

Abstract
Key points
Multiple vasodilators have been identified as being important in matching blood flow to the metabolism of contracting skeletal muscle and it has been hypothesized that this process is governed by redundancy between vasodilators, where one vasodilator can compensate for the loss of another.
In the present study, we aimed to determine whether redundancy between vasodilators exists by investigating whether vasodilators relevant to skeletal muscle contraction can inhibit the effects of other vasodilators.
We show that potassium can inhibit vasodilatations induced by adenosine and nitric oxide, and also that adenosine and nitric oxide can interact in a way that changes over time.
Furthermore, we show that inward rectifying potassium channels and Na+/K+ATPase are partially mechanistically responsible for the interaction between potassium and adenosine and nitric oxide.
Our data provide proof of principle that vasodilators relevant to muscle contraction interact and also that redundancy may govern the processes of active hyperaemia.
Abstract
Redundancy, in active hyperaemia, where one vasodilator can compensate for another if the first is missing, would require that one vasodilator inhibits the effects of another; therefore, if the first vasodilator is inhibited, its inhibitory influence on the second vasodilator is removed and the second vasodilator exerts a greater vasodilatory effect. We aimed to determine whether vasodilators relevant to skeletal muscle contraction [potassium chloride (KCl), adenosine (ADO) and nitric oxide] inhibit one another and, in addition, to investigate the mechanisms for this interaction. We used the hamster cremaster muscle and intravital microscopy to directly visualize 2A arterioles when exposed to a range of concentrations of one vasodilator [10−8 to 10−5 m S‐nitroso‐N‐acetyl penicillamine (SNAP), 10−8 to 10−5 m ADO, 10 and 20 mm KCl] in the absence and then in the presence of a second vasodilator (10−7 m ADO, 10−7 m SNAP, 10 mm KCl). We found that KCl significantly attenuated SNAP‐induced vasodilatations by ∼65.8% and vasodilatations induced by 10−8 to 10−6 m ADO by ∼72.8%. Furthermore, we observed that inhibition of KCl vasodilatation, by antagonizing either Na+/K+ ATPase using ouabain or inward rectifying potassium channels using barium chloride, could restore the SNAP‐induced vasodilatation by up to ∼53.9% and 30.6%, respectively, and also restore the ADO‐induced vasodilatations by up to ∼107% and 76.7%, respectively. Our data show that vasodilators relevant to muscle contraction can interact in a way that alters the effectiveness of other vasodilators. These data suggest that active hyperaemia may be the result of complex interactions between multiple vasodilators via a redundant control paradigm.
Abbreviations
- ADO
adenosine
- BaCl
barium chloride
- KIR
inward rectifying potassium channels
- NO
nitric oxide
- Oua
ouabain
- PKA
protein kinase A
- PSS
physiological salt solution
- SNAP
S‐nitroso‐N‐acetyl penicillamine
- VSM
vascular smooth muscle
Introduction
Skeletal muscle blood flow can increase over 100‐fold during the transition from rest to strenuous exercise via a multitude of processes that mediate active hyperaemia. Active hyperaemia is largely mediated by vasoactive factors derived locally from active skeletal muscle fibres and functions to couple blood flow with metabolism (Clifford & Hellsten, 2004; Rowell, 2004; Joyner & Wilkins, 2007; Sarelius & Pohl, 2010). Despite their importance, the identification of the critical communicating factors between skeletal muscle and the vasculature has led to a list of putative vasoactive substances where the identity of any one, single predominant regulator of active hyperaemia has not yet been identified.
Historically, experiments in this area have relied on pharmacological antagonism to assess the influences of individual vasoactive factors during the active hyperaemic response. Such an experimental design presumes that any resultant change in vessel diameter, or blood flow, in the presence of an antagonist is indicative of the contribution of individual vasodilators to mediating the hyperaemic response. However, despite the assessment of numerous vasodilators, no single vasodilator has been identified as being uniquely important to the process. For example, studies investigating adenosine (ADO) as the predominant regulator of active hyperaemia began in the late 1970s and early 1980s (Tabaie et al. 1977; Honig & Frierson, 1980; Proctor & Duling, 1982; Kille & Klabunde, 1984), yet, despite these and the many studies that followed, there is still little consensus regarding the role of ADO in the blood flow response associated with muscle contraction (Marshall, 2007). A similar history exists for other putative vasodilators involved in active hyperaemia, again, with no clear identity of a single critical vasodilator responsible for this important phenomenon (Clifford & Hellsten, 2004; Sarelius & Pohl, 2010; Mortensen & Saltin, 2014). Thus, the current methodological approaches used to investigate individual influences of vasoactive factors have failed to identify any uniting mechanism, or predominating factor, in the regulation of active hyperaemia. Given the critical nature of active hyperaemia, current thinking has led to the idea of a more complex, integrated, redundant control system (Chilian & Koshida, 2001; Rowell, 2004; Joyner & Wilkins, 2007; Hellsten et al. 2012). Importantly, this redundant model has been suggested not as a result of strong evidence for the redundant control system but because of the lack of support for a predominant single vasodilator.
Redundancy describes the operation of a system where one component has the same function as another and exists to engage if the first component fails. When part of a complex system, redundancy is a critical operational element for preventing failure. Clearly, it would be advantageous for physiologically critical systems, such as active hyperaemia, to evolve to include redundancy. Redundancy in the active hyperaemic response would necessitate that, when one vasodilator is inhibited, another would take its place (Rowell, 2004).This requires that multiple vasodilators are present at the same time during muscle contraction and could be achieved via inhibitory interactions between vasodilators where one vasodilator attenuates the effects of other vasodilators. Therefore, when the first vasodilator is inhibited (either pharmacologically or physiologically), its inhibition on the second vasodilator is removed and the effects of the second vasodilator will be amplified.
The literature contains many descriptions of vasodilators and interacting vasodilator processes. Flow‐induced vasodilatations have been shown to be potentiated by ADO (Kuo & Chancellor, 1995) and oxygen tension (Frisbee & Lombard, 1999; Frisbee et al. 2000) and to interact with myogenic constriction (Sun et al. 1995). There are reports that transmural pressure increases the response of arterioles to noradrenaline (Lombard et al. 1990) and enhances arteriolar vasodilatation to ADO (Zhang et al. 2000). Frisbee (2002) reported an interplay between ADO, oxygen tension, noradrenaline, vascular pressure and wall shear stress in establishing vascular diameter (Frisbee, 2002). It has been suggested that nitric oxide (NO) may inhibit endothelial derived hyperpolarizing factor responses (Olmos et al. 1995; Bauersachs et al. 1996; Kessler et al. 1999; Nishikawa et al. 2000; Huang et al. 2001), although not invariably (Schildmeyer & Bryan, 2002; Watanabe et al. 2005). Furthermore, there is evidence to support interactions between NO and prostaglandins (Boushel et al. 2002; Schrage et al. 2004; Mortensen et al. 2007; Markwald et al. 2011), NO and ADO (Casey et al. 2013), and between ADO, NO and potassium (K+) (Dua et al. 2009). However, whether vasodilators interact in an inhibitory manner, and whether there is redundant control over vasodilatation, remains to be tested directly.
We aimed to obtain direct evidence for the presence of redundant control over vasodilatation; specifically, whether vasodilators relevant to muscle contraction, K+, ADO and NO, can inhibit the ability of each other to cause vasodilator to cause vasodilatation. These vasodilators are known to be present contemporaneously at the onset of muscle contraction (Armstrong et al. 2007; Dua et al. 2009; Ross et al. 2013) and therefore represent prime candidates for testing these interactions. Furthermore, we aimed to explore possible mechanisms for this interaction. We hypothesized that K+ will inhibit the ability of NO and ADO to vasodilate. We tested this hypothesis at the level of the terminal microvasculature, comprising the arteriolar levels responsible for distribution of blood flow within the tissue, as distinct from larger arteries and arterioles that are responsible for gross blood flow delivery to the tissue. Using the in situ blood perfused hamster cremaster muscle and intravital microscopy to visualize arterioles directly in vivo, we applied a range of concentrations of NO in the absence and presence of physiologically relevant concentrations of K+(10 mm KCl) (Hnik et al. 1972; Vyskocil et al. 1983; Nordsborg et al. 2003; Clausen, 2011, 2013) and vice versa to determine whether K+ had the ability to inhibit NO vasodilatation and also whether NO had the ability to inhibit K+ vasodilatation. This was repeated with ADO and K+, as well as ADO and NO. We anticipate that the data obtained from these studies will provide proof of principle that vasodilators involved in active hyperaemia interact in a redundant manner, thereby significantly shifting the direction of future work in the area of blood flow regulation.
Methods
All experiments were approved by the Institutional Animal Care Committee Review Board at the University of Guelph and were conducted in accordance with the guidelines of the Canadian Council on Animal Care (CCAC). Following all experimental protocols, animals were killed with an overdose of sodium pentobarbital (0.26 mg ml−1 i.v. to effect).
General protocol: preparing the cremaster muscle for experimentation
Adult male Golden Syrian hamsters (100–140 g) (n = 114) were anaesthetized with sodium pentobarbital (70 mg kg−1, i.p.), tracheotomized and catheterized using polyethylene catheters (outer tip diameter ∼0.5 mm) placed in the left femoral artery (to monitor mean arterial pressure) and the left femoral vein for supplemental sodium pentobarbital infusion (10 mg ml−1 saline, 0.56 ml h−1) throughout the experimental protocol. The animal was placed on an acrylic platform. Oesophageal temperature was maintained at 37°C via convective heat from a coiled water‐filled glass tube (42°C) secured under the hamster. The right cremaster was prepared for in situ microscopy as described originally (Baez, 1973) and subsequently modified (Murrant, 2005). Briefly, a lateral longitudinal cut was made in the scrotum. The skin and fascia were separated from the cremaster muscle. The isolated cremaster was then cut longitudinally and separated from both the testis and epididymis. After separation, the testicle was pushed into the abdominal cavity. The cremaster muscle was spread over a semicircular lucite plate. The edges of the cremaster were secured to the lucite plate by insect pins to maintain muscle tension. Throughout the cremaster isolation surgery and all of the experimental protocols, the cremaster muscle was constantly superfused with a physiological salt solution (PSS) containing (in mm L–1) 131.9 NaCl, 4.7 KCl, 2.0 CaCl2, 1.2 MgSO4 and 30 NaHCO3, and 0.3 mg l−1 tubocurarine hydrochloride pentahydrate. A physiological pH of PSS was attained (pH 7.35–7.45) and maintained by aeration with 5%CO2 and 95%N2 gas. Cremaster muscle temperature was maintained by heating the superfusion solution to 42°C and adjusting the drip rate to achieve 34 ± 0.5°C. After the cremaster isolation surgery, the hamster was transferred onto the microscope stage and allowed to equilibrate for 45–60 min prior to data collection.
Visualization of the microvasculature of the cremaster muscle was achieved via transillumination with a tungsten lamp and a BX51WI microscope (Olympus Canada Inc., Richmond Hill, ON, Canada) using a 40× long working distance water immersion objective (numerical aperture 0.80). The microscope image of the cremaster microvasculature was displayed on a video monitor using a video camera (DC220; Dage‐MTI Inc., Michigan, MI, USA) and digitized using EZ Grabber video compression software (Geniatech, Guangdong, China). The approximate final magnification of the site was 2000×. Diameter measurements were reproducible to within ±0.3 μm (n = 10). Transverse arterioles (2A arterioles) of ∼40 μm maximum diameter were used for observations. These arterioles were identified by counting up three branch orders from capillary vessels as described previously (Sweeney and Sarelius, 1989; Murrant and Sarelius, 2000).
Protocol 1: Assessing the potential for interactions between ADO, K+ and NO: sequential addition of vasodilators
To determine whether one vasodilator can affect the ability of another vasodilator to evoke vasodilatation, we tested the effects of each of K+, ADO and NO on the ability of each to vasodilatate. Generally, we first established a dose–response for each vasodilator independently, and then the dose–response was repeated in the presence of a second vasodilator.
To test whether K+ can alter the vasodilator ability of NO, the cremaster was exposed to a range of concentrations of the NO donor, S‐nitroso‐N‐acetylpenicillamine (SNAP) (10−8 to 10−5 m) via addition to the PSS superfusing the entire preparation. Each concentration of SNAP was added in sequential order from lowest to highest concentration at 2 min intervals. Following the final concentration, the superfusate was returned to normal PSS and the effects of SNAP were washed out for 20 min. Following washout, 10 mm KCl was added to the PSS (equimolar NaCl removed to preserve the osmolarity of the superfusate), 5 min prior to the re‐application of the same concentrations of SNAP (10−8 to 10−5 m), in the presence of 10 mm KCl in the PSS.
To test the hypothesis that NO can alter the vasodilator ability of K+, the above protocol was repeated using 10 and 20 mm KCl added in the absence and presence of 10−7 m SNAP. To test for interactions between the remaining vasodilators, this protocol was repeated using 10−8 to 10−5 m ADO in the absence and presence of 10 mm KCl, 10 and 20 mm KCl in the absence and presence of 10−7 m ADO, 10−8 to 10−5 m SNAP in the absence and presence of 10−7 m ADO, and 10−8 to 10−5 m ADO in the absence and presence of 10−7 m SNAP.
Protocol 2: Assessing the potential for interactions between ADO, K+ and NO and the simultaneous addition of vasodilators
In protocol 1, the vasodilators were added sequentially to determine potential interactions. During muscle contraction, ADO, NO and K+ have been shown to be present at the onset of contraction (Armstrong et al. 2007; Dua et al. 2009; Ross et al. 2013) and therefore would be present simultaneously. To test for simultaneous interaction between NO and K+ the cremaster was exposed to 10−7 m SNAP for 10 min. The effects of SNAP were then washed out with 20 min of superfusion with PSS. Following washout, 10−7 m SNAP and 10 mm KCl were superfused together over the preparation for 10 min. To test for simultaneous interactions between other vasodilators, the above protocol was repeated with an initial 10 min exposure of 10−7 m ADO in the absence and presence of 10 mm KCl and an initial exposure of 10−7 m ADO or 10−7 m SNAP for 10 min (chosen randomly) followed by superfusion of 10−7 m ADO and 10−7 m SNAP together. In a separate set of experiments, 10 mm KCl was superfused over the preparation for 10 min for comparison with the 10 min exposure of 10−7 m SNAP and 10−7 m ADO.
Protocol 3: Testing for the mechanism of K+ interaction with ADO and NO
K+ has been shown to cause arteriolar vasodilatation through inward rectifying K+ channels (KIR) (Loeb et al. 2000; Burns et al. 2004) and the sodium‐potassium pump (Na+/K+ATPase) (Haddy, 1983; Burns et al. 2004). We tested whether these mechanisms were responsible for K+‐mediated inhibition of NO and ADO‐induced vasodilatation. We inhibited NO and ADO vasodilatation with K+, and then inhibited the effects of K+ with either KIR or Na+/K+ATPase antagonists barium chloride (BaCl) or ouabain (Oua), respectively, to determine whether inhibiting K+ could restore the NO and ADO‐mediated vasodilatation.
To ensure the efficacy of the inhibitors, we tested the ability of 5 × 10−5 m BaCl (n = 6) and, separately, 10−4 m Oua (n = 6) to attenuate 10 mm KCl‐induced vasodilatations. The cremaster was exposed to 10 mm KCl for 10 min followed by a 20 min washout. The preparation was then superfused with either 5 × 10−5 m BaCl or 10−4 m Oua for 30 min. Next, 10 mm KCl was re‐applied for 10 min in the presence of BaCl or Oua. Application of 5 × 10−5 m BaCl significantly attenuated the peak vasodilator response of KCl by 43.5%, from 11.1 ± 1.6 μm to 6.3 ± 1.6 μm, whereas exposure to 10−4 m Oua significantly attenuated the peak vasodilator response of KCl by 40.1% from 9.3 ± 1.5 μm to 5.5 ± 1.8 μm. These data confirm that the chosen concentration of each antagonist was sufficient with respect to antagonizing KCl‐induced vasodilatation.
To test the hypothesis that KIR and/or Na+/K+ATPase were partly responsible for the K+‐induced attenuation of the SNAP or ADO‐induced vasodilatation, the cremaster was exposed to a 5 min application of 10 mm KCl, followed by a range of SNAP concentrations (10−8 to 10−5 m) or ADO (10−8 to 10−5 m) in the presence 10 mm KCl. Drugs were then washed out for 20 min. Immediately following washout, either 5 × 10−5 m BaCl or 10−4 m Oua was superfused over the preparation for 30 min. The SNAP‐KCl or the ADO‐KCl exposure was then repeated in the presence of the blocker (BaCl or Oua).
Following each experiment within a protocol, maximal arteriolar diameters were recorded after 2 min of superfusion with 10−2 m sodium nitroprusside (NO donor; Sigma‐Aldrich, St Louis, MO, USA), which is considered to produce maximal vasodilatation (Murrant et al. 2014).
Data analysis and statistics
Protocol 1
Arteriolar diameter at the observation site was continuously recorded for 1 min prior to the initial dose–response, during the dose–response, and for the first 2 min of the washout period. Arteriolar diameter was then recorded 1 min prior to the addition of the interacting vasodilator, during the 5 min application of the interacting vasodilator, during the subsequent dose–response, and for the first 2 min of the washout period. Arteriolar diameter was measured every 10 s during the recording period. For protocol 1, control baseline diameters were defined as the diameter of the arteriole prior to the application of the first vasodilator dose–response. Experimental baseline diameter was defined as arteriolar diameter after the second, interacting vasodilator was added and just prior to re‐application of the dose–response.
Protocol 2
Arteriolar diameter at the observation site was continuously recorded for 1 min prior to the drug (or drugs) being added, during the 10 min drug exposure, and for the first 2 min of the washout. For protocol 2, the control baseline diameter was defined as the diameter just prior to the initial drug application. Experimental baseline diameter was defined as the diameter just prior to the second drug application.
Protocol 3
Arteriolar diameter at the observation site was continuously recorded for 1 min prior to the application of KCl, during the dose–response, and for the first 2 min of the washout period in the absence and presence of each blocker. For protocol 3, control baseline diameter was defined as the diameter just prior to the vasodilator dose–response and experimental baseline diameter was the diameter of the arteriole in the presence of the antagonist just prior to re‐application of the vasodilator dose–response.
Only one arteriole was observed per cremaster preparation (with n indicating the number of arterioles observed). All experiments were analysed offline. For each protocol, arteriolar diameter was measured every 10 s during the recording period. Still images from digitized recording were captured every 10 ± 1 s (mean ± SE) using FrameShot software (EoF Productions, Sacramento, CA, USA) and arteriolar diameters were measured using ImageJ (NIH, Bethesda, MD, USA).
Data are reported as the mean ± SE. Where only two group means were compared (i.e. effectiveness of K+ blockers), Student's t test was used to compare responses. Baseline and maximal diameters were compared using an ANOVA. Group means in dose–responses and data shown over time were compared with a repeated measures ANOVA. When the ANOVA identified significant differences, a protected least square difference test was used post hoc to determine whether changes in diameter were significantly different. Because of differences observed in baseline diameters in Table 3, linear regression analysis was used to determine correlations between initial baseline diameter and the response to the lowest dose of each drug under control conditions. P < 0.05 was considered statistically significant.
Table 3.
Average baseline and maximum arteriolar diameter for experiments in protocol 3
| Baseline diameter (μm) | ||||
|---|---|---|---|---|
| Protocol | Control | Experimental | Maximum diameter (μm) | n |
| SNAP + KCl + BaCl | 19.0 ± 2.0 | 18.0 ± 2.7 | 30.6 ± 2.6 | 7 |
| ADO + KCl + BaCl | 18.9 ± 2.3 | 16.1 ± 2.6 | 39.2 ± 2.6 | 9 |
| SNAP + KCl + Oua | 16.8 ± 1.4 | 14.8 ± 2.4 | 38.2 ± 2.4 | 6 |
| ADO + KCl + Oua | 16.3 ± 2.1 | 12.2 ± 1.1* | 36.7 ± 2.0 | 9 |
Data are reported as the mean ± SE. *Experimental baseline significantly different from control baseline.
Results
Protocol 1: Assessing the potential for interactions between ADO, K+ and NO: sequential addition of vasodilators
Overall, there were no differences between control and experimental baseline diameters (Table 1). On assaying the effects of K+ on NO and vice versa, the range of SNAP concentrations produced the expected increase in arteriolar diameter (Fig. 1 A). Upon addition of 10 mm KCl, we observed a transient vasodilatation peaking at 5.9 ± 1.7 μm between 2 and 4 min followed by a return to baseline by ∼5 min (an example of the transient nature of the KCl vasodilatation is shown in Fig. 4 A and B). Re‐application of the SNAP dose–response in the presence of 10 mm KCl resulted in a blunted vasodilatation for all concentrations of SNAP compared to SNAP alone (Fig. 1 A). Conversely, the vasodilator response produced by KCl was not affected by the addition of SNAP. KCl alone at 10 and 20 mm produced the expected arteriolar vasodilatations (Fig. 1 B). Following washout, 10−7 m SNAP was applied and produced a peak change in vasodilatation of 18.7 ± 2.8 μm at ∼160 s before returning to baseline. When KCl was re‐applied in the presence of SNAP, the KCl‐induced vasodilatations at each concentration remained unchanged (Fig. 1 B).
Table 1.
Average baseline and maximum arteriolar diameter for the experiments in protocol 1
| Baseline diameter (μm) | ||||
|---|---|---|---|---|
| Protocol | Control | Experimental | Maximum diameter (μm) | n |
| NO effects on K+ | 24.0 ± 1.9 | 27.1 ± 1.6 | 44.8 ± 3.0 | 8 |
| K+ effects on NO | 17.3 ± 1.7 | 21.0 ± 3.3 | 38.2 ± 2.6 | 11 |
| ADO effects on K+ | 13.6 ± 2.0 | 17.1 ± 3.9 | 32.0 ± 2.5 | 7 |
| K+ effects on ADO | 15.6 ± 1.5 | 22.1 ± 4.3 | 38.4 ± 2.9 | 6 |
| NO effects on ADO | 20.9 ± 2.5 | 19.6 ± 4.5 | 50.2 ± 4.3 | 7 |
| ADO effects on NO | 18.2 ± 1.8 | 17.9 ± 1.7 | 41.0 ± 2.2 | 9 |
Data are reported as the mean ± SE.
Figure 1. KCl attenuated the magnitude of vasodilatation produced by SNAP but SNAP did not affect the magnitude of KCl‐induced vasodilatation .

A, change in diameter in response to incremental concentrations of SNAP in the absence (■) and presence (▲) of 10 mm KCl. B, change in diameter in response to incremental doses of KCl in the absence (■) and presence (▲) of 10−7 m SNAP. *SNAP + KCl differs significantly from SNAP alone.
Figure 4. KCl attenuated the vasodilatations induced by ADO and SNAP when added simultaneously, and the simultaneous addition of SNAP and ADO showed complex interactions over time .
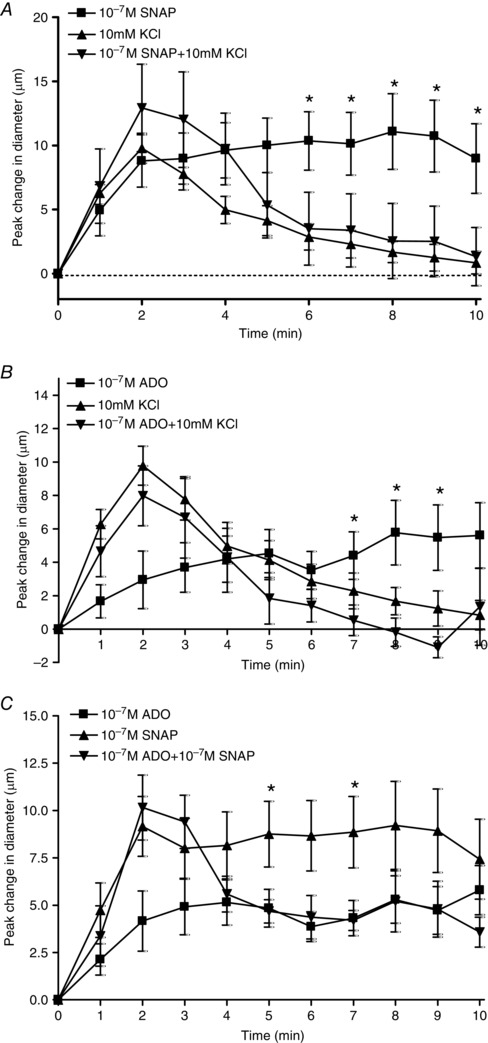
A, change in diameter in response to 10−7 m SNAP alone (■), 10 mm KCl alone (▲) and 10−7 m SNAP + 10 mm KCl added together simultaneously (▼). * SNAP alone differed significantly from SNAP + KCl. B, change in diameter in response to 10−7 m ADO alone (■), 10 mm KCl alone (▲) and 10−7 m ADO + 10 mm KCl added together simultaneously (▼). * ADO alone differed significantly from ADO + KCl. C, change in diameter in response to 10−7 m ADO alone (■),10−7 m SNAP alone (▲) and 10−7 m ADO + 10−7 m SNAP added together simultaneously (▼). * SNAP alone differed significantly from ADO + SNAP.
In experiments aiming to determine whether ADO and K+ interact, we observed that the presence of 10 mm KCl significantly attenuated the vasodilator response to 10−8 to 10−6 m ADO (Fig. 2 A). Conversely, the presence of ADO did not affect the vasodilator response induced by either 10 mm or 20 mm KCl (Fig. 2 B). In these experiments, 10 and 20 mm KCl alone produced the expected arteriolar vasodilatations (Fig. 2 B). Following washout, 10−7 m ADO was applied and produced a peak change in vasodilatation of 3.3 ± 1.5 μm before returning to baseline. The KCl concentrations were then re‐applied in the presence of ADO, which had no effect on the ability of either 10 mm or 20 mm KCl to vasodilate (Fig. 2 B).
Figure 2. KCl attenuated the magnitude of vasodilatation produced by ADO but ADO did not affect the magnitude of KCl‐induced vasodilatation .
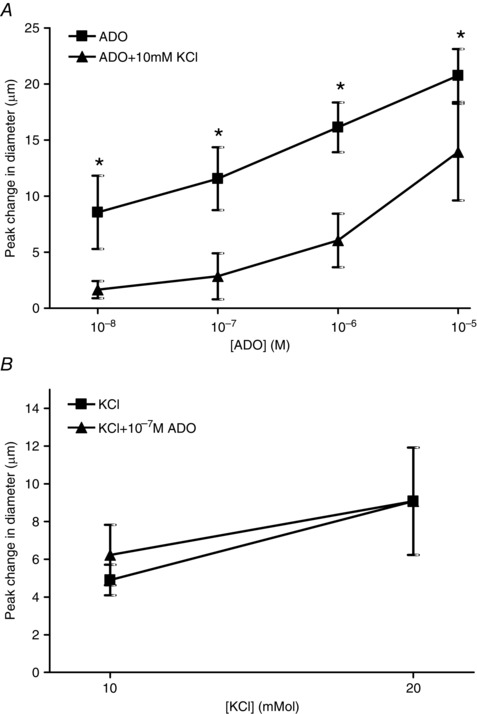
A, change in diameter in response to incremental doses of ADO in the absence (■) and presence (▲) of 10 mm KCl. B, change in diameter in response to incremental doses of KCl in the absence (■) and presence (▲) of 10−7 m ADO. *ADO + KCl differs significantly from ADO alone.
In experiments aiming to determine whether NO and ADO interact, we added a range of concentrations of SNAP and observed increases in arteriolar diameter (Fig. 3 A). Following washout, 10−7 m ADO was applied and produced a peak change in vasodilatation of 8.3 ± 2.2 μm at ∼60 s before returning to baseline. The SNAP doses were then re‐applied in the presence of ADO and we observed that the presence of ADO had no effect on the ability of NO to cause vasodilatation at any concentration tested. Similarly, ADO did not affect the vasodilator response to NO. We added a range of concentrations of ADO and observed increases in arteriolar diameter (Fig. 3 B). Following washout, the application of 10−7 m SNAP produced a peak change in diameter of 11.0 ± 4.0 μm at ∼1 min before returning to baseline. The ADO concentrations were then re‐applied in the presence of SNAP and we observed that the presence of SNAP had no effect on the ability of ADO to cause vasodilatation at any concentration tested.
Figure 3. SNAP did not affect the vasodilator response of ADO and ADO did not affect the magnitude of SNAP‐induced vasodilatation .
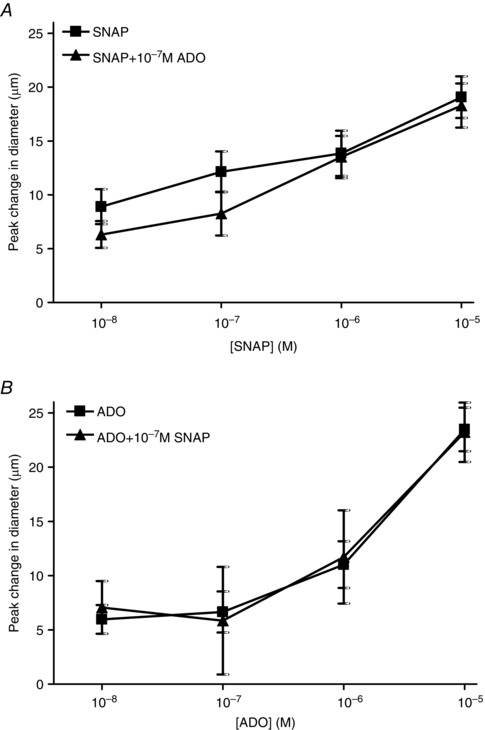
A, change in diameter in response to incremental doses of SNAP in the absence (■) and presence (▲) of 10−7 m ADO. B,change in diameter in response to incremental doses of ADO in the absence (■) and presence (▲) of 10−7 m SNAP.
Protocol 2: Assessing the potential for interactions between ADO, K+ and NO: simultaneous addition of vasodilators
Baseline and maximal diameters for the simultaneous experiments are reported in Table 2. The addition of 10 mm KCl alone produced a transient vasodilatation of 9.4 ± 0.6 μm that peaked at ∼2 min and then returned to baseline (Fig. 4 A). SNAP alone at 10−7 m produced a sustained vasodilatation of 9.0 ± 2.7 μm that peaked at ∼2 min and was sustained throughout the 10 min duration of the application period.When SNAP and KCl were added simultaneously, the vasodilator response was similar to the response elicited by KCl alone; it was transient, peaking at 12.9 ± 3.4 μm by 2 min and then recovered to baseline. There was no evidence for summation of the two vasodilators and no indication of a sustained NO vasodilatation.
Table 2.
Average baseline and maximum arteriolar diameter for experiments in protocol 2
| Baseline diameter (μm) | ||||
|---|---|---|---|---|
| Protocol | Control | Experimental | Maximum diameter (μm) | n |
| Simultaneous application of 10−7 m SNAP and 10 mm KCl | 19.0 ± 2.1 | 22.0 ± 2.2 | 42.0 ± 3.2 | 8 |
| Simultaneous application of 10−7 m ADO and 10 mm KCl | 15.8 ± 1.4 | 16.7 ± 1.2 | 34.2 ± 1.8 | 7 |
| Simultaneous application of 10−7 m SNAP and 10−7 m ADO | 16.8 ± 0.9 | 16.1 ± 2.0 | 38.1 ± 1.6 | 8 |
Data are reported as the mean ± SE.
Similarly, we assayed vasodilatation evoked by simultaneous addition of KCl and ADO. Initially, as described above, the addition of 10 mm KCl alone produced a transient vasodilatation with a peak change in diameter of 9.4 ± 0.6 μm by∼2 min and then returned to baseline (Fig. 4 B). ADO alone at 10−7 m produced a sustained vasodilatation of 5.6 ± 2.0 μm that peaked at ∼2 min and was sustained throughout the 10 min duration of the application period.When ADO and KCl were added simultaneously, the vasodilator response mimicked that of KCl alone, where the vasodilatation was transient, peaking at 8.0 ± 1.8 μm by 2 min before recovering to baseline. There was no evidence for summation of the two vasodilators and no indication of a sustained ADO vasodilatation.
The vasodilator response evoked by simultaneous addition of ADO and SNAP was also investigated (Fig. 4 C). The addition of 10−7 m SNAP alone produced an increase in vessel diameter of 7.1 ± 1.9 μm that was sustained throughout the 10 min exposure. Similarly, the addition of 10−7 m ADO alone also produced a sustained increase in vessel diameter of ∼5.8 ± 1.3 μm throughout the 10 min exposure. When both 10−7 m SNAP and 10−7 m ADO were added simultaneously, the response over the first 3 min was not significantly different from the SNAP response alone. However, from 4–10 min, the vasodilatation decreased and was not significantly different from the ADO response alone (Fig. 4 C). Again, there was no evidence of summation of the two vasodilators and a complex interaction was demonstrated, where it appears that NO was responsible for the initial vasodilatation but, over time, its effects were less influential and those of ADO became more pronounced.
Protocol 3: Testing for the mechanism of K+ interaction with ADO and NO
Baseline and maximal diameters for this protocol are reported in Table 3. Baseline diameters did not significantly differ between the control and experimental conditions within this protocol, with the exception of SNAP + KCl in the presence of Oua. To determine whether baseline diameter alters the reactivity of the arterioles, we plotted the baseline diameter versus the change in diameter under the control conditions from each of our three protocols. We found no linear correlation between the two variables (average r 2 = 0.12 ± 0.05), indicating that the baseline diameter had no significant impact on the ability of the arteriole to vasodilatate. We have previously shown this lack of relationship in both hamster (Armstrong et al. 2007) and mouse (Leonard et al. 2011) microvasculature. Therefore, the differences in the control and experimental baseline diameters in the SNAP + KCl in the presence of Oua protocol did not influence the ability of the arterioles to respond, nor influence the conclusions drawn from these experiments.
In the presence of the KIR antagonist BaCl, the vasodilator responses to 10−6 and 10−5 m SNAP + 10 mm KCl were significantly amplified compared to 10−6 and 10−5 m SNAP + 10 mm KCl alone (Fig. 5 A). Therefore, in the presence of BaCl, the KCl inhibition of SNAP was partially removed. Similarly, the addition of Oua significantly amplified the vasodilator response to 10−7 and 10−6 m SNAP + 10 mm KCl compared to the response elicited by SNAP + KCl alone (Fig. 5 B) indicating that inhibiting KIR channels and Na+/K+ ATPase could rescue the vasodilator component of SNAP that was inhibited by KCl. Oua partially restored the KCl‐induced SNAP inhibition at 10−7 m SNAP and was able to completely restore the vascular response to 10−6 m SNAP.
Figure 5. KCl attenuation of SNAP vasodilatation is reversed through the inhibition of KIR channels and Na+/K+ATPase .
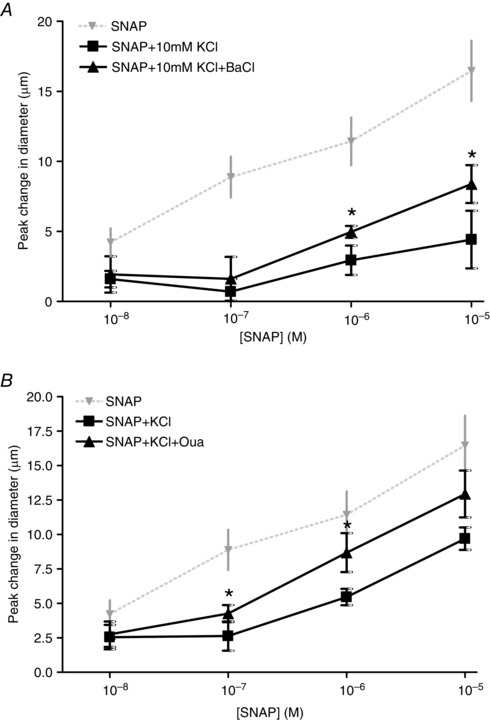
A, change in diameter in response to KCl + SNAP in the absence (■) and presence (▲) of BaCl. * SNAP + KCl differed significantly from SNAP + KCl + BaCl. For comparison, the diameter change in response to SNAP only (▼) is shown (data from Fig. 1). B, change in diameter in response to KCl + SNAP in the absence (■) and presence (▲) of 10−4 m Oua. *SNAP + KCl differed significantly from SNAP + KCl + Oua. For comparison, the diameter change in response to SNAP only (▼) is shown (data from Fig. 1).
In the presence of ADO + KCl, inhibiting the effects of K+ by inhibition of KIR channels with BaCl increased the vasodilator response elicited by 10−8 and 10−7 m ADO + 10 mm KCl (Fig. 6 A); therefore, BaCl abolished the inhibition of K+ on the ADO‐induced vasodilatation. Similarly, Oua was able to enhance the vasodilator response of 10−8 and 10−7 m ADO in the presence of KCl but not at higher concentrations (Fig. 6 B). Thus, inhibition (using Oua) of the inhibitor (K+) was able to restore the ADO‐induced vasodilatation in a manner that was dependent on the concentration of ADO. Thus, inhibiting KIR channels and Na+/K+ATPase could rescue the vasodilator component of ADO that was inhibited by KCl.
Figure 6. KCl inhibition of ADO vasodilatation is reversed through the inhibition of KIR channels and Na+/K+ATPase .
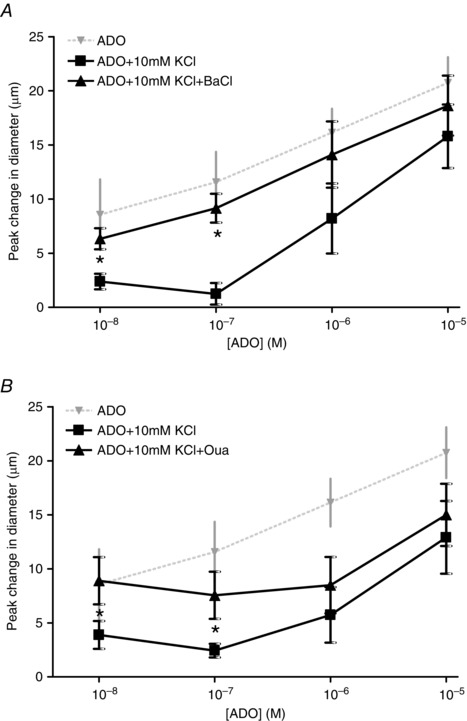
A, change in diameter in response to ADO + KCl in the absence (■) and presence (▲) of BaCl. *ADO + KCl differed significantly from ADO + KCl + BaCl. For comparison, the diameter change in response to ADO only (▼) is shown (data from Fig. 2). B, change in diameter in response to ADO + KCl in the absence (■) and presence (▲) of 10−4 m Oua. *ADO + KCl differed significantly from ADO + KCl + Oua. For comparison, the diameter in response to ADO only (▼) is shown (data from Figure 2).
Discussion
The primary purpose of the present study was to determine whether three prominent vasodilators released as products of muscle contraction, NO, K+ and ADO, were able to interact and influence the ability of each other to cause vasodilatation. The primary, novel finding of the present study was that KCl was able to significantly attenuate the magnitude of vasodilatation elicited by a wide range of NO and ADO concentrations. Furthermore, we show that we can reverse the inhibition, by K+, of ADO and NO by inhibiting the signalling mechanisms through which K+ causes vasodilatation. Thus, mechanistically, both KIR channels and Na+/K+ATPase are partly responsible for the ability of K+ to attenuate NO‐ and ADO‐induced vasodilatation. Our findings offer proof of principle for the existence of redundancy between physiologically relevant vasodilators and may provide a viable control paradigm that determines the amount of vasodilatation occurring during active hyperaemia. This implies that the magnitude of vasodilatation responsible for the increase in blood flow in response to exercise is not determined by the sum of the effects of individual vasodilators; rather, vasodilatation is the manifestation of a complex set of interactions between multiple vasodilators.
Vasodilator interactions
Our data demonstrate that vasodilator inhibition can occur immediately and can also occur over time where the effects of one vasodilator initially dominate and the actions of a second are expressed over time. Each of these types of interaction scenarios has relevance to muscle contraction‐induced vasodilatation. At the onset of contraction, vasodilatation occurs immediately (Mihok & Murrant, 2004), partly as a result of the immediate production of vasodilators resulting from skeletal muscle activation. For example, both K+ and ADO have been shown to be important within a single contraction (Armstrong et al. 2007; Ross et al. 2013). The source of K+ is likely the result of the generation of action potentials in skeletal muscle and ADO has been shown to be produced extracellularly from AMP during muscle contraction (Hellsten & Frandsen, 1997; Armstrong et al. 2007; Ross et al. 2013). NO is proposed to be released from skeletal muscle during contraction as a consequence of Ca2+‐dependent stimulation of NO synthase (Balon & Nadler, 1994; Kobzik et al. 1994). Through our simultaneous addition of vasodilators (protocol 2), we show that, if vasodilators were present at the same time, then interactions can occur sufficiently fast to foster redundancy.
As muscle contraction continues, there is build‐up of vasodilators in the interstitium. Our finding that interactions occur when the vasodilators are added sequentially (Protocol 1) is important here: this suggests that, if K+ is already present, and the concentrations of NO and ADO increase as contraction continues, the inhibition on NO and ADO‐induced vasodilatation will be maintained. Given that K+ was able to inhibit a large range of concentrations of ADO and NO, the redundancy between K+ and ADO, as well as that between K+ and NO, will continue to exist even though the concentration of the inhibited vasodilators is changing.
Our data also indicate that inhibitory interactions may be long‐lasting.We observed that, when SNAP or ADO was applied following exposure to K+ but after K+ had been washed out, the dilator responses evoked by ADO or SNAP lasted only 2–4 min. Similarly, when SNAP or ADO was applied after ADO or SNAP, respectively, had been applied but washed out, the dilator responses again lasted only 2–4 min. This behaviour raises the possibility that exposure to each of the agonists caused a slowly developing and persistent effect on the intracellular mechanisms for vasodilatation, which affects both the duration and magnitude of vasodilatation evoked by K+, ADO and/or NO. Maimon et al. (2014) have demonstrated that pre‐exposure to ADO can affect the mechanisms by which ADO produces vasodilatation: they suggest that pre‐exposure to ADO induces long‐lasting protein kinase A (PKA)‐dependent phosphorylation of KATP channels such that subsequent application of ADO causes dilatation independently of PKA via a mechanism dependent on endothelial cell Ca2+ and cyclic nucleotide‐gated channels. This pre‐exposure phenomena does not help to explain our results because, in protocols with multiple applications of vasodilators, the inhibitory effects were vasodilator‐dependent and not dependent simply on whether there was pre‐exposure to ADO. However, it is possible that a pre‐exposure phenomena occurs within our experiments where pre‐exposure may have altered the duration of the responses. Further studies will be required to confirm whether this is a viable explanation.
Mechanistic basis for vasodilator inhibition
We have implicated both KIR channels and Na+/K+ATPase in the signalling pathway for K+ as part of the pathway through which K+ suppresses ADO and NO vasodilatation. K+ is hypothesized to cause hyperpolarization of vascular smooth muscle (VSM) directly by opening KIR channels and increasing the Na+/K+ATPase (Knot et al. 1996; Edwards et al. 1998; Burns et al. 2004) or by hyperpolarizing endothelial cells that can transmit the hyperpolarization to VSM through myoendothelial gap junctions (Emerson & Segal, 2000; Busse et al. 2002; Garland et al. 2011). Altering the membrane potential may change the signalling pathway mechanisms of other vasoactive compounds, especially if voltage‐gated channels are involved. However, our data indicate a mechanism that downplays a central role for hyperpolarization. Our sequential experiments show that adding K+ to the preparation 5 min prior to re‐exposing the preparation to a range of concentrations of either NO or ADO produced a transient vasodilatation, where the diameter recovered back to baseline levels, and therefore we assume that the hyperpolarization associated with this vasodilatation has also recovered. Thus, the inhibition of ADO and NO by K+ occurred in the absence of a changed membrane potential: a maintained, hyperpolarized state does not appear to be integral to the inhibition by K+.
ADO can cause endothelium‐dependent vasodilatation via A2A receptors (Marshall, 2007; Maimon et al. 2014) and also increase cAMP to open KATP channels (Kleppisch & Nelson, 1995; Danialou et al. 1997; Hein & Kuo, 1999) on either VSM or endothelial cells to promote hyperpolarization and VSM relaxation. How K+ stimulation of KIR channels or increased Na+/K+ATPase activity interferes with this pathway is not known. Confounding the determination of the cellular pathways that promote vasodilator interaction is the potential for signalling pathways to change in response to the same vasodilator (Maimon et al. 2014). For example, in our experiments, K+ inhibited ADO vasodilatation during the re‐application of ADO and therefore inhibited the PKA‐independent pathway induced by ADO, rather than the PKA‐dependent pathway that would have been induced by the initial ADO application. Given the potential for multiple signalling pathways originating from a single vasodilator, the determination of how cell signalling pathways interact will require very careful study.
Interactions between ADO and NO implicates potential cross‐talk between cAMP and cGMP‐dependent pathways. In VSM of arteries and arterioles, the effects of cross‐talk between cGMP‐dependent and cAMP‐dependent pathways has been shown to amplify vasodilatation (Grace et al. 1988; de Wit et al. 1994). Interestingly, using the same preparation as that employed in the present study, DeWit et al. (1994) showed that the vasodilatation produced by an increase in cGMP via sodium nitroprusside was amplified in the presence of increasing cAMP via prostacyclin. We did not observe this amplification when we increased cGMP via SNAP and cAMP via ADO. Therefore, it is also possible that the mechanism by which cAMP and cGMP are increased is an important variable, implicating other signalling intermediates as being important in the interactions that take place.
The importance of vascular level in vasodilator interactions
The interaction of vasodilators may vary depending on the level of the vasculature stimulated. We demonstrate interactions between K+ with ADO and NO on 2A arterioles, which are considered to be important in the distribution of blood flow within the tissue (Klitzman et al. 1982; Lindbom & Arfors, 1984; Sarelius, 1986; Sweeney & Sarelius, 1989). These arterioles differ functionally from larger arteries and arterioles that are more responsible for delivery of blood flow to a tissue. They also differ from other arteriolar levels in their membrane receptor populations and reactivity to vasodilators. For example, 1A arterioles are more reactive to NO than 2A arterioles (Hester et al. 1993). Thus, if a redundant pathway between K+ and NO exists at the 1A level, it may be different from the redundant relationship found at the 2A level, indicating that the predominant combination of interacting vasodilators may differ depending on the vascular level being studied. The interactions that occur at the 1A arteriolar level and their contributions to blood flow regulation deserve investigation in future studies.
Redundancy
The inhibition of one vasodilator by another provides the necessary framework for redundancy within the blood flow control system. The extent of redundant control over blood flow will depend on the extent of the interactions between vasodilators. Figure 7 shows different inhibitory paradigms that would achieve redundancy between vasodilators. Figure 7A is the simplest paradigm: a linear inhibitory relationship where A inhibits B, which inhibits C such that, if A is inhibited, then B increases its effects, and so on. The interaction between vasodilators could also be more complex, where one vasodilator could inhibit the effects of more than one vasodilator (Fig. 7 B). The paradigm could be circular, where each vasodilator has a redundant relationship with another vasodilator (Fig. 7 C). Furthermore, the paradigm could comprise a more complex network with multiple vasodilators, with each being critical and each having their own network, and where networks interact (Fig. 7 D). Whether K+, ADO and NO fit into a simple system or are part of a more complex network of interacting vasodilators will remain unknown until we understand the number of dilators involved and the extent of the redundant networks. For example, during muscle contraction, we do not know whether K+, ADO and NO act as a small redundant network with K+ as the primary inhibiting vasodilator or whether they are part of a larger set of networks where K+ may be inhibited by another vasodilator. The limitations of the redundancy will also be dictated by how complete the inhibition is. In some redundant relationships, there appears to be full restoration of the dilatation or blood flow when one vasodilator is inhibited, as suggested with individual inhibition of either NO or prostaglandin in humans (Hellsten et al. 2012). In other instances, a single inhibitor can show 20–40% inhibition of flow (adenosine inhibition: Marshall 2007; KIR channel inhibition in humans: Crecelius et al. 2014), indicating that the redundant system may not be able to compensate fully from the inhibition of some vasodilators. Redundancy, in these cases, may prevent blood flow for diminishing <40%. To better understand the role of redundancy in active hyperaemia, both the networks of interactions and the efficiencies of these interactions will need to be defined.
Figure 7. Potential vasodilator interactions to describe different redundancy paradigms .
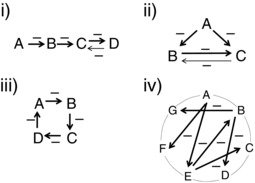
Different paradigms represented in (i) to (iv) show different ways in which vasodilators can inhibit each other to create redundancy. i), a more singular relationship between vasodilator interactions where (A) inhibits (B), which can inhibit (C), which can inhibit (D). (D) is shown to be able to inhibit a vasodilator (C) because, if it did not, then inhibiting (D) alone should show a significant decrease in vasodilatation because there would be no redundant system to fill in for (D) if it was inhibited. ii), shows a scenario where one vasodilator can inhibit more than one vasodilator: (A) can inhibit both (B) and (C). (B) and (C) are shown to interact as well. If they did not, there would be no redundant system to replace the effects of (B) or (C) and inhibiting either (B) or (C) would result in a significant decrease in vasodilatation. iii), a slightly more integrated system of inhibition that is a variation of (A) where each vasodilator interacts with another vasodilator. iv), a more integrated network of vasodilator interactions where one group of interacting vasodilators can also interact with another group of interacting vasodilators. These are just a few potential relationships out of many, depending on how many vasodilators are involved and also on the complexity of the redundant networks.
Conclusions
We have shown that vasodilators relevant to active hyperaemia have the ability to interact with each other, where K+ is able to attenuate the vasodilatation produced by either ADO or NO. The present study provides proof of principle that redundancy through inhibition among vasodilators exists, thus implicating redundancy as a possibile control paradigm in active hyperaemia. Accordingly, redundancy would explain the inability to identify a single vasodilator as being critically important to the process and also explain how such a critical process is maintained in the absence of individual vasodilators. Our data implies that the vasodilatation during active hyperaemia may be the result of complex interactions between vasodilator substances. Many complex, biological systems are governed by redundancy and the critical process of active hyperaemia may be one such process.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
IL was responsible for the conception and design of the experiments, data collection, analysis and interpretation of the data, and reviewing the manuscript. CM was responsible for the conception and experimental design, data analysis and interpretation of the data, and writing the manuscript. Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was funded by NSERC, Canada.
References
- Armstrong ML, Dua AK & Murrant CL (2007). Potassium initiates vasodilatation induced by a single skeletal muscle contraction in hamster cremaster muscle. J Physiol 581, 841–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baez S (1973). An open cremaster muscle preparation for the study of blood vessels by in vivo microscopy. Microvasc Res 5, 384–394. [DOI] [PubMed] [Google Scholar]
- Balon TW & Nadler JL (1994). Nitric oxide release is present from incubated skeletal muscle preparations. J Appl Physiol 77, 2519–2521. [DOI] [PubMed] [Google Scholar]
- Bauersachs J, Popp R, Hecker M, Sauer E, Fleming I & Busse R (1996). Nitric oxide attenuates the release of endothelium‐derived hyperpolarizing factor. Circulation 94, 3341–3347. [DOI] [PubMed] [Google Scholar]
- Boushel R, Langberg H, Gemmer C, Olesen J, Crameri R, Scheede C, Sander M & Kjaer M (2002). Combined inhibition of nitric oxide and prostaglandins reduces human skeletal muscle blood flow during exercise. J Physiol 543, 691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns WR, Cohen KD & Jackson WF (2004). K+‐induced dilation of hamster cremasteric arterioles involves both the Na+/K+‐ATPase and inward‐rectifier K+ channels. Microcirculation 11, 279–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse R, Edwards G, Feletou M, Fleming I, Vanhoutte PM & Weston AH (2002). EDHF: bringing the concepts together. Trends Pharmacol Sci 23, 374–380. [DOI] [PubMed] [Google Scholar]
- Casey DP, Mohamed EA & Joyner MJ (2013). Role of nitric oxide and adenosine in the onset of vasodilation during dynamic forearm exercise. Euro J Appl Physiol 113, 295–303. [DOI] [PubMed] [Google Scholar]
- Chilian WM & Koshida R (2001). EDHF and NO: different pathways for production – similar actions. Circ Res 89, 648–649. [PubMed] [Google Scholar]
- Clausen T (2011). In isolated skeletal muscle, excitation may increase extracellular K+ 10‐fold; how can contractility be maintained? Exp Physiol 96, 356–368. [DOI] [PubMed] [Google Scholar]
- Clausen T (2013). Excitation‐induced exchange of Na+, K+, and Cl– in rat EDL muscle in vitro and in vivo: physiology and pathophysiology. J Gen Physiol 141, 179–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford PS & Hellsten Y (2004). Vasodilatory mechanisms in contracting skeletal muscle. J Appl Physiol 97, 393–403. [DOI] [PubMed] [Google Scholar]
- Crecelius AR, Luckasen GJ, Larson DG & Dinenno FA (2014). KIR channel activation contributes to onset and steady‐state exercise hyperemia in humans. Am J Physiol Heart Circ Physiol 307, H782–H791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danialou G, Vicaut E, Sambe A, Aubier M & Boczkowski J (1997). Predominant role of A1 adenosine receptors in mediating adenosine induced vasodilatation of rat diaphragmatic arterioles: involvement of nitric oxide and the ATP‐dependent K+ channels. Br J Pharmacol 121, 1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit C, von Bismarck P & Pohl U (1994). Synergistic action of vasodilators that increase cGMP and cAMP in the hamster cremaster microcirculation. Cardiovasc Res 28, 1513–1518. [DOI] [PubMed] [Google Scholar]
- Dua AK, Dua N & Murrant CL (2009). Skeletal muscle contraction‐induced vasodilator complement production is dependent on stimulus and contraction frequency. Am J Physiol Heart Circ Physiol 297, H433–H442. [DOI] [PubMed] [Google Scholar]
- Edwards G, Dora KA, Gardener MJ, Garland CJ & Weston AH (1998). K+ is an endothelium‐derived hyperpolarizing factor in rat arteries. Nature 396, 269–272. [DOI] [PubMed] [Google Scholar]
- Emerson GG & Segal SS (2000). Electrical coupling between endothelial cells and smooth muscle cells in hamster feed arteries: role in vasomotor control. Circ Res 87, 474–479. [DOI] [PubMed] [Google Scholar]
- Frisbee JC (2002). Regulation of in situ skeletal muscle arteriolar tone: interactions between two parameters. Microcirculation 9, 443–462. [DOI] [PubMed] [Google Scholar]
- Frisbee JC & Lombard JH (1999). Elevated oxygen tension inhibits flow‐induced dilation of skeletal muscle arterioles. Microvasc Res 58, 99–107. [DOI] [PubMed] [Google Scholar]
- Frisbee JC, Roman RJ, Falck JR, Linderman JR & Lombard JH (2000). Impairment of flow‐induced dilation of skeletal muscle arterioles with elevated oxygen in normotensive and hypertensive rats. Microvasc Res 60, 37–48. [DOI] [PubMed] [Google Scholar]
- Garland CJ, Hiley CR & Dora KA (2011). EDHF: spreading the influence of the endothelium. Br J Pharmacol 164, 839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace GC, Macdonald PS & Dusting GJ (1988). Cyclic nucleotide interactions involved in endothelium‐dependent dilatation in rat aortic rings. Euro J Pharmacol 148, 17–24. [DOI] [PubMed] [Google Scholar]
- Haddy FJ (1983). Potassium effects on contraction in arterial smooth muscle mediated by Na+, K+‐ATPase. Federation Proc 42, 239–245. [PubMed] [Google Scholar]
- Hein TW & Kuo L (1999). cAMP‐independent dilation of coronary arterioles to adenosine: role of nitric oxide, G proteins, and K(ATP) channels. Circ Res 85, 634–642. [DOI] [PubMed] [Google Scholar]
- Hellsten Y & Frandsen U (1997). Adenosine formation in contracting primary rat skeletal muscle cells and endothelial cells in culture. J Physiol 504 ( Pt 3 ), 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellsten Y, Nyberg M, Jensen LG & Mortensen SP (2012). Vasodilator interactions in skeletal muscle blood flow regulation. J Physiol 590, 6297–6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hester RL, Eraslan A & Saito Y (1993). Differences in EDNO contribution to arteriolar diameters at rest and during functional dilation in striated muscle. Am J Physiol Heart Circ Physiol 265, H146–H151. [DOI] [PubMed] [Google Scholar]
- Hnik P, Vyskocil F, Kriz N & Holas M (1972). Work‐induced increase of extracellular potassium concentration in muscle measured by ion‐specific electrodes. Brain Res 40, 559–562. [DOI] [PubMed] [Google Scholar]
- Honig CR & Frierson JL (1980). Role of adenosine in exercise vasodilation in dog gracilis muscle. Am J Physiol 238, H703‐715. [DOI] [PubMed] [Google Scholar]
- Huang A, Sun D, Carroll MA, Jiang H, Smith CJ, Connetta JA, Falck JR, Shesely EG, Koller A & Kaley G (2001). EDHF mediates flow‐induced dilation in skeletal muscle arterioles of female eNOS‐KO mice. Am J Physiol Heart Circ Physiol 280, H2462–H2469. [DOI] [PubMed] [Google Scholar]
- Joyner MJ & Wilkins BW (2007). Exercise hyperaemia: is anything obligatory but the hyperaemia? J Physiol 583, 855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler P, Popp R, Busse R & Schini‐Kerth VB (1999). Proinflammatory mediators chronically downregulate the formation of the endothelium‐derived hyperpolarizing factor in arteries via a nitric oxide/cyclic GMP‐dependent mechanism. Circulation 99, 1878–1884. [DOI] [PubMed] [Google Scholar]
- Kille JM & Klabunde RE (1984). Adenosine as a mediator of postcontraction hyperemia in dog gracilis muscle. Am J Physiol Heart Circ Physiol 246, H274–H282. [DOI] [PubMed] [Google Scholar]
- Kleppisch T & Nelson MT (1995). Adenosine activates ATP‐sensitive potassium channels in arterial myocytes via A2 receptors and cAMP‐dependent protein kinase. Proc Nat Acad Sci U S A 92, 12441–12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klitzman B, Damon DN, Gorczynski RJ & Duling BR (1982). Augmented tissue oxygen supply during striated muscle contraction in the hamster. Relative contributions of capillary recruitment, functional dilation, and reduced tissue PO2 . Circ Res 51, 711–721. [DOI] [PubMed] [Google Scholar]
- Knot HJ, Zimmermann PA & Nelson MT (1996). Extracellular K(+)‐induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K(+) channels. J Physiol 492, 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobzik L, Reid MB, Bredt DS & Stamler JS (1994). Nitric oxide in skeletal muscle. Nature 372, 546–548. [DOI] [PubMed] [Google Scholar]
- Kuo L & Chancellor JD (1995). Adenosine potentiates flow‐induced dilation of coronary arterioles by activating KATP channels in endothelium. Am J PhysiolHeart Circ Physiol 269, H541–H549. [DOI] [PubMed] [Google Scholar]
- Leonard S, Croy BA & Murrant CL (2011). Arteriolar reactivity in lymphocyte‐deficient mice. Am J Physiol Heart Circ Physiol 301, H1276–H1285. [DOI] [PubMed] [Google Scholar]
- Lindbom L & Arfors KE (1984). Non‐homogeneous blood flow distribution in the rabbit tenuissimus muscle. Differential control of total blood flow and capillary perfusion. Acta Physiol Scand 122, 225–233. [DOI] [PubMed] [Google Scholar]
- Loeb AL, Godeny I & Longnecker DE (2000). Functional evidence for inward‐rectifier potassium channels in rat cremaster muscle arterioles. Microvasc Res 59, 1–6. [DOI] [PubMed] [Google Scholar]
- Lombard JH, Eskinder H, Kauser K, Osborn JL & Harder DR (1990). Enhanced norepinephrine sensitivity in renal arteries at elevated transmural pressure. Am J Physiol 259, H29‐33. [DOI] [PubMed] [Google Scholar]
- Maimon N, Titus PA & Sarelius IH (2014). Pre‐exposure to adenosine, acting via A(2A) receptors on endothelial cells, alters the protein kinase A dependence of adenosine‐induced dilation in skeletal muscle resistance arterioles. J Physiol 592, 2575–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markwald RR, Kirby BS, Crecelius AR, Carlson RE, Voyles WF & Dinenno FA (2011). Combined inhibition of nitric oxide and vasodilating prostaglandins abolishes forearm vasodilatation to systemic hypoxia in healthy humans. J Physiol 589, 1979–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JM (2007). The roles of adenosine and related substances in exercise hyperaemia. J Physiol 583, 835–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihok ML & Murrant CL (2004). Rapid biphasic arteriolar dilations induced by skeletal muscle contraction are dependent on stimulation characteristics. Can J Physiol Pharmacol 82, 282–287. [DOI] [PubMed] [Google Scholar]
- Mortensen SP, Gonzalez‐Alonso J, Damsgaard R, Saltin B & Hellsten Y (2007). Inhibition of nitric oxide and prostaglandins, but not endothelial‐derived hyperpolarizing factors, reduces blood flow and aerobic energy turnover in the exercising human leg. J Physiol 581, 853–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen SP & Saltin B (2014). Regulation of the skeletal muscle blood flow in humans. Exp Physiol 99, 1552–1558. [DOI] [PubMed] [Google Scholar]
- Murrant CL (2005). Stimulation characteristics that determine arteriolar dilation in skeletal muscle. Am J Physiol Regul Integr Comp Physiol 289, R505–R513. [DOI] [PubMed] [Google Scholar]
- Murrant CL, Dodd JD, Foster AJ, Inch KA, Muckle FR, Ruiz DA, Simpson JA & Scholl JH (2014). Prostaglandins induce vasodilatation of the microvasculature during muscle contraction and induce vasodilatation independent of adenosine. J Physiol 592, 1267–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrant CL & Sarelius IH (2000). Local and remote arteriolar dilations initiated by skeletal muscle contraction. Am J Physiol Heart Circ Physiol 279, H2285–H2294. [DOI] [PubMed] [Google Scholar]
- Nishikawa Y, Stepp DW & Chilian WM (2000). Nitric oxide exerts feedback inhibition on EDHF‐induced coronary arteriolar dilation in vivo. Am J Physiol Heart Circ Physiol 279, H459–H465. [DOI] [PubMed] [Google Scholar]
- Nordsborg N, Mohr M, Pedersen LD, Nielsen JJ, Langberg H & Bangsbo J (2003). Muscle interstitial potassium kinetics during intense exhaustive exercise: effect of previous arm exercise. Am J Physiol Regul Integr Comp Physiol 285, R143–R148. [DOI] [PubMed] [Google Scholar]
- Olmos L, Mombouli JV, Illiano S & Vanhoutte PM (1995). cGMP mediates the desensitization to bradykinin in isolated canine coronary arteries. Am J PhysiolHeart Circ Physiol 268, H865–H870. [DOI] [PubMed] [Google Scholar]
- Proctor KG & Duling BR (1982). Adenosine and free‐flow functional hyperemia in striated muscle. Am J PhysiolHeart Circ Physiol 242, H688–H697. [DOI] [PubMed] [Google Scholar]
- Ross GA, Mihok ML & Murrant CL (2013). Extracellular adenosine initiates rapid arteriolar vasodilation induced by a single skeletal muscle contraction in hamster cremaster muscle. Acta Physiol 208, 74–87. [DOI] [PubMed] [Google Scholar]
- Rowell LB (2004). Ideas about control of skeletal and cardiac muscle blood flow (1876‐2003): cycles of revision and new vision. J Appl Physiol 97, 384–392. [DOI] [PubMed] [Google Scholar]
- Sarelius I & Pohl U (2010). Control of muscle blood flow during exercise: local factors and integrative mechanisms. Acta Physiol 199, 349–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarelius IH (1986). Cell flow path influences transit time through striated muscle capillaries. Am J PhysiolHeart Circ Physiol 250, H899–H907. [DOI] [PubMed] [Google Scholar]
- Schildmeyer LA & Jr Bryan, RM . (2002). Effect of NO on EDHF response in rat middle cerebral arteries. Am J Physiol Heart Circ Physiol 282, H734–H738. [DOI] [PubMed] [Google Scholar]
- Schrage WG, Joyner MJ & Dinenno FA (2004). Local inhibition of nitric oxide and prostaglandins independently reduces forearm exercise hyperaemia in humans. J Physiol 557, 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Huang A, Koller A & Kaley G (1995). Flow‐dependent dilation and myogenic constriction interact to establish the resistance of skeletal muscle arterioles. Microcirculation 2, 289–295. [DOI] [PubMed] [Google Scholar]
- Sweeney TE & Sarelius IH (1989). Arteriolar control of capillary cell flow in striated muscle. Circ Res 64, 112–120. [DOI] [PubMed] [Google Scholar]
- Tabaie HM, Scott JB & Haddy FJ (1977). Reduction of exercise dilation by theophylline. Proc Soc Exp Biol Med 154, 93–97. [DOI] [PubMed] [Google Scholar]
- Vyskocil F, Hnik P, Rehfeldt H, Vejsada R & Ujec E (1983). The measurement of K+e concentration changes in human muscles during volitional contractions. Pflügers Archiv 399, 235–237. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Yashiro Y, Mizuno R & Ohhashi T (2005). Involvement of NO and EDHF in flow‐induced vasodilation in isolated hamster cremasteric arterioles. J Vasc Res 42, 137–147. [DOI] [PubMed] [Google Scholar]
- Zhang C, Hein TW & Kuo L (2000). Transmural difference in coronary arteriolar dilation to adenosine: effect of luminal pressure and K(ATP) channels. Am J Physiol Heart Circ Physiol 279, H2612–H2619. [DOI] [PubMed] [Google Scholar]