

Abstract
Key points
Dystrophin deficiency disrupts sarcolemmal targeting of neuronal nitric oxide synathase, resulting in functional muscle ischaemia.
Chronic treatment of dystrophic mice with an inorganic nitric oxide (NO) donor alleviates this ischaemia and improves many features of the dystrophic phenotype.
The present study translates this preclinical work by showing that a single oral dose of sodium nitrate,which serves as a NO donor when reduced to circulating nitrite by the commensal bacteria in the oral cavity, alleviates functional muscle ischaemia and restores normal blood flow regulation in human patients with dystrophinopathy.
The results of the present study further support the mechanistic hypothesis that circulating nitrite serves as an alternative NO donor when reduced by deoxyhaemoglobin and/or deoxymyoglobin in exercising muscle.
Abstract
Becker muscular dystrophy (BMD) is a progressive X‐linked muscle wasting disease for which there is no treatment. BMD is caused by in‐frame mutations in the gene encoding dystrophin, a structural cytoskeletal protein that also targets other proteins to the sarcolemma. Among these is neuronal nitric oxide synthase mu (nNOSμ), which requires specific spectrin‐like repeats (SR16/17) in dystrophin's rod domain and the adaptor protein α‐syntrophin for sarcolemmal targeting. When healthy skeletal muscle is exercised, sarcolemmal nNOSμ‐derived nitric oxide (NO) attenuates α‐adrenergic vasoconstriction, thus optimizing perfusion. In the mdx mouse model of dystrophinopathy, this protective mechanism (functional sympatholysis) is defective, resulting in functional muscle ischaemia. Treatment with a NO‐donating non‐steroidal anti‐inflammatory drug (NSAID) alleviates this ischaemia and improves the murine dystrophic phenotype. In the present study, we report that, in 13 men with BMD, sympatholysis is defective mainly in patients whose mutations disrupt sarcolemmal targeting of nNOSμ, with the vasoconstrictor response measured as a decrease in muscle oxygenation (near infrared spectroscopy) to reflex sympathetic activation. Then, in a single‐arm, open‐label trial in 11 BMD patients and a double‐blind, placebo‐controlled cross‐over trial in six patients, we show that acute treatment with oral sodium nitrate, an inorganic NO donor without a NSIAD moiety, restores sympatholysis and improves post‐exercise hyperaemia (Doppler ultrasound). By contrast, sodium nitrate improves neither sympatholysis, nor hyperaemia in healthy controls. Thus, a simple NO donor recapitulates the vasoregulatory actions of sarcolemmal nNOS in BMD patients, and constitutes a putative novel therapy for this disease.
Abbreviations
- BMD
Becker muscular dystrophy
- deoxyHb
deoxyhaemoglobin
- deoxyMb
deoxymyoglobin
- DMD
Duchenne muscular dystrophy
- LBNP
lower body negative pressure
- MVC
maximal voluntary contraction
- NIRS
near infrared spectroscopy
- NO
nitric oxide
- NO2−
nitrite
- NO3−
nitrate
- nNOSμ
neuronal nitric oxide synthase mu
- NSAID
non‐steroidal anti‐inflammatory drug
- PDE5
phosphodiesterase 5
- TLS
total labile signal
Introduction
Mutations in the gene encoding the cytoskeletal protein dystrophin cause Duchenne and Becker muscular dystrophy (Bushby et al. 2010 a; Bushby et al. 2010 b; Emery, 2002), X‐linked muscle wasting diseases for which there is as yet no disease‐specific therapy. Duchenne muscular dystrophy (DMD) is caused by out‐of‐frame mutations yielding no functional dystrophin, whereas Becker muscular dystrophy (BMD) is caused by in‐frame mutations yielding truncated dystrophin (Monaco, 1989). Dystrophin deficiency leads to progressive muscle fibre atrophy, culminating in a loss of ambulation and death (Bushby et al. 2010 a; Bushby et al. 2010 b). Although glucocorticoids can prolong ambulation by a few years in patients with DMD, this is by no means a cure and, currently, there is no therapy to offer patients with BMD.
The nitric oxide (NO)‐cGMP pathway constitutes a putative new drug target for patients with dystrophinopathy. Dystrophin is both a structural protein that stabilizes the sarcolemma and a scaffolding protein that targets other proteins to the sarcolemma (Emery, 2002). Among these is a muscle‐specific splice variant of neuronal nitric oxide synthase mu (nNOSμ), which requires specific spectrin‐like repeats (SR16/17) in dystrophin's rod domain and the adaptor protein α‐syntrophin for sarcolemmal targeting (Brenman et al. 1995; Kameya et al. 1999; Lai et al. 2009). Previous work from our group, as well as other studies, suggests that, when healthy skeletal muscle is exercised, sarcolemmal nNOSμ‐derived NO attenuates α‐adrenergic vasoconstriction, thus optimizing perfusion (Sander et al. 2000; Thomas et al. 1998; Chavoshan et al. 2002; Hansen et al. 1996; Thomas et al. 1994; Thomas & Victor, 1998; Thomas et al. 2001; Thomas et al. 2003; Vongpatanasin et al. 2011) This protective mechanism, termed functional sympatholysis, is defective in mdx mice (the standard murine model of DMD) (Thomas et al. 1998; Thomas et al. 2012), nNOS null mice (Thomas et al. 1998), paediatric patients with DMD (Sander et al. 2000; Nelson et al. 2014) and many but not all adult patients with BMD (Martin et al. 2012).
Although sympatholysis is critically dependent on sarcolemmal targeting nNOS by R16,17 in transgenic mdx mouse studies (Lie et al. 2009), clinical translation of this hypothesis is limited to two cases of BMD (Martin et al. 2012). Thus, the first aim of the present study was to rigorously test the hypothesis that sympatholysis is defective in men with BMD mutations specifically interrupting sarcolemmal anchoring of nNOS. Accordingly, we conducted mutational analysis, immunohistochemistry and western blot experiments on fresh muscle biopsy tissue, as well as sympatholysis studies on 13 BMD patients. The patients’ sympatholysis data were compared with normative values obtained from healthy age‐matched male controls.
We previously showed that sympatholysis is rescued in patients with DMD and BMD by phosphodiesterase 5 (PDE5) inhibition, which prolongs the half‐life of cGMP, the downstream target of NO in vascular smooth muscle (Martin et al. 2012; Nelson et al. 2014). PDE5 inhibition also has been shown in mdx mice to prevent exercise‐induced spasm of skeletal muscle microvessels that can cause post‐exercise fatigue, and to benefit respiratory and cardiac muscle dysfunction (Asai et al. 2007; Adamo et al. 2010; Khairallah et al. 2008; Percival et al. 2012; Kobayashi et al. 2008). However, PDE5 inhibition cannot rescue NO production in dystrophic muscle, and thus may not fully recapitulate all the cytoprotective effects of the missing sarcolemmal nNOS. Although our group has implicated a major role for sarcolemmal‐archored nNOS in normal muscle blood flow regulation, other studies have implicated major cytoprotective roles with respect to muscle inflammation, oxidative stress, fat deposition (Cordani et al. 2014; Buono et al. 2012), fibrosis (Brunelli et al. 2007; Sciorati et al. 2011; Tidball & Wehling‐Henricks, 2004) and muscle regeneration through activation of satellite cells (Anderson, 2000; Brunelli et al. 2007).
Recently, chronic treatment of mdx mice with a NO‐donating non‐steroidal anti‐inflammatory drug (NSAID) was shown to rescue functional sympatholysis (Thomas et al. 2012), as well as to increase muscle mass and grip strength, decrease inflammation and improve cardiac function (Uaesoontrachoon et al. 2014). Thus, the second aim of the present study was to begin to translate this promising preclinical work to human muscular dystrophy patients. Accordingly, we conducted a single‐arm, open‐label treatment trial, and then a double‐blind, placebo‐controlled cross‐over trial, aiming to determine whether sodium nitrate (a simple inorganic NO donor without a NSAID moiety) would restore functional sympatholysis in BMD patients lacking sarcolemmal nNOS.
In a subset of patients with BMD, we tested whether sodium nitrate also might improve post‐exercise hyperaemia, which is severely blunted in mdx mice and nNOS null mice as a result of post‐exercise microvessel spasm (Kobayashi et al. 2008). When ingested, commensal bacteria in the oral cavity convert nitrate (NO3 −) to nitrite (NO2 −), which permeates the gastrointestinal mucosa to increase systemic bioavailability (Lundberg et al. 2008). Circulating NO2 −, in turn, can serve as an alternative NO donor but only when reduced by deoxyhaemoglobin (deoxyHb) and/or deoxymyoglobin (deoxyMb) (Lundberg et al. 2008). Because the intramuscular concentrations of deoxyHb and deoxyMb are low at rest and rise when muscles are exercised, we hypothesized that treatment with oral sodium nitrate would improve exercise‐induced hyperaemia in BMD skeletal muscle at the same time as having little, if any, effect on blood flow in resting BMD skeletal muscle. Finally, we challenged healthy controls with sodium nitrate to test whether the NO donor would have any effect on either functional sympatholysis or post‐exercise hyperaemia when sarcolemmal nNOS is plentiful.
Methods
Ethical approval
The present study was approved by the Institutional Review Board at Cedars‐Sinai Medical Center, and conformed with the standards set by the Declaration of Helsinki. All subjects provided their written informed consent before participating.
Subjects
We recruited ambulatory adult male patients aged 18–45 years with a pre‐existing clinical diagnosis of BMD. Mutational analysis with direct sequencing of dsytrophin was performed using multiplex ligation‐dependent probe amplification of genomic DNA isolated from whole blood (University of Utah Genome Centre, Salt Lake City, UT, USA) (Flanigan et al. 2003). We also studied thirteen healthy male controls matched for age and body mass index. Potential subjects (both cases and controls) were excluded if they had a history of hypertension or measured blood pressure >140/90 mmHg; a history of diabetes mellitus or fasting glucose >200 mg dL−1; heart failure by history, physical examination, elevated brain natriuretic peptide, left ventricular ejection fraction ≤50% by echocardiography; arrhythmia by 12‐lead electrocardiogram; nocturnal ventilator support; or any contraindication to nitrate supplementation (preexisting use of nitrates, α‐adrenergic blockers, PDE5 inhibitors, l‐arginine or potent inhibitors of cytochrome P450 3A4).
Measurements
Immunohistochemistry and western blot experiments
Open muscle biopsies from the biceps brachii of the non‐dominant arm (n = 7) or punch needle muscle biopsies from the vastus lateralis (n = 8) were obtained using a standard technique. Two of the biopsies from the vastus lateralis, P5 and P8, have been reported previously (Table 1) (Martin et al. 2012). Fresh muscle samples were mounted in optimal cutting temperature compound and frozen in isopentane cooled in liquid nitrogen. Cryosections (6 mm) were cut and mounted onto SuperFrost Plus slides (Fisher Scientific Co., Pittsburgh, PA, USA).
Table 1.
Patient characteristics
| Grip strength | ||||||||
|---|---|---|---|---|---|---|---|---|
| Table | Deletion/ | Age at | Age at onset of | Right | Left | Creatinekinase | Frequency wheelchair/ scooter | |
| ID | mutation | study | symptoms | arm | arm | (U L–1) | use | Medications |
| 1 | 3–7 | 21 | 6 | 8 | 9 | 2522 | Frequently | Chromium, creatine, enalapril |
| 2 | 3–7 | 24 | 12 | 34 | 33 | 3863 | Rarely | None |
| 3 | 45–47 | 18 | 12 | 29 | 35 | 1510 | Never | None |
| 4 | 45–47 | 40 | 4 | 14 | 18 | 1473 | Frequently | Lisinopril, simvastatin |
| 5 | 45–48 | 44 | 31 | 25 | 32 | 616 | Rarely | None |
| 6 | 45–48 | 41 | 35 | 40 | 43 | 657 | Never | None |
| 7 | 45–48 | 44 | 30 | 39 | 41 | 455 | Never | None |
| 8 | 14–44 | 51 | 47 | 20 | 18 | 917 | Never | None |
| 9 | Splice donor Ex46 | 13 | 25 | 44 | 46 | 3574 | Never | CoQ10, protein supplement, royal jelly |
| 10 | Splice donor Ex25 | 51 | 26 | 22 | 22 | 444 | Rarely | Dermasil |
| 11 | Nonsense Mutation Ex29 | 24 | 4 | 32 | 40 | 651 | Never | Lisinopril, CoQ10, multivitamin |
| 12 | 48–55 | 43 | 38 | 46 | 43 | 554 | Never | Prednisone |
| 13 | Splice Donor Ex46 | 21 | 12 | 39 | 41 | 5104 | Never | None |
| 14 | 45–55 | 40 | 30 | 23 | 20 | 1552 | Never | None |
| 15 | 45–53 | 35 | 7 | 11 | – | 2282 | Never | None |
| 16 | Splice Acceptor Ex26 | 25 | 18 | 17 | 15 | 2766 | Never | Detrol |
Normal creatinine kinase range: 0–171 U L–1.
Sections were incubated with monoclonal antibodies for nNOS (NCL‐NOS‐1; Novocastra, Leica Microsystems, Wetzlar, Germany; dilution 1:400) and dystrophin C‐terminus (NCLDYS2; Novocastra; dilution 1:1000). Immunodetection was carried out with a sensitive detection protocol (X‐Cell‐Plus HRP Detection; MP‐XCPDAB‐U100; Menapath, Menarini Diagnostics, Woking, UK) in accordance with the manufacturer's instructions. Sections were visualized with Liquid Stable DAB (Menapath), counterstained in Carazzi's haematoxylin, dehydrated and permanently mounted. Primary antibody was omitted in negative controls. Patient biopsies were compared with a stored muscle sample showing normal histology and protein expression, which was obtained from a healthy individual. Haematoxylin and eosin staining was performed in accordance with a standard protocol.
Whole‐muscle lysate was extracted and western blotting was performed in accordance with a previously reported protocol (Anderson & Davison, 1999). The membranes were probed with antibodies for the dystrophin C‐terminus (dilution 1:100), dystrophin rod domain (NCL‐DYS1; Novocastra; dilution 1:300), and anti‐desmin (Dako, Glostrup, Denmark; dilution 1:400) as indicators of muscle proteins. Membranes were then incubated in HRP conjugated rabbit anti‐mouse IgG secondary antibody (Dako; dilution 1:300), followed by chemiluminescent detection (SuperSignalWest Pico Chemiluminescent Substrate, Pierce, Rockford, IL, USA).
Muscle blood flow regulation
Two indices of muscle blood flow regulation were assessed: (1) functional sympatholysis, which refers specifically to exercise‐induced attenuation of reflex vasoconstriction (the study's primary end‐point), and (2) post‐exercise hyperaemia (a secondary end‐point), which is a measure of the exercise‐induced increase in bulk muscle blood flow. Both are impaired in the mdx mouse and the nNOS null mouse (Kobayashi et al. 2008; Thomas et al. 1998; Thomas & Victor, 1998) and also in boys with DMD (Nelson et al. 2014; Sander et al. 2000).
Functional sympatholysis
Exercise‐induced attenuation of reflex vasoconstriction was assessed by applying the same reflex sympathetic stimulus (lower body negative pressure; LBNP) with the subjects’ forearm muscles either resting or performing light rhythmic handgrip exercise. With the subjects supine and the lower body enclosed in an airtight chamber, LBNP was applied at −20 to −30 mmHg, which simulates mild orthostatic stress (transition from supine to seated position) and engages mainly cardiac mechanoreceptors to trigger reflex sympathetic activation to the skeletal muscle circulation. Sympathetic constriction of skeletal muscle microvessels was measured as the LBNP‐induced decrease in forearm muscle oxygenation (oxygenated haemoglobin plus oxygenated myoglobin; HbO2 + MbO2), as measured by near infrared (NIR) spectroscopy (Hansen et al. 1996).
Skeletal muscle oxygenation with NIR spectroscopy
Forearm muscle oxygenation was measured with NIR spectroscopy, which is based on the principle that laser light with wavelengths in the 700–900 nm range easily penetrates skeletal muscle, and is absorbed by the iron‐porphyrin moieties in haemoglobin and myoglobin. Changes in NIR light absorption are proportional to changes in the relative concentrations of oxygenated hemoglobin and myoglobin (HbO2 + MbO2). Because of their almost identical absorption spectra, individual contributions of HbO2 and MbO2 cannot be determined. The NIR signals reflect changes in oxygenation occurring mainly in the microvasculature because vessels >1 mm in diameter are maximal absorbers of photons as a result of the high extinction coefficient of blood. Thus, NIR spectroscopy provides continuous measurement of the adequacy of tissue oxygen delivery relative to its use. At a constant level of oxygen demand, a decrease in HbO2 + MbO2 with LBNP is a valid measure of reflex sympathetic vasoconstriction in the skeletal muscle microcirculation (Hansen et al. 1996).
To monitor the tissue absorption of NIR light, five optical fibre bundles (four emitting bundles and one detector bundle connected to a photomultiplier tube) housed in a customized flexible rubber casing were placed with adhesive on the skin over the flexor digitorum profundus muscle, the main muscle recruited during handgrip. Four fixed emitter‐detector distances (1.5, 2.0, 2.5 and 3.0 cm) allowed direct calculation, and thus subtraction of scatter from skin and subcutaneous non‐muscle tissue (Gratton et al. 1997). Each emitting bundle contained two laser‐diode light sources: one at 830 nm, the wavelength at which the oxygenated and deoxygenated Hb/Mb species exhibit similar absorption coefficients, and the other at 690 nm, the wavelength at which light is absorbed primarily by the deoxygenated species. The difference between absorption at the two wavelengths is the HbO2 + MbO2 (Jobsis, 1977). The NIR signals were sampled at a rate of 5 Hz, converted to HbO2 + MbO2 concentration using validated algorithms, displayed as the running average of 50 consecutive samples, and stored digitally for analysis (OxiplexTS, ISS, Inc., Champaign, IL, USA). Before each experiment, absorption and scattering coefficients at each wavelength were calibrated against an external standard. After each experiment, a cuff was inflated on the upper arm to suprasystolic pressure of 250 mmHg to establish the total labile signal (TLS) (i.e. the difference between the baseline and nadir in muscle tissue oxygenation). Changes in forearm tissue oxygenation were expressed as a percentage of TLS.
Handgrip exercise
Handgrip exercise was performed using a dynamometer (Smedley Hand Dynamometer modified by Stoelting, Wood Dale, IL, USA). To determine maximal voluntary contraction (MVC), each subject was asked to grip the dynamometer as hard as possible. Force output was displayed on a computer screen to provide visual feedback for subjects. Subjects performed intermittent isometric handgrip (20 handgrips min–1, 50% duty cycle) at 20% MVC for 7 min. This mild level of handgrip exercise alone does not activate sympathetic outflow to skeletal muscle (Hansen et al. 1996). LBNP was applied only during minutes 4–5 of each exercise bout. Then, subjects continued performing rhythmic handgrip for minutes 6–7 with LBNP turned off.
Post‐exercise hyperaemia
The exercised‐induced increase in muscle blood flow was measured as the difference between brachial artery blood flow at rest and immediately after cessation of the 7 min rhythmic handgrip protocol. Brachial artery mean blood velocity (MBV) was measured using pulsed‐Doppler ultrasonography (iE33; Siemens AG, Berlin, Germany). Data were acquired continuously via a 9 MHz probe with 60 deg of insonation. The ultrasound gate was optimized to ensure complete insonation of the entire vessel cross‐section with constant intensity. The Doppler audio signal was converted to a real‐time flow velocity signal using a validated Doppler audio converter (Herr et al. 2010) and recorded using a PowerLab data acquisition system (ADInstruments, Boulder, CO, USA). Brachial artery diameter was measured by B‐mode imaging. Volumetric brachial artery blood flow was calculated as MBV (cm s–1) × πr 2 × 60, where r is radius of the brachial artery.
Exercise‐induced increase in deoxyHb + deoxyMb
NIR spectroscopy also was used to quantify the peak increase in skeletal muscle deoxyHb and deoxyMb concentration [deoxyHb + deoxyMb] during the first 3 min of rhythmic handgrip at 20% MVC alone, without LBNP.
Plasma [NO3 −] and [NO2 −]
Venous blood samples were drawn into lithium‐heparin tubes. Care was taken to avoid all contact with residual NO3 − and NO2 − (e.g. surfaces, pipets and tubes). Samples were centrifuged at 2455 g and 4°C for 10 min, within 1 min of collection. Plasma was subsequently extracted and immediately frozen at −80°C for later analysis.
As reported previously (Kenjale et al. 2011), plasma NO3 − and NO2 − were measured (within 30 min of thawing) by chemiluminescence using Ionics/Sievers NO analyser (NOA 280i) in accordance with the manufacturer's instructions (Sievers Instruments, Boulder, CO, USA). The reductant used for nitrite analysis was potassium iodide in acetic acid, which has the reduction potential to convert nitrite to nitric oxide but is insufficient to reduce any higher oxides of nitrogen (e.g. nitrate) and thus is relatively specific for nitrite. To obtain concentrations of total plasma nitrogen oxides, we used the same apparatus with a stronger reductant, vanadium chloride in hydrochloric acid at 94°C. This stronger reductant reduces the sum of all nitrogen oxides with an oxidation state of +2 or higher, which is predominantly nitrate [μm] but also includes both nitrite [nm] and nitrosothiols [nm].
Blood pressure regulation
To determine whether oral sodium nitrate affects blood pressure regulation in men with BMD, arm blood pressure was measured using a validated oscillometric monitor (Vital Signs Monitor 300 Series; Welch Allyn, Beaverton, OR, USA) with the subject in each of three positions: supine, seated and standing.
Data analysis
All data were analysed offline by a single observer (RR), who was blinded to condition assignment. Exercise‐induced attenuation of reflex vasoconstriction (i.e. functional sympatholysis) was systematically assessed using PowerLab software. Using the pressure inside the LBNP chamber as a trigger, the software was programed to average the HbO2 + MbO2 signal for 20 s before the onset of LBNP and for 20 s before the offset of LBNP; the difference between these mean values was taken as the LBNP‐induced change in forearm muscle tissue oxygenation. Similarly, exercise‐induced hyperaemia was measured as the change in brachial artery blood flow from rest to immediately post‐exercise, using the onset of LBNP at rest as the marker for rest (i.e. 60 s before the onset of LBNP) and the cessation of handgrip exercise as the marker for post‐exercise (i.e. 60 s mean value immediately after exercise cessation). Finally, to assess the change in deoxyHb and dexoyMb concentration in response to handgrip exercise, the deoxy NIR signal was averaged for 20 s before the onset of exercise, and 20 s before the onset of exercise + LBNP. The change in deoxyHb + deoxyMb from rest to exercise was expressed in absolute terms, as well as a percentage of TLS.
Specific protocols
Protocol 1: Relationship between BMD mutation, sarcolemmal nNOS and functional sympatholysis before treatment with sodium nitrate
As part of the initial visit,we quantified functional sympatholysis by NIR spectroscopy before treatment with sodium nitrate in all patients with BMD. Eligible patients had blood drawn for dystrophin sequencing, and, on a subsequent visit, underwent skeletal muscle biopsy for subsequent immunohistochemistry and western blot experiments to determine the presence or absence of nNOS staining at the sarcolemma, nNOS protein content, and staining for the C‐ and N‐terminus of dystrophin. We hypothesized that sympatholysis would be impaired in BMD patients with essentially no sarcolemmal staining for nNOS but well preserved in those with positive sarcolemmal nNOS staining. We compared the patients’ NIR responses with those of 13 healthy male controls to bracket the range of normal.
Protocol 2: Open‐label treatment trial in patients with BMD
As an initial test of the hypothesis that sodium nitrate can restore functional sympatholysis in BMD, 11 patients with BMD and impaired sympatholysis prior to treatment repeated the handgrip/sympatholysis protocol 3 h after ingesting 140 ml of concentrated nitrate rich beetroot juice, containing 8.4 mmol of inorganic nitrate (James White Drinks, Ipswich, UK). Three hours represents the time for peak absorption of sodium nitrate (Wylie et al. 2013). This dose of sodium nitrate was shown previously to produce a consistent increase in muscle oxygenation and exercise performance in patients with peripheral artery disease (Kenjale et al. 2011). Venous blood was drawn for plasma concentrations of NO3 − and NO2 − at the same time as the pre‐treatment and post‐treatment NIR measurements. To assess test–retest reproducibility, a subset of patients (n = 4) returned to the laboratory several months later to repeat the open‐label trial.
Protocol 3: Placebo‐controlled cross‐over trial in patients with BMD
To confirm positive results of the open‐label trial with a more rigorous experimental design, a subset of six BMD patients then completed a randomized, double‐blind, placebo‐controlled cross‐over trial with a 2 week or greater washout period before cross‐over. Patients drank 140 ml of either: (1) concentrated sodium‐nitrate rich juice or (2) placebo, comprising the same juice depleted of sodium nitrate (James White Drinks). The order of sodium nitrate/placebo was random. Plasma [NO3 −] and [NO2 −] were measured immediately before and 3 h after treatment. We tested the effect of sodium nitrate vs. placebo on both functional sympatholysis with NIR spectroscopy and on post‐exercise hyperaemia with brachial artery Doppler velocimetry, with the latter aiming to translate previous preclinical studies showing post‐exercise vasospasm in the mdx and nNOS null mouse (Kobayashi et al. 2008).
Protocol 4: Open‐label treatment trial in healthy controls
To test whether NO‐mediation of sympatholysis and post‐exercise hyperaemia is already saturated when sarcolemmal nNOS is plentiful and thus cannot be made super‐normal by treatment sodium nitrate, eight healthy controls performed the same open‐label treatment trial; in addition to sympatholysis by NIR spectroscopy, we also measured brachial artery blood flow by Doppler velocimetry at rest and immediately after the handgrip protocol both before and 3 h after sodium nitrate.
Statistical analysis
Muscle biopsy samples were assessed for either the presence or absence of sarcolemmal nNOS by one expert (RB). Exercise‐induced attenuation of reflex vasoconstriction (functional sympatholysis) was assessed by comparing the LBNP‐induced ΔHbO2 + MbO2 at rest vs. the LBNP‐induced decrease in ΔHbO2 + MbO2 during handgrip. To compare group differences, or to compare treatment effect, we performed a two‐way ANOVA, followed by Tukey's multiple comparison test. The exercise‐induced change in brachial artery blood flow was assessed by comparing blood flow at rest with blood flow immediately after exercise. To assess treatment effect, we performed a two‐way ANOVA with Tukey's multiple comparison test. The change in [deoxyHb + deoxyMb] in response to handgrip exercise was assessed by comparing the deoxyHb + deoxyMb concentration at rest and after 3 min of handgrip exercise using a paired samples t test. The influence of sodium nitrate (or placebo) on arterial blood pressure was compared within each posture (ie. standing, sitting and supine) by assessing the change from baseline to 3 h post‐ingestion using a two‐way ANOVA with Tukey's multiple comparison test. Data are expressed as the mean ± SEM, unless specified otherwise. P < 0.05 was considered statistically significant.
Results
A total of 21 patients were screened for eligibility and five were excluded: three did not have BMD by mutational analysis, one was too weak to perform handgrip and one had severe upper arm contractures that precluded blood flow measurements. The dystrophin mutations and clinical indices of disease severity of the remaining 16 BMD patients are shown in Table 1. All were ambulatory, although two patients frequently required a scooter for long distances. The most common mutation, seen in five patients, was deletion of exons 45–47 or 45–48. Three patients were receiving an angiotensin‐converting enzyme inhibitor for cardiac prophylaxis. Only one patient was receiving glucocorticoid therapy. Patients were well matched with 13 healthy male controls except, as expected, for higher resting heart rate and lower maximum grip strength (maximum voluntary contraction, MVC) (Table 2).
Table 2.
Baseline characteristics: patients vs. controls
| MVC | ||||||||
|---|---|---|---|---|---|---|---|---|
| Group | n | Age (years) | BMI (kg m–2) | SBP (mmHg) | DBP (mmHg) | HR (bpm) | Right arm (kg) | Left arm (kg) |
| Patients with BMD | 16 | 34 ± 12 | 25 ± 5 | 117 ± 11 | 71 ± 11 | 75 ± 12* | 28 ± 12* | 30 ± 12* |
| Healthy Controls | 13 | 33 ± 8 | 23 ± 2 | 115 ± 7 | 67 ± 8 | 63 ± 9 | 49 ± 7 | 47 ± 6 |
BMI, body mass index; SBP, systolic blood pressure; DBP, diastolic blood pressure; HR, heart rate.
*P < 0.05, patients vs. controls.
BMD mutations that interrupt sarcolemmal anchoring of nNOS impair functional sympatholysis
Viable muscle biopsy samples were obtained from 13 of 15 patients with BMD (Table 3). In the absence of treatment with sodium nitrate, we found a close relationship between BMD mutation, sarcolemmal anchoring of nNOS and functional sympatholysis (Fig. 1 and Table 3).
Table 3.
Relationship between sarcolemmal nNOS and functional sympatholysis
| BMD patients | Healthy controls | |||||
|---|---|---|---|---|---|---|
| nNOS | ||||||
| Protein | Functional | Functional | ||||
| Sarcolemmal | content | Sympatholysis | sympatholysis | |||
| Patient # | Dystrophin mutation | (Yes/No) | (% of control) | (% attenuation) | Subject # | (% attenuation) |
| 1 | Deletion exons 3–7 | No | 24 | 21 | 1 | 33 |
| 2 | Deletion exons 3–7 | No | 4 | 16 | 2 | 33 |
| 3 | Deletion exons 45–77 | No | 15 | 13 | 3 | 45 |
| 4 | Deletion exons 45–47 | No | 21 | 15 | 4 | 50 |
| 5 | Deletion exons 45–48 | No | 3 | 22 | 5 | 52 |
| 6 | Deletion exons 45–48 | No | 5 | 6 | 6 | 52 |
| 7 | Deletion exons 45–48 | No | 13 | −7 | 7 | 57 |
| 8 | Deletion exons 14–44 | Yes | 6 | 53 | 8 | 60 |
| 9 | Splice donor mutation exon 46 | Yes | 117 | 87 | 9 | 64 |
| 13 | Splice donor mutation exon 46 | Yes | 11 | 23 | 10 | 67 |
| 10 | Splice donor mutation exon 25 | Yes | 53 | 80 | 11 | 71 |
| 11 | Nonsense mutation exon 29 | Yes | 16 | 65 | 12 | 71 |
| 12 | Deletion exons 48–55 | Yes | 54 | −12 | 13 | 106 |
Figure 1. BMD mutations that interrupt sarcolemmal anchoring of nNOS impair functional sympatholysis .
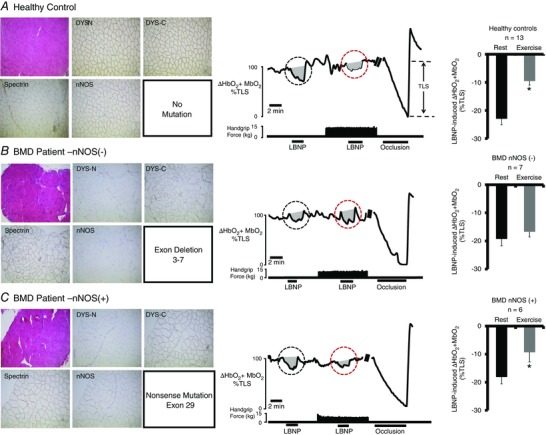
Left: representative immunohistochemistry from muscle biopsies from a healthy control subject (A) and two BMD patients (B and C). Muscle biopsy sections stained with haematoxylin and eosin (upper left) from the two BMD patients show typical dystrophic hallmarks, including variation in muscle fibre size, internal nuclei, fibrosis and an increase in fat tissue. Staining for dystrophin C‐terminus (DYS‐C) is present in both BMD patients, although no staining for dystrophin N‐terminus (DYS‐N) is present (evidence for truncated dystrophin). Sarcolemmal nNOS staining is absent in (B) but present in (C). Middle: representative tracing from a healthy control subject (A), a BMD patient lacking sarcolemmal nNOS (B, P2) and a BMD patient with nNOS retained at the sarcolemma (C, P11). A black circle highlights the LBNP‐induced change in forearm muscle oxygenation at rest, whereas a red circle highlights the LBNP‐induced change in forearm muscle oxygenation during exercise. Note that, in healthy controls, the decrease in forearm muscle oxygenation is greatly attenuated (termed functional sympatholysis). At the end of each experiment, an arm cuff was inflated to suprasystolic pressure to occlude the forearm circulation, producing a maximal decrease in forearm muscle oxygenation to calculate the TLS. Right: summary forearm muscle oxygenation data from 13 healthy subjects (A), seven BMD patients in whom sarcolemmal nNOS was absent (B) and six BMD patients in whom sarcolemmal nNOS was present (C). Data are reported as the mean + SE.
Illustrative immunohistochemistry experiments and group means for sympatholysis are shown in Fig. 1. In the healthy controls, when LBNP was superimposed on mild handgrip, the reflex decrease in muscle oxygenation was attenuated by 59 ± 6% compared to the LBNP‐induced reflex decrease at rest (ΔHbO2 + MbO2: −22.9 ± 2.2% vs. –9.5 ± 1.4%, rest vs. exercise, P < 0.05), demonstrating functional sympatholysis. Consistent with our hypothesis, sympatholysis was: (a) impaired in BMD patients lacking sarcolemmal nNOS because handgrip failed to attenuate the LBNP‐induced decrease in muscle oxygenation (ΔHbO2 + MbO2: −19.3 ± 2.4% vs. –16.7 ± 1.8%, rest vs. handgrip, not statistically significant) but (b) preserved in BMD patients with sarcolemma nNOS in whom handgrip attenuated the LBNP‐induced decrease in muscle oxygenation by 50 ± 16% (ΔHbO2 + MbO2: −18.2 ± 2.4% vs. –9.3 ± 3.4%, rest vs. handgrip, P < 0.05).
The subject‐specific data for 13 patients with BMD and 13 healthy controls are shown in Table 3. Data from P5 and P8 have been reported previously (Martin et al. 2012; Nelson et al. 2014). Sarcolemmal staining for nNOS was absent in seven BMD patients (including all five patients with deletion of exons 45–47 or 45–48, and both patients with deletion of exons 3–7) but present in the other six patients. Sympatholysis was greatly impaired in all seven patients lacking sarcolemmal nNOS but well preserved in four of six patients in whom nNOS was detected at the sarcolemma. The nNOS protein content was severely reduced in all seven BMD patients lacking sarcolemmal nNOS but not consistently reduced in the six patients with sarcolemmal nNOS. Two first‐cousins with the same splice donor mutation in exon 46 both had positive staining for sarcolemmal nNOS; sympatholysis was well preserved in the cousin with normal nNOS protein (P9) but impaired in the other cousin with markedly reduced nNOS protein (P13).
Treatment with sodium nitrate restores functional sympatholysis in BMD patients lacking sarcolemmal nNOS
Open‐label trial
The summary data (n = 10 patients) for the open‐label trial are shown in Fig. 2. Before treatment, sympatholysis was impaired (ΔHbO2 + MbO2: −16.2 ± 1.2% vs. –15.9 ± 1.6%, rest vs. exercise, P < 0.05). Sodium nitrate restored functional sympatholysis (ΔHbO2 + MbO2: −17.6 ± 1.2% vs. –10.5 ± 0.9%, rest vs. exercise, P < 0.05) but had no effect on the LBNP‐induced reflex decrease in muscle oxygenation when the forearm muscles were resting (ΔHbO2 + MbO2: −16.4 ± 1.2% vs. −17.3 ± 1.3%, untreated rest vs. treated rest, not statistically significant).
Figure 2. Sodium nitrate alleviates functional muscle ischaemia in BMD: acute treatment trial .
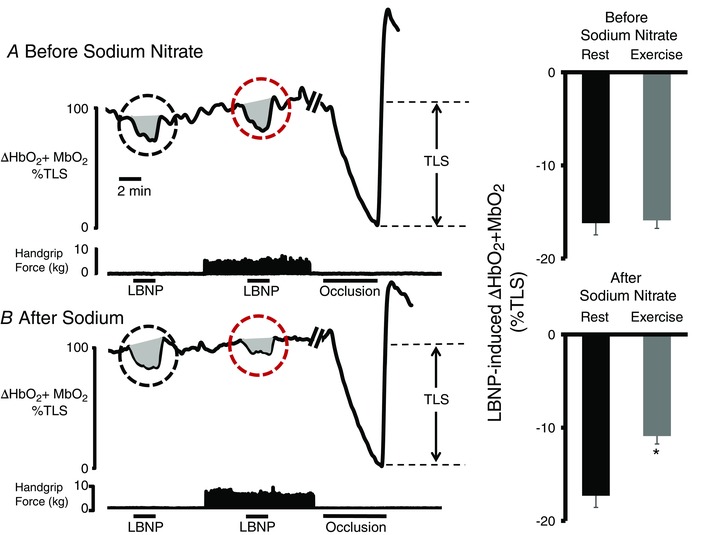
A, representative tracing (left) from a patient with BMD, showing that sympatholysis is impaired because handgrip fails to attenuate the LBNP‐induced reflex decrease in forearm muscle oxygenation (black circle). At the end of each experiment, an arm cuff was inflated to suprasystolic pressure to occlude the forearm circulation, producing a maximal decrease in forearm muscle oxygenation to calculate the TLS. Right: summary forearm muscle oxygenation data from the 10 patients with BMD studied at rest (black bar) and during exercise (grey bar), expressed as a percentage of TLS. B, representative tracing from the same BMD subject after oral treatment with sodium nitrate, showing that the LBNP‐induced decrease in forearm muscle oxygenation is greatly attenuated during mild handgrip exercise (grey circle). Summary data are shown on the right (n = 10). Of note, we observed no difference between the LBNP response at rest before and after sodium nitrate (not statistically significant). Data are reported as the mean + SE.
The patient‐specific data for the open‐label trial are shown in Fig. 3. Treatment with sodium nitrate restored functional sympatholysis in eight of the 10 BMD patients lacking sympatholysis before treatment. The two non‐responders (P1 and P13) had the largest treatment‐induced increases in plasma [NO2 –]. An eleventh patient (P5) with impaired sympatholysis prior to treatment showed: (a) no initial treatment response because he had brushed his teeth before ingesting the sodium nitrate, resulting in a trivial increase in plasma [NO2 −], but (b) clear restoration of sympatholysis when he repeated the study with the same dose of oral sodium nitrate and refrained from using toothpaste, resulting in a much larger increase in plasma [NO2 −].
Figure 3. Patient‐specific data from the acute treatment trial .
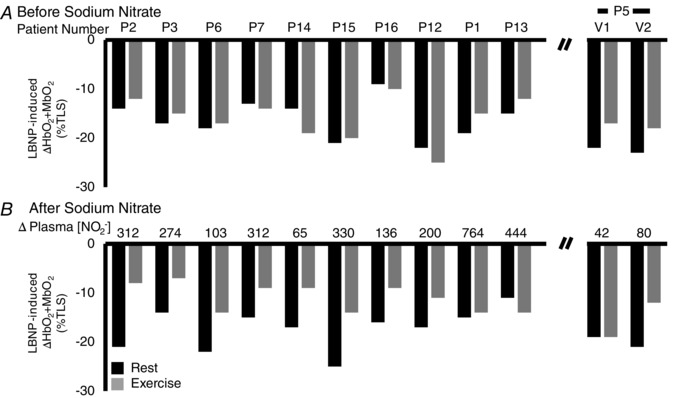
A, at baseline, the LBNP‐induced decrease in forearm muscle oxygenation (HbO2 + MbO2) is very consistent at rest (black bar) and during exercise (grey bar) for each patient. B, sodium nitrate restored functional sympatholysis in all but two subjects (P1 and P13). Note that Patient 5 (P5) deviated from the protocol by brushing his teeth during this initial study visit (V1). Accordingly, his data were removed from the analysis. When we repeated the experiment in P11 several months later (V2) (i.e. when the subject was reminded to avoid using toothpaste and mouthwash), the change in plasma nitrite concentration doubled from the first visit, and sympatholysis was improved. Data are expressed as a percentage of the TLS.
In four of the BMD patients (P2, P3, P6 and P14), several weeks after washout of the initial sodium nitrate treatment, we established within‐subject test–retest reproducibility of the sodium nitrate treatment effect (Fig. 4).
Figure 4. Test–retest reproducibility .
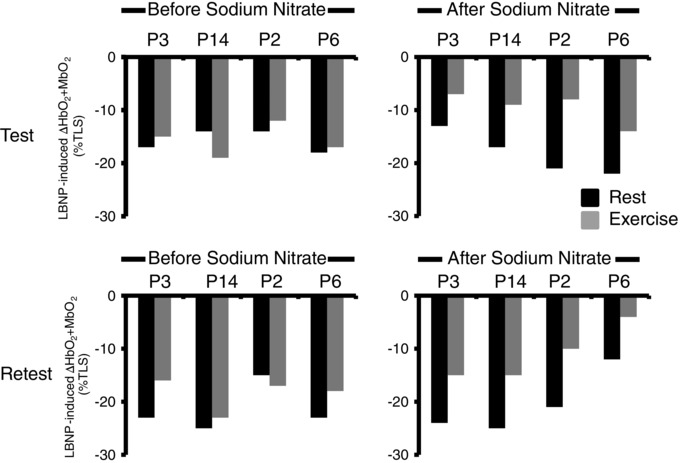
Four BMD subjects were studied several weeks apart. The data show excellent test–retest reproducibility for functional sympatholysis before sodium nitrate ingestion (showing that functional muscle ischaemia is consistently impaired in four of four patients) and after sodium nitrate ingestion (showing consistent restoration of functional sympatholysis in four of four patients).
Double‐blind, placebo‐controlled cross‐over trial
In the subsequent placebo‐controlled trial performed in six of the 11 patients from the open‐label trial (P2, P3, P7, P12, P14 and P16), we confirmed that sodium nitrate restores functional sympatholysis, whereas placebo was without effect (Fig. 5). Sodium nitrate also augmented the exercise‐induced increase in forearm muscle blood flow compared to placebo, despite having no effect on blood flow in resting muscle (Fig. 5).
Figure 5. Placebo‐controlled cross‐over trial: sodium nitrate alleviates functional muscle ischaemia and increased exercise‐induced hyperaemia in BMD .
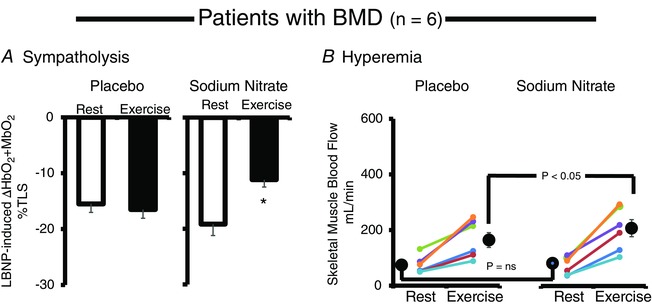
A, sodium nitrate supplementation restored functional sympatholysis (exercise‐induced attenuation of reflex vasoconstriction, measured by NIR) in men with BMD (n = 6), whereas placebo had no such effect. B, sodium nitrate supplementation does not affect resting skeletal muscle blood flow (measured by brachial artery Doppler ultrasound) but significantly increases exercise‐induced hyperaemia compared to placebo (n = 6). Data are expressed as the mean ± SE.
Treatment with sodium nitrate does not enhance functional sympatholysis or post‐exercise hyperaemia beyond the physiological range in healthy controls
By contrast to the robust effects of sodium nitrate on vascular regulation in patients with BMD lacking sarcolemmal nNOS, the same NO donor under the same experimental conditions had no significant effect on either functional sympatholysis or post‐exercise hyperaemia in eight healthy controls (Fig. 6). Indeed, the tendency was for both indices of vascular regulation to be somewhat attenuated after sodium nitrate; however, in neither case did this tendency achieve statistical significance. In addition, we treated one BMD patient (P11) with sarcolemmal nNOS and preserved functional sympatholysis before treatment: acute treatment with sodium nitrate had no effect on sympatholysis in this patient (pretreatment: LBNP‐induced ΔHbO2 + MbO2: −19% vs. −7%, rest vs. exercise; post‐treatment LBNP‐induced ΔHbO2 + MbO2: −21% vs. −6%, rest vs. exercise).
Figure 6. Sodium nitrate does not improve functional sympatholysis and does not increase exercise‐induced hyperaemia in healthy volunteers .

A, sodium nitrate supplementation failed to improve functional sympatholysis (exercise‐induced attenuation of reflex vasoconstriction, measured by NIR) in healthy male volunteers (n = 8). B, sodium nitrate supplementation did not affect resting skeletal muscle blood flow (measured by brachial artery Doppler ultrasound), nor did it affect exercise‐induced hyperaemia (n = 8). Data are expressed as the mean ± SE.
Mechanistic insights
During the first 3 min of the mild rhythmic handgrip (prior to onset of LBNP), intramuscular [deoxyHb + deoxyMb] increased significantly (Fig. 7 A), reflecting increased oxygen consumption by the metabolically active muscle. The exercise‐induced increase [deoxyHb + deoxyMb] tended to be somewhat larger after treatment with oral sodium nitrate (7.9 ± 2.9% vs. 14.1 ± 3.1% ∆TLS, placebo vs. sodium nitrate, respectively). As expected, sodium nitrate ingestion raised plasma [NO2 −] by five‐fold above placebo (Fig. 7 B). The combination of increased intramuscular [deoxyHb + deoxyMb] and increased circulating [NO2 −] fulfills the two conditions needed to generate NO from NO2 − in exercising dystrophic skeletal muscle (Fig. 7 C).
Figure 7. Circulating nitrite serves as an alternative nitric oxide donor only in the presence of deoxyHb and/or deoxyMb .

A, rhythmic handgrip exercise significantly increases skeletal muscle deoxyHb and deoxyMb content, as measured by NIR over the flexor digitorum profundus muscle (n = 6). Insert: original NIR record, showing a steep increase in deoxyHb content during the transition from rest to exercise. All data are expressed as a percentage of the TLS. B, sodium nitrate significantly increases circulating plasma nitrite concentration in patients with BMD (n = 6). C, circulating NO2 −is reduced to NO via deoxyHb in the microvascular space, and by deoxyMb in skeletal myocytes, leading to improvements in skeletal muscle blood flow regulation. Data are reported as the mean + SE.
Safety and tolerability
Sodium nitrate was well tolerated by all subjects (cases and controls), with no adverse events. Several subjects reported red coloured urine and stool after ingestion of both sodium nitrate and placebo (an expected side effect associated with beetroot juice). Sodium nitrate had almost no effect on resting blood pressure in the patients with BMD in the supine, seated or standing position (Table 4).
Table 4.
Effect of sodium nitrate on systemic blood pressure and heart rate in patients with BMD
| Open‐label trial (n = 11) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Sitting | Supine | Standing | P | ||||||
| Before | After | Before | After | Before | After | ||||
| treatment | treatment | treatment | treatment | treatment | treatment | Posture | Treatment | Interaction | |
| Systolic blood pressure (mmHg) | 119 ± 4 | 117 ± 3 | 121 ± 4 | 116 ± 3 | 120 ± 3 | 116 ± 2 | 0.969 | 0.073 | 0.059 |
| Diastolic blood pressure (mmHg) | 78 ± 4 | 75 ± 3 | 71 ± 5 | 73 ± 4 | 73 ± 4 | 73 ± 3 | 0.058 | 0.015 | 0.678 |
| Heart rate (bpm) | 87 ± 2 | 80 ± 1 | 80 ± 1 | 76 ± 1 | 91 ± 1 | 82 ± 1 | 0.004 | 0.014 | 0.422 |
| Placebo‐controlled trial (n = 6) | |||||||||
| Placebo | |||||||||
| Systolic blood pressure (mmHg) | 120 ± 4 | 115 ± 5 | 123 ± 5 | 116 ± 6 | 118 ± 7 | 118 ± 5 | 0.955 | 0.110 | 0.042 |
| Diastolic blood pressure (mmHg) | 74 ± 3 | 70 ± 5 | 71 ± 7 | 72 ± 6 | 70 ± 6 | 74 ± 7 | 0.583 | 0.200 | 0.472 |
| Heart rate (bpm) | 80 ± 3 | 75 ± 2 | 74 ±3 | 72 ± 3 | 87 ± 2 | 81 ± 2 | 0.004 | 0.355 | 0.447 |
| Sodium nitrate | |||||||||
| Systolic blood pressure (mmHg) | 119 ± 3 | 116 ± 5 | 124 ± 6 | 119 ± 5 | 121 ± 7 | 116 ± 5 | 0.598 | 0.809 | 0.980 |
| Diastolic blood pressure (mmHg) | 80 ± 4 | 77 ± 5 | 85 ± 3 | 73 ± 6 | 76 ± 5 | 74 ± 4 | 0.137 | 0.832 | 0.516 |
| Heart rate (bpm) | 93 ± 4 | 83 ± 4 | 87± 4 | 77 ± 3 | 93 ± 3 | 83 ± 3 | 0.098 | 0.030 | 0.999 |
Data reported as the mean ± SE.
Discussion
A growing concern in the muscular dystrophy field is that experimental therapies that are highly effective in preclinical mouse studies, including NO donors (Thomas et al. 2012; Uaesoontrachoon et al. 2014), are lost in clinical translation (Duan, 2006). The present clinical study addresses this concern in two ways. First, by utilizing the natural mutational heterogeneity that exists in BMD, we provide the strongest clinical evidence to date that sarcolemmal nNOS is essential for normal vasomodulation of exercising skeletal muscle. Then, we show that a single oral dose of a simple inorganic NO donor, sodium nitrate, alleviates functional muscle ischaemia in men with BMD mutations that disrupt sarcolemmal anchoring of nNOS. Specifically, the NO donor both restores functional sympatholysis in exercising dystrophic skeletal muscle and improves post‐exercise hyperaemia with no discernable effect on blood flow in resting muscle.
The most common dystrophin mutations causing BMD, present in 30% of our present subjects and in 40% of our previous BMD cohort (Martin et al. 2012), delete exons 45–47 or 45–48 that encode the SR 16,17 spectrin‐like repeats in the mid‐portion of dystrophin's rod domain that are essential for sarcolemmal anchoring of nNOS (Chao et al. 1996; Torelli et al. 2004). As expected, in the present study, nNOS was absent from the sarcolemma in all these patients. The finding that nNOS also was absent from the sarcolemma in both patients with deletion of exons 3–7 is consistent with similar findings in two other patients with the same mutation reported previously (Chao et al. 1996) and suggests that this mutation may directly or indirectly affect the R16/17 nNOS binding site.
Functional sympatholysis was markedly impaired in all seven BMD patients lacking sarcolemmal nNOS, all of whom also had markedly reduced nNOS expression, but well preserved in four of the six patients with nNOS staining at the sarcolemma. This comprises further evidence in humans that sarcolemmal nNOSμ is essential for the normal modulation of reflex sympathetic vasoconstriction in active skeletal muscle. In the absence of such modulation, sympathetic vasoconstriction is unopposed in forearm muscles when lightly exercised, simulating the common condition of a BMD patient performing repetitive arm activities of daily living. These new data both confirm and extend our prior observations in which muscle biopsy tissue was obtained on only two patients with BMD (also included for completeness in the present study): sympatholysis was preserved in the patient with sarcolemmal nNOS (P8) and absent in the other patient lacking sarcolemmal nNOS (P5) (Martin et al. 2012).
Interestingly, in the present study, two first‐cousins with the identical splice donor mutation (not deletion) of exon 46 both stained positive for sarcolemmal nNOS but were discordant for sympatholysis, which was indistinguishable from normal in the cousin with completely normal nNOS expression (P9) and blunted, although not totally absent, in the other cousin with greatly reduced expression (P13). In addition to reduced NO production, increased NO destruction by reactive oxygen species, which are generated excessively in contracting mdx mouse muscle (Prosser et al. 2013), constitutes another possible mechanism of impaired sympatholysis in two of six BMD patients with sarcolemmal nNOS (P12 and P13). Sympatholysis is impaired by overproduction of reactive oxygen species in common acquired pathophysiological states in which the dystrophin–glycoprotein complex is fully intact (Thomas et al. 2001; Zhao et al. 2006; Price et al. 2013). We consider that this is the most plausible explanation for impaired sympatholysis in the patient with deletion of exons 48–53 (P12) because his nNOS protein level was not severely reduced. Although he was the only patient being treated with prednisone, we previously found that sympatholysis is consistently impaired in boys with DMD in both the presence and absence of background glucocorticoid therapy (Nelson et al. 2014).
The seminal finding of the present study (i.e. that oral sodium nitrate restores functional sympatholysis in patients with BMD mutations that disrupt sarcolemmal targeting of nNOS) provides key additional evidence that defective NO signalling indeed causes the impaired vasomodulation. Previous studies using non‐specific and sometimes weak NOS inhibitors in healthy humans have provided conflicting evidence as to whether NO plays a major or minor role in mediating functional sympatholysis (Chavoshan et al. 2002; Dinenno & Joyner, 2003). By contrast, the marked and immediate effect of the simple NO donor to restore sympatholysis in nine of 11 BMD patients indicates that NO generation constitutes a fundamental mechanism by which exercising human skeletal muscle regulates its own perfusion and further implicates the NO pathway as a putative new drug target for many patients with dystrophinopathy. In addition, under the same experimental conditions, sodium nitrate did not enhance sympatholysis beyond the normal physiological range in healthy controls (or in one BMD patient with preserved sarcolemmal nNOS and normal sympatholysis), indicating that exercise‐induced modulation of reflex vasoconstriction is already optimized when sarcolemmal nNOS is plentiful.
We do not know why sodium nitrate failed to restore sympatholysis in two of the 11 patients because both had very large increases in plasma [NO2 –]. Compared with our other BMD patients, one non‐responder, the patient with deletion of exons 3–7 (P1), had by far the most advanced disease and also was the one non‐responder in our tadalafil study (Martin et al. 2012). Because his maximal grip is so weak, handgrip at only 20% MVC many not generate sufficient force to effectively constitute exercise, the sine qua non for sympatholysis. The other non‐responder in the present study was one of the cousins with the splice donor mutation in exon 46; the one with impaired sympatholysis despite preserved sarcolemmal nNOS (P13). Increased NO destruction by hyperstimulation of reactive oxygen species could potentially explain how his sympatholysis was both impaired and unresponsive to a NO donor.
The present clinical study translates our recent preclinical data (Thomas et al. 2012) by showing that an inorganic NO donor is a highly effective means of counteracting functional muscle ischaemia not only in mdx mice, but also in human patients with BMD, specifically those with exon deletions that disrupt the nNOS binding site. Moreover, the clinical data extend the mouse data, which showed dose‐dependent correction of sympatholysis after 30–90 days of treatment with a NO‐donating NSAID, in three important ways. First, NO donation alone, without a NSAID moiety, which theoretically could increase NO bioavailability by reducing reactive oxygen species, is sufficient to rescue functional sympatholysis. Second, a single dose of the NO donor (and not continuous daily dosing for 30–90 days) is all that is required to produce the effect, clearly implicating NO donation as the mechanism of action. Third, this mechanism is strictly dependent upon an increased plasma concentration of nitrite (the putative NO donor) because functional ischaemia was unaffected by the nitrate‐depleted placebo. The dependence on plasma nitrite is further supported by the one patient in whom the same dose of oral sodium nitrate either fully corrected or had no effect on abnormal vasomodulation depending on whether or not he brushed his teeth, thus disrupting the oral flora needed to convert nitrate to nitrite, the actual NO donor (Govoni et al. 2008).
Additional studies combining Doppler velocimetry and NIR spectroscopy provide important clues about the underlying molecular mechanism by which circulating nitrite serves as an alternative NO donor to correct the blood flow abnormality in dystrophic muscle. The Doppler data show that the effect of increased circulating nitrite is specific for exercising muscle with no effect on blood flow in resting muscle. The NIR data link this specificity to the exercise‐induced increase in the intramuscular [deoxyHb + deoxyMb], which is required to reduce nitrite to NO. Because deoxyMb generates NO from nitrite 36‐fold faster than deoxyHb (Shiva et al. 2007), we postulate that this rapid chemical reaction largely explains how in patients with BMD oral sodium nitrate recapitulates the vasomodulation normally produced by sarcolemmal‐targeted nNOS. Because Mb is expressed solely in skeletal and cardiac muscle, we postulate that most of the nitrite reduction occurs in these tissues, which cause the morbidity and mortality in BMD and other forms of dystrophinopathy.
In men with BMD studied under the present experimental conditions, sodium nitrate exerted a large improvement in functional sympatholysis but a small (but detectable) effect on post‐exercise hyperaemia. We suggest two plausible explanations. First, when nitrite is reduced by deoxyHb + deoxyMb, NO is released at the level of the small intramuscular microvessels: the site of the vascular tree interrogated by NIR spectroscopy (Hansen et al. 1996). By contrast, brachial artery Doppler is a relatively blunt tool because the conduit artery sample volume is far proximal from the downstream site of action NO2 – and thus the greatest concentration of released NO in the microcirculation. Second, exercise‐induced attenuation of LBNP‐induced reflex vasoconstriction (i.e. functional sympatholysis) constitutes a highly specific action of NO for modulatingα‐adrenergic signalling via a post‐receptor mechanism (Thomas et al. 1994; Thomas et al. 1998; Thomas & Victor, 1998; Thomas et al. 2001; Thomas et al. 2003). By contrast, brachial artery Doppler provides a measure of exercise‐induced (active) hyperaemia, which is probably driven both by NO and by other mediators that remain incompletely understood (Clifford & Hellsten, 2004).
The lack of any detectable effect of circulating nitrite on systemic blood pressure in our BMD patients argues against widespread dilatation in other vascular beds. We interpret these data to suggest that, with the dose used in the present study, generation of NO from nitrite occurs mainly at the level of the most distal microvessels mediating nutrient exchange in contracting skeletal muscle rather than at the level of the more proximal resistance vessels regulating blood pressure. Whether chronic daily dosing would cause an unsafe drop in blood pressure cannot be determined by the present data, although this is unexpected. A recent meta‐analysis showed that daily dosing with sodium nitrate of 5–44 mmol day−1 causes a small reduction in blood pressure averaging −4/−1 mmHg in normotensive and hypertensive adults (Siervo et al. 2013). Moreover, 30 day feeding studies with the DASH (Dietary Approach to Stop Hypertension) diet, which delivers two‐to three‐fold more nitrate than the 8.4 mmol dose of sodium nitrate used in the present study, also produces a small reduction in blood pressure averaging −5/−3 mmHg (Eckel et al. 2014).
The present study has several limitations. As an initial approach, we utilized an open‐label study design. However, the beneficial effects of the NO donor on muscle blood flow regulation were: (1) evident in nine of the 11 patients lacking sympatholysis at baseline; (2) reproducible when patients were studied repeatedly; (3) confirmed in a randomized, double‐blind, placebo‐controlled cross‐over trial; and (4) dependent on the conversion of ingested NO3 − to plasma NO2 −. Because forearm muscle oxygenation was the primary end‐point and blood flow a secondary end‐point of the present study, the data neither define the contribution of microvascular ischaemia to the pathogenesis of BMD, nor address whether restoring muscle perfusion with sodium nitrate can improve the clinical phenotype. Because this was an acute treatment trial, we do not know whether our positive results can be sustained safely with chronic therapy. Inorganic NO donors do not engender nitrate tolerance, which is an advantage over organic nitrates; in healthy subjects, nitrate tolerance produces reactive oxygen species that destroy NO and impair sympatholysis(Fadel et al. 2012).
A potential safety concern is that daily nitrate therapy might exacerbate the chronic nitrosoactive stress in dystrophic muscle that occurs when nNOS is mislocalized to the cytosol instead of the sarcolemma (Li et al. 2011). Cytosolic nNOS inhibits force production in dystrophin‐null mice (Li et al. 2011) and is correlated with adverse hypernitrosylation of the calcium release channel ryanodine receptor and more severe clinical phenotypes in patients with BMD (Gentil et al. 2012). A key distinction is that cytosolic nNOS is assumed to continually generate a low‐level of NO, whereas our data indicate that NO2 – mainly generates NO when the muscle is exercised; a phasic rather than continuous pattern that more closely mimics the normal production of NO by sarcolemmal nNOS.
Despite these limitations, our data show that a simple NO donor constitutes a remarkably effective strategy for alleviating functional muscle ischaemia in human dystrophinopathy patients. Although there is an emerging body of literature on oral sodium nitrate to treat common acquired forms of cardiovascular disease such as hypertension, peripheral artery disease and coronary disease (Kenjale et al. 2011; Hendgen‐Cotta et al. 2012; Larsen et al. 2006), the present study is the first to examine its potential therapeutic benefit for human muscular dystrophy. Future studies will be needed to determine whether inorganic NO donors could be more effective on clinically meaningful outcomes than PDE5 inhibitors, which currently are undergoing evaluation in a Phase 3 clinical trial for DMD (NCT01865084). In the present study, the observed effect of sodium nitrate on vascular regulation is comparable to that of PDE5 inhibition in our recent studies of adult patients with BMD (Martin et al. 2012) and paediatric patients with DMD (Nelson et al. 2014). Moreover, as suggested by recent studies in mouse models of muscular dystrophy, NO donating drugs, such as PDE5 inhibitors, normalize muscle blood flow regulation (Thomas et al. 2012) and decrease muscle inflammation (Uaesoontrachoon et al. 2014) but, unlike PDE5 inhibitors, they also increase muscle mass and thus grip strength (Uaesoontrachoon et al. 2014), presumably by stimulation of muscle regeneration through activation of satellite cells (Anderson, 2000; Brunelli et al. 2007; Marques et al. 2005). Further clinical research is warranted.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
MDN, FR, XT, JDA and RGV conceived and designed the experiments. MDN, ET, RB, FR, XT, OM, AS, TS, SS, RR, NM, SH, RE, JDA and RGV were responsible for collection, analysis and interpretation of data. MDN, ET, RB, FR, XT, OM, AS, TS, SS, RR, NM, SH, RE, JDA and RGV drafted the article or revised it critically for important intellectual content.All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
The present study was supported by research funding to Dr R. G. Victor from Coalition Duchenne, the National Centre for Advancing Translational Sciences (UL 1TR000124) and the Burns and Allen Chair in Cardiology Research at Cedars‐Sinai Medical Centre. Dr M. D. Nelson was supported by a research fellowship grant from the Canadian Institutes for Health Research.
References
- Adamo CM, Dai DF, Percival JM, Minami E, Willis MS, Patrucco E, Froehner SC & Beavo JA (2010). Sildenafil reverses cardiac dysfunction in the mdx mouse model of Duchenne muscular dystrophy. Proc Natl Acad Sci USA 107, 19079–19083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JE (2000). A role for nitric oxide in muscle repair: nitric oxide‐mediated activation of muscle satellite cells. Mol Biol Cell 11, 1859–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson LVB & Davison K (1999). Multiplex western blotting system for the analysis of muscular dystrophy proteins. Am J Pathol 154, 1017–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai A, Sahani N, Kaneki M, Ouchi Y, Martyn JAJ & Yasuhara SE (2007). Primary role of functional ischemia, quantitative evidence for the two‐hit mechanism, and phosphodiesterase‐5 inhibitor therapy in mouse muscular dystrophy. PLoS ONE 2, e806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Xia H, Aldape K & Bredt DS (1995). Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 82, 743–752. [DOI] [PubMed] [Google Scholar]
- Brunelli S, Sciorati C, D'Antona G, Innocenzi A, Covarello D, Galvez BG, Perrotta C, Monopoli A, Sanvito F, Bottinelli R, Ongini E, Cossu G & Clementi E (2007). Nitric oxide release combined with nonsteroidal antiinflammatory activity prevents muscular dystrophy pathology and enhances stem cell therapy. Proc Natl Acad Sci USA 104, 264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buono R, Vantaggiato C, Pisa V, Azzoni E, Bassi MT, Brunelli S, Sciorati C & Clementi E (2012). Nitric oxide sustains long‐term skeletal muscle regeneration by regulating fate of satellite cells via signaling pathways requiring Vangl2 and cyclic GMP. Stem Cells 30, 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, Kaul A, Kinnett K, McDonald C, Pandya S, Poysky J, Shapiro F, Tomezsko J & Constantin C (2010. a). Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 9, 77–93. [DOI] [PubMed] [Google Scholar]
- Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, Kaul A, Kinnett K, McDonald C, Pandya S, Poysky J, Shapiro F, Tomezsko J & Constantin C (2010. b). Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol 9, 177–189. [DOI] [PubMed] [Google Scholar]
- Chao DS, Gorospe JR, Brenman JE, Rafael JA, Peters MF, Froehner SC, Hoffman EP, Chamberlain JS & Bredt DS (1996). Selective loss of sarcolemmal nitric oxide synthase in Becker muscular dystrophy.J Exp Med 184, 609–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavoshan B, Sander M, Sybert TE, Hansen J, Victor RG & Thomas GD (2002). Nitric oxide‐dependent modulation of sympathetic neural control of oxygenation in exercising human skeletal muscle. J Physiol 540, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford PS & Hellsten Y (2004). Vasodilatory mechanisms in contracting skeletal muscle. J Appl Physiol 97, 393–403. [DOI] [PubMed] [Google Scholar]
- Cordani N, Pisa V, Pozzi L, Sciorati C & Clementi E (2014). Nitric oxide controls fat deposition in dystrophic skeletal muscle by regulating fibro‐adipogenic precursor differentiation. Stem Cells 32, 874–885. [DOI] [PubMed] [Google Scholar]
- Dinenno FA & Joyner MJ (2003). Blunted sympathetic vasoconstriction in contracting skeletal muscle of healthy humans: is nitric oxide obligatory? J Physiol 553, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D (2006). Challenges and opportunities in dystrophin‐deficient cardiomyopathy gene therapy. Hum Mol Genet 15, R253–R261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckel RH, Jakicic JM, Ard JD, de Jesus JM, Houston Miller N, Hubbard VS, Lee IM, Lichtenstein AH, Loria CM, Millen BE, Nonas CA, Sacks FM, Smith J, Svetkey LP, Wadden TA & Yanovski SZ (2014). 2013 AHA/ACC guideline on lifestyle management to reduce cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force onPractice Guidelines. J Am Coll Cardiol 63, 2960–2984. [DOI] [PubMed] [Google Scholar]
- Emery AE (2002). The muscular dystrophies. Lancet 359, 687–695. [DOI] [PubMed] [Google Scholar]
- Fadel PJ, Farias M III, Gallagher KM, Wang Z & Thomas GD (2012). Oxidative stress and enhanced sympathetic vasoconstriction in contracting muscles of nitrate‐tolerant rats and humans. J Physiol 590, 395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanigan KM, Niederhausern AV, Dunn DM, Alder J, Mendell JR & Weiss RB (2003). Rapid direct sequence analysis of the dystrophin gene. Am J Hum Genet 72, 931–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentil C, Leturcq F, Ben Yaou R, Kaplan JC, Laforet P, Penisson‐Besnier I, Espil‐Taris C, Voit T, Garcia L & Pietri‐Rouxel F (2012). Variable phenotype of del45‐55 Becker patients correlated with nNOSμ mislocalization and RYR1 hypernitrosylation. Hum Mol Genet 21, 3449–3460. [DOI] [PubMed] [Google Scholar]
- Govoni M, Jansson E, Weitzberg E & Lundberg JO (2008). The increase in plasma nitrite after a dietary nitrate load is markedly attenuated by an antibacterial mouthwash. Nitric Oxide 19, 333–337. [DOI] [PubMed] [Google Scholar]
- Gratton E, Fantini S, Franceschini MA, Gratton G & Fabiani M (1997). Measurements of scattering and absorption changes in muscle and brain. Philos Trans R Soc Lond B Biol Sci 352, 727–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen J, Thomas GD, Harris SA, Parsons WJ & Victor RG (1996). Differential sympathetic neural control of oxygenation in resting and exercising human skeletal muscle. J Clin Invest 98, 584–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendgen‐Cotta UB, Luedike P, Totzeck M, Kropp M, Schicho A, Stock P, Rammos C, Niessen M, Heiss C, Lundberg JO, Weitzberg E, Kelm M & Rassaf T (2012). Dietary nitrate supplementation improves revascularization in chronic ischemia. Circulation 126, 1983–1992. [DOI] [PubMed] [Google Scholar]
- Herr MD, Hogeman CS, Koch DW, Krishnan A, Momen A & Leuenberger UA (2010). A real‐time device for converting Doppler ultrasound audio signals into fluid flow velocity. Am J Physiol Heart Circ Physiol 298, H1626–H1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobsis FF (1977). Noninvasive, infrared monitoring of cerebral and myocardial oxygen sufficiency and circulatory parameters. Science 198, 1264–1267. [DOI] [PubMed] [Google Scholar]
- Kameya S, Miyagoe Y, Nonaka I, Ikemoto T, Endo M, Hanaoka K, Nabeshima YI & Takeda S (1999). Alpha 1‐syntrophin gene disruption results in the absence of neuronal‐type nitric‐oxide synthase at the sarcolemma but does not induce muscle degeneration. J Biol Chem 274, 2193–2200. [DOI] [PubMed] [Google Scholar]
- Kenjale AA, Ham KL, Stabler T, Robbins JL, Johnson JL, VanBruggen M, Privette G, Yim E, Kraus WE & Allen JD (2011). Dietary nitrate supplementation enhances exercise performance in peripheral arterial disease. J Appl Physiol 110, 1582–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khairallah M, Khairallah RJ, Young ME, Allen BG, Gillis MA, Danialou G, Deschepper CF, Petrof BJ & Des Rosiers C (2008). Sildenafil and cardiomyocyte‐specific cGMP signaling prevent cardiomyopathic changes associated with dystrophin deficiency. Proc Natl Acad Sci USA 105, 7028–7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi YM, Rader EP, Crawford RW, Iyengar NK, Thedens DR, Faulkner JA, Parikh SV, Weiss RM, Chamberlain JS, Moore SA & Campbell KP (2008). Sarcolemma‐localized nNOS is required to maintain activity after mild exercise. Nature 456, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Thomas GD, Yue Y, Yang HT, Li D, Long C, Judge L, Bostick B, Chamberlain JS, Terjung RL & Duan D (2009). Dystrophins carrying spectrin‐like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest 119, 624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen FJ, Ekblom BR, Sahlin K, Lundberg JO & Weitzberg E (2006). Effects of dietary nitrate on blood pressure in healthy volunteers. N Engl J Med 355, 2792–2793. [DOI] [PubMed] [Google Scholar]
- Li D, Yue Y, Lai Y, Hakim CH & Duan D (2011). Nitrosative stress elicited by nNOS delocalization inhibits muscle force in dystrophin‐null mice. J Pathol 223, 88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg JO, Weitzberg E & Gladwin MT (2008). The nitrate‐nitrite‐nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov 7, 156–167. [DOI] [PubMed] [Google Scholar]
- Marques MJ, Luz MAM, Minatel E & Neto HS (2005). Muscle regeneration in dystrophic mdx mice is enhanced by isosorbide dinitrate. Neurosci Lett 382, 342–345. [DOI] [PubMed] [Google Scholar]
- Martin EA, Barresi R, Byrne BJ, Tsimerinov EI, Scott BL, Walker AE, Gurudevan SV, Anene F, Elashoff RM, Thomas GD & Victor RG (2012). Tadalafilalleviates muscle ischemia in patients with becker muscular dystrophy. Sci Trans Med 4, 162ra155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco AP (1989). Dystrophin, the protein product of the Duchenne/Becker muscular dystrophy gene. Trends Biochem Sci 14, 412–415. [DOI] [PubMed] [Google Scholar]
- Nelson MD, Rader F, Tang X, Tavyev J, Nelson SF, Miceli MC, Elashoff RM, Sweeney HL & Victor RG (2014). PDE5 inhibition alleviates functional muscle ischemia in boys with Duchenne muscular dystrophy. Neurology 82, 2085–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Percival JM, Whitehead NP, Adams ME, Adamo CM, Beavo JA & Froehner SC (2012). Sildenafil reduces respiratory muscle weakness and fibrosis in the mdx mouse model of Duchenne muscular dystrophy. J Pathol 228, 77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price A, Raheja P, Wang Z, Arbique D, Adams‐Huet B, Mitchell JH, Victor RG, Thomas GD & Vongpatanasin W (2013). Differential effects of nebivolol versus metoprolol on functional sympatholysis in hypertensive humans. Hypertension 61, 1263–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser BL, Khairallah RJ, Ziman AP, Ward CW & Lederer WJ (2013). X‐ROS signaling in the heart and skeletal muscle: stretch‐dependent local ROS regulates [Ca2+]i. J Mol Cell Cardiol 58, 172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander M, Chavoshan B, Harris SA, Iannaccone ST, Stull JT, Thomas GD & Victor RG (2000). Functional muscle ischemia in neuronal nitric oxide synthase‐deficient skeletal muscle of children with Duchenne muscular dystrophy.Proc Natl Acad Sci USA 97, 13818–13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciorati C, Miglietta D, Buono R, Pisa V, Cattaneo D, Azzoni E, Brunelli S & Clementi E (2011). A dual acting compound releasing nitric oxide (NO) and ibuprofen, NCX 320, shows significant therapeutic effects in a mouse model of muscular dystrophy. Pharmacol Res 64, 210–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiva S, Huang Z, Grubina R, Sun J, Ringwood LA, MacArthur PH, Xu X, Murphy E, Darley‐Usmar VM & Gladwin MT (2007). Deoxymyoglobinis a nitrite reductase that generates nitric oxide and regulates mitochondrial respiration. Circ Res 100, 654–661. [DOI] [PubMed] [Google Scholar]
- Siervo M, Lara J, Ogbonmwan I & Mathers JC (2013). Inorganic nitrate and beetroot juice supplementation reduces blood pressure in adults: a systematic review and meta‐analysis. J Nutr 143, 818–826. [DOI] [PubMed] [Google Scholar]
- Thomas GD, Hansen J & Victor RG (1994). Inhibition of alpha 2‐adrenergic vasoconstriction during contraction of glycolytic, not oxidative, rat hindlimb muscle. Am J Physiol Heart Circ Physiol 266, H920–H929. [DOI] [PubMed] [Google Scholar]
- Thomas GD, Sander M, Lau KS, Huang PL, Stull JT & Victor RG (1998). Impaired metabolic modulation of alpha‐adrenergic vasoconstriction in dystrophin‐deficient skeletal muscle.Proc Natl Acad Sci USA 95, 15090–15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GD, Shaul PW, Yuhanna IS, Froehner SC & Adams ME (2003). Vasomodulation by skeletal muscle‐derived nitric oxide requires alpha‐syntrophin‐mediated sarcolemmal localization of neuronal nitric oxide synthase. Circ Res 92, 554–560. [DOI] [PubMed] [Google Scholar]
- Thomas GD & Victor RG (1998). Nitric oxide mediates contraction‐induced attenuation of sympathetic vasoconstriction in rat skeletal muscle. J Physiol 506, 817–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GD, Ye J, De Nardi C, Monopoli A, Ongini E & Victor RG (2012). Treatment with a nitric oxide‐donating NSAID alleviates functional muscle ischemia in the mouse model of Duchenne muscular dystrophy. PLoS ONE 7, e49350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GD, Zhang W & Victor RG (2001). Impaired modulation of sympathetic vasoconstriction in contracting skeletal muscle of rats with chronic myocardial infarctions: role of oxidative stress. Circ Res 88, 816–823. [DOI] [PubMed] [Google Scholar]
- Tidball JG & Wehling‐Henricks M (2004). Expression of a NOS transgene in dystrophin‐deficient muscle reduces muscle membrane damage without increasing the expression of membrane‐associated cytoskeletal proteins. Mol Genet Metab 82, 312–320. [DOI] [PubMed] [Google Scholar]
- Torelli S, Brown S, Jimenez‐Mallebrera C, Feng L, Muntoni F & Sewry C (2004). Absence of neuronal nitric oxide synthase (nNOS) as a pathological marker for the diagnosis of Becker muscular dystrophy with rod domain deletions. Neuropathol Appl Neurobiol 30, 540–545. [DOI] [PubMed] [Google Scholar]
- Uaesoontrachoon K, Quinn JL, Tatem KS, Van Der Meulen JH, Yu Q, Phadke A, Miller BK, Gordish‐Dressman H, Ongini E, Miglietta D & Nagaraju K (2014). Long‐term treatment with naproxcinod significantly improves skeletal and cardiac disease phenotype in the mdx mouse model of dystrophy. Hum Mol Genet 23, 3239–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vongpatanasin W, Wang Z, Arbique D, Arbique G, Adams‐Huet B, Mitchell JH, Victor RG & Thomas GD (2011). Functional sympatholysis is impaired in hypertensive humans. J Physiol 589, 1209–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie LJ, Kelly J, Bailey SJ, Blackwell JR, Skiba PF, Winyard PG, Jeukendrup AE, Vanhatalo A & Jones AM (2013). Beetroot juice and exercise: pharmacodynamic and dose–response relationships. J Appl Physiol 115, 325–336. [DOI] [PubMed] [Google Scholar]
- Zhao W, Swanson SA, Ye J, Li X, Shelton JM, Zhang W & Thomas GD (2006). Reactive oxygen species impair sympathetic vasoregulation in skeletal muscle in angiotensin II‐dependent hypertension. Hypertension 48, 637–643. [DOI] [PubMed] [Google Scholar]