine the interaction between inhibition of DPP4 with sitagliptin and NPY1–36 and PYY1–36 on proliferation of and collagen production by cardiac fibroblasts.
MATERIALS AND METHODS
Animals.
The present study used 12-wk-old male SHR and WKY rats obtained from Taconic Farms (Germantown, NY). Mean arterial blood pressures in SHR and WKY rats were ∼150 and 105 mmHg, respectively. The Institutional Animal Care and Use Committee approved all procedures. This investigation conformed with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Pub. No. 85-23, Revised 1996).
Peptides, BIBP3226, and signaling inhibitors.
Peptides, BIBP3226, pertussis toxin, U73122, and GF109203X were obtained from Sigma-Aldrich (St. Louis, MO). Because we found that PP-fold peptides are easily denatured, PP-fold peptide solutions were made freshly each day and were not subjected to either freezing or vigorous agitation (gentle shaking only).
Isolation, culture, and characterization of cardiac fibroblasts.
A complete description of our method for isolating and culturing rat cardiac fibroblasts can be found in Dubey et al. (14). The purity and morphology of both SHR and WKY cardiac fibroblasts were determined by immunofluorescence staining for the cardiac fibroblast markers vimentin (12) and discoidin domain-containing receptor (DDR)2 (13) using anti-vimentin rabbit monoclonal antibody (catalog no. 5741, Cell Signaling Technology, Danvers, MA) and anti-DDR2 rabbit polyclonal antibody (catalog no. bs-4194R, Bioss Antibodies, Woburn, MA). Immunofluorescence was accomplished using our previously described method (49). All experiments used cardiac fibroblasts in early passage (passages 2–5).
Western blot analysis for DPP4, Y1 receptor, and receptor for activated C kinase 1.
A description of our methods for detecting DPP4, Y1 receptor, and receptor for activated C kinase (RACK)1 protein by Western blot analysis can be found in Jackson et al. (24), Dubinion et al. (15), and Cheng et al. (10), respectively. Quantitative densitometry was accomplished using UN-SCAN-IT gel digitizing software (Silk Scientific, Orem, UT).
Determination of RACK1 protein expression in membrane and cytosolic cellular compartments.
Membrane and cytosolic fractions were isolated from SHR and WKY cardiac fibroblasts using the Mem-PER Plus Membrane Protein Extraction Kit (Thermo Scientific, Rockford, IL). Four preparations of WKY and SHR cardiac fibroblasts at the same passage were plated into T-75 flasks and maintained in DMEM-F-12 containing 10% FBS under standard tissue culture conditions. At confluency (∼7 million cells), cells were dislodged with trypsin and washed twice with cell wash solution. Cells were centrifuged, and the supernatant was replaced with permeabilization buffer, vortexed, and incubated at 4°C for 10 min with constant mixing. Permeabilized cells were centrifuged for 15 min at 16,000 g. The supernatant containing cytosolic proteins was collected. Solubilization buffer was then added to the pellet, resuspended, and incubated at 4°C for 30 min with constant mixing. Samples were centrifuged for 15 min at 16,000 g, and the supernatant containing solubilized membrane and membrane-associated proteins was collected. Samples were stored at −20°C until analyzed. RACK1 expression in total cell extracts and in membrane and cytosolic fractions of cardiac fibroblasts were determined as previously described (10) and normalized to α-tubulin.
Assay for DPP4 activity.
DPP4 activity was measured in cellular extracts using the BioVision DPP4 Fluorometric Assay Kit (BioVision, Milpitas, CA), which measures the release of 7-amino-4-methyl coumarin (AMC) from glycine-proline-AMC (substrate for DPP4).
Assay for PYY3–36.
PYY3–36 levels in the medium were measured using a radioimmunoassay kit (catalog no. PYY-67HK, Millipore, Billerica, MA) that recognizes only PYY3–36 and does not cross react with PYY1–36.
Real-time quantitative PCR for collagen mRNA.
For assessment of collagen mRNA expression, cardiac fibroblasts were cultured to confluence in DMEM-F-12 containing 10% FBS under standard tissue culture conditions. Cells were rendered quiescent with DMEM containing 0.4% BSA for 48 h. Collagen synthesis was initiated by treating growth-arrested cells for 48 h with culture medium supplemented with PDGF (25 ng/ml) and containing either no treatments or 10 nmol/l of either NPY1–36 or PYY1–36. RNA was extracted, and real-time quantitative PCR was performed as previously described (10) using primers for rat collagen type IA1 (forward: 5′-CAGGCTGGTGTGATGGGATT-3′ and reverse: 5′-CGGGACCTTGTTCACCTCTC-3′), collagen type IA2 (forward: 5′-AGGGTGTTCAAGGTGGCAAA-3′ and reverse: 5′-CCACGTTCTCCTCTTGGACC-3′), collagen type IIIA1 (forward: 5′-TTCCTGGGAGAAATGGCGAC-3′ and reverse: 5′-GATAGCCACCCATTCCTCCG-3′), and β-actin (forward: 5′-ACTCTTCCAGCCTTCCTTC-3′ and reverse: 5′-ATCTCCTTCTGCATCCTGTC-3′).
Extraction of sitagliptin.
Tablets containing sitagliptin phosphate monohydrate (Januvia) were purchased from the University of Pittsburgh Medical Center hospital pharmacy. Tablets were pulverized using a mortar and pestle and extracted with water, and the extract was passed through a 2-μm filter as previously described (24).
Lentivirus RACK1 short hairpin RNA production.
Human embryonic kidney (HEK)-293T cells were plated at a density of 7 × 106 cells into T-225 flasks and maintained in DMEM-F-12 containing 10% FBS under standard tissue culture conditions to reach 50–70% confluence the following day. A third-generation lentivirus pGFP-C-shLenti vector encoding puromycin resistance marker, turbo green fluorescent protein (turboGFP), and either RACK1 29-bp short hairpin (sh)RNA or nontargeting control shRNA (OriGene, Rockville, MD) were incubated along with MegaTran transfection reagent, packaging plasmids (OriGene), and placed into Opti-MEM for 15 min at room temperature to allow complex formation. Growth media was replaced with fresh growth media mixed with the transfection complex. The next day, robust GFP expression seen under a fluorescent microscope confirmed plasmid uptake and virus production, at which point the viral supernatant was collected and replaced with fresh growth media. Viral supernatant was again collected the following day. The combined viral supernatant was filtered through a 0.45-μm filter to remove cellular debris and concentrated in Centricon Plus-70 centrifugal filter units (EMD Millipore). The viral concentrate was spun down in an ultracentrifuge at 26,000 rpm for 100 min at 4°C. The supernatant was aspirated, and the remaining viral pellet was resuspended in 25–50 μl sterile DMEM-F-12 and stored at −80°C in aliquots.
Knockdown of RACK1.
SHR cardiac fibroblasts were plated at a density of 75,000 cells/well in a 12-well tissue culture dish and maintained in DMEM-F-12 containing 10% FBS under standard tissue culture conditions. Cells reached 60–70% confluence the next day, and the growth media was replaced with fresh media containing 8 μg/ml polybrene to increase lentivirus transduction efficiency. RACK1 shRNA lentivirus (1 μl) was then pipetted into each of six wells, and 1 μl nontargeting control lentivirus was pipetted into each of the other six wells and incubated overnight at 37°C. Growth media containing lentivirus was replaced with fresh media and incubated overnight at 37°C. Cardiac fibroblasts showed robust GFP expression under a fluorescent microscope 48 h posttransduction, confirming the expression of pGFP-C-shLenti vector elements. Growth media was replaced with fresh media containing 5 μg/ml puromycin and replaced every 24 h for 2 days to select for only transduced cardiac fibroblasts. Puromycin media was then replaced with fresh growth media to allow recovery and incubated overnight at 37°C. Cardiac fibroblasts were serum starved for 48 h in DMEM-F-12 containing 0.1% FBS and then treated every 24 h for 3 days with either PDGF (25 ng/ml) alone or a combination of PDGF (25 ng/ml), NPY1–36 (10 nmol/l), and sitagliptin (1 μmol/L). Cells were then dislodged, counted, and lysed to confirm RACK1 knockdown by Western blot analysis.
Assessment of cell proliferation and collagen production.
Cardiac fibroblasts were maintained in DMEM-F-12 containing 10% FBS under standard tissue culture conditions. For cell number experiments, subconfluent cultures were growth arrested for 48 h in DMEM containing 0.4% BSA and then treated every 24 h for 4 days without or with various treatments. In the presence of FBS, cardiac fibroblasts proliferate very rapidly. Therefore, to examine the effects of receptor agonists on proliferation, the assay must be performed in the absence of serum. However, cardiac fibroblasts, like most cells, will not tolerate more than 24–48 h without serum unless a small concentration of a growth factor is added. Therefore, these experiments were conducted in the presence of a low concentration of PDGF-BB (25 ng/ml). This low concentration was selected to maintain the viability of cells without stimulating proliferation so that the effects of treatments on cell proliferation could be observed. Treatments were dissolved directly in the culture medium to avoid any influences of other solvents. After 4 days, cells were dislodged and counted using a state-of-the-art Nexcelom Cellometer Auto T4 cell counter (Nexcelom Bioscience, Lawrence, MA). For assessment of collagen production, cardiac fibroblasts were allowed to grow to confluence in DMEM-F-12 containing 10% FBS under standard tissue culture conditions. Cells were rendered quiescent with DMEM containing 0.4% BSA for 48 h. Collagen synthesis was initiated by treating growth-arrested cells for 48 h with culture medium supplemented with PDGF (25 ng/ml) and l-[3H]proline (2 μCi/ml) and containing or lacking the various treatments. Experiments were terminated, and the radioactivity in cells was counted in a liquid scintillation counter. We found that terminating the experiments at 48 h prevented overgrowth of cells yet permitted proline incorporation to occur. Because collagen synthesis was assessed in confluent cells but not in overgrown cells, changes in collagen levels reflected increased production per cell rather than simply increased cell number.
Statistical analysis.
Data were analyzed by two-factor and three-factor ANOVA. Single comparisons were conducted with unpaired Student's t-tests. If interaction terms were significant, post hoc comparisons were performed using Fisher's least-significant-difference tests. The criterion of significance was P < 0.05. Data are presented as means ± SE.
RESULTS
Characterization of cardiac fibroblasts.
As shown in Fig. 1, the morphologies of SHR and WKY cardiac fibroblasts were similar, and all cells demonstrated high expression of the cardiac fibroblast markers vimentin and DDR2.
Fig. 1.

Morphology of spontaneously hypertensive rat (SHR; A and C) and Wistar-Kyoto (WKY; B and D) cardiac fibroblasts (CFs) and expression of the fibroblast markers vimentin (A and B; green) and discoidin domain-containing receptor 2 (DDR2; C and D; red) by SHR and WKY CFs. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; blue).
Effects of NPY1–36 and PYY1–36 on the proliferation of cardiac fibroblasts.
Our first objective was to determine whether NPY1–36 and PYY1–36 concentration dependently increase the proliferation of cardiac fibroblasts and, if so, whether this response is augmented by inhibition of DPP4 and greater in cardiac fibroblasts from genetically hypertensive (SHR) versus cardiac fibroblasts from normotensive rats (WKY rats). Accordingly, we examined the concentration-dependent effects of both NPY1–36 and PYY1–36 in both SHR and WKY cardiac fibroblasts and in the absence and presence of sitagliptin (a selective DPP4 inhibitor). Although there are many commonly used surrogates for cell proliferation, in this study rather than use a surrogate for cell proliferation, we chose to measure cell number (the gold standard for cell proliferation) using a state-of-the-art Nexcelom Cellometer Auto T4 cell counter (Bioscience). Importantly, NPY1–36 and PYY1–36 did indeed concentration dependently (P < 0.0001 by two-factor ANOVA) increase cell proliferation in both SHR and WKY cardiac fibroblasts (Fig. 2), and these effects were augmented by sitagliptin [P < 0.0001 for the NPY1–36 × sitagliptin interaction in SHR cardiac fibroblasts, P = 0.0003 for the NPY1–36 × sitagliptin interaction in WKY cardiac fibroblasts, P < 0.0001 for the PYY1–36 × sitagliptin interaction in SHR cardiac fibroblasts, and P = 0.0604 (near significant) for the PYY1–36 × sitagliptin interaction in WKY cardiac fibroblasts, all by two-factor ANOVAs; Fig. 2]. Analysis of these data by three-factor ANOVA (Table 1) corroborated interactions between 1) sitagliptin and NPY1–36 (P < 0.0001), 2) sitagliptin and PYY1–36 (P < 0.0001), 3) SHR/WKY cardiac fibroblasts and NPY1–36 (P < 0.0001), and 4) SHR/WKY cardiac fibroblasts and PYY1–36 (P < 0.0001). Our data show conclusively that the endogenous Y1 agonists NPY1–36 and PYY1–36 stimulate proliferation of cardiac fibroblasts and that these effects are augmented by inhibition of DPP4 and are greater in cardiac fibroblasts from genetically hypertensive versus genetically normotensive animals.
Fig. 2.

Effects of neuropeptide Y (NPY)1–36 (A and B) and peptide (PYY)1–36 (C and D) in the absence or presence of sitagliptin (1 μmol/l) on cell number in SHR (A and C) and WKY (B and D) CFs. Cells were treated for 4 days with the indicated treatments. a,bSignificantly different [by Fisher's least-significant-difference (LSD) test] from corresponding basal (a) or corresponding concentration of peptide in the absence of sitagliptin (b). Sample sizes (n individual experiments) are shown.
Table 1.
Three-factor ANOVAs
| P Value | |
|---|---|
| Effects of strain, sitagliptin, and NPY1–36 on the proliferation of SHR and WKY cardiac fibroblasts | |
| Strain | <0.0001 |
| Sitagliptin | <0.0001 |
| Strain × sitagliptin | 0.0024 |
| NPY1-36 | <0.0001 |
| Strain × NPY1–36 | <0.0001 |
| Sitagliptin × NPY1–36 | <0.0001 |
| Strain × sitagliptin × NPY1–36 | 0.0823 |
| Effects of strain, sitagliptin, and PYY1–36 on the proliferation of SHR and WKY cardiac fibroblasts | |
| Strain | <0.0001 |
| Sitagliptin | <0.0001 |
| Strain × sitagliptin | 0.0039 |
| PYY1–36 | <0.0001 |
| Strain × PYY1–36 | <0.0001 |
| Sitagliptin × PYY1–36 | <0.0001 |
| Strain × sitagliptin × PYY1–36 | 0.1446 |
| Effects of strain, sitagliptin, and NPY1–36 on proline incorporation by SHR and WKY cardiac fibroblasts | |
| Strain | <0.0001 |
| Sitagliptin | <0.0001 |
| Strain × sitagliptin | <0.0001 |
| NPY1–36 | <0.0001 |
| Strain × NPY1–36 | <0.0001 |
| Sitagliptin × NPY1–36 | <0.0001 |
| Strain × sitagliptin × NPY1–36 | 0.0004 |
| Effects of strain, sitagliptin, and PYY1–36 on proline incorporation by SHR and WKY cardiac fibroblasts | |
| Strain | 0.9287 |
| Sitagliptin | <0.0001 |
| Strain × sitagliptin | 0.0194 |
| PYY1–36 | <0.0001 |
| Strain × PYY1–36 | <0.0001 |
| Sitagliptin × PYY1–36 | 0.0051 |
| Strain × sitagliptin × PYY1-36 | 0.2590 |
NPY, neuropeptide Y; PYY, peptide YY; SHR, spontaneously hypertensive rat; WKY, Wistar-Kyoto.
Effects of Y1 receptor antagonism on NPY1–36- and PYY1–36-induced proliferation of cardiac fibroblasts and effects of NPY3–36 and PYY3–36 on the proliferation of cardiac fibroblasts.
We hypothesized that the reason DPP4 inhibition augments the ability of NPY1–36 and PYY1–36 to concentration dependently increase the proliferation of cardiac fibroblasts is because DPP4 inhibition reduces the metabolism of NPY1–36 and PYY1–36 (which are Y1 receptor agonists) to NPY3–36 and PYY3–36 (which are not Y1 receptor agonists). We reasoned that if this hypothesis is correct, antagonism of Y1 receptors should block the proliferative effects of NPY1–36 and PYY1–36 and neither NPY3–36 nor PYY3–36 should stimulate cell proliferation regardless of the absence or presence of a DPP4 inhibitor and regardless of whether the cells were from SHR or WKY rats. To test these predictions, we repeated all of the experiments shown in Fig. 2 but in the presence of BIBP3226, a highly selective Y1 receptor antagonist. Also, we repeated all of the experiments shown in Fig. 2 but with NPY3–36 and PYY3–36 instead of NPY1–36 and PYY1–36. As predicted, BIBP3226 abolished the ability of both NPY1–36 and PYY1–36 to concentration dependently increase the proliferation of cardiac fibroblasts (Fig. 3). Moreover, BIBP3226 abolished these responses regardless of whether the cardiac fibroblasts were from SHR or WKY rats and regardless of whether cells were treated or not with sitagliptin. In addition, regardless of the absence or presence of sitagliptin and regardless of whether cells were from SHR or WKY rats, neither NPY3–36 nor PYY3–36 affected the proliferation of cardiac fibroblasts (Fig. 4). These results are entirely consistent with the concept that DPP4 inhibition augments the ability of NPY1–36 and PYY1–36 to concentration dependently increase the proliferation of cardiac fibroblasts because DPP4 inhibition reduces the metabolism of NPY1–36 and PYY1–36 to NPY3–36 and PYY3–36.
Fig. 3.

Effects of NPY1–36 (A and B) and PYY1–36 (C and D) in the absence or presence of sitagliptin (1 μmol/l) on cell number in SHR (A and C) and WKY (B and D) CFs pretreated with BIBP3226 (1 μmol/l). Cells were treated for 4 days with the indicated treatments. Sample sizes (n individual experiments) are shown.
Fig. 4.

Effects of NPY3–36 (A and B) and PYY3–36 (C and D) in the absence or presence of sitagliptin (1 μmol/l) on cell number in SHR (A and C) and WKY (B and D) CFs. Cells were treated for 4 days with the indicated treatments. Sample sizes (n individual experiments) are shown.
Expression of DPP4 and Y1 receptor protein in SHR and WKY cardiac fibroblasts, DPP4 activity in cardiac fibroblasts, and conversion of PYY1–36 to PYY3–36 in cardiac fibroblasts.
Our hypothesis that DPP4 modulates the ability of NPY1–36 and PYY1–36 to concentration dependently increase the proliferation of cardiac fibroblasts by altering the metabolism of NPY1–36 and PYY1–36 (Y1 receptor agonists) to NPY3–36 and PYY3–36 (not Y1 receptor agonists) also predicts that 1) DPP4 and Y1 receptors are expressed in cardiac fibroblasts, 2) DPP4 activity is expressed in cardiac fibroblasts, and 3) cardiac fibroblasts can metabolize PP-fold peptides and this is blocked by DPP4 inhibition. Accordingly, we examined each of these predictions, as described below.
Monomeric DPP4 is a 110- to 120-kDa glycoprotein; however, DPP4 is biologically active only as a tightly associated dimer that exists as a 220- to 240-kDa glycoprotein (19, 32). Published Western blots for DPP4 have indicated that DPP4 can be detected as a 220- to 240-kDa band (26), 110- to 120-kDa band (34), and as a lower-molecular-weight band (4, 24), with the latter likely representing a DPP4 cleavage product. In SHR and WKY cardiac fibroblasts, we detected all three forms of DPP4, and all three forms were similarly abundant in SHR versus WKY cardiac fibroblasts (Fig. 5). Moreover, both SHR and WKY cardiac fibroblasts expressed DPP4 activity (9.7 ± 3.0 and 2.9 ± 0.4 pmol AMC·30 min−1·2 million cells−1, respectively, n = 6 for each group). DPP4 activity tended to be higher in SHR cardiac fibroblasts (P = 0.073). These data indicate that DPP4 is expressed and is active in cardiac fibroblasts.
Fig. 5.

A: dipeptidyl peptidase 4 (DPP4) protein expression in SHR and WKY CFs. Western blot analysis revealed the expression of DPP4 protein in SHR and WKY CFs, as evidenced by the detection of previously reported DPP4 bands (220 kDa, DPP4 dimer; 110 kDa, DPP4 monomer; 70 kDa, lower-molecular-weight DPP4 fragment). Expression of DPP4 protein was similar in WKY and SHR CFs, as quantified by densitometric analysis of Western blots (normalized to α-tubulin as a loading control). B: Y1 receptor protein expression in SHR and WKY CFs. Western blot analysis revealed the expression of Y1 receptor protein in WKY and SHR CFs, as evidenced by detection of previously reported Y1 receptor band at 70 kDa. Expression of Y1 receptor protein was similar in WKY and SHR CFs, as quantified by densitometric analysis of Western blots (normalized to α-tubulin as a loading control). Sample sizes (n individual samples) are shown.
Published Western blots for Y1 receptor protein show that this protein is detected at 66–73 kDa (15, 43, 53), and in cardiac fibroblasts, we detected a Western blot band at 70 kDa that was similarly expressed in SHR versus WKY cardiac fibroblasts (Fig. 5). These data indicate that Y1 receptors, along with DPP4, are expressed in cardiac fibroblasts.
Confluent monolayers of SHR cardiac fibroblasts were incubated in PBS for 1 or 4 h with 10 nmol/l PYY1–36 without or with sitagliptin, and the medium was assayed for PYY3–36. In the absence of added PYY1–36 (either with or without sitagliptin), PYY3–36 was below the detection limit. Incubation with exogenous PYY1–36 for either 1 or 4 h increased PYY3–36 levels in the medium, and sitagliptin significantly attenuated the increase in PYY3–36 levels (Fig. 6); however, sitagliptin did not abolish the PYY1–36-induced increase in PYY3–36 levels. These data indicate DPP4 in cardiac fibroblasts does indeed metabolize PP-fold peptides.
Fig. 6.
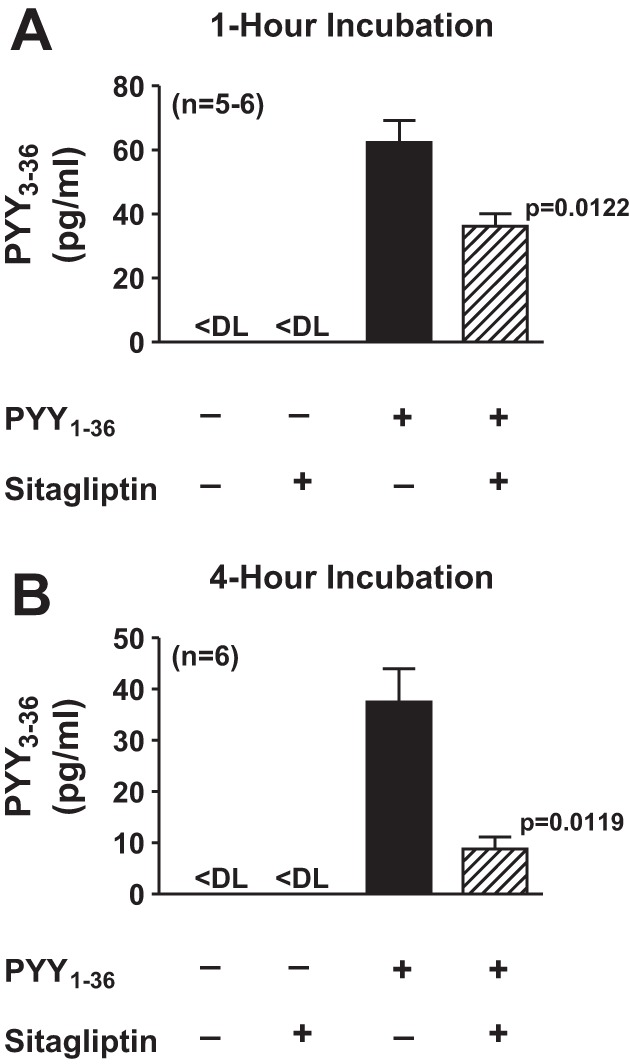
Levels of PYY3–36 in the medium after incubation of SHR CFs with PYY1–36 in the absence and presence of sitagliptin. CFs were incubated with PYY1–36 (10 nmol/l) for 1 h (A) or 4 h (B), and medium was collected and assayed for PYY3–36. P values are for effects of sitagliptin (by an unpaired Student's t-test). Sample sizes (n individual samples) are shown. DL, detection limit.
Effects of NPY1–36, PYY1–36, and sitagliptin on [3H]proline incorporation in SHR and WKY cardiac fibroblasts.
As shown in Fig. 2, both NPY1–36 and PYY1–36 concentration dependently increase the proliferation of cardiac fibroblasts in SHR and WKY cardiac fibroblasts, and these effects were augmented by DPP4 inhibition and greater in cells from SHRs compared with cells from WKY rats. Because cardiac fibroblasts can induce cardiac dysfunction both by overproliferating and overproducing the extracellular matrix, we wondered whether the effects we observed on cardiac fibroblast proliferation could be extrapolated to cardiac fibroblast production of collagen. Therefore, we examined the effects of both NPY1–36 and PYY1–36 on an index of collagen production, namely, [3H]proline incorporation. These experiments were performed in confluent cardiac fibroblasts so that changes in [3H]proline incorporation indicated changes in collagen production rather than changes in cell number. In both SHR and WKY cardiac fibroblasts, NPY1–36 and PYY1–36 significantly (P < 0.0001 by two-factor ANOVA) enhanced [3H]proline incorporation (Fig. 7), and this effect was augmented by sitagliptin [P < 0.0001 for the NPY1–36 × sitagliptin interaction in SHR cardiac fibroblasts, P = 0.0230 for the NPY1–36 × sitagliptin interaction in WKY cardiac fibroblasts, P = 0.0267 for the PYY1–36 × sitagliptin interaction in SHR cardiac fibroblasts, and P = 0.0849 (near significant) for the PYY1–36 × sitagliptin interaction in WKY cardiac fibroblasts, all by two-factor ANOVAs; Fig. 7]. Moreover, analysis of these data by three-factor ANOVA (Table 1) corroborated an interaction between sitagliptin and NPY1–36 (P < 0.0001) and between sitagliptin and PYY1–36 (P = 0.0051) and further demonstrated an interaction between SHR/WKY cardiac fibroblasts and NPY1–36 (P < 0.0001) and SHR/WKY cardiac fibroblasts and PYY1–36 (P < 0.0001), i.e., the effects of NPY1–36 and PYY1–36 were greater in SHR cardiac fibroblasts. In addition, there was a significant (P = 0.0004) three-way interaction among SHR/WKY cardiac fibroblasts, NPY1–36, and sitagliptin, indicating that the interaction between NPY1–36 and sitagliptin was greater in SHR cells.
Fig. 7.

Effects of NPY1–36 (10 nmol/l; A and B) and PYY1–36 (10 nmol/l; C and D) in the absence or presence of sitagliptin (1 μmol/l) on [3H]proline incorporation in SHR (A and C) and WKY (B and D) CFs. a, bSignificant (by Fisher's LSD test) effect of agonist compared with the corresponding value in the absence of agonist (a) and corresponding value of agonist in the absence of sitagliptin (b). Sample sizes (n individual experiments) are shown.
Effects of NPY1–36 and PYY1–36 on collagen mRNA in SHR and WKY cardiac fibroblasts.
To determine whether NPY1–36 and PYY1–36 increase collagen production by increasing collagen mRNA levels, we examined, by real-time quantitative PCR, the steady-state levels of mRNA for collagen type IA1, type IA2, and type IIIA1 after 48 h of treatment with 10 nmol/l of either NPY1–36 or PYY1–36. Neither peptide affected collagen mRNA levels (Fig. 8).
Fig. 8.

Effects of NPY1–36 and PYY1–36 on the expression of collagen mRNA in WKY (A–C) and SHR (D–F) CFs. CFs were cultured to confluence in DMEM-F-12 containing 10% FBS under standard tissue culture conditions. Cells were rendered quiescent with DMEM containing 0.4% BSA for 48 h. Collagen synthesis was initiated by treating growth-arrested cells for 48 h with culture medium supplemented with PDGF (25 ng/ml) and containing either no treatments or 10 nmol/l of either NPY1–36 or PYY1–36. RNA was extracted, and real-time quantitative PCR was performed. Sample sizes (n individual samples) are shown. Ct, threshold cycle.
The mechanism by which PP-fold peptides stimulate cardiac fibroblast proliferation and why this effect is greater in SHR cardiac fibroblasts.
Our previous findings demonstrated that 1) NPY1–36 stimulates the proliferation of rat VSMCs by engaging the Y1 receptor/Gi/PLC/PKC pathway (10); 2) in VSMCs, RACK1 scaffolds Gi, PLC, and PKC in a signaling complex that increases signaling efficiency (10); 3) NPY1–36 and PYY1–36 stimulate proliferation of SHR VSMCs more so than WKY VSMCs (10); and 4) the greater effects of NPY1–36 and PYY1–36 in SHR versus WKY VSMCs is due to increased membrane expression of RACK1 in SHR VSMCs (10). Therefore, we hypothesized that similar mechanisms may explain the growth-promoting effects of NPY1–36 and PYY1–36 in cardiac fibroblasts and may explain why these peptides have a greater effect on proliferation of SHR cardiac fibroblasts.
Figure 9 shows that RACK1 is highly expressed in both SHR and WKY cardiac fibroblasts and that SHR cardiac fibroblasts have ∼20-fold higher levels of RACK1 in the membrane compartment (i.e., the organizational site for the Gi/PLC/PKC signaling pathway) compared with WKY cardiac fibroblasts, which express only trace amounts of membrane-localized RACK1. Moreover, the ratio of cytosolic RACK1 (normalized to the loading control) to total RACK1 (normalized to the loading control) was 0.89 ± 0.05 in WKY cardiac fibroblasts versus 0.73 ± 0.06 in SHR cardiac fibroblasts (P = 0.0426).
Fig. 9.

Expression of receptor for activated C kinase (RACK)1 in WKY and SHR CFs. A: Western blots for RACK1 (36 kDa) and α-tubulin (50 kDa; loading control) expression in WKY and SHR CFs. Subcellular fractions from SHR versus WKY CFs were processed on the same gel with the same amount of total protein. Bar graphs show densitometry measurements for total RACK1 (B), membrane-localized RACK1 (C), and cytosolic RACK1 (D) normalized to α-tubulin. Sample sizes (n individual samples) are shown.
To determine whether RACK1 participates in the proliferative effects of NPY1–36, we developed a RACK1 shRNA-expressing lentiviral vector and used this to knock down RACK1 in SHR cardiac fibroblasts. As shown in Fig. 10, RACK1 shRNA nearly abolished RACK1 expression and attenuated the proliferative effects of NPY1–36 in SHR cardiac fibroblasts. Moreover, the proliferative effects of NPY1–36 in SHR cardiac fibroblasts were attenuated by inhibition of Gi with pertussis toxin, blockade of PLC with U73122, or blockade of PKC with GF109203X (Fig. 11).
Fig. 10.

Knockdown of RACK1 in SHR CFs. A: Western blots for RACK1 (36 kDa) and α-tubulin (50 kDa; loading control) expression in SHR CFs subjected to either a nontargeting short hairpin (sh)RNA lentiviral vector or a RACK1 shRNA lentiviral vector. Some CFs were pretreated (Tx) with NPY1–36 (10 nmol/l) plus sitagliptin (1 μmol/l) for 3 days, whereas other CFs were not pretreated (No Tx). B: densitometry results from the blots shown in A. C: effects of NPY1–36 (10 nmol/l) plus sitagliptin (1 μmol/l) for 3 days on SHR CF number in cells pretreated with either nontargeting shRNA or RACK1 targeting shRNA. a,bSignificantly different (by Fisher's LSD test) from the corresponding nontargeting shRNA (a) or No Tx (b). Sample sizes (n individual experiments) are given shown.
Fig. 11.
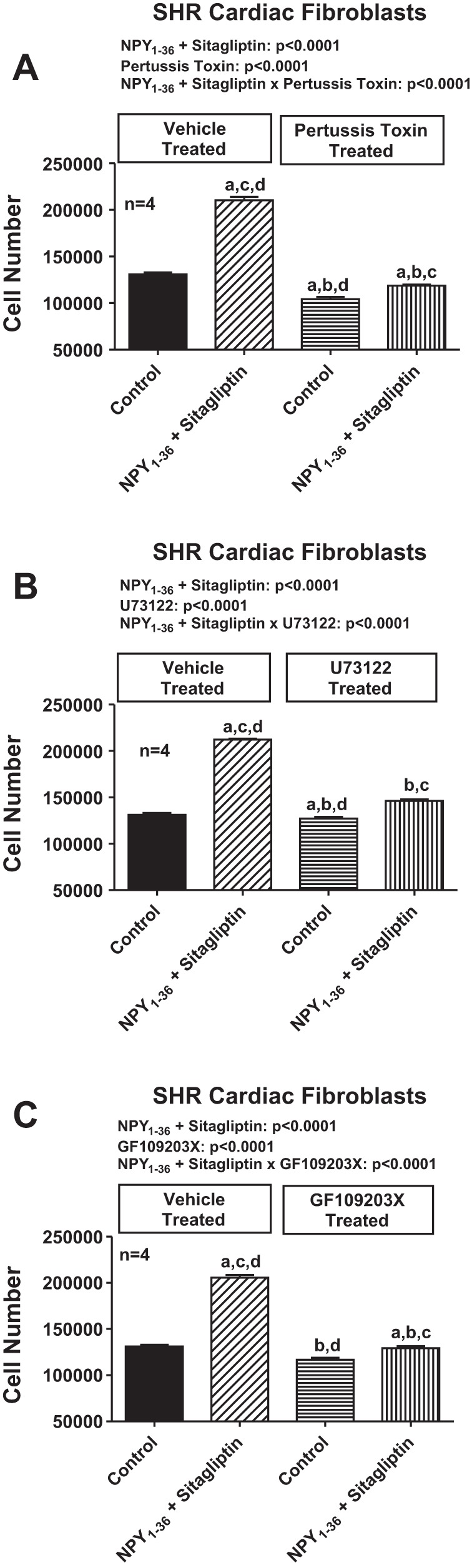
Effects of inhibition of the Gi/phospholipase C/PKC pathway on NPY1–36/sitagliptin-induced proliferation of SHR CFs. Bar graphs show the effects of NPY1–36 (10 nmol/l) plus sitagliptin (1 μmol/l) on the number of SHR CFs in the absence and presence of pertussis toxin (100 ng/ml; A), U73122 (10 μmol/l; B), or GF109203X (10 μmol/l; C). a-dSignificantly different (by Fisher's LSD test) from the group without inhibitor and without NPY1–36/sitagliptin (a), group without inhibitor but with NPY1–36/sitagliptin (b), group with inhibitor but without NPY1–36/sitagliptin (c), or group with inhibitor and with NPY1–36/sitagliptin (d). Sample sizes (n individual experiments) are shown.
Potential significance of the proliferative and collagen-inducing effects of NPY1–36 and PYY1–36 in cardiac fibroblasts.
To our knowledge, this is the first report of stimulation of cardiac fibroblast proliferation and collagen production by either NPY1–36 or PYY1–36. Is this biologically or pathophysiologically significant? ANG II is widely viewed as a peptide that participates directly in stimulating cardiac fibroblasts and inducing cardiac fibrosis (11, 30, 37, 54, 56–58). Therefore, we performed a head-to-head comparison of the concentration-dependent effects of ANG II versus NPY1–36 versus PYY1–36 on cell proliferation and collagen production in both SHR and WKY cardiac fibroblasts. As shown in Fig. 12, both NPY1–36 and PYY1–36 were >10- to 100-fold more potent than ANG II with regard to stimulating the proliferation of and collagen production by SHR or WKY cardiac fibroblasts. In this regard, the threshold concentration for effects of NPY1–36 and PYY1–36 in SHR cells was 1–3 nmol/l and the threshold concentration in WKY cells was 3–10 nmol/l. In contrast, even 100 nmol/l ANG II had no effect in either SHR or WKY cells.
Fig. 12.

Comparison of the effects of ANG II, NPY1–36, and PYY1–36 on cell number and collagen production in SHR and WKY CFs. A: ANG II versus NPY1–36 on cell number in SHR CFs. B: ANG II versus PYY1–36 on cell number in SHR CFs. C: ANG II versus NPY1–36 on cell number in WKY CFs. D: ANG II versus PYY1–36 on cell number in WKY CFs. E: ANG II versus NPY1–36 on [3H]proline incorporation in SHR CFs. F: ANG II versus PYY1–36 on [3H]proline incorporation in SHR CFs. G: ANG II versus NPY1–36 on [3H]proline incorporation in WKY CFs. H: ANG II versus PYY1–36 on [3H]proline incorporation in WKY CFs. Cells were treated for 4 days (cell number) or 2 days ([3H]proline incorporation) with the indicated treatments. a, bSignificantly different (by Fisher's LSD test) from corresponding basal (a) or corresponding concentration of ANG II (b). Sample sizes (n individual experiments) are shown.
DISCUSSION
In the present study, we report the novel finding that both NPY1–36 and PYY1–36 stimulate proliferation of and collagen production by cardiac fibroblasts. These new results are potentially important because the myocardium is exposed to NPY1–36 and PYY1–36 and proliferation of and collagen production by cardiac fibroblasts participate in pathological myocardial remodeling (such as cardiac hypertrophy) and heart failure (7). The fact that both NPY1–36 and PYY1–36 have much greater effects on cardiac fibroblast biology than even ANG II, the quintessential profibrotic hormone (11, 30, 37, 54, 56–58), indicates the potential pathophysiological significance of the present findings.
A noteworthy observation is that the effects of NPY1–36 and PYY1–36 on proliferation and collagen production are greater in SHR versus WKY cardiac fibroblasts. This finding has important implications. Hypertension is associated with myocardial fibrosis and abnormal changes in left ventricular (LV) geometry leading to diastolic abnormalities, such as impaired relaxation and filling of the LV (18, 48, 50). Clinically, this is known as LV diastolic dysfunction (LVDD). In hypertensive patients, LVDD often precedes alterations in LV systolic function and may precipitate symptoms of heart failure even in the presence of a normal ejection fraction (18, 48, 50). Indeed, the prevalence of LVDD in hypertensive patients is an increasingly important public health problem. The concept that NPY1–36 and PYY1–36 stimulate pathological behavior of cardiac fibroblasts more aggressively in genetic hypertension could lead to a better understanding of the mechanisms of hypertension-induced LVDD and diastolic heart failure.
Yet another novel finding of the present study is that cardiac fibroblasts rely, at least in vitro, on DPP4 to metabolize and inactivate PP-fold peptides. The evidence for this conclusion is that cardiac fibroblasts express DPP4 protein and activity and inhibition of DPP4 attenuates the metabolism of PYY1–36 to PYY3–36. Because we do not have an assay that distinguishes NPY1–36 from NPY3–36 yet we do have an assay that detects PYY3–36 without cross reacting with PYY1–36, in the present study, we assessed the ability of DPP4 to metabolize PP-fold peptides by measuring the production of PYY3–36 from PYY1–36. Likely, both peptides are metabolized similarly by DPP4, so the results for PYY1–36 metabolism should reflect the ability of DPP4 to metabolize NPY1–36.
The concept that DPP4 metabolizes PP-fold peptides in cardiac fibroblasts has medical implications. In this regard, the present study shows that although both NPY1–36 and PYY1–36 enhance proliferation of and collagen production by cardiac fibroblasts, NPY3–36 and PYY3–36 do not share this activity with their parent peptides. This suggests that DPP4 activity, by converting NPY1–36 and PYY1–36 to NPY3–36 and PYY3–36, respectively, may be important for moderating the progrowth effects of PP-fold peptides on cardiac fibroblasts. In support of this conclusion is our unequivocal observation that inhibition of DPP4 with sitagliptin augments the ability of both NPY1–36 and PYY1–36 to enhance both proliferation of and collagen production by cardiac fibroblasts.
Because enhanced proliferation of and collagen production by cardiac fibroblasts could lead to cardiac fibrosis and heart failure, it is important to consider whether DPP4 inhibitors (gliptins; for example, alogliptin, linagliptin, saxagliptin, sitagliptin, and vildagliptin), which are used for the management of type 2 diabetes, may increase the risk of cardiac hypertrophy and heart failure. Although gliptins may be cardioprotective under some circumstances (39), and although the long-term effects of gliptins on organ function (particularly in the heart, kidney, and pancreas) remain to be established (39), there is increasing preclinical and clinical evidence that gliptins can indeed cause cardiac hypertrophy and heart failure. For example, our previous study (47) showed that chronic treatment with sitagliptin (40 mg·kg−1·day−1 for 21 days) enhances ANG II-induced cardiac hypertrophy in SHRs. Consistent with our finding is a recent preclinical study by Hemmeryckx et al. (22) demonstrating that in diabetic Akita mice chronically fed a high-fat and high-cholesterol diet, 3 mo of treatment with sitagliptin (50 mg·kg−1·day−1) induces LV hypertrophy.
The conclusions from these preclinical studies appear to extrapolate to patients. A recent study involving 16,492 patients randomized to placebo or saxagliptin and followed for 2.1 yr (median) did not show any cardiovascular benefits of DPP4 inhibition and in fact demonstrated a statistically significant (P = 0.007) increase in the number of patients hospitalized for heart failure (40). Recently, Wang et al. (51) analyzed 8,288 matched pairs of diabetic patients who were treated or not with sitagliptin. During a median of 1.5 yr, there was a significant (P = 0.017) increase in the risk of hospitalization for heart failure in patients taking sitagliptin, and the risk for heart failure was proportional to exposure to sitagliptin (51). A very recent meta-analysis involving 94 trials enrolling 85,224 diabetic patients noted a significantly (P = 0.034) increased risk (15.8%) of heart failure with long-term use of DPP4 inhibitors (38). In a large (5,380 patients) randomized clinical trial, alogliptin did not increase hospitalized for heart failure in patients with a history of heart failure at baseline but nearly doubled those hospitalized for heart failure (hazard ratio: 1.76, P = 0.026) in patients without a history of heart failure (59). This may be explained by the fact that patients with a history of heart failure and enrolled in a clinical trial would be optimally managed for their disease and optimal management may compensate for the adverse cardiac effects of DPP4 inhibitors. Consistent with this concept, the TECOS study involving 14,671 patients randomized to sitagliptin or placebo did not detect an increase in hospitalized for heart failure. However, the majority of patients received β-blockers, renin-angiotensin system inhibitors, statins, and aspirin and therefore had “background care” that would have attenuated any heart failure signal (21).
Our findings in the present report strongly suggest that augmentation of the effects of NPY1–36 and PYY1–36 on cardiac fibroblasts contributes to gliptin-induced heart failure. However, given the range of peptides processed by DPP4, it is also possible that additional DPP4 substrates stimulate cardiac fibroblast proliferation/collagen production and are involved in gliptin-induced cardiac hypertrophy/heart failure. We are actively investigating this possibility. It is also conceivable that under some circumstances, gliptins are cardioprotective, yet in selected patients, for example, those with elevated NPY1–36 and PYY1–36, the opposite could be true. The present findings underscore the need for continued evaluation of this novel and important class of antidiabetic agents.
Perhaps the most important finding of the present study is that the selective Y1 receptor antagonist BIBP3226 completely blocks the ability of both NPY1–36 and PYY1–36 to stimulate cardiac fibroblast proliferation. This finding is consistent with the expression of Y1 receptors in cardiac fibroblasts and with the well-known fact that NPY1–36 and PYY1–36 are Y1 receptor agonists. Importantly, BIBP3226 blocks the progrowth effects of NPY1–36 and PYY1–36 regardless of whether DPP4 is active or inhibited with sitagliptin. These findings suggest that antagonists of Y1 receptors might be effective therapeutics to prevent or treat heart failure, particularly in patients with high levels of cardiac sympathetic activation, increased levels of circulating PYY1–36, or who are taking DPP4 inhibitors for the management of type 2 diabetes.
How do PP-fold peptides stimulate the proliferation of cardiac fibroblasts and why are these effects greater in SHR cardiac fibroblasts? RACK1 is a seven-sided propeller protein with seven WD40 repeats, and RACK1 participates in cell signaling by functioning as a scaffolding protein that interacts with G protein βγ subunits, PLC, and PKC (8, 45) and therefore organizes and modulates the Gi/PLC/PKC pathway. Y1 receptors are coupled to Gi (33), and recent studies from our laboratory (9, 10) have indicated that membrane-localized RACK1 participates in the proliferation of preglomerular VSMCs. Therefore, we hypothesize that in cardiac fibroblasts, the Gi/PLC/PKC pathway mediates the biological effects of PP-fold peptides, that this pathway is organized by RACK1, and that increased membrane localization of RACK1 explains the greater effects of PP-fold peptides in SHR cardiac fibroblasts. Consistent with this hypothesis, we observe that membrane-localized RACK1 is 20-fold greater in SHR compared with WKY cardiac fibroblasts and that shRNA knockdown of RACK1 or inhibition of Gi, PLC, or PKC attenuates the proliferative response to NPY1–36 in SHR cardiac fibroblasts. There is likely a greater translocation of RACK1 from the cytosol to the membrane in SHR cardiac fibroblasts. However, because most of the total pool of RACK1 is in the cytosol (not in the membrane), a translocation to the membrane of a small amount from the large cytosolic pool results in a large percent increase in the membrane but only a small percentage decrease in the cytosol. Nonetheless, the ratio of cytosolic RACK1 to total RACK1 is significantly decreased in SHR cardiac fibroblasts, indicating a significant, but small, decrease in the distribution of RACK1 to the cytosol in SHR cardiac fibroblasts. Since membrane-localized RACK1 is greater both in SHR preglomerular VSMCs (10) and cardiac fibroblasts (present study), it is likely that this is a genetic trait and may underlie much of the cardiovascular pathology, including hypertension, in SHRs. We are actively pursuing this hypothesis.
Although beyond the scope of the present study, the mechanism by which DPP4 inhibitors increase the risk of heart failure requires the development of a reliable preclinical animal model of DPP4-induced heart failure. Such a model may require combining chronic (months or years) DPP4 inhibition with underlying heart disease/hypertension. We are currently attempting to develop such an animal model of gliptin-induced heart dysfunction so that we can test whether receptor antagonists of DPP4 substrates (for example, Y1 antagonists) can attenuate gliptin-induced heart failure.
The present study demonstrates that NPY1–36 and PYY1–36 are more potent agonists with regard to stimulating proliferation of adult rat cardiac fibroblasts than is ANG II. Indeed, we observed that ANG II is a very weak stimulant of proliferation of adult rat cardiac fibroblasts, a finding consistent with the discordant results reported for the in vitro effects of ANG II on the proliferation of adult rat cardiac fibroblasts (28) and with another study (6) that reported no effect of ANG II on the proliferation of cardiac fibroblasts isolated from adult rats. Although there is a report (36) of ANG II stimulating proliferation of adult cardiac fibroblasts, in general, high concentrations (i.e., 100-1,000 nmol/l) of ANG II were used. That PP-fold peptides stimulate proliferation at 1 nmol/l underscores the potential significance of the present findings.
In conclusion, cardiac sympathetic nerves release NPY1–36, and the intestines secrete PYY1–36. It is likely, therefore, that these PP-fold peptides are elevated in the hearts of selected patients. The present study establishes that low concentrations of NPY1–36 and PYY1–36 stimulate proliferation of and collagen production by cardiac fibroblasts and that these effects are augmented by blockade of DPP4. Importantly, a recent study (25) has demonstrated that in SHRs, chronic inhibition of DPP4 increases arterial blood pressure. Thus, in some patients, DPP4 inhibitors may enhance the direct profibrotic effects of PP-fold peptides in the heart and simultaneously worsen hypertension. These actions could synergize to induce cardiac fibrosis leading to heart failure. Because the progrowth effects of NPY1–36 and PYY1–36 are enhanced in genetic hypertension, patients with a genetic predisposition to hypertension may be particularly at risk. Although DPP4 inhibitors may be quite safe in most patients with type 2 diabetes, physicians should be aware of the possibility of unintended consequences of blocking the cardiac metabolism of PP-fold peptides. Finally, the present study raises the proposition of preventing and treating LVDD and heart failure with Y1 receptor antagonists and the concept that RACK1 may participate in the pathophysiology of heart failure and hypertension.
GRANTS
This work was supported by National Institutes of Health Grants DK-091190, HL-109002, HL-069846, DK-068575, and DK-079307. X. Zhu is a Howard Hughes Medical Institute Research Fellow.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: X.Z. and D.G.G. performed experiments; X.Z., D.G.G., and E.K.J. edited and revised manuscript; X.Z., D.G.G., and E.K.J. approved final version of manuscript; E.K.J. conception and design of research; E.K.J. analyzed data; E.K.J. interpreted results of experiments; E.K.J. prepared figures; E.K.J. drafted manuscript.
REFERENCES
- 1.Anini Y, Fu-Cheng X, Cuber JC, Kervran A, Chariot J, Roz C. Comparison of the postprandial release of peptide YY and proglucagon-derived peptides in the rat. Pflügers Arch 438: 299–306, 1999. [DOI] [PubMed] [Google Scholar]
- 2.Ballantyne GH. Peptide YY1–36 and peptide YY3–36: part I. Distribution, release and actions. Obes Surg 16: 651–658, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Barnett A. DPP-4 inhibitors and their potential role in the management of type 2 diabetes. Int J Clin Pract 60: 1454–1470, 2006. [DOI] [PubMed] [Google Scholar]
- 4.Bauvois B. A collagen-binding glycoprotein on the surface of mouse fibroblasts is identified as dipeptidyl peptidase IV. Biochem J 252: 723–731, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berglund MM, Hipskind PA, Gehlert DR. Recent developments in our understanding of the physiological role of PP-fold peptide receptor subtypes. Exp Biol Med (Maywood) 228: 217–244, 2003. [DOI] [PubMed] [Google Scholar]
- 6.Brilla CG, Zhou G, Matsubara L, Weber KT. Collagen metabolism in cultured adult rat cardiac fibroblasts: response to angiotensin II and aldosterone. J Mol Cell Cardiol 26: 809–820, 1994. [DOI] [PubMed] [Google Scholar]
- 7.Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol 45: 657–687, 2005. [DOI] [PubMed] [Google Scholar]
- 8.Chen S, Spiegelberg BD, Lin F, Dell EJ, Hamm HE. Interaction of Gβγ with RACK1 and other WD40 repeat proteins. J Mol Cell Cardiol 37: 399–406, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Cheng D, Zhu X, Barchiesi F, Gillespie DG, Dubey RK, Jackson EK. Receptor for activated protein kinase C1 regulates cell proliferation by modulating calcium signaling. Hypertension 58: 689–695, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng D, Zhu X, Gillespie DG, Jackson EK. Role of RACK1 in the differential proliferative effects of neuropeptide Y1–36 and peptide YY1–36 in SHR vs. WKY preglomerular vascular smooth muscle cells. Am J Physiol Renal Physiol 304: F770–F780, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chung CC, Hsu RC, Kao YH, Liou JP, Lu YY, Chen YJ. Androgen attenuates cardiac fibroblasts activations through modulations of transforming growth factor-β and angiotensin II signaling. Int J Cardiol 176: 386–393, 2014. [DOI] [PubMed] [Google Scholar]
- 12.Colombo F, Gosselin H, El-Helou V, Calderone A. β-Adrenergic receptor-mediated DNA synthesis in neonatal rat cardiac fibroblasts proceeds via a phosphatidylinositol 3-kinase dependent pathway refractory to the antiproliferative action of cyclic AMP. J Cell Physiol 195: 322–330, 2003. [DOI] [PubMed] [Google Scholar]
- 13.Cowling RT, Yeo SJ, Kim IJ, Park JI, Gu Y, Dalton ND, Peterson KL, Greenberg BH. Discoidin domain receptor 2 germline gene deletion leads to altered heart structure and function in the mouse. Am J Physiol Heart Circ Physiol 307: H773–H781, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dubey RK, Gillespie DG, Mi MZ, Jackson EK. Exogenous and endogenous adenosine inhibits fetal calf serum-induced growth of rat cardiac fibroblasts: role of A2B receptors. Circulation 96: 2656–2666, 1997. [DOI] [PubMed] [Google Scholar]
- 15.Dubinion JH, Mi Z, Zhu C, Gao L, Jackson EK. Pancreatic polypeptide-fold peptide receptors and angiotensin II-induced renal vasoconstriction. Hypertension 47: 545–551, 2006. [DOI] [PubMed] [Google Scholar]
- 16.Freire RD, Cardoso MA, Gimeno SG, Ferreira SR; Japanese-Brazilian Diabetes Study Group. Dietary fat is associated with metabolic syndrome in Japanese Brazilians. Diabetes Care 28: 1779–1785, 2005. [DOI] [PubMed] [Google Scholar]
- 17.Fu-Cheng X, Anini Y, Chariot J, Castex N, Galmiche JP, Roze C. Mechanisms of peptide YY release induced by an intraduodenal meal in rats: neural regulation by proximal gut. Pflügers Arch 433: 571–579, 1997. [DOI] [PubMed] [Google Scholar]
- 18.Galderisi M. Diagnosis and management of left ventricular diastolic dysfunction in the hypertensive patient. Am J Hypertens 24: 507–517, 2011. [DOI] [PubMed] [Google Scholar]
- 19.Gorrell MD. Dipeptidyl peptidase IV and related enzymes in cell biology and liver disorders. Clin Sci 108: 277–292, 2005. [DOI] [PubMed] [Google Scholar]
- 20.Gouya G, Voithofer P, Neuhold S, Storka A, Vila G, Pacher R, Wolzt M, Hülsmann M. Association of nutritional risk index with metabolic biomarkers, appetite-regulatory hormones and inflammatory biomarkers and outcome in patients with chronic heart failure. Int J Clin Pract 68: 1293–1300, 2014. [DOI] [PubMed] [Google Scholar]
- 21.Green JB, Bethel MA, Armstrong PW, Buse JB, Engel SS, Garg J, Josse R, Kaufman KD, Koglin J, Korn S, Lachin JM, McGuire DK, Pencina MJ, Standl E, Stein PP, Suryawanshi S, Van de Werf F, Peterson ED, Holman RR TECOS. Study Group. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med 373: 232–242, 2015. [DOI] [PubMed] [Google Scholar]
- 22.Hemmeryckx B, Swinnen M, Gallacher DJ, Rong Lu H, Roger Lijnen H. Effect of sitagliptin treatment on metabolism and cardiac function in genetic diabetic mice. Eur J Pharmacol 723: 175–180, 2014. [DOI] [PubMed] [Google Scholar]
- 23.Hulting J, Sollevi A, Ullman B, Franco-Cereceda A, Lundberg JM. Plasma neuropeptide Y on admission to a coronary care unit: raised levels in patients with left heart failure. Cardiovasc Res 24: 102–108, 1990. [DOI] [PubMed] [Google Scholar]
- 24.Jackson EK, Kochanek SJ, Gillespie DG. Dipeptidyl peptidase IV regulates proliferation of preglomerular vascular smooth muscle and mesangial cells. Hypertension 60: 757–764, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jackson EK, Mi Z, Tofovic SP, Gillespie DG. Effect of dipeptidyl peptidase 4 inhibition on arterial blood pressure is context dependent. Hypertension 65: 238–249, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jackson EK, Zhang M, Liu W, Mi Z. Inhibition of renal dipeptidyl peptidase IV enhances peptide YY1–36-induced potentiation of angiotensin II-mediated renal vasoconstriction in spontaneously hypertensive rats. J Pharmacol Exp Ther 323: 431–437, 2007. [DOI] [PubMed] [Google Scholar]
- 27.Kaye DM, Lambert GW, Lefkovits J, Morris M, Jennings G, Esler MD. Neurochemical evidence of cardiac sympathetic activation and increased central nervous system norepinephrine turnover in severe congestive heart failure. J Am Coll Cardiol 23: 570–578, 1994. [DOI] [PubMed] [Google Scholar]
- 28.Lijnen P. Angiotensin II-induced proliferation of neonatal and adult rat cardiac fibroblasts. Hypertension 51: e50, 2008. [DOI] [PubMed] [Google Scholar]
- 29.Manning S, Batterham RL. The role of gut hormone peptide YY in energy and glucose homeostasis: twelve years on. Annu Rev Physiol 76: 585–608, 2014. [DOI] [PubMed] [Google Scholar]
- 30.Marko L, Henke N, Park JK, Spallek B, Qadri F, Balogh A, Apel IJ, Oravecz-Wilson KI, Choi M, Przybyl L, Binger KJ, Haase N, Wilck N, Heuser A, Fokuhl V, Ruland J, Lucas PC, McAllister-Lucas LM, Luft FC, Dechend R, Muller DN. Bcl10 mediates angiotensin II-induced cardiac damage and electrical remodeling. Hypertension 64: 1032–1039, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McIntosh CH, Demuth HU, Pospisilik JA, Pederson R. Dipeptidyl peptidase IV inhibitors: how do they work as new antidiabetic agents? Regul Pept 128: 159–165, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Mentlein R. Dipeptidyl-peptidase IV (CD26)–role in the inactivation of regulatory peptides. Regul Pept 85: 9–24, 1999. [DOI] [PubMed] [Google Scholar]
- 33.Michel MC, Beck-Sickinger A, Cox H, Doods HN, Herzog H, Larhammar D, Quirion R, Schwartz T, Westfall TX 4th. International Union of Pharmacology recommendations for the nomenclature of neuropeptide Y, peptide YY, and pancreatic polypeptide receptors. Pharmacol Rev 50: 143–150, 1998. [PubMed] [Google Scholar]
- 34.Moran GW, O'Neill C, Padfield P, McLaughlin JT. Dipeptidyl peptidase-4 expression is reduced in Crohn's disease. Regul Pept 177: 40–45, 2012. [DOI] [PubMed] [Google Scholar]
- 35.Nyquist-Battie C, Cochran PK, Sands SA, Chronwall BM. Development of neuropeptide Y and tyrosine hydroxylase immunoreactive innervation in postnatal rat heart. Peptides 15: 1461–1469, 1994. [DOI] [PubMed] [Google Scholar]
- 36.Olson ER, Shamhart PE, Naugle JE, Meszaros JG. Angiotensin II-induced extracellular signal-regulated kinase 1/2 activation is mediated by protein kinase Cδ and intracellular calcium in adult rat cardiac fibroblasts. Hypertension 51: 704–711, 2008. [DOI] [PubMed] [Google Scholar]
- 37.Qi GM, Jia LX, Li YL, Li HH, Du J. Adiponectin suppresses angiotensin II-induced inflammation and cardiac fibrosis through activation of macrophage autophagy. Endocrinology 155: 2254–2265, 2014. [DOI] [PubMed] [Google Scholar]
- 38.Savarese G, Perrone-Filardi P, D'Amore C, Vitale C, Trimarco B, Pani L, Rosano GM. Cardiovascular effects of dipeptidyl peptidase-4 inhibitors in diabetic patients: a meta-analysis. Int J Cardiol 181: 239–244, 2015. [DOI] [PubMed] [Google Scholar]
- 39.Scheen AJ. Cardiovascular effects of gliptins. Nat Rev Cardiol 10: 73–84, 2013. [DOI] [PubMed] [Google Scholar]
- 40.Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, Ohman P, Frederich R, Wiviott SD, Hoffman EB, Cavender MA, Udell JA, Desai NR, Mosenzon O, McGuire DK, Ray KK, Leiter LA, Raz I; SAVOR-TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med 369: 1317–1326. [DOI] [PubMed] [Google Scholar]
- 41.Shanks J, Herring N. Peripheral cardiac sympathetic hyperactivity in cardiovascular disease: role of neuropeptides. Am J Physiol Regul Integr Comp Physiol 305: R1411–R1420, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shanks J, Manou-Stathopoulou S, Lu CJ, Li D, Paterson DJ, Herring N. Cardiac sympathetic dysfunction in the prehypertensive spontaneously hypertensive rat. Am J Physiol Heart Circ Physiol 305: H980–H986, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sheikh SP, Williams JA. Structural characterization of Y1 and Y2 receptors for neuropeptide Y and peptide YY by affinity cross-linking. J Biol Chem 265: 8304–8310. [PubMed] [Google Scholar]
- 44.Siddesha JM, Valente AJ, Sakamuri SSVP, Yoshida T, Gardner JD, Somanna N, Takahashi C, Noda M, Chandrasekar B. Angiotensin II stimulates cardiac fibroblast migration via the differential regulation of matrixins and RECK. J Mol Cell Cardiol 65: 9–18, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sklan EH, Podoly E, Soreq H. RACK1 has the nerve to act: structure meets function in the nervous system. Prog Neurobiol 78: 117–134, 2006. [DOI] [PubMed] [Google Scholar]
- 46.Sosunov AA, Hassall CJ, Loesch A, Turmaine M, Feher E, Burnstock G. Neuropeptide Y-immunoreactive intracardiac neurones, granule-containing cells and nerves associated with ganglia and blood vessels in rat and guinea-pig heart. Cell Tissue Res 289: 445–454, 1997. [DOI] [PubMed] [Google Scholar]
- 47.Tofovic DS, Bilan V, Jackson EK. Sitagliptin enhances angiotensin II-induced hypertension and cardiac hypertrophy in spontaneously hypertensive rats. Hypertension 54: E64–E65, 2009. [Google Scholar]
- 48.Verma A, Solomon SD. Diastolic dysfunction as a link between hypertension and heart failure. Med Clin North Am 93: 647–664, 2009. [DOI] [PubMed] [Google Scholar]
- 49.Verrier JD, Jackson TC, Gillespie DG, Janesko-Feldman K, Bansal R, Goebbels S, Nave KA, Kochanek PM, Jackson EK. Role of CNPase in the oligodendrocytic extracellular 2′,3′-cAMP-adenosine pathway. Glia 61: 1595–1606, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Volpe M, McKelvie R, Drexler H. Hypertension as an underlying factor in heart failure with preserved ejection fraction. J Clin Hypertens (Greenwich) 12: 277–283, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang KL, Liu CJ, Chao TF, Huang CM, Wu CH, Chen SJ, Yeh CM, Chen TJ, Lin SJ, Chiang CE. Sitagliptin and the risk of hospitalization for heart failure: a population-based study. Int J Cardiol 177: 86–90, 2014. [DOI] [PubMed] [Google Scholar]
- 52.Warner MR, Senanayake PD, Ferrario CM, Levy MN. Sympathetic stimulation-evoked overflow of norepinephrine and neuropeptide Y from the heart. Circ Res 69: 455–465, 1991. [DOI] [PubMed] [Google Scholar]
- 53.Wieland HA, Eckard CP, Doods HN, Beck-Sickinger AG. Probing of the neuropeptide Y-Y1-receptors interaction with anti-receptor antibodies. Eur J Biochem 255: 595–603, 1998. [DOI] [PubMed] [Google Scholar]
- 54.Williams SM, Golden-Mason L, Ferguson BS, Schuetze KB, Cavasin MA, Demos-Davies K, Yeager ME, Stenmark KR, McKinsey TA. Class I HDACs regulate angiotensin II-dependent cardiac fibrosis via fibroblasts and circulating fibrocytes. J Mol Cell Cardiol 67: 112–125, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang T, Levy MN. Sequence of excitation as a factor in sympathetic-parasympathetic interactions in the heart. Circ Res 71: 898–905, 1992. [DOI] [PubMed] [Google Scholar]
- 56.Yu L, Yang G, Weng X, Liang P, Li L, Li J, Fan Z, Tian W, Wu X, Xu H, Fang M, Ji Y, Li Y, Chen Q, Xu Y. Histone methyltransferase SET1 mediates angiotensin II-induced endothelin-1 transcription and cardiac hypertrophy in mice. Arterioscler Thromb Vasc Biol 35: 1207–1217, 2015. [DOI] [PubMed] [Google Scholar]
- 57.Yu N, Jiang J, Yu Y, Li H, Huang X, Ma Y, Zhang L, Zou J, Zhang B, Chen S, Liu P. SLC41A1 knockdown inhibits angiotensin II-induced cardiac fibrosis by preventing Mg2+ efflux and Ca2+ signaling in cardiac fibroblasts. Arch Biochem Biophys 564: 74–82, 2014. [DOI] [PubMed] [Google Scholar]
- 58.Yu Y, Chen S, Xiao C, Jia Y, Guo J, Jiang J, Liu P. TRPM7 is involved in angiotensin II induced cardiac fibrosis development by mediating calcium and magnesium influx. Cell Calcium 55: 252–260, 2014. [DOI] [PubMed] [Google Scholar]
- 59.Zannad F, Cannon CP, Cushman WC, Bakris GL, Menon V, Perez AT, Fleck PR, Mehta CR, Kupfer S, Wilson C, Lam H, White WB; EXAMINE Investigators. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet 385: 2067–2076, 2015. [DOI] [PubMed] [Google Scholar]