

Abstract
Key points
Deficiency of dysferlin causes limb‐girdle muscular dystrophy 2B and Miyoshi myopathy with cardiac involvement that leads to dilated cardiomyopathy and heart failure. The pathogenesis and pathophysiology of dysferlin cardiomyopathy are not fully understood.
We studied cardiac phenotypes of young dysferlin gene knockout mice to investigate the primary pathological and pathophysiological changes.
In comparison with wild‐type controls, dysferlin‐deficient cardiomyocytes showed slower Ca2+ re‐sequestration, and dysferlin deficiency blunted the β‐adrenergic effect on relaxation and pumping function of ex vivo working hearts.
Dysferlin deficiency increased phosphorylation of ventricular myosin light chain 2, suggesting a compensatory response to the impaired cardiac lusitropic function.
The data suggest that delayed calcium re‐sequestration and post‐translational modification of myofilament proteins may provide potential targets to develop new treatments for dysferlin cardiomyopathy.
Abstract
Dysferlin is a cell membrane bound protein with a role in the repair of skeletal and cardiac muscle cells. Deficiency of dysferlin leads to limb‐girdle muscular dystrophy 2B (LGMD2B) and Miyoshi myopathy. In cardiac muscle, dysferlin is located at the intercalated disc and transverse tubule membranes. Loss of dysferlin causes death of cardiomyocytes, notably in ageing hearts, leading to dilated cardiomyopathy and heart failure in LGM2B patients. To understand the primary pathogenesis and pathophysiology of dysferlin cardiomyopathy, we studied cardiac phenotypes of young adult dysferlin knockout mice and found early myocardial hypertrophy with largely compensated baseline cardiac function. Cardiomyocytes isolated from dysferlin‐deficient mice showed normal shortening and re‐lengthening velocities in the absence of external load with normal peak systolic Ca2+ but slower Ca2+ re‐sequestration than wild‐type controls. The effects of isoproterenol on relaxation velocity, left ventricular systolic pressure and stroke volume were blunted in dysferlin‐deficient mouse hearts compared with that in wild‐type hearts. Young dysferlin‐deficient mouse hearts expressed normal isoforms of myofilament proteins whereas the phosphorylation of ventricular myosin light chain 2 was significantly increased, implying a molecular response to the impaired lusitropic function. These early phenotypes of diastolic cardiac dysfunction and blunted lusitropic response of cardiac muscle to β‐adrenergic stimulation indicate a novel pathogenic mechanism of dysferlin cardiomyopathy.
Abbreviations
- CaMKII
calmodulin kinase II
- cMyBP‐C
cardiac myosin binding protein‐C
- LGMD2B
limb‐girdle muscular dystrophy 2B
- DTT
dithiothreitol
- LVPmax
left ventricular systolic peak pressure
- LVPmin
left ventricular end diastolic pressure
- mAb
monoclonal antibody
- MHC
myosin heavy chain
- MLC2v
ventricular myosin light chain 2
- MM
Myoshimyopathy
- TnI
troponin I
- TnT
troponin T
- TP
time for reaching peak cytosolic calcium
- TR25
time for 25% calcium re‐sequestration
- TR75
time for 75% calcium re‐sequestration
Introduction
Dysferlin, an ∼230 kDa protein, is a member of the ferlin family and is expressed in adult mammalian cardiac and skeletal muscles, as well as multiple non‐muscle tissues including kidney, brain and lung (Han & Campbell, 2007). Dysferlin is associated with cell membrane through a C‐terminal transmembrane domain. Dysferlin contains multiple C2 domains, which enables interactions with calcium, phospholipids and partner proteins such as annexin(Lennon et al. 2003), calpain‐3, L‐type calcium channel (Ampong et al. 2005) and caveolin‐3 (Matsuda et al. 2001), to function in membrane trafficking and fusion, as well as cell signalling(Glover & Brown, 2007).
In mammalian skeletal muscle, dysferlin is predominantly associated with plasma membrane and enriched in the sub‐sarcolemma vesicles (Bansal et al. 2003). Mutations in the dysferlin gene cause three types of muscular dystrophies: limb girdle muscular dystrophy type 2B (LGMD2B), Myoshi myopathy (MM) and a distal anterior myopathy, which are collectively named dysferlinopathy. Dysferlinopathy presents clinically as an autosomal recessive disease of muscle weakness, often evident in young adulthood and slowly progressing during ageing and advancing under the stress of physical activities (Bashir et al. 1998; Liu et al. 1998).
Studies using various dysferlin‐deficient mouse models have suggested that defective myocyte membrane repair and attenuated muscle regeneration are the major pathological mechanisms that cause dysferlinopathy. Dysferlin was found to accumulate at injury sites of the plasma membrane of muscle fibres (Bansal et al. 2003). Dysferlin‐deficient mouse muscle fibres were defective with regard to calcium‐dependent membrane repair following laser‐induced membrane injury (Bansal et al. 2003; Doherty & McNally, 2003; Bansal & Campbell, 2004).
In cardiac muscle, dysferlin is found at the sarcolemma of cardiomyocytes and the intercalated discs (Chase et al. 2009). Some LGMD2B patients progressively develop dilated cardiomyopathy with reduced left ventricular ejection fraction and interstitial fibrosis (Kuru et al. 2004; Wenzel et al. 2007). In LGMD2B patients without cardiac symptoms, mild left ventricular hypertrophy is observed (Wenzel et al. 2007). A recent study using two‐dimensional strain echocardiography found left ventricular hypertrophy and decreased peak systolic longitudinal strain in some MM patients whose overall left ventricular systolic function was preserved (Choi et al. 2010).
Studies of dysferlin‐deficient animal models have found that cardiac muscle contractility and overall gene expression patterns were affected (Wenzel et al. 2007). Echocardiographic studies on dysferlin‐deficient SJL/J mice detected impaired left ventricular contractile function (Wenzel et al. 2007). It has been demonstrated that the loss of dysferlin led to defective repair of the cardiomyocyte membrane and cell death following β‐adrenergic stimulation‐induced increase of cardiac workload, resulting in loss of myocardial contractility and ventricular dilatation (Han et al. 2007). Significant interstitial fibrosis was found in ageing SJL/Jmouse hearts (Han et al. 2007), while ageing dysferlin‐deficient A/J mice also develop cardiomyopathy (Chase et al. 2009).
Although the evidence from clinical and animal studies clearly demonstrated significant heart involvement in dysferlin deficiency, the primary changes in cardiac muscle contractility in dysferlinopathy remain to be investigated. To understand the pathogenesis and pathophysiology of dysferlin cardiomyopathy, here we studied the function of ex vivo working hearts and isolated cardiomyocytes of young adult dysferlin gene knockout mice. The results showed early cardiac hypertrophy and delayed Ca2+ re‐sequestration, while the baseline cardiac function was largely compensated for. Young adult dysferlin knockout mouse hearts had a significantly blunted lusitropic response to β‐adrenergic stimulation, exhibiting reduced increases in left ventricular pressure development and stroke volume. Dysferlin‐deficient cardiac muscle showed an increased phosphorylation of regulatory myosin light chain, implying a molecular response to the impaired lusitropic function. The early phenotype of cardiac diastolic dysfunction indicates a novel pathogenic mechanism of dysferlin cardiomyopathy.
Materials and methods
Ethical approval
Animal studies were carried out under protocols approved by the Institutional Animal Care and Use Committee of Wayne State University.
Dysferlin knockout mice
Dysferlin gene targeted mice of strain C57BL/6, in which exons 53–55 are deleted, were purchased from The Jackson Laboratory (Bar Harbor, ME, USA; B6.129‐Dysftm1Kcam/J) (Bansal et al. 2003). Wild‐type C57BL/6 mice were purchased from Jackson Lab. as control. 12–16 weeks old mice of both sexes were used in the study. At this age, the dysferlin knockout mice showed only mild regeneration activity in skeletal muscles.
SDS‐PAGE and Western blotting
SDS‐PAGE and Western blotting were performed as described previously (Wei et al. 2010, 2013, 2014). Briefly, mouse cardiac muscle were rapidly dissected post‐mortem and frozen on dry ice for storage at −80°C. Muscle samples were homogenized in 40 volumes of SDS‐gel sample buffer containing 50 mmTris‐HCl, 2% SDS and 150 mmdithiothreitol (DTT), pH8.8, using a high speed mechanical homogenizer (Pro Scientific Inc., Oxford, CT, USA). The SDS‐gel samples were heated at 80°C for 5 min, clarified by centrifugation in a microcentrifuge at 17,000 g for 5 min, and loaded to 14% or 2–12% gradient SDS‐gel with an acrylamide/bis‐acrylamide ratio of 180:1 in Laemmli buffer at pH 8.8 for both stacking and resolving gels. Protein bands resolved in the gel were stained with Coomassie Blue R250 or blotted on a nitrocellulose membrane using a semidry electrical transfer device (Bio‐Rad, Hercules, CA, USA). The nitrocellulose membranes were blocked with 1% BSA in Tris‐buffered saline (TBS) and incubated with anti‐dysferlin monoclonal antibody (mAb) Ham1/7B6 (Abcam Inc., Cambridge, MA, USA), anti‐cardiac troponin T (TnT) mAb CT3 (Chong & Jin, 2009), anti‐troponin I (TnI) mAb TnI‐1 (Jin et al. 2001), an anti‐ventricular myosin light chain 2 (MLC2v) rabbit mAb (Abcam), an anti‐Ser282 phosphorylated cardiac myosin binding protein‐C (cMyBP‐C) antibody (Enzo Lifesciences, New York, USA), an anti‐calmodulin kinase II (CaMKII) rabbit mAb (Abcam), or an anti‐Thr286 phosphorylated CaMKII rabbit antiserum (Abcam), diluted in TBS containing 0.1% BSA, at 4°C overnight. The membranes were then washed with TBS containing 0.5% Triton X‐100 and 0.05% SDS and incubated with alkaline phosphatase‐conjugated anti‐mouse IgG or anti‐rabbit IgG second antibody (Santa Cruz Biotechnology, Dallas, TX, USA). Washed again, the membranes were developed in 5‐bromo‐4‐chloro‐3‐indolyl phosphate/nitro blue tetrazolium substrate solution to visualize the protein bands identified by the specific antibodies. The SDS‐gels and Western blots were scanned at 600 d.p.i. resolution and quantified by densitometry using NIH ImageJ software.
Glycerol SDS‐PAGE of myosin heavy chain isoforms
Cardiac myosin heavy chain (MHC) isoforms in wild‐type and dysferlin knockout mouse hearts were examined using glycerol‐SDS‐PAGE as described previously (Feng et al. 2009). Briefly, SDS‐gel samples were diluted to a final concentration of 1:24,000 (mg of muscle weight per μl). Five microlitres per lane was loaded on the SDS‐gel containing 30% glycerol, 0.4% SDS, 200 mmTris‐HCl and 100 mm glycine (pH8.8), and 8% acrylamide/bis‐acrylamide at a ratio of 50:1. The glycerol‐SDS‐gels were run at 100 V at 4°C for 24 h with the upper anode buffer containing 100 mm Tris base, 150 mm glycine, 0.1% SDS and 10 mm β‐mercaptoethanol and the lower cathode buffer being a 1:2 dilution of the upper buffer without β‐mercaptoethanol. The gels were stained with Coomassie Blue R250 to visualize the MHC isoform bands.
Histological studies
Wild‐type and dysferlin knockout mouse hearts were retrogradely perfused with Krebs–Henseleit buffer containing 50 mmKCl at 37°C to relax the cardiac muscle. After blotting on a paper towel to remove excessive buffer, the hearts were embedded in O.C.T. compound (Sakura Finetek USA, Inc., Torrance, CA, USA) and snap frozen in liquid‐nitrogen‐chilled 2‐methylbutane at −159°C for 30 s and then stored at −20°C overnight before cryosectioning. Cryosectionsof 5 μm were cut using a Leica CM1950 Cryostat (Leica Microsystems Inc., Buffalo Grove, IL, USA). The sections were plated on glass slides and stored at −80°C.
The sections were fixed in a solution containing 75% acetone and 25% ethanol for 5 min and washed with distilled water briefly to remove O.C.T. compound. The sections were stained with 0.1% (w/v) Mayer's haematoxylin (Electron Microscopy Sciences, Hatfield, PA, USA) solution containing 0.5% (w/v) ammonium aluminum sulphate for 15 min, washed with running tap water and dehydrated in 80% ethanol for 2 min twice. The dehydrated sections were stained with 0.25% Eosin Y (Sigma Aldrich, St Louis, MO, USA) in 80% ethanol for 30 s and then washed with 80% ethanol for 2 min twice and 100% ethanol once. The H&E‐stained sections were cleared with xylene, mounted in Permount solution (Fisher Scientific, Pittsburgh, PA, USA), examined using a Zeiss Axiovert 100 microscope and photographed.
Immunofluorescence microscopy
To localize dysferlin in cardiac muscle sections and in isolated cardiomyocytes, we performed double‐immunolabelling of dysferlin and desmin (a Z‐disc and intercalated disc protein). Cardiac muscle sections and cardiomyocytes were fixed with 50% acetone and 50% methanol for 5 min, blocked with 3% BSA in PBS containing 0.05% Tween‐20 (PBS‐T) and stained with rabbit anti‐dysferlin mAb Romeo (Abcam) and mouse anti‐desmin mAb D1033 (Sigma) at 4°C overnight, followed by PBS‐T washes and incubation with fluorescein isothiocyanate‐ or tetramethylrhodamine‐conjugated second antibody. Fluorescence images were taken using a Leica spinning disc confocal microscope.
Isolation of adult mouse cardiomyocytes
As described previously (Wei & Jin, 2014), mice were heparinized by i.p. injection of 100 units of heparin. Thirty minutes later, the mice were anaesthetized by i.p. injection of sodium pentobarbital (100 mg kg−1 body weight). Hearts were quickly dissected and cannulated through the aorta for retrograde Langendorff perfusion with a modified Krebs–Henseleit buffer (120 mmNaCl, 5.4 mmKCl, 1.2 mm MgSO4, 1.2 mm NaH2PO4, 5.6 mm glucose, 20 mm NaHCO3, oxygenated with 95% O2 and 5% CO2, 37°C) at constant flow of 3 ml min−1 for 30 min. The perfusion was switched to a circulating digestion solution containing 12.5 μm CaCl2 0.25 mg ml−1LiberaseBlendzyme I (Roche) and 0.014% trypsin (Invitrogen, Carlsbad, CA, USA) at 37°C for 15–20 min until the hearts turned pale and flaccid. The ventricles were dissected and disaggregated with gentle pipetting using a wide pore transfer pipette in a 10 cm Petri dish containing enzyme stopping buffer (Krebs–Henseleit buffer containing 2.5% BSA and 12.5 μm CaCl2) and the cell suspension was filtered through a 100 μm nylon mesh to remove tissue debris. The isolated cardiomyocytes were settled by gravity in a 15 ml conical tube for 15 min and re‐suspended in 12 ml fresh stopping buffer. Extracellular calcium was added gradually to reach 1 mm from a 100 mm stock in four steps at 20 min intervals. The cardiomyocytes were then photographed using a Zeiss Axiovert 100 microscope to measure resting cell length, cell width and sarcomere length using NIH ImageJ software.
Measurements of cardiomyocyte contractility and intracellular calcium transient
As described previously (Wei & Jin, 2014), cardiomyocytes were loaded into a perfusion chamber mounted on the stage of a Nikon Eclipse ST100 inverted microscope via a heating adaptor and perfused with oxygenated buffer containing 132 mmNaCl, 4.8 mmKCl, 1.2 mm MgCl2, 10 mm HEPES, 15 mm glucose, 2 mm sodium pyruvate and 1.8 mm CaCl2, pH7.4, at 37°C with constant flow of 1 ml min−1. Rod‐shaped cardiomyocytes with clear edges and sarcomeric striations were chosen for contractility studies under 2 Hz field pacing with 10 V pulses of 4 ms duration using a Myopacer stimulator (IonOptix, Milton, MA, USA) in the absence or presence of 3, 10 or 30 nm isoproterenol. Cell shortening and re‐lengthening were recorded using a CCD camera (IonOptix) at a sampling frequency of 240 Hz and analysed using the SoftEdge program (IonOptix).
Isolated cardiomyocytes were loaded with Fura‐2AM (Invitrogen) in the presence of 1.25 mm CaCl2 at 37°C in the dark for 10 min and washed with Joklik medium containing 1.25 mm CaCl2 and 500 μm probenecid at room temperature for 30 min. Cells were then transferred to the perfusion chamber and contractions were stimulated as above. The transient of cytosolic‐free calcium was measured by 510 nm excitation of Fura fluorescence and recording of the ratio of 360/380 nm emission using a photomultiplier tube (PMT‐300, IonOptix).
Ex vivo mouse working heart preparation and functional measurements
As described previously (Feng et al. 2008; Wei et al. 2010), wild‐type and dysferlin‐deficient mice were heparinized and anaesthetized, and the heart was rapidly isolated as above and cannulated using a modified 18G needle 6 mm long to start retrograde Langendorff perfusion within 3 min after opening the thoracic cavity. The pulmonary vein and pulmonary artery were cannulated using a 16G needle and a bevelled PE‐50 tubing, respectively. A 1.2 French pressure–volume catheter (model FTS‐1212B‐3518, Transonic Sciences Inc., London, ON, Canada) with 3.5 mm electrode spacing was calibrated in Krebs–Henseleit buffer at 37°C and inserted through the apex into the left ventricle of the mouse heart via a path made using a 30G needle. The aortic cannula was connected to an MLT844 pressure transducer (Capto, Horten, Norway). A 0.5 ml air bubble was introduced in the aortic outflow track to mimic the in vivo aortic compliance.
After establishing the cannulations, the heart was switched to working mode with antegrade perfusion and allowed to stabilize for 10 min under 8 Hz (480 beats per minute) supraventricular pacing using an isolated stimulator (A365, World Precision Instruments, Sarasota, FL, USA) via a pair of platinum electrodes. Aortic pressure and left ventricular pressure and volume data were recorded at a sampling rate of 1000 Hz using a Powerlab 16‐channel digital data acquiring system and analysed using Chart 5 software (ADInstruments, Colorado Springs, CO, USA). Aortic output and pulmonary effluent from coronary flow were recorded by direct drop counting using a pair of polarized copper electrodes connected to the data acquisition system. Baseline heart function was recorded at 10 mmHg preload and 55 mmHg afterload. Stroke volume was measured as the sum of aortic and coronary flows and normalized to heart weight. Left ventricular maximum and minimum pressures and changes (±dP/dt), and pressure–volume loop were calculated using Chart 5 software.
Responses to stepwise alterations of preload between 5 and 20 mmHg were tested at 55 mmHg afterload. Responses to stepwise increases of afterload up to 90 mmHg were tested at 10 mmHg preload. Heart function was allowed to stabilize for 3 min before sampling data at each preload and afterload. Isoproterenol (10 nm; Sigma Aldrich) was given via antegrade perfusion to examine the effects on cardiac function.
Pro‐Q Diamond phosphoprotein staining
Protein extracts from cardiac muscle were resolved on SDS‐PAGE as above for staining of phosphoprotein bands with Pro‐Q Diamond reagent (Invitrogen) according to the manufacturer's protocol. The gel was fixed in 50% methanol and 10% acetic acid overnight, washed with deionized water three times, 10 min each, and incubated in Pro‐Q Diamond staining solution with shaking in the dark for 1 h. The gel was de‐stained in the dark with 20% acetonitrile, 50 mm sodium acetate at pH 4.0 three times, 30 min each. After further washes with deionized water three times, 5 min each, the gel was imaged using a Typhoon 9410 fluorescence scanner (GE Healthcare) with excitation at 532 nm, recording the emission at 560 nm, and analysed using NIH ImageJ software. The gel was then re‐stained with Coomassie Blue R‐250 and scanned to quantify the input of proteins.
Measurement of MLC2v phosphorylation
Urea‐glycerol‐PAGE was used to examine the phosphorylation of cardiac MLC2v as previously described (Huang et al. 2004). Briefly, proteins in the cardiac muscle lysates were precipitated using 10% trichloroacetic acid (Sigma) containing 10 mm DTT on ice for 30 min. The precipitates were collected by centrifugation at 14,000 g at 4°C for 10 min, followed by washing with 10% trichloroacetic acid, 10 mm DTT three times and 100% ice cold acetone twice. The protein precipitates were air‐dried at room temperature for 15 min and re‐suspended in a gel sample buffer containing 8 m urea, 10 mm DTT, 22 mm Tris‐HCl, pH 8.6, 22 mm glycine, 5% (v/v) glycerol and 0.1% (w/v) bromphenol blue. The samples were incubated at room temperature for 45 min to dissolve the proteins and run on a 12% gel with acrylamide/bis‐acrylamide at a ratio of 29:1 containing 40% (v/v) glycerol, 22 mm Tris‐HCl, pH 8.6, and 22 mm glycine at 200 V for 5 h. The running buffer contained 22 mm Tris‐HCl, pH 8.6, 22 mm glycine, 10 mm DTT and 0.1% thioglycolic acid. The resulting gel was electrically transferred as above to blot the nitrocellulose membrane. Non‐phosphorylated and phosphorylated MLC2v bands with distinct gel mobility were quantified by densitometry scan using NIH ImageJ software.
Data analysis
The SDS‐PAGE gel and Western blot images were scanned at 600 d.p.i. for quantitative studies. All data are expressed as mean ± SEM. Two‐way ANOVA and Student's t test were used to determine the statistical significance of differences.
Results
Early and up‐regulated expression of dysferlin in mouse heart during postnatal development
Previous studies have detected dysferlin in cardiac muscle in intracellular vesicles, sarcoplasmic membrane (Han et al. 2007), at the transverse tubules (Ampong et al. 2005) and intercalated discs (Chase et al. 2009), as confirmed in the C57BL/6 mouse strain studied here with similar levels in the free walls of left and right ventricles, the septum and atria (Figs. 1 A and 2).
Figure 1. Expression of dysferlin in mouse cardiac muscle and up‐regulation during postnatal development .
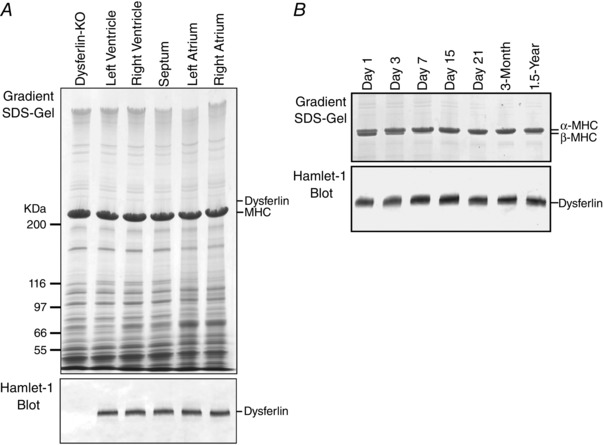
A,representative gradient SDS‐PAGE gel and Western blot showed that dysferlin is expressed at similar levels in left, right ventricular and atrial muscles of adult mouse heart. B, Western blot detected a significant amount of dysferlin in ventricular muscle at birth, which was visibly up‐regulated after 7 days during postnatal development. The 1.5‐year‐old mouse heart remained with high level of dysferlin, whereas 2–12% gradient SDS‐gel detected a small amount β‐myosin heavy chain (MHC), indicating an ageing‐related adaptation.
Figure 2. Localization of dysferlin at intercalated disc and transverse tubular membrane of young adult mouse heart .
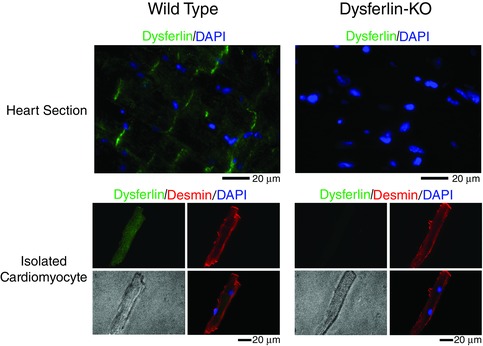
Immunofluorescence microscopic images of cardiac muscle sections and isolated cardiomyocytes confirmed the localization of dysferlin at intercalated discs and transverse tubular membranes of C57BL/6 mice. Desmin and cell nuclei were stained as structural references.
We examined the expression of dysferlin in cardiac muscle during development. The results in Fig. 1 B showed the expected switching of β‐ to α‐MHC in day 1–7 postnatal mouse hearts. Dysferlin was expressed at a significant level in newborn hearts with visible increases during postnatal development. The most notable increase was from postnatal day 3 to day 7 when the expression of β‐myosin was diminished. The level of dysferlin in wild‐type mouse cardiac muscle showed no significant change from 2 weeks to 1.5 years of age, despite a detectable re‐expression of β‐myosin in the 1.5‐year‐old heart reflecting ageing‐related adaptation (Compagno et al. 2001). The plateaued expression of dysferlin in normal young mouse cardiac muscle validated the use of ∼3‐month‐old dysferlin knockout and control mice to study cardiomyopathy resulting from dysferlin deficiency.
Dysferlin‐deficient mouse hearts had detectable hypertrophy at 3 months of age
Some LGMD2B patients without overt cardiac symptoms had mild left ventricular hypertrophy (Wenzel et al. 2007). Normalized to body weight, dysferlin‐deficient mice also exhibited mild cardiac hypertrophy at 3 months of age in comparison with wild‐type control, indicating an early adaptation (Fig. 3 B). Body weight and tibial length of dysferlin knockout mice were normal as compared with wild‐type mice, indicating that dysferlin deficiency did not hinder overall development and growth.
Figure 3. Detectable hypertrophy of dysferlin‐deficient mouse hearts at 3 months of age .
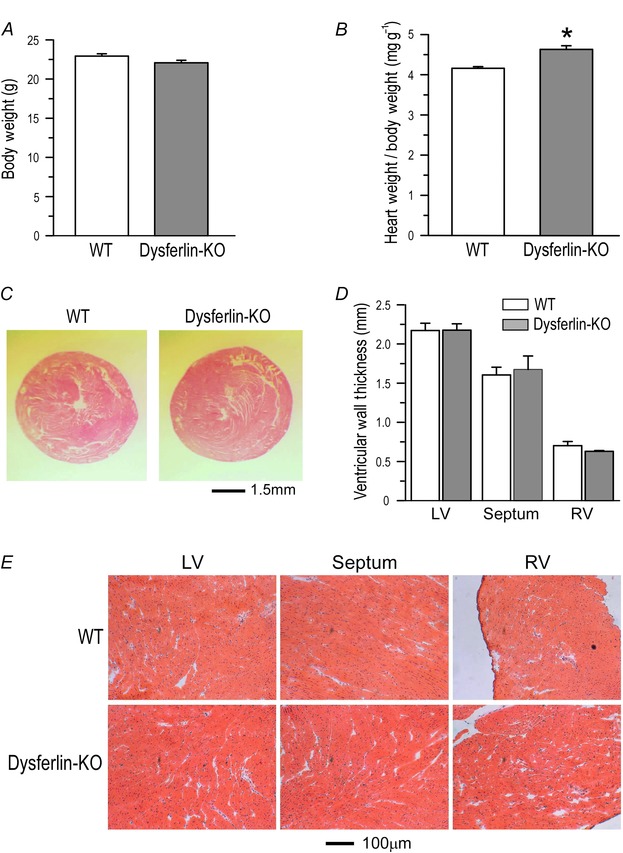
A, at 3 months, body weight and tibial lengthof dysferlin knockout mice showed no difference from wild‐type controls. B, heart weight/body weight ratio of dysferlin knockout mice (4.63±0.09) was higher than that of wild‐type mice (4.16±0.04) (*P<0.05 in Student's t test). C, H&E‐stained cross‐sections showed no ventricular dilatation of dysferlin knockout mouse heart in comparison with that of wild‐type heart. D, thicknesses of left and right ventricular wall and septum of dysferlin knockout mouse hearts showed no difference from wild‐type controls. E, H&E‐stained sections showed no fibrosis in 3‐month‐old dysferlin knockout mouse hearts.
Histological studies showed that dysferlin‐deficient mouse hearts had normal morphology at 3 months of age (Fig. 3 C), without detectable change in chamber size and wall thickness (Fig. 3 D) or fibrosis (Fig. 3 E).
Cardiomyocytes isolated from young adult dysferlin‐deficient mice showed no significant change in morphology and sarcomere length
Cardiomyocytes isolated from ∼3‐month‐old dysferlin knockout and wild‐type mice showed similar morphology (Fig. 4 A). Dysferlin‐deficient cardiomyocytes showed similar length (133.26±5.04 μm) and width (27.15±1.89 μm) to that of wild‐type cells (126.26±3.29 and 24.53±1.47 μm, respectively; Fig. 4 B). The slack sarcomere length of dysferlin‐deficient cardiomyocytes (1.82±0.003 μm) was not different from wild‐type control (1.81±0.009 μm) (Fig. 4 B). The absence of significant remodelling in cellular structure indicates a compensated state of the young adult dysferlin‐deficient mouse hearts studied.
Figure 4. Cardiomyocytes isolated from dysferlin knockout mouse hearts showed no significant change in morphology .
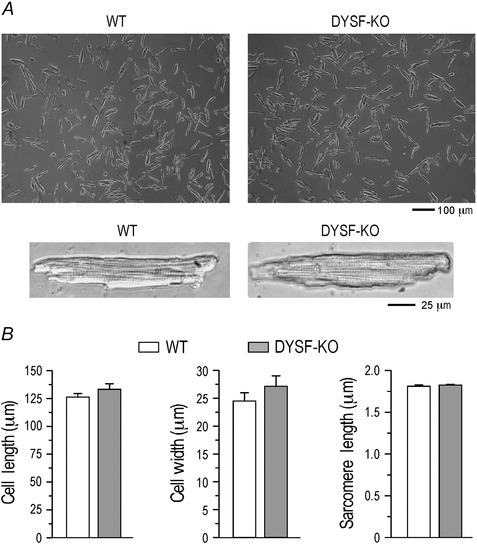
A,representative images of cardiomyocytes isolated from 3‐month‐old dysferlin knockout mice showed normal morphology and pattern of striation. B,the length and width of dysferlin‐deficient cardiomyocytes showed a slight trend of increases from that of wild‐type control, reflecting the hypertrophic phenotype. However, no statistical significance was established at the young age. Resting sarcomere length was not different in dysferlin knockout and wild‐type cardiomyocytes.
Dysferlin‐deficient mouse hearts express normal isoforms of myofilament proteins
Western blots showed that dysferlin‐deficient mouse hearts had normal compositions of the thin filament proteins cardiac TnT and cardiac TnI at ∼3 months of age (Fig. 5 A). Glycerol‐SDS‐PAGE revealed that dysferlin‐deficient mouse hearts contained exclusively α‐MHC (Fig. 5 B), indicating preserved normal isoforms of thick filament proteins. These findings further demonstrate a compensated state of the young adult dysferlin‐deficient mouse hearts.
Figure 5. Dysferlin‐deficient young adult mouse hearts expressed normal isoforms of myofilament proteins .
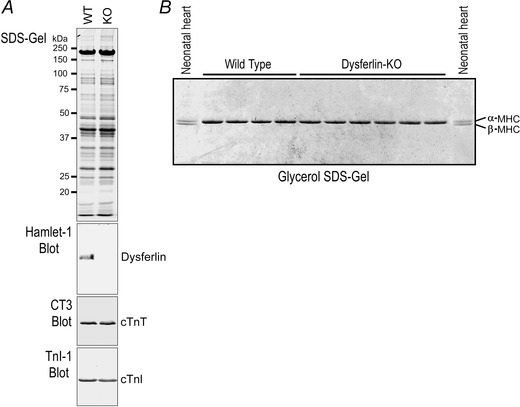
A, SDS‐PAGE gel and Western blots showed that at ∼3 months old, dysferlin knockout mouse cardiac muscle expressed normal cardiac TnT and cardiac TnI. B, glycerol‐SDS‐gel showed that ventricular muscles of ∼3‐month‐old dysferlin knockout mice expressed exclusively α‐MHC, as that in wild‐type control hearts. Neonatal wild‐type mouse heart expressing both α‐ and β‐MHC was included as control.
Comparable contractility of cardiomyocytes isolated from dysferlin‐deficient and wild‐type mice
Paced at 2 Hz, isolated dysferlin‐deficient mouse cardiomyocytes showed shortening amplitudes similar to that of wild‐type cells (Fig. 6 A). Shortening and re‐lengthening velocities were similar between dysferlin‐deficient and wild‐type cardiomyocytes (Fig. 6 B and C). Isoproterenol at 3–30 nm produced comparable increases in contractile amplitude and shortening and re‐lengthening velocities in the two groups (Fig. 6). The results demonstrate that in the absence of external load, cardiomyocyte contractility and β‐adrenergic responses were not affected by dysferlin deficiency at the young age.
Figure 6. Contractility of cardiomyocytes isolated from young adult dysferlin‐deficient and wild‐type mice .
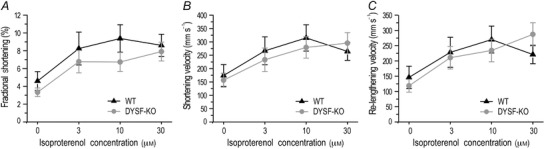
In the absence of external load, dysferlin knockout cardiomyocytes exhibited shortening amplitude (A), shortening (B) and re‐lengthening (C) velocities similar to wild‐type controls at baseline and under isoproterenol stimulation.
Effects of dysferlin deficiency on calcium transient in cardiomyocytes
It has been suggested that dysferlin is enriched at the transverse tubules of skeletal muscle cells (Klinge et al. 2010; Kerr et al. 2013). Through interaction with L‐type calcium channel and other proteins within the transverse tubule membrane (Matsuda et al. 2001; Ampong et al. 2005), dysferlin may play a role in the development and maintenance of the transverse tubule network and intracellular calcium homeostasis under stress conditions (Kerr et al. 2013). Our results showed that paced at 2 Hz, resting cytosolic calcium in the dysferlin‐deficient cardiomyocytes was lower than the wild‐type control level in the presence of 3 and 10 nm isoproterenol, although statistical significance was not established (Fig. 7 A). The peak calcium level was not different between the two groups (Fig. 7 B).
Figure 7. Altered cytosolic calcium transient in dysferlin‐deficient cardiomyocytes .
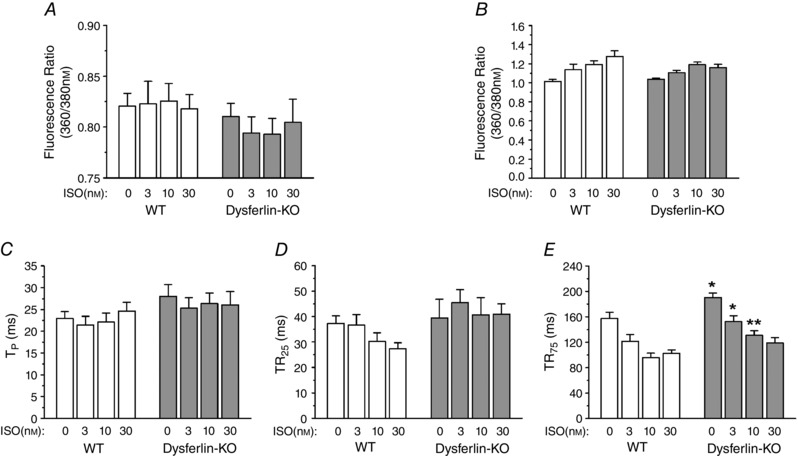
A, when stimulated with 3 or 10 nm isoproterenol, the level of resting cytosolic calcium in dysferlin knockout cardiomyocytes during paced contractions showed a trend of lower levels as compared with that in wild‐type cells, although statistical significance was not established. B–D, the peak cytosolic calcium (B), time to peak calcium (TP) (C) and time to 25% calcium re‐sequestration (TR25) (D) were not different between dysferlin knockout and wild‐type cardiomyocytes. E, the time to 75% calcium re‐sequestration (TR75) was significantly longer in dysferlin knockout cardiomyocytes than that in wild‐type cells at baseline and under 3 or 10 nm isoproterenol treatment (*P<0.05 in Student's t test). Isoproterenol at 30 nm diminished the difference.
The calcium transient in dysferlin‐deficient and wild‐type cardiomyocytes showed no significant difference in the time to reach peak cytosolic calcium (TP, Fig. 7 C) and the time for 25% re‐sequestration (Fig. 7 D). However, the time for 75% calcium re‐sequestration was significantly longer in dysferlin‐deficient cardiomyocytes than in wild‐type cells (Fig. 7 E). β‐Adrenergic stimulation accelerated calcium re‐sequestration in both groups, as shown by the shortened TR75, of which 30 nm isoproterenol produced a complete correction in dysferlin‐deficient cardiomyocytes. The results indicate that dysferlin deficiency causes an impairment of calcium homeostasis, especially calcium re‐sequestration, in young adult cardiomyocytes.
Dysferlin deficiency impairs lusitropic function of ex vivo working hearts
Heart function was affected in patients with LGMD2B (Wenzel et al. 2007; Choi et al. 2010) and dysferlin‐deficient mice (Wenzel et al. 2007). Using ex vivo working hearts in the absence of systemic influence, our present study compared the ventricular contractile and relaxation velocities, systolic peak pressure and end diastolic pressure, and stroke volume between dysferlin‐deficient and wild‐type mouse hearts under various levels of preload, afterload and β‐adrenergic stimulation.
The results are summarized in Table 1. At baseline, young adult dysferlin‐deficient and wild‐type mouse hearts showed similar left ventricular contractile and relaxation velocities, left ventricular systolic peak pressure (LVPmax) and end diastolic pressure (LVPmin). Dysferlin‐deficient mouse hearts had a trend of lower baseline stroke volume than that of wild‐type hearts, although statistical significance was not reached (P = 0.074 in two‐tailed Student's t test). These findings indicate a partially compensated baseline function at young age in dysferlin‐deficient mice.
Table 1.
Baseline cardiac function and β‐adrenergic response of ex vivo working mouse hearts
| Wild‐type (n = 7) | Dysf‐KO (n=6) | ||
|---|---|---|---|
| dP/dt (mmHg s–1) | Baseline | 3862.44 ± 127.56 | 3750.34 ± 131.66 |
| 10 nm ISO | 7222.41 ± 192.09# | 7064.61 ± 120.33# | |
| –dP/dt (mmHg s–1) | Baseline | −2754.35 ± 130.03 | −2826.79 ± 67.28 |
| 10 nmISO | −4955.45 ± 188.07# | −4000.33 ± 140.37#, * | |
| LVPmax (mmHg) | Baseline | 75.15 ± 2.41 | 73.54 ± 2.01 |
| 10 nmISO | 98.55 ± 3.44# | 88.84 ± 0.99#, * | |
| LVPmin (mmHg) | Baseline | 5.02 ± 0.58 | 3.96 ± 0.78 |
| 10 nmISO | 2.67 ± 0.87# | 2.26 ± 0.571# | |
| Stroke volume (μl mg–1) | Baseline | 0.14 ± 0.01 | 0.12 ± 0.01& |
| 10 nmISO | 0.20 ± 0.01# | 0.14 ± 0.01#, * |
Functions of ex vivo working mouse hearts were measured at 10 mmHg preload, 55 mmHg afterload and 8 Hz pacing. At baseline, dysferlin knockout (Dysf‐KO) mouse hearts showed a trend of lower stroke volume than wild‐type controls (& P = 0.074 in two‐tailed Student's t test). Isoproterenol at 10 nm increased the function of both groups (# P<0.05 compared with baseline in Student's t test), in which dysferlin knockout hearts responded with lower increases than that of wild‐type hearts (*P<0.05 compared with wild‐typegroup in Student's t test).
Isoproterenol at 10 nm produced positive effects on both dysferlin‐deficient and wild‐type mouse hearts (Table 1). However, relaxation velocity, LVPmax and stroke volume of dysferlin‐deficient hearts showed blunted β‐adrenergic responses whereas the contractile velocity response was normal (Table 1, Fig. 8).
Figure 8. Decreased lusitropic response of dysferlin‐deficient mouse hearts to β‐adrenergic stimulation .
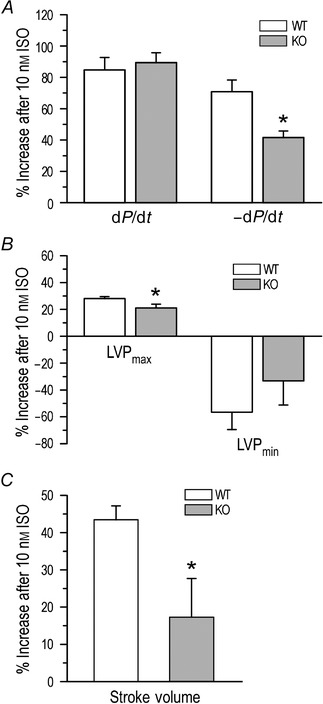
Ex vivo working heart studies demonstrated that in contrast to that of wild‐type hearts, dysferlin‐deficient mouse hearts had blunted responses to 10 nm isoproterenol. A, lower increase in relaxation velocity (41.57±4.18% vs. 70.81±7.50%) with no difference in contractile velocity; B, lower increase in left ventricular peak systolic pressure (LVPmax, 21.14±2.76% vs. 28.08±1.45%) with no difference in left ventricular end diastolic pressure (LVPmin); and C, lower increase in stroke volume (17.03±10.29%vs. 43.03%±3.37%). *P<0.05 in Student's t test.
The impaired function of dysferlin‐deficient mouse hearts is independent of changes in afterload and preload
The ex vivo working heart approach readily allows adjustments of preload and afterload while measuring multiple functional responses in real time (Feng et al. 2008; Wei et al. 2010). The results confirmed the decreased stroke volume of dysferlin‐deficient hearts, which remained lower than wild‐type controls at afterload ranging between 55 and 90 mmHg (Fig. 9, measured at 10 mmHg preload). Similarly, the stroke volume of dysferlin‐deficient hearts remained lower than that of wild‐type hearts when preload was altered between 5 and 20 mmHg (Fig. 10, measured at 55 mmHg preload). Nonetheless, the Frank–Starling response to the increases in preload was preserved in dysferlin‐deficient hearts (Fig. 10).
Figure 9. Decreased function of dysferlin‐deficient mouse hearts was independent of afterload .
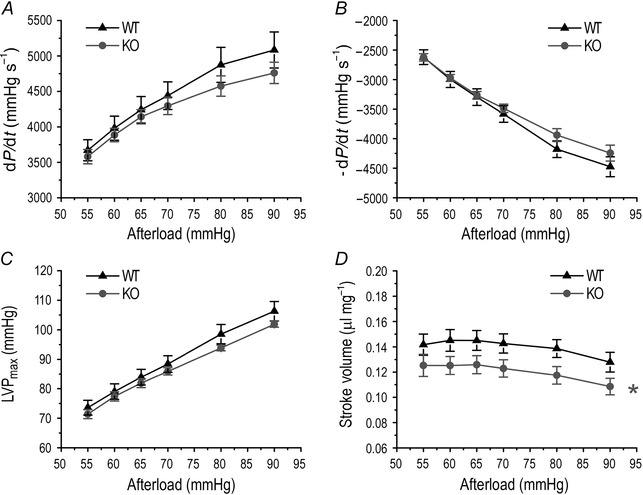
Ex vivo working heart studies showed that when afterload was altered between 55 and 90 mmHg, contractile (A) and relaxation (B) velocities and maximum left ventricular pressure (LVPmax) (C) increased, whereas the stroke volume showed a trend of decrease (D). Similar responses to the change in afterload were seen for dysferlin knockout and wild‐type mouse hearts. The stroke volume of dysferlin knockout mouse hearts was lower than wild‐type controls at all afterload levels studied (D, *P<0.05 in two‐way ANOVA).
Figure 10. Decreased function of dysferlin‐deficient mouse hearts was independent of preload with preserved Frank‐Starling response .
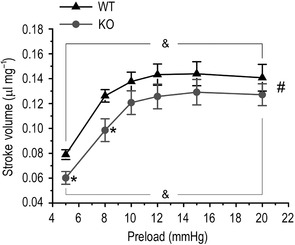
Ex vivo working heart study showed that when preload was altered between 5 and 20 mmHg, the stroke volume of dysferlin knockout mouse hearts was lower than that of wild‐type hearts at all preload levels examined (*P<0.05 in Student's t‐test and # P<0.05 in two‐way ANOVA). However, dysferlin knockout and wild‐type groups exhibited parallel Frank–Starling responses to increases in preload (& P<0.05 when stroke volumes were compared with that at preload of 5 mmHg in two‐way ANOVA).
Increased phosphorylation of ventricular myosin light chain 2 in young adult dysferlin‐deficient mouse hearts
Pro‐Q Diamond staining showed that in isoproterenol‐treated working hearts and rapidly isolated hearts of dysferlin‐deficient mice, the phosphorylation of thin filament proteins cardiac TnT and cardiac TnI was not different from that in wild‐type control hearts (Fig. 11). Pro‐Q staining of SDS‐gel showed that dysferlin‐deficientex vivo working hearts treated with 10 nm isoproterenol had a trend of higher level of phosphorylation of MyBP‐C than wild‐type control (Fig. 11 A), but this was not seen in hearts rapidly isolated from anaesthetized mice (Fig. 11 B). MyBP‐C phosphorylation is protein kinase A (PKA)‐dependent (Kuster et al. 2012). However, phosphorylation of cardiac TnI was unchanged in dysferlin‐deficient mouse hearts (Fig. 11), precluding an overall increase in β‐adrenergic tone.
Figure 11. Increased phosphorylation of MLC2v in young adult dysferlin‐deficient mouse hearts .
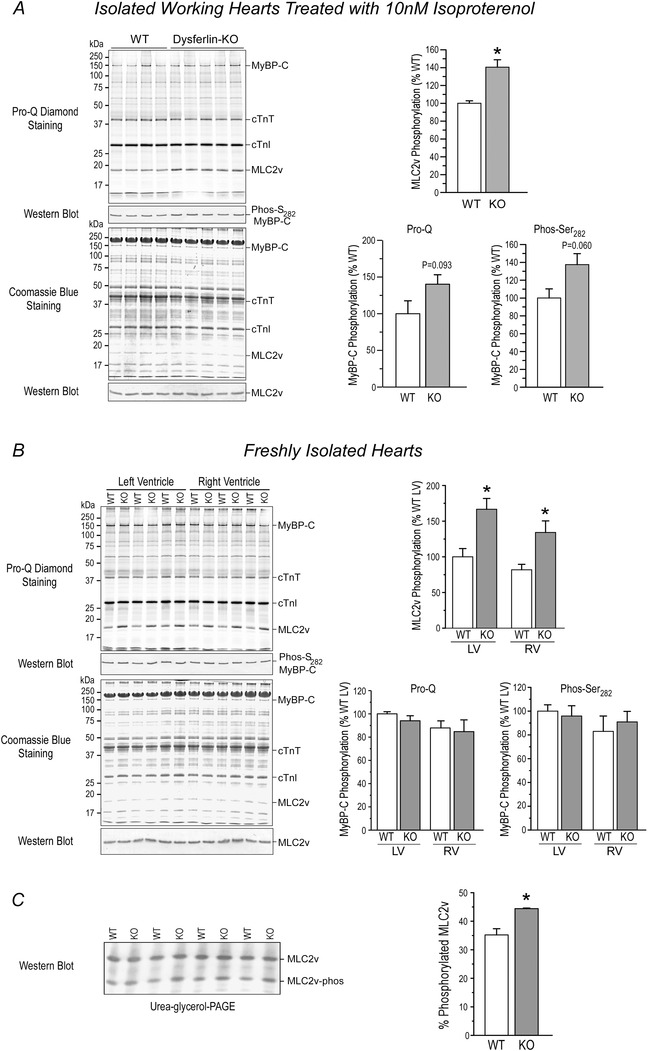
A, representative Pro‐Q‐stained gel of total protein extracts from isolated working mouse hearts treated with 10 nm isoproterenol and densitometry quantification showed a 52.10±4.1% higher phosphorylation of MLC2v in dysferlin knockout hearts as compared with wild‐type controls (*P<0.05 in Student's t test). Phosphorylation of cardiac TnT and cardiac TnI was not different in the two groups. Pro‐Q diamond stain detected a trend of increase in the phosphorylation of MyBP‐C in dysferlin knockout hearts (40.16±12.61% higher than wild‐type control, P = 0.093 in two‐tailed Student's t test). Western blot further showed a similar trend of increased MyBP‐C phosphorylation at Ser282 (37.47±12.36% higher than wild‐type control, P = 0.060 in two‐tailed Student's t test). B, representative Pro‐Q‐stained gel of rapidly isolated cardiac muscle and densitometry analysis confirmed the increased phosphorylation of MLC2v in dysferlin knockout mouse hearts in vivo as compared with that in wild‐type mice (increased by 58.42±5.28% in left ventricle and 54.16±2.72% in right ventricle). In contrast to ex vivo working hearts treated with 10 nm isoproterenol, dysferlin knockout mouse hearts rapidly isolated under anaesthesia did not show the trend of in vivo increases in MyBP‐C phosphorylation. C, representative Western blot of urea‐glycerol‐PAGE gel and densitometry analysis confirmedthe increased phosphorylation of MLC2v in dysferlin knockout mouse working hearts treated with 10 nm isoproterenol (44.43±0.18% of total MLC2v vs. 35.19±2.20% in wild‐type control hearts). *P<0.05 in Student's t‐test.
Anti‐phospho‐Ser282blot detected a similar increase in the phosphorylation of MyBP‐C (Fig. 11). Previous studies showed that MyBP‐C phosphorylation at Ser282 was also catalysed by CaMKII (Gautel et al. 1995). However, the protein level and Thr286 phosphorylation of CaMKII were not altered in the cardiac muscle of young dysferlin‐deficient mice (Fig. 12).
Figure 12. Protein level and Thr286 phosphorylation of CaMKII were not altered in the cardiac muscle of young dysferlin‐deficient mice .
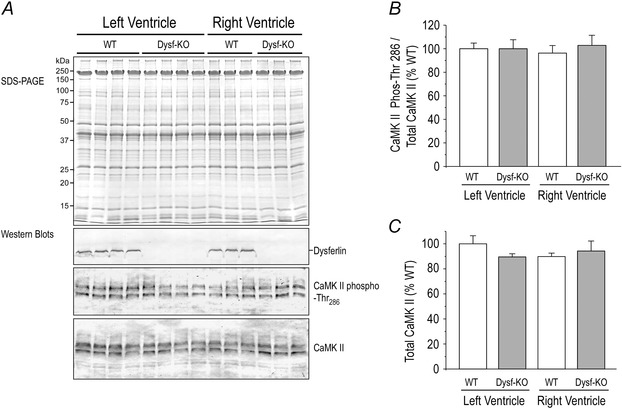
Representative Western blot (A) and densitometry analysis showed that compared with wild‐type controls, total CaMKII protein (B) and phosphorylation at Thr286 (C) were not altered in left and right ventricular cardiac muscles of 3‐month‐old dysferlin knockout mice.
In contrast, Pro‐Q Diamond staining showed that dysferlin‐deficientyoung adult mouse hearts had significant increases in the phosphorylation of MLC2v (Fig. 11 A and B), which was confirmed using urea‐glycerol‐PAGE (Fig. 11 C).
Discussion
Dysferlin is found at the sarcoplasmic membrane of skeletal and cardiac muscles (Chase et al. 2009; Roche et al. 2011). In addition to skeletal muscle phenotypes (Liu et al. 1998; Illarioshkin et al. 2000), impaired cardiac function and myocardial hypertrophy were seen in LGMD2B patients. The lesions of dysferlin knockout mouse hearts, such as interstitial fibrosis and ventricular dilatation, became obvious in old age due to insufficient membrane repair (Han et al. 2007). To understand the primary pathology and pathophysiology of cardiac dysferlinopathy, our present study examined the hearts of young adult dysferlin knockout mice and provided the following mechanistic insights.
Myocardial hypertrophy with compensated cardiac function
An earlier study reported concentric ventricular hypertrophy in human patients with LGMD2B at middle age (Wenzel et al. 2007). Ventricular dilatation and interstitial fibrosis were found in ageing dysferlin knockout mice (Han et al. 2007). Our present study showed emerging cardiac hypertrophy in dysferlin knockout mice at ∼3 months of age. This result indicates that dysferlin deficiency causes early myocardial remodelling before overt degeneration of cardiac muscle. Therefore, the pathogenesis of cardiac dysferlinopathy may involve primary changes in cardiomyocyte function in addition to accumulated muscle membrane injury like that seen in skeletal muscle dysferlinopathy. While cardiac hypertrophy was found at the organ level in 3‐month‐old dysferlin‐deficient mice (Fig. 3), isolated cardiomyocytes only showed a slight trend of increase in size (Fig. 4), consistent with an early stage of hypertrophy.
Delayed re‐sequestration of cytosolic calcium in cardiomyocytes
In cardiac muscle, dysferlin has been shown to interact with the L‐type calcium channel and other proteins within the transverse tubule membrane region (Ampong et al. 2005). Dysferlin is also involved in the development and maintenance of the transverse tubule network and regulating intracellular calcium homeostasis in skeletal muscle cells, especially under stress conditions (Kerr et al. 2013). In cardiomyocytes isolated from young adult dysferlin‐deficient mice, we found that the cytosolic calcium re‐sequestration, but not release, was significantly delayed (Fig. 7). This suggests that dysferlin is involved in calcium homeostasis in cardiomyocytes and the delayed cytosolic calcium re‐sequestration may be an underlying mechanism for cardiomyopathies seen in LGMD2B patients. Note that some LGMD2B patients showed abnormal ECG, including delay of intraventricular conduction and abnormal repolarization (Wenzel et al. 2007). Together with our result, this evidence suggests that dysferlin deficiency may lead to calcium homeostasis‐related electrophysiological abnormalities in dysferlin cardiomyopathy.
In cardiomyocytes, calcium re‐sequestration is largely governed by the phospholamban‐regulated function of the SERCA (sarcoendoplasmic reticulum (SR) calcium transport ATPase)pump, of which defects could prolong the late phase (TR75) of calcium re‐sequestration. However, Western blotting and densitometry analysis showed that the protein level, phosphorylation and subcellular distribution of phospholamban in dysferlin knockout cardiomyocytes were not different from that in wild‐type cells (Fig. 13). The observation that the delayed calcium re‐sequestration in dysferlin‐deficient cardiomyocytes was not due to a defect in SERCA pump activity or capacity is supported by the lack of elevation of resting cytosolic calcium and the correction of TR75 by high dose of isoproterenol (Fig. 7). Further studies are required to identify the mechanism for the altered calcium homeostasis in dysferlin‐deficient cardiomyocytes.
Figure 13. No change in phospholamban in young adult dysferlin‐deficient mouse cardiomyocytes .
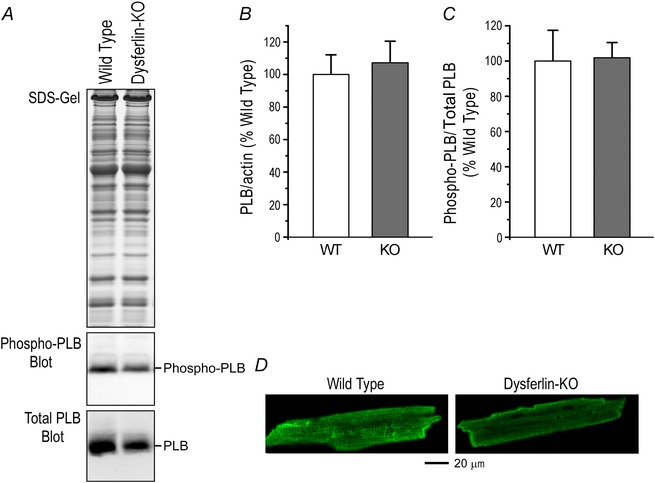
Representative Western blot (A) and densitometry analysis showed that the protein level (B) and Ser16 phosphorylation (C) of phospholamban were not altered in 3‐month‐old dysferlin knockout mouse cardiomyocytes. D, immunofluorescence microscopy showed similar subcellular distribution of phospholamban in dysferlin knockout and wild‐type mouse cardiomyocytes.
Changes in calcium homeostasis in cardiomyocytes may affect the calcium–calmodulin signalling pathway to lead to cardiac hypertrophy and heart failure (Maier & Bers, 2002; Zhang & Brown, 2004). CaMKII plays a significant role in this signalling pathway and phosphorylation at Thr286 is a regulatory site (Anderson et al. 2011). However, SDS‐PAGE and Western blot results in Fig. 12 showed no changes in the protein level and phosphorylation of CaMKII at Thr286 in the cardiac muscle of young dysferlin knockout mice, consistent with a compensated state.
Decreased lusitropic response to β‐adrenergic stimulation
Clinical studies have reported impaired cardiac function in patients with LGMD2B and MM (Wenzel et al. 2007; Choi et al. 2010). Our ex vivo working heart data showed that baseline cardiac contractile and relaxation functions were both preserved in the dysferlin‐deficient mouse heart. However, β‐adrenergic stimulation produced significantly lower increases in relaxation velocity, left ventricular pressure development and stroke volume (Table 1 and Fig. 8). The impaired lusitropic function indicates a phenotype of diastolic heart failure in dysferlin‐deficient mice.
The decreased lusitropy of dysferlin knockout cardiac muscle with delayed calcium re‐sequestration and blunted β‐adrenergic response implies an impaired β‐adrenergic signalling. As the phosphorylation of cardiac TnI and myosin binding protein C, two known PKA substrates under β‐adrenergic signalling, was not decreased in dysferlin knockout hearts (Fig. 11), upstream mechanisms such as internalization of betareceptors is less likely to be the determinant. The level of phospholamban was also unchanged in dysferlin knockout hearts (Fig. 13). Therefore, further studies are needed to explore the molecular mechanism for the impaired lusitropy and blunted β‐adrenergic response of the dysferlin‐deficient heart.
Although the decreased lusitropic function would contribute to decreased cardiac performance by negatively impacting on the Frank–Starling response, the impaired functions of dysferlin‐deficient mouse hearts were independent of changes in afterload and preload (Figs. 9 and 10), and the Frank–Starling response was preserved although at a lower level (Fig. 10). By contrast, the blunted lusitropic response to β‐adrenergic stimulation was not seen in isolated cardiomyocytes examined in the absence of external load (Fig. 6). The correlation of cardiac muscle lusitropic dysfunction in dysferlin deficiency with mechanical loads may underlie the primary pathology and early pathophysiology of cardiac dysferlinopathy as a case of diastolic cardiac dysfunction and merits further investigation.
Differentially increased phosphorylation of MLC2v and MyBP‐C
Young adult dysferlin‐deficient mouse hearts had normal isoform composition of MHC, cardiac TnT and cardiac TnI, consistent with the compensated state of cardiac function. A primary mechanism regulating cardiac muscle contractility is the phosphorylation of various thin and thick filament proteins, including cardiac TnI (Solaro et al. 1976; Moir & Perry, 1980; Garvey et al. 1988; Zhang et al. 1995), MyBP‐C (Garvey et al. 1988; Gautel et al. 1995; Weisberg & Winegrad, 1996; Kunst et al. 2000) and MLC2v (Frearson & Perry, 1975; Holroyde et al. 1979; Resink et al. 1981). Our study of young adult dysferlin‐deficient mouse hearts did not detect significant changes in the phosphorylation of cardiac TnIand cardiac TnT, but MyBP‐C showed a trend of higher phosphorylation in 10 nm isoproterenol‐treated ex vivodysferlin knockout mouseworking hearts (Fig. 11).
β‐Adrenergic/PKA‐dependent phosphorylation of MyBP‐C at Ser282 (Fig. 11 A) is known to accelerate the force generation of cardiac muscle (Edes et al. 1995; Tong et al. 2004; Solaro et al. 2013). MyBP‐C phosphorylation was decreased in hypertrophic and failing hearts (Copeland et al. 2010). While the increased phosphorylation of MyBP‐C inyoungadult dysferlin‐deficient mouse hearts may be an adaptive and compensatory response, the phosphorylation of cardiac TnI, which is also β‐adrenergicPKA‐dependent, in dysferlin‐deficient mouse hearts was similar to that in wild‐type hearts (Fig. 11), precluding an overall increase in β‐adrenergic tone.
An interesting finding in our study was the significant increase in the phosphorylation of MLC2v in dysferlin knockout mouse hearts (Fig. 11). Phosphorylation of MLC2v was increased in swimming‐induced physiological cardiac hypertrophy (Warren et al. 2012) and loss of MLC2v phosphorylation produced early impairment of cardiac muscle relaxation (Sheikh et al. 2012). Increased phosphorylation of MLC2v has been shown to increase the rate of cross‐bridge cycling by increasing the rate constant for the transition from non‐force‐generating cross‐bridges to a force‐generating state whereas the reverse rate constant is unaffected (Sweeney & Stull, 1990;Sweeney et al. 1993). Therefore, the increased phosphorylation of MLC2v in young adult dysferlin knockout mouse hearts may enhance lusitropy in adaptation to the prolonged decay in cytosolic Ca2+ transient as an early mechanism of functional compensation.
MLC2v is the sole known substrate of the cardiac isoform of myosin light chain kinase, cMLCK (High & Stull, 1980; Kamm & Stull, 2011). Recent studies demonstrated that in cardiac muscle, at least half of the MLC2v protein was constitutively phosphorylated by cMLCK (Chang et al. 2015), which plays a role in preventing cardiac hypertrophy (Yuan et al. 2015). Consistent with the idea that MLC2v phosphorylation is necessary for normal cardiac performance (Ding et al. 2010), conditional knockout of cMLCK in mouse cardiac muscle caused heart failure (Chang et al. 2015). Although the 40–70% increases of MLC2v phosphorylation in the hearts of young dysferlin‐deficient mice was on top of a significant level of constitutive phosphorylation, its role in cardiac muscle adaptation to the impaired diastolic function merits further investigation.
Adding new insights into the early pathology and pathophysiology of cardiac dysferlinopathy, the impaired calcium re‐sequestration and possibly compensatory increases in MLC2v and MyBP‐C phosphorylation suggest potentially therapeutic targets for correcting the diastolic dysfunction of dysferlin cardiomyopathies. Our present study has laid a foundation for follow‐up studies on the underlying mechanism that mediates dysferlin deficiency‐induced delay of calcium re‐sequestration and myofilament protein modifications.
Additional information
Competing interests
None
Author contributions
The work was performed at Wayne State University School of Medicine in Detroit, Michigan, USA. B.W.: collection, assembly, analysis and interpretation of data; drafting and revising the article critically for important intellectual content. H.W.: collection, assembly, analysis and interpretation of data; drafting the article critically for important intellectual content. J.‐P.J.: conception and design of the experiments; interpretation of data; drafting and revising the article critically for important intellectual content. All authors approved the final version of the manuscript, all persons designated as authors qualify for authorship and all those who qualify for authorship are listed.
Funding
This study was supported by National Institutes of Health grants AR048816 and HL‐098945 to J.P.J.
Translational perspective
Deficiency of dysferlin leads to limb‐girdle muscular dystrophy 2B and Miyoshi myopathy with cardiac involvement that leads to dilated cardiomyopathy and heart failure. Our study of dysferlin gene knockout mice demonstrated early myocardial hypertrophy when baseline cardiac function was compensated. Dysferlin‐deficient cardiomyocytes exhibited significantly slower Ca2+ re‐sequestration than that in wild‐type cells. The diastolic function of dysferlin knockout mouse hearts showed blunted β‐adrenergic response. Phosphorylation of ventricular myosin light chain 2 was significantly increased, implying a molecular response to the impaired diastolic function. These early phenotypes of impaired diastolic cardiac function indicate a pathogenic mechanism of dysferlin cardiomyopathy and potentially novel targets for the development of therapeutic approaches.
Acknowledgements
We thank Ms. Hui Wang for technical assistance and Dr. Steven Cala for sharing the phospholamban antibody.
References
- Ampong BN, Imamura M, Matsumiya T, Yoshida M & Takeda S (2005). Intracellular localization of dysferlin and its association with the dihydropyridine receptor. Acta Myol 24, 134–144. [PubMed] [Google Scholar]
- Anderson ME, Brown JH & Bers DM (2011). CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol 51, 468–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal D & Campbell KP (2004). Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol 14, 206–213. [DOI] [PubMed] [Google Scholar]
- Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, McNeil PL & Campbell KP (2003). Defective membrane repair in dysferlin‐deficient muscular dystrophy. Nature 423, 168–172. [DOI] [PubMed] [Google Scholar]
- Bashir R, Britton S, Strachan T, Keers S, Vafiadaki E, Lako M, Richard I, Marchand S, Bourg N, Argov Z, Sadeh M, Mahjneh I, Marconi G, Passos‐Bueno MR, Moreira Ede S, Zatz M, Beckmann JS & Bushby K (1998). A gene related to Caenorhabditis elegans spermatogenesis factor fer‐1 is mutated in limb‐girdle muscular dystrophy type 2B. Nat Genet 20, 37–42. [DOI] [PubMed] [Google Scholar]
- Chang AN, Battiprolu PK, Cowley PM, Chen G, Gerard RD, Pinto JR, Hill JA, Baker AJ, Kamm KE & Stull JT (2015). Constitutive phosphorylation of cardiac myosin regulatory light chain in vivo . J Biol Chem 290, 10703–10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase TH, Cox GA, Burzenski L, Foreman O & Shultz LD (2009). Dysferlin deficiency and the development of cardiomyopathy in a mouse model of limb‐girdle muscular dystrophy 2B. Am J Pathol 175, 2299–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi ER, Park SJ, Choe YH, Ryu DR, Chang SA, Choi JO, Lee SC, Park SW, Kim BJ, Kim DK & Oh JK (2010). Early detection of cardiac involvement in Miyoshi myopathy: 2D strain echocardiography and late gadolinium enhancement cardiovascular magnetic resonance. J CardiovascMagnReson 12, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong SM & Jin JP (2009). To investigate protein evolution by detecting suppressed epitope structures. J Mol Evol 68, 448–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compagno V, Di Liegro I, Cestelli A & Donatelli M (2001). Effect of ageing and hypertension on β‐myosin heavy chain in heart of spontaneously hypertensive rats. Int J Mol Med 7, 507–508. [DOI] [PubMed] [Google Scholar]
- Copeland O, Sadayappan S, Messer AE, Steinen GJ, van der Velden J&Marston SB (2010). Analysis of cardiac myosin binding protein‐C phosphorylation in human heart muscle. J Mol Cell Cardiol 49, 1003–1011. [DOI] [PubMed] [Google Scholar]
- Ding P, Huang J, Battiprolu PK, Hill JA, Kamm KE & Stull JT (2010). Cardiac myosin light chain kinase is necessary for myosin regulatory light chain phosphorylation and cardiac performance in vivo . J Biol Chem 285, 40819–40829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty KR & McNally EM (2003). Repairing the tears: dysferlin in muscle membrane repair. Trends Mol Med 9, 327–330. [DOI] [PubMed] [Google Scholar]
- Edes I, Kiss E, Kitada Y, Powers FM, Papp JG, Kranias EG & Solaro RJ (1995). Effects of Levosimendan, a cardiotonic agent targeted to troponin C, on cardiac function and on phosphorylation and Ca2+ sensitivity of cardiac myofibrils and sarcoplasmic reticulum in guinea pig heart. Circ Res 77, 107–113. [DOI] [PubMed] [Google Scholar]
- El‐Armouche A, Boknik P, Eschenhagen T, Carrier L, Knaut M, Ravens U & Dobrev D (2006). Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation 114, 670–680. [DOI] [PubMed] [Google Scholar]
- Feng HZ, Biesiadecki BJ, Yu ZB, Hossain MM & Jin JP (2008). Restricted N‐terminal truncation of cardiac troponin T: a novel mechanism for functional adaptation to energetic crisis. J Physiol 586, 3537–3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng HZ, Wei B & Jin JP (2009). Deletion of a genomic segment containing the cardiac troponin I gene knocks down expression of the slow troponin T gene and impairs fatigue tolerance of diaphragm muscle. J Biol Chem 284, 31798–31806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frearson N & Perry SV (1975). Phosphorylation of the light‐chain components of myosin from cardiac and red skeletal muscles. Biochem J 151, 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvey JL, Kranias EG & Solaro RJ (1988). Phosphorylation of C‐protein, troponin I and phospholamban in isolated rabbit hearts. Biochem J 249, 709–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautel M, Zuffardi O, Freiburg A & Labeit S (1995). Phosphorylation switches specific for the cardiac isoform of myosin binding protein‐C: a modulator of cardiac contraction? EMBO J 14, 1952–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover L & Brown RH, Jr (2007). Dysferlin in membrane trafficking and patch repair. Traffic 8, 785–794. [DOI] [PubMed] [Google Scholar]
- Han R, Bansal D, Miyake K, Muniz VP, Weiss RM, McNeil PL & Campbell KP (2007). Dysferlin‐mediated membrane repair protects the heart from stress‐induced left ventricular injury. J Clin Invest 117, 1805–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han R & Campbell KP (2007). Dysferlin and muscle membrane repair. CurrOpin Cell Biol 19, 409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- High CW & Stull JT (1980). Phosphorylation of myosin in perfused rabbit and rat hearts. Am J Physiol 239, H756–764. [DOI] [PubMed] [Google Scholar]
- Holroyde MJ, Small DA, Howe E & Solaro RJ (1979). Isolation of cardiac myofibrils and myosin light chains with in vivo levels of light chain phosphorylation. Biochim Biophys Acta 587, 628–637. [DOI] [PubMed] [Google Scholar]
- Huang QQ, Fisher SA & Brozovich FV (2004). Unzipping the role of myosin light chain phosphatase in smooth muscle cell relaxation. J Biol Chem 279, 597–603. [DOI] [PubMed] [Google Scholar]
- Illarioshkin SN, Ivanova‐Smolenskaya IA, Greenberg CR, Nylen E, Sukhorukov VS, Poleshchuk VV, Markova ED & Wrogemann K (2000). Identical dysferlin mutation in limb‐girdle muscular dystrophy type 2B and distal myopathy. Neurology 55, 1931–1933. [DOI] [PubMed] [Google Scholar]
- Jin JP, Yang FW, Yu ZB, Ruse CI, Bond M & Chen A (2001). The highly conserved COOH terminus of troponin I forms a Ca2+‐modulated allosteric domain in the troponin complex. Biochemistry 40, 2623–2631. [DOI] [PubMed] [Google Scholar]
- Kamm KE & Stull JT (2011). Signaling to myosin regulatory light chain in sarcomeres. J BiolChem 286, 9941–9947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JP, Ziman AP, Mueller AL, Muriel JM, Kleinhans‐Welte E, Gumerson JD, Vogel SS, Ward CW, Roche JA & Bloch RJ (2013). Dysferlin stabilizes stress‐induced Ca2+ signaling in the transverse tubule membrane. Proc Natl Acad Sci USA 110, 20831–20836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinge L, Harris J, Sewry C, Charlton R, Anderson L, Laval S, Chiu YH, Hornsey M, Straub V, Barresi R, Lochmuller H & Bushby K (2010). Dysferlin associates with the developing T‐tubule system in rodent and human skeletal muscle. Muscle Nerve 41, 166–173. [DOI] [PubMed] [Google Scholar]
- Kunst G, Kress KR, Gruen M, Uttenweiler D, Gautel M & Fink RH (2000). Myosin binding protein C, a phosphorylation‐dependent force regulator in muscle that controls the attachment of myosin heads by its interaction with myosin S2. Circ Res 86, 51–58. [DOI] [PubMed] [Google Scholar]
- Kuru S, Yasuma F, Wakayama T, Kimura S, Konagaya M, Aoki M, Tanabe M & Takahashi T (2004). [A patient with limb girdle muscular dystrophy type 2B (LGMD2B) manifesting cardiomyopathy]. RinshoShinkeigaku 44, 375–378. [PubMed] [Google Scholar]
- Kuster DW, Bawazeer AC, Zaremba R, Goebel M, Boontje NM, van der Velden J (2012) Cardiac myosin binding protein C phosphorylation in cardiac disease. J Muscle Res Cell Motil 33, 43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon NJ, Kho A, Bacskai BJ, Perlmutter SL,Hyman BT & Brown RH, Jr (2003). Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound‐healing. J Biol Chem 278, 50466–50473. [DOI] [PubMed] [Google Scholar]
- Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C, Serrano C, Urtizberea JA, Hentati F, Hamida MB, Bohlega S, Culper EJ, Amato AA, Bossie K, Oeltjen J, Bejaoui K, McKenna‐Yasek D, Hosler BA, Schurr E, Arahata K, de Jong PJ & Brown RH, Jr (1998). Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet 20, 31–36. [DOI] [PubMed] [Google Scholar]
- Maier LS & Bers DM (2002). Calcium, calmodulin, and calcium‐calmodulin kinase II: heartbeat to heartbeat and beyond. J Mol Cell Cardiol 34, 919–939. [DOI] [PubMed] [Google Scholar]
- Matsuda C, Hayashi YK, Ogawa M, Aoki M, Murayama K, Nishino I, Nonaka I, Arahata K & Brown RH, Jr (2001). The sarcolemmal proteins dysferlin and caveolin‐3 interact in skeletal muscle. Hum Mol Genet 10, 1761–1766. [DOI] [PubMed] [Google Scholar]
- Moir AJ & Perry SV (1980). Phosphorylation of rabbit cardiac‐muscle troponin I by phosphorylase kinase.The effect of adrenaline. Biochem J 191, 547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resink TJ, Gevers W & Noakes TD (1981). Effects of extracellular calcium concentrations on myosin P light chain phosphorylation in hearts from running‐trained rats. J Mol Cell Cardiol 13, 753–765. [DOI] [PubMed] [Google Scholar]
- Roche JA, Ru LW, O'Neill AM, Resneck WG, Lovering RM & Bloch RJ (2011). Unmasking potential intracellular roles for dysferlin through improved immunolabeling methods. J Histochem Cytochem 59, 964–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh F, Ouyang K, Campbell SG, Lyon RC, Chuang J, Fitzsimons D, Tangney J, Hidalgo CG, Chung CS, Cheng H, Dalton ND, Gu Y, Kasahara H, Ghassemian M, Omens JH, Peterson KL, Granzier HL, Moss RL, McCulloch AD & Chen J. (2012). Mouse and computational models link Mlc2v dephosphorylation to altered myosin kinetics in early cardiac disease. J Clin Invest 122, 1209–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solaro RJ, Henze M & Kobayashi T (2013). Integration of troponin I phosphorylation with cardiac regulatory networks. Circ Res 112, 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solaro RJ, Moir AJ & Perry SV (1976). Phosphorylation of troponin I and the inotropic effect of adrenaline in the perfused rabbit heart. Nature 262, 615–617. [DOI] [PubMed] [Google Scholar]
- Sweeney HL & Stull JT (1990). Alteration of cross‐bridge kinetics by myosin light chain phosphorylation in rabbit skeletal muscle: implications for regulation of actin‐myosin interaction. Proc Natl Acad Sci USA 87, 414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney HL, Bowman BF & Stull JT (1993). Myosin light chain phosphorylation in vertebrate striated muscle: regulation and function. Am J Physiol 264, C1085–1095. [DOI] [PubMed] [Google Scholar]
- Tong CW, Gaffin RD, Zawieja DC & Muthuchamy M (2004). Roles of phosphorylation of myosin binding protein‐C and troponin I in mouse cardiac muscle twitch dynamics. J Physiol 558, 927–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh MP, Vallet B, Autric F & Demaille JG (1979). Purification and characterization of bovine cardiac calmodulin‐dependent myosin light chain kinase. J Biol Chem 254, 12136–12144. [PubMed] [Google Scholar]
- Warren SA, Briggs LE, Zeng H, Chuang J, Chang EI, Terada R, Li M, Swanson MS, Lecker SH, Willis MS, Spinale FG, Maupin‐Furlowe J, McMullen JR, Moss RL & Kasahara H (2012). Myosin light chain phosphorylation is critical for adaptation to cardiac stress. Circulation 126, 2575–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei B, Dui W, Liu D, Xing Y, Yuan Z & Ji G (2013). MST1, a key player, in enhancing fast skeletal muscle atrophy. BMC Biol 11, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei B, Gao J, Huang XP & Jin JP (2010). Mutual rescues between two dominant negative mutations in cardiac troponin I and cardiac troponin T. J Biol Chem 285, 27806–27816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei B, Lu Y & Jin JP (2014). Deficiency of slow skeletal muscle troponin T causes atrophy of type I slow fibers and decreases tolerance to fatigue. J Physiol 592, 1367–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H & Jin JP (2014). A dominantly negative mutation in cardiac troponin I at the interface with troponin T causes early remodeling in ventricular cardiomyocytes. Am J Physiol Cell Physiol 307, C338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg A & Winegrad S (1996). Alteration of myosin cross bridges by phosphorylation of myosin‐binding protein C in cardiac muscle. Proc Natl Acad Sci USA 93, 8999–9003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel K, Geier C, Qadri F, Hubner N, Schulz H, Erdmann B, Gross V, Bauer D, Dechend R, Dietz R, Osterziel KJ, Spuler S & Ozcelik C (2007). Dysfunction of dysferlin‐deficient hearts. J Mol Med (Berl) 85, 1203–1214. [DOI] [PubMed] [Google Scholar]
- Yuan CC, Muthu P, Kazmierczak K, Liang J, Huang W, Irving TC, Kanashiro‐Takeuchi RM, Hare JM & Szczesna‐Cordary D (2015). Constitutive phosphorylation of cardiac myosin regulatory light chain prevents development of hypertrophic cardiomyopathy in mice. Proc Natl Acad Sci USA 112, E4138–E4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Zhao J, Mandveno A & Potter JD (1995). Cardiac troponin I phosphorylation increases the rate of cardiac muscle relaxation. Circ Res 76, 1028–1035. [DOI] [PubMed] [Google Scholar]
- Zhang T & Brown JH (2004). Role of Ca2+/calmodulin‐dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc Res 63, 476–486. [DOI] [PubMed] [Google Scholar]