

Abstract
Cholestatic‐liver diseases (CLDs) arise from diverse causes ranging from genetic factors to drug‐induced cholestasis. The so‐called diseases of civilization (obesity, diabetes, metabolic disorders, non‐alcoholic liver disease, cardiovascular diseases, etc.) are intricately implicated in liver and gall bladder diseases. Although CLDs have been extensively studied, there seem to be important gaps in the understanding of human disease. Despite the fact that many animal models exist and substantial clinical data are available, translation of this knowledge towards therapy has been disappointingly limited. Recent advances in liver cell culture such as in vivo‐like 3D cultivation of human primary hepatic cells, human induced pluripotent stem cell‐derived hepatocytes; and cutting‐edge analytical techniques such as ‘omics’ technologies and high‐content screenings could play a decisive role in deeper mechanistic understanding of CLDs. This Topical Review proposes a roadmap to human biology‐based research using omics technologies providing quantitative information on mechanisms in an adverse outcome/disease pathway framework. With modern sensitive tools, a shift in paradigm in human disease research seems timely and even inevitable to overcome species barriers in translation.
Abbreviations
- ABC
ATP‐binding cassette
- AOP
adverse outcome pathway
- BA
bile acids
- BDL
bile duct ligation
- BSEP
bile salt export pump
- CAR
constitutive androstane/active receptor
- CLD
cholestatic‐liver disease
- FXR
farnesoid X receptor
- hiPSC
human induced pluripotent stem cells
- NTCP
Na+‐dependent taurocholate cotransporting polypeptide
- OATP
organic anion transporting polypeptide
- PBC
primary biliary cholangitis
- PSC
primary sclerosing cholangitis
- PXR
pregnane X receptor
Introduction
Liver and gall bladder diseases are very common all over the world, posing a significant health burden worldwide (Shaheen et al. 2006; Williams, 2006). It is now known that diseases related to modern lifestyle such as obesity, diabetes, non‐alcoholic fatty liver disease and other nutritional/metabolic disorders are either the cause or the consequences of liver and gall bladder diseases (Fig. 1). Liver is a very complex vital organ performing a diverse range of metabolic functions including the regulation of carbohydrate metabolism; lipid synthesis and secretion of plasma lipoproteins; cholesterol metabolism; synthesis and secretion of bile salts; digestion; storage of nutrients, vitamins and minerals; synthesis and secretion of serum albumin, clotting factors, enzymes and other proteins; ammonia detoxification through urea and glutamine formation; and biotransformation/detoxification of drugs and other xenobiotics. Liver disorders can result from various insults such as infections, drugs, toxins, ischaemia and autoimmune disorders. Persisting disturbances in liver functions due to resulting hepatocellular injury lead to chronic liver disease(s). The diverse functions of liver are performed by parenchymal (hepatocytes) and non‐parenchymal cells (mainly Kupffer cells, stellate cells, sinusoidal endothelial cells and biliary epithelial cells) communicating and working together. The liver parenchyma accounts for approximately 60% of total liver mass with non‐parenchymal cells making up the rest.
Figure 1.
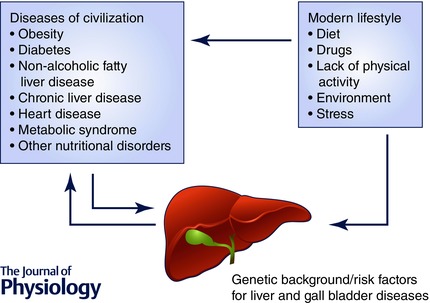
Diseases of civilization are usually a cause or consequence of liver and gall bladder diseases
Biliary epithelial cells or cholangiocytes constitute around 5% of liver cells (Sirica et al. 2008). These form an intricate network of tiny channels (bile canaliculi) that merge to form the bile ducts channeling and collecting bile from liver lobules for storage in the gall bladder and subsequent intestinal secretion. Cholangiocytes that line the bile ducts have secretory functions and those that line the smaller bile ducts and canaliculi play roles in inflammatory and proliferative responses.
Bile mainly comprises the bile acids (BAs), which are the end products of cholesterol metabolism. From cholesterol to BAs, there are 17 energy consuming enzymatic reactions (Russell, 2009); and hence a very efficient and controlled recycling system for BAs exists in humans. About 95% of BAs are reabsorbed through the enterohepatic circulation. The functional BA pool is maintained by an extended system of transporters (Thomas et al. 2008) as shown in Fig. 2. As hepatocytes in the liver are organized over the sinusoids, specific hepatic transporters are expressed at the polarized membranes (Table 1). These transporters are involved in adaptive response to BAs overload and accumulation e.g. in disease conditions such as cholestasis. BAs, having hormonal functions; exert effects via the nuclear receptors (Fig. 3) in the regulation of a variety of metabolic effects – including glucose, lipid and energy metabolism (Watanabe et al. 2006; Lefebvre et al. 2009; Wei et al. 2009; Torres et al. 2012; Li & Chiang, 2015); cholesterol uptake, metabolism and secretion (De Fabiani et al. 2003); xenobiotic metabolism (Hofmann & Hagey, 2008; Zollner & Trauner, 2009); endocrine (Houten et al. 2006; Keitel et al. 2008) and immunological signalling (Ishizawa et al. 2008; Makishima et al. 2002) – and have antimicrobial effects in the digestive tract (Begley et al. 2005; Kurdi et al. 2006).
Figure 2. Bile acid transport system in hepatocytes .
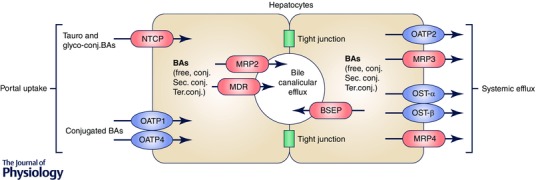
After bile acid (BA) synthesis in hepatocytes, BAs are mono‐conjugated and are excreted through the bile salt export pump (BSEP) into the bile canaliculi. The divalent BAs are excreted by the multidrug resistance‐associated protein 2 (MRP2) and the multidrug export pump (MDR). These bile acids are secreted as bile into the ileum where they may be further conjugated and metabolized. BAs are then recycled into the liver via the portal vein, being taken up mainly by the Na+–taurocholate co‐transporting polypeptide (NTCP) and to a lesser extent by the organic anion transporter proteins (OATP1 and OATP4). During bile acid overload or cholestasis, BAs can be secreted into the systemic circulation through MRP3 and MRP4 and also to some extent via OATP2 and OST‐α and ‐β. Figure adapted from Thomas et al. (2008) with permission from Macmillan Publishers Ltd.
Table 1.
Bile acid transporters in human hepatocytes
| Uptake | Canalicular efflux (apical side) | Basolateral efflux (basolateral side) |
|---|---|---|
| NTCP (SLC10A1) | BSEP (ABCB11) | MRP3 (ABCC3) |
| OATP1A2 (SLCO1A2; OATPA) | MRP2 (ABCC2) | MRP4 (ABCC4) |
| OATP1B1 (SLCO1B1; OATP2) | MDR1 (ABCB1) | OSTα/OSTβ |
| OATP1B3 (SLCO1B3; OATP8) | BCRP (ABCG2) | |
| MDR3 (ABCB4) | ||
| ABCG5/ABCG8 |
Abbreviations, where an asterisk represents a number: ABCB, ATP‐binding cassette sub‐family B member*; ABCC, ATP‐binding cassette sub‐family C member*; ABCG, ATP‐binding cassette sub‐family G member*; BCRP, breast cancer resistance protein; BSEP, bile salt export pump; MDR, multidrug‐resistance*; MRP, multidrug resistance protein*; OATP, organic anion‐transporting protein*; OST, organic solute transporter (α or β); SLC: solute carrier*; SLCO, solute carrier organic anion*.
Figure 3. Bile acids, via their direct effects on nuclear receptors (FXR, PXR, CAR and PPARα), not only regulate their own synthesis, metabolism and clearance, but also play a significant role in glucose, lipid and cholesterol metabolism and xenobiotic (phase 0‐III) metabolism .
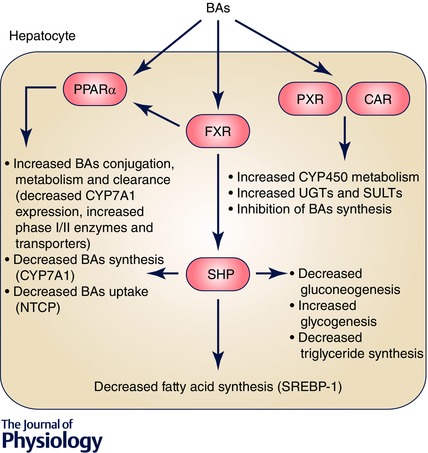
PPARα, peroxisome proliferator‐activated receptor α. SHP, small heterodimer partner; UGT, uridine 5'‐diphospo‐glucuronosyltransferase; SULTs, sulfotransferases.
Cholestatic‐liver diseases (CLDs), also called hepatobiliary diseases, include a range of clinical, biochemical and histological manifestations due to cholestasis. Cholestasis is derived from Greek chole meaning bile and stasis meaning halting/stopping. Cholestasis refers to obstruction of bile flow leading to the accumulation of bile in the liver and spillage of bile components into the systemic circulation. Cholestasis often involves inflammatory and autoimmune processes affecting the intrahepatic or extrahepatic biliary tree.
CLDs are diverse in occurrence; some are common in children, others in adults; and some occur predominately in men while others in women (Carbone et al. 2013). The underlying causes are also diverse including congenital disease, genetic predisposition, drug‐induced cholestasis and infections (Table 2). Up to 30% cases of drug‐induced liver injury are associated with cholestasis with a mortality rate of 8% (Bjornsson & Olsson, 2005). Cholestasis is often chronic in nature and ultimately leads to fibrosis and liver cirrhosis. In the case of autoimmune CLDs, in some patients especially those with primary sclerosing cholangitis (PSC; see Table 2), the risk for developing cholangiocarcinoma is high (Bergquist & von Seth, 2015).
Table 2.
Cholestatic liver diseases
| Disorder | Explanation |
|---|---|
| Progressive familial intrahepatic cholestasis (PFIC) | Genetic disorders associated with mutations in the genes ATP8B1 (PFIC type 1), ABCB11 (PFIC type 2) and ABCB4 (PFIC type 3). These genes encode trans‐membrane transporters involved in the transport of aminophospholipids (FIC1), bile salts (BSEP) and phosphatidylcholine (MDR3), respectively. |
| Biliary atresia | An idiopathic inflammatory disorder characterized by absence of lumen in part or all of extrahepatic biliary tract and often affecting the intrahepatic bile ducts. It is the most frequent cause of chronic cholestasis in infants and children and the reason for liver transplantation. |
| Alagille syndrome | A congenital deficiency in interlobular bile ducts associated with mutations in human JAG1 encoding a ligand in the NOTCH signalling pathway. This autosomal dominant multi‐system disorder varies greatly in clinical phenotype even in monozygotic twins. |
| Arthrogryposis, renal dysfunction and cholestasis syndrome (ARC) | Genetic disorder associated with mutations in genes VPS33 and VIPAR (in some patients). VPS33B‐VIPAR complex plays a role in apical‐basolateral polarity in liver and kidney. |
| Primary biliary cirrhosis (PBC) | An autoimmune disease characterized by chronic small bile duct cholangitis occurring predominantly in women above 40 years of age. The disease is associated with an autoantibody against the mitochondrial pyruvate dehydrogenase and/or the E3 binding proteins. In severe cases, liver transplantation is the only lifesaving option. |
| Primary sclerosing cholangitis (PSC) | A chronic cholestatic syndrome of unknown aetiology characterized by progressive fibrosis of the biliary tree. It occurs predominantly in men with an average onset age of 40 years. It is a premalignant disease of the biliary tract and may lead to cholangiocarcinoma. Currently, liver transplantation is the only lifesaving option for patients with end stage PSC with 5 year post‐transplant survival of 75–85%. |
| Cholangiocarcinoma (CC) | Malignant hepatobiliary cancer arising from the cholangiocytes often diagnosed at a late stage with very poor prognosis. |
| Intrahepatic cholestasis of pregnancy (ICP) | Reversible cholestasis during pregnancy with incidence ranging from 0.1 to 15 %. The multifactorial pathogenesis includes genetic, hormonal and environmental factors. Mutations in the gene ABCB4 encoding the MDR3 transporter protein are reported as a major cause, in combination with hormones like oestrogen and progesterone. |
| Cholestasis associated with total parenteral nutrition (TPN) | TPN is administered in cases of intestinal failure and is associated with cholestasis. TNP‐cholestasis is common in children and infants due to prematurity and short bowel length. The cause is excess of glucose and lipids in the parenteral nutrition. The high glucose levels result in increased insulin release, which in turn activates fatty acid synthesis and inhibits fatty acid breakdown leading to steatosis and cholestasis. |
| Cholestasis associated with infections | Infections such as cytomegalovirus, rubella and syphilis may cause cholangitis leading to chronic cholestasis. Inflammation of the gut can result in leaking of bacterial endotoxins and bacterial contamination of the biliary tree leading to cholestasis. |
| Drug‐induced cholestasis | Drugs may interfere with bile acid synthesis, metabolism and transport leading to cholestasis. Although usually reversible upon withdrawal of the drug, drugs may cause chronic cholestasis resulting in hepatobiliary damage requiring liver transplantation. |
Therapeutic options for CLDs are still very limited. Many mechanistic studies are based on animal data. On the other hand, substantial clinical data are also available, but the clinical translation of this knowledge in therapy has been disappointingly limited. This is partly due to the lack of validation of animal results in humans and the lack of integrated use of clinical data in the understanding of the disease.
This underlines the urgent need to understand the mechanistic details of physiological functions and pathobiology of the liver in humans and to apply this knowledge in diagnosis and therapy. Taking the example of cholestatic‐liver diseases, this Topical Review brings into focus the limitations of animal models and proposes a roadmap to a human biology‐based research paradigm using modern ‘omics’ technology, advanced in vitro human cell models with better physiological resemblance to in vivo and other scientific tools within an overarching disease pathway framework rooted in systems biology.
From animal models to human disease
Animal models have been and are still used in the mechanistic investigations of pathobiology of human CLDs. The general reasons for the use of animals (especially rodents) are usually the low costs, ease of breeding and the possibility of genetic manipulation in mice. The other advantage is the fact that effects can be monitored/measured in intact whole organisms allowing studies on inter‐organ effects as well as overall biological homeostasis. Animal models allow experiments that cannot be conducted in humans for ethical reasons. The possibility of genetic manipulation resulted in an increase in the number of animal models for the study of CLDs. Besides genetic modifications such as knock‐out animals, e.g. mdr2 −/− mice, other CLD animal models are established by dietary manipulation, chemical treatment, xenobiotic induction, bile duct ligation (BDL), immunization and infections. Bile duct injury and cholestasis can be provoked by the use of the chemicals such as 3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine or α‐naphthylisothiocyanate in rodents and these models have been widely applied for the study of xenobiotic‐induced cholangiopathies. Detailed characteristics and limitations of animal models of hepatobiliary disease are described in recent reviews (Ueno et al. 2010; Osterreicher & Trauner, 2012; Halilbasic et al. 2013; Liu et al. 2013; Pollheimer et al. 2014). Although animal studies have provided mechanistic insights into CLDs, it is clear that direct extrapolation of animal data to human physiology is very challenging. The major reasons for this are species‐specific physiological differences in the gut and the liver, the BA pool composition and characteristics, the circadian rhythms and feeding behaviour, diet, and immunological and CYP 450 systems, in addition to differences in the onset and progression of cholestasis.
Human BAs are more hydrophobic (Heuman, 1989) and therefore more toxic. Elevated serum BAs in BDL mice are mouse specific and do not correspond to the elevated bile acids (such as chenodeoxycholic acid and cholic acid) in humans. Therefore, direct cytotoxic effect of BAs in the mouse BDL model is of very limited relevance (Zhang et al. 2012). Abcb11 −/− (mouse gene for bile salt export pump; BSEP) knock‐out mice show a very mild cholestatic phenotype as compared to humans (Wang et al. 2001). This is mainly due to compensatory up‐regulation of an ATP‐binding cassette (ABC) transporter, called mdr1 or abcb1a (Lam et al. 2005). In patients, such compensatory expression of MDR1 or ABCB1 (Keitel et al. 2005) is not observed. In mice the BAs can be excreted via other canalicular transporters and therefore BSEP transporter inhibition results in only mild cholestasis (Wang et al. 2001; Lam et al. 2005). In humans mutations in ABCB11 or inhibition of BSEP, via for example drugs, may lead to very serious consequences. As for another ABC transporter, the multidrug resistance associated protein (MDR2), heterozygous mice (mdr2 −/+) do not develop cholestatic liver injury, whereas heterozygous mutations in humans are reported to cause cholestasis (Jacquemin & Hadchouel, 1999). In addition, rats have very high basolateral bile salt efflux, which protects them from hepatic injury (Jemnitz et al. 2010). Human organic anion transporting polypeptides (OATPs) belonging to the SLC (solute carrier) family and located at the basolateral membranes of the hepatocytes are electrogenic and facilitate diffusion of bile salts down their electrochemical gradients (Martinez‐Becerra et al. 2011), whereas the rat oatp1a1 is electroneutral, suggesting that different members of the OATP family have different mechanisms of action. Moreover, significant species‐dependent differences in the inhibition of Na+‐dependent taurocholate cotransporting polypeptide (NTCP), another SLC transporter, have been reported explaining the lack of hepatotoxicity of the model drug bosentan in rats (Leslie et al. 2007). Nuclear receptors play an essential role in BA homeostasis by controlling the synthesis, metabolism and transport of BAs. Transcriptional regulation playing a key role in development of CLDs is different in rodents and other model organisms as compared to humans, e.g. feed‐forward regulation of CYP7A1 via the liver X‐receptor is limited to rodents (Goodwin et al. 2003).
Significant differences in the inflammatory responses between rats and humans (Seok et al. 2013) are also reported. Immune responses are different in rodents than in humans (Oertelt et al. 2006; Khanna & Burrows, 2011). In humans, anti‐mitochondrial antibodies are shown to play an essential role in the pathogenesis of primary biliary cirrhosis (Kaplan & Gershwin, 2005). In animal models, immunity to mitochondrial antigens is not sufficient to elicit hepatobiliary injury (Poupon et al. 2000). In humans, immunoglobulin (Ig) A binds to the polymeric Ig receptors located on the basolateral membranes of cholangiocytes and has a protective role in biliary mucosal immune defence (Mantis & Forbes, 2010). Mice do not have polymeric IgA receptors (Oertelt et al. 2006) and the immune mechanisms are different. Until now, no established animal model shows all the attributes of the two human auto‐immune diseases, namely primary biliary cholangitis (PBC) and PSC (Pollheimer et al. 2014; Tsuneyama et al. 2012).
Another very important consideration is that drug metabolism and elimination are different in animals and humans (Martignoni et al. 2006), not only in CYP 450‐mediated biotransformation but also in Phase II metabolism (conjugation reactions) and transport of the drug metabolites (elimination) into the bile. BAs cause apoptotic injury to liver parenchyma in rodents whereas in humans BAs induce necrosis providing further evidence that the mechanisms of obstructive cholestasis in humans are different from animals (Woolbright et al. 2015).
In addition, diet (fundamentally different in humans, an omnivore, from rodents, which are ganivores) plays a role in defining the gut microbiota of different species (Karasov et al. 2011). The most abundant bacterial genus in humans Bifidobacterium, does not colonize rodent gut and therefore such animal models are of very low relevance to humans (Pang et al. 2007). The gut microbiota plays an important role in BA homeostasis (Jones et al. 2014), energy regulation and metabolism (Nieuwdorp et al. 2014), immunity (Mann et al. 2013) and pathogenesis (Bourzac, 2014). Intestinal inflammation and immune response are modulated by gut microbiota and show significant differences between humans and rodents (Mann et al. 2013).
Summarizing, although a variety of animal models are available, significant species–specific differences in liver immunology and biliary physiology exist and these lead to differences in pathogenesis and progression of CLDs in humans as compared with animals. Species–specific differences often pose an insurmountable challenge in the translation of animal results into clinical practice (Pound & Bracken, 2014). It is most likely that studies with experimental animal models will continue but the 21st century scientific goal should be a move towards human systems, as explained below, in order to surmount the inevitable species barriers.
The new paradigm: understanding human organ physiology and disease pathways
Advances in cell culture methods and analytical techniques are now allowing study of disease mechanisms in vitro using human derived cells such as primary hepatocytes, genetically modified human cell lines and human induced pluripotent cell (hiPSC)‐derived liver models. These are expected to play a major role in the study of human CLDs and screening of therapeutic agents.
Human‐specific models and tools: advances in liver cell culture and techniques
In vitro studies are traditionally based on monolayer cultures of cells where the 3D architecture of the tissue is lost. In in vivo liver, cell‐to‐cell contacts and communication across the extracellular matrix is ensured within a three‐dimensional arrangement. The extracellular matrix regulates cell morphology and gene expression in vivo (Bissell et al. 1982; Le Beyec et al. 2007). A three‐dimensional environment influences the epigenetic plasticity of the cells (Spencer et al. 2007; Xu et al. 2007). Conventional 2D hepatic cultures rapidly lose liver‐like functionality (Paine & Andreakos, 2004; Godoy et al. 2013) leading to poor concordance between experimental in vitro data and in vivo data, especially with respect to xenobiotic metabolism and transporter activities. Using growth factors supplemented medium, primary hepatocytes can be maintained viable and functional for longer periods of time (Mueller et al. 2012). Primary human hepatocytes although offering the advantage of providing a palette of genetic backgrounds, are limited in their availability; disease aetiology and therapy of donors; and viability. As such, cell lines are still used in drug development and screenings (Gomez‐Lechon et al. 2014). However, hepatic cell line(s) with functional hepatocytes and co‐cultures will be essential in the study of human disease pathogenesis.
The differentiated human HepaRG cells, consisting of hepatocytes and biliary like cells, are commonly used in drug uptake, metabolism and elimination studies due to their primary hepatocyte‐like metabolic competence and transporter activities (Kanebratt and Andersson, 2008 a,b). HepaRG 3D cultures show a network of bile canaliculi and harbour functional apical and basolateral transporters (Guillouzo et al. 2007; Gunness et al. 2013; Klein, 2015; Mueller et al. 2014). This cell line alone or in co‐culture with other non‐parenchymal cells could be an important tool in the study of hepatobiliary diseases.
Other very significant progress in the area of hiPSCs (Asgari et al. 2010; Schwartz et al. 2014) is making the application of patient‐ and disease‐specific hiPS cells a reality (Ghodsizadeh et al. 2010; Siller et al. 2013; McCracken et al. 2014). These in vitro models are also expected to be useful in the screening of compounds for personalized therapy.
Much development effort is underway for high throughput generation of the 3D cultures as aggregates (Gevaert et al. 2014), micro‐patterned co‐cultures (Khetani & Bhatia, 2008) and 3D printing (Billiet et al. 2014). High‐content platforms are designed and are already in use in drug development for screening compounds (Bale et al. 2014; Tolosa et al. 2014). At the same time, highly advanced imaging and other techniques (including automated methods for assessing multiple readouts such as cell viability, shape of the nuclei, cell area, mitochondrial membrane potential, phospholipid accumulation, cytoskeleton integrity and apoptosis) are playing an important role in the study of biological pathways (Ramaiahgari et al. 2014; Sirenko et al. 2014). Such an advanced technology has opened up a great opportunity to study human disease in vitro as it enables analysis of biochemical and metabolic activities of living cells in functional tissue and organ contexts, at the same time allowing high‐resolution real‐time imaging (Bhatia & Ingber, 2014). These high content and high throughput platforms are already changing the toxicity screening paradigm (Patlewicz et al. 2013) and allowing pathway‐based in vitro‐only safety assessment (Adeleye et al. 2014; Kleensang et al. 2014).
Omics technologies for understanding disease pathobiology
Recent advances in technology along with high‐throughput, high‐content analyses and great leaps in computational power have played a key role in slowly but surely shifting the paradigm of understanding organ physiology, disease, target and biomarker identification, development of therapeutics and toxicology from the traditional reductionist approaches to a more holistic approach. In the past decade, the omics technologies have provided tremendous opportunities in discovery biology especially in the understanding of human disease.
A number of genome‐wide association studies (GWAS) on CLDs have appeared during the last few years and have been excellently reviewed (Suter et al. 2004; Krawczyk et al. 2010; Mullenbach & Lammert, 2011; Mells et al. 2013). These studies have provided important insights into the pathogenesis of hereditary (such as progressive familial intrahepatic cholestasis types I–III), autoimmune (PBC and PSC) as well as drug‐induced cholestasis. Genome‐wide association studies have also provided evidence of genetic heterogeneity of PBC and PSC based on ethnicity. Environmental factors and the epigenome are reported to have a profound impact on the development and progression of polygenic CLDs (Krawczyk et al. 2010; Mells et al. 2013). Trimethylation of the histone H3K4 is reported to be essential for the activation of the BSEP, NTCP and MRP2 genes by nuclear receptors (Ananthanarayanan et al. 2011). In the case of PBC, sex‐dependent epigenetic factors have also been reported (Selmi et al. 2004). An excellent review on the influence of epigenetic factors in bile acid homeostasis was recently published (Smith et al. 2013).
The bile proteome is becoming a major focus of interest due to its relatively easier application in a clinical set‐up and the possibility of discovering biomarkers for biliary disease (Farina et al. 2014). Bile proteomics was effectively applied to distinguish patients with PSC and cholangiocarcinoma (Lankisch et al. 2011). In another recent study, bile proteomics was used to recognize benign from the malignant biliary strictures in patients (Navaneethan et al. 2015). Liver biopsies can also be used for proteomics analysis for protein biomarkers for perturbed biological function. The lysosomal‐associated membrane protein‐2 from liver samples from PBC patients was identified as a marker for the prognosis of the disease (Wang et al. 2013).
Metabolomics includes a comprehensive qualitative and quantitative analysis of low molecular weight metabolites in a cell, organ or whole organism (Fiehn, 2002). Metabolomics sums up all the upstream effects of genomics, transcriptomics and proteomics; and represents the actual phenotype (Mueller et al. 2012; Klein and Heinzle, 2012; Ramirez et al. 2013). Metabolomics was used to distinguish between patients of PBC and PSC (Trottier et al. 2012) and has been valuable in defining a ‘core metabolomics phenotype’ for hepatobiliary diseases (Beyoglu & Idle, 2013).
Systems biology tools are no doubt expanding the understanding of human physiology and disease. However, there are yet unmet requirements for a wide application of the omics technologies. The most important limitations of these methods are the complexity of the methods and analytical techniques, standardization and validation. Omics technologies often generate huge datasets for which powerful bioinformatics tools are needed. Public databases are needed to store and exchange information and facilitate integrated systems analysis. Computational models incorporating mechanistic information as well as genetic information are required for prediction. Systems biology tools are aimed at very early indication of perturbation patterns in physiology that may lead to adverse outcomes/diseases. As such, many detected changes may not be biologically or pathologically relevant. However, integration of multi‐omics on different scales using mathematical models will allow identification of patterns of gene transcripts, proteins and metabolites and link them to an adverse outcome/disease pathway. In addition, omics technologies provide biomarkers at different levels of biological organization that are often non‐specific. It is expected that a set of biomarkers will provide better and more reliable information than single biomarkers. Finally, the successful implementation of systems biology tools in healthcare and industry is still a challenge.
In future, it is expected that human in vitro models based on human liver cells and patient‐specific iPSC‐derived hepatocytes will be preferentially used. These will yield rich human‐relevant information at different levels of omics (Fig. 4) not only for understanding the disease but also for identification of the most suitable therapeutic options under specific conditions.
Figure 4. Advanced human in vitro models, e.g. primary cells or those derived from hiPSC maintained in 3D, will provide high content and data‐rich ‘omics’ information and biomarkers for diagnosis; and can be used in screening for novel therapy options .
3D hepatic organoid by Daniel Mueller and Patrina Gunness; fluorescence image taken at the Karolinska Institute, Stockholm, Sweden.
Adverse outcome/disease pathway(s)
The concept of the adverse outcome pathway (AOP) was recently developed in the field of risk assessment for chemicals (Landesmann et al. 2013) and ecotoxicology (Ankley et al. 2010). An AOP is aimed at describing the link between a molecular initiating event (e.g. receptor binding) and an adverse outcome (e.g. cholestasis), with a number of intermediary ‘key’ events (Garcia‐Reyero, 2015). AOPs have been described for skin sensitization, liver cholestasis, liver steatosis and fibrosis (OECD, 2012; Vinken et al. 2013; Willett et al. 2014). Recently, an AOP for drug‐induced cholestasis has been described with BSEP inhibition as the molecular initiating event (Vinken et al. 2013). Inhibition of BSEP should result in BA accumulation, which leads to inflammation and activation of nuclear receptors farnesoid X receptor (FXR), pregnane X receptor (PXR) and constitutive androstane/active receptor (CAR). These in turn lead to adaptive changes in BA transporters and metabolism. However, persisting stress and BA load results in altered liver functions and ultimately cholestasis.
Components of the system including mechanistic details could be described in detail using a pathway framework as it is based on measurable changes in the biological state (Villeneuve et al. 2014 a,b). It is highly recommended for an AOP to have direct human relevance, and an AOP based on only animal data is not sufficient. Biological systems are highly complex and interconnected in addition to being very robust, showing adaptive response to stress stimuli. Biological processes are non‐linear and highly ‘wired’ together with feed‐back loops and cross regulation. An AOP should not only give information about the structure of the system but also provide clues to the dynamics of the system by temporal description of the biological processes. Although the idea of the AOP originated in the field of toxicology, a broader application to describing disease mechanisms is possible as a framework for the organization and linking of biological information at different biological levels (cells, tissues, organs).
Roadmap for cholestatic‐liver diseases research and perspectives for clinical translation and personalized medicine
Although toxicology is embracing technology and benefiting from a systems biology approach, research on disease is lagging behind despite the availability of human clinical data at the genome, proteome and metabolome levels. CLDs result from a diverse range of hepatobiliary dysfunction with causes ranging from genetic predisposition to life style. Various pathways are involved in liver injury and repair. Mechanistic understanding of these pathways will provide biomarkers for diagnosis and targets for new therapies. Integrated data analysis from traditional histopathology to transcriptomics, proteomics and metabolomics is expected to improve the understanding of the pathogenesis of the CLDs in humans. Systems analysis would allow the prediction of risk for developing these diseases. In addition, these methods could also be applied for monitoring disease progression and therapy success.
In a disease pathway framework, the effects at molecular, cellular and tissue levels should provide novel targets for therapy. Systems information can also be derived at the organ and organism levels allowing reliable and early diagnosis as well as guiding symptomatic therapy (Fig. 5).
Figure 5. An adverse outcome/disease pathway framework to scrutinize systems information (from ‘omics’) for the identification of novel targets for therapy and biomarkers for early diagnosis at different levels of biological organization .
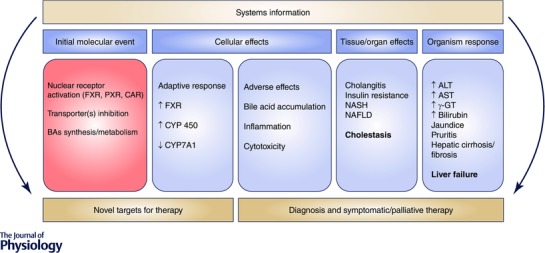
NAFLD, non‐alcoholic fatty liver disease; NASH, non‐alcoholic steatohepatitis; ALT, alanine transaminase; AST, aspartate transaminase; γ‐GT‐gamma glutamyl transferase. An up arrow indicates increase and down arrow indicates decrease.
Currently, the therapeutic options especially for inflammatory cholestasis are very limited. Ursodeoxycholic acid is the only approved treatment for PBC, with little effectiveness in PSC. Immunosuppressive drugs are generally ineffective. The lack of therapeutic options is mainly due to the gaps in the understanding of human disease. A systems approach to the understanding of the pathogenesis of cholestasis is expected to provide opportunities for therapeutic interventions, as new targets could be discovered. The understanding of the genetic causes and the possibility of sequencing genomes could play a role in the prediction of an individual's risk of developing a CLD. Knowledge of the underlying causes will certainly help in the assessment of the prognosis of the disease. Temporal monitoring of the system (patient) will provide insights into the dynamics of the disease guiding the therapy.
Conclusion
CLDs are complex and although manifest as cholestasis resulting from perturbed bile acid homeostasis, they are intertwined with glucose, lipid and energy metabolism as well as the immune response of the patient. Such complex pathogenesis requires a systems understanding leaning on new technologies. Although animal studies have advanced our knowledge of CLDs, there has not been significant clinical translation of that knowledge in the treatment or prevention of these diseases. There is a huge amount of clinical and animal data available on cholestasis. Modern in vitro methods based on human cells (and co‐cultures) maintained in in vivo‐like conditions provide an invaluable tool for the investigation and validation of human‐relevant mechanisms involved in the development of cholestasis and its progression. Omics technologies and computational modelling will enhance the knowledge and allow prediction. The shift in paradigm towards a human‐relevant systems approach to the understanding of cholestasis seems essential to bring a breakthrough that will pave the way for new therapeutic options for CLDs and eventually personalized therapy.
Additional information
Competing interests
There are no conflicts of interest.
Funding
Humane Society International (HSI) provided support for the writing of this article. The author is a researcher working on the SEURAT‐1 NOTOX project funded by the European Community's Seventh Framework Programme (FP7/2007‐2013) under grant agreement N° 267038 and Cosmetics Europe.
Acknowledgements
Dr G. Langley is thanked for her valuable suggestions. Prof. F. Lammert and Dr Christoph Jüngst, medical faculty of the Saarland University, are especially thanked for their comments.
Biography
Fozia Noor graduated summa cum laude from Heidelberg University at the Institute of Pharmacy and Molecular Biotechnology obtaining her PhD with Nils Metzler‐Nolte. She joined Elmar Heinzle's group at the Biochemical Engineering Institute of Saarland University where she is currently finalizing her Habilitation as a group leader of cell culture and systems toxicology laboratory. Her research focuses on the development and application of in vitro methods including 3D cultivation systems of liver and heart for toxicological and mechanistic studies in combination with in vitro metabolomics.

References
- Adeleye Y, Andersen M, Clewell R, Davies M, Dent M, Edwards S, Fowler P, Malcomber S, Nicol B, Scott A, et al (2014). Implementing toxicity testing in the 21st century (TT21C): Making safety decisions using toxicity pathways, and progress in a prototype risk assessment. Toxicology 332, 102–111. [DOI] [PubMed] [Google Scholar]
- Ananthanarayanan M, Li Y, Surapureddi S, Balasubramaniyan N, Ahn J, Goldstein JA & Suchy FJ (2011). Histone H3K4 trimethylation by MLL3 as part of ASCOM complex is critical for NR activation of bile acid transporter genes and is downregulated in cholestasis. Am J Physiol Gastrointest Liver Physiol 300, G771–G781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ankley GT, Bennett RS, Erickson RJ, Hoff DJ, Hornung MW, Johnson RD, Mount DR, Nichols JW, Russom CL, Schmieder PK, et al (2010). Adverse outcome pathways: a conceptual framework to support ecotoxicology research and risk assessment. Environ Toxicol Chem 29, 730–741. [DOI] [PubMed] [Google Scholar]
- Asgari S, Pournasr B, Salekdeh GH, Ghodsizadeh A, Ott M & Baharvand H (2010). Induced pluripotent stem cells: a new era for hepatology. J Hepatol 53, 738–751. [DOI] [PubMed] [Google Scholar]
- Bale SS, Vernetti L, Senutovitch N, Jindal R, Hegde M, Gough A, McCarty WJ, Bakan A, Bhushan A, Shun TY, et al (2014). In vitro platforms for evaluating liver toxicity. Exp Biol Med (Maywood) 239, 1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley M, Gahan CG & Hill C (2005). The interaction between bacteria and bile. FEMS Microbiol Rev 29, 625–651. [DOI] [PubMed] [Google Scholar]
- Bergquist A & von Seth E (2015). Epidemiology of cholangiocarcinoma. Best Pract Res Clin Gastroenterol 29, 221–232. [DOI] [PubMed] [Google Scholar]
- Beyoglu D & Idle JR (2013). The metabolomic window into hepatobiliary disease. J Hepatol 59, 842–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia SN & Ingber DE (2014). Microfluidic organs‐on‐chips. Nat Biotechnol 32, 760–772. [DOI] [PubMed] [Google Scholar]
- Billiet T, Gevaert E, De Schryver T, Cornelissen M & Dubruel P (2014). The 3D printing of gelatin methacrylamide cell‐laden tissue‐engineered constructs with high cell viability. Biomaterials 35, 49–62. [DOI] [PubMed] [Google Scholar]
- Bissell MJ, Hall HG & Parry G (1982). How does the extracellular matrix direct gene expression? J Theor Biol 99, 31–68. [DOI] [PubMed] [Google Scholar]
- Bjornsson E & Olsson R (2005). Outcome and prognostic markers in severe drug‐induced liver disease. Hepatology 42, 481–489. [DOI] [PubMed] [Google Scholar]
- Bourzac K (2014). Microbiome: the bacterial tightrope. Nature 516, S14–S16. [DOI] [PubMed] [Google Scholar]
- Carbone M, Mells GF, Pells G, Dawwas MF, Newton JL, Heneghan MA, Neuberger JM, Day DB, Ducker SJ, Sandford RN, et al (2013). Sex and age are determinants of the clinical phenotype of primary biliary cirrhosis and response to ursodeoxycholic acid. Gastroenterology 144, 560–569; quiz e13–e14. [DOI] [PubMed] [Google Scholar]
- De Fabiani E, Mitro N, Gilardi F, Caruso D, Galli G & Crestani M (2003). Coordinated control of cholesterol catabolism to bile acids and of gluconeogenesis via a novel mechanism of transcription regulation linked to the fasted‐to‐fed cycle. J Biol Chem 278, 39124–39132. [DOI] [PubMed] [Google Scholar]
- Farina A, Delhaye M, Lescuyer P & Dumonceau JM (2014). Bile proteome in health and disease. Compr Physiol 4, 91–108. [DOI] [PubMed] [Google Scholar]
- Fiehn O (2002). Metabolomics—the link between genotypes and phenotypes. Plant Mol Biol 48, 155–171. [PubMed] [Google Scholar]
- Garcia‐Reyero N (2015). Are adverse outcome pathways here to stay? Environ Sci Technol 49, 3–9. [DOI] [PubMed] [Google Scholar]
- Gevaert E, Dolle L, Billiet T, Dubruel P, van Grunsven L, van Apeldoorn A & Cornelissen R (2014). High throughput micro‐well generation of hepatocyte micro‐aggregates for tissue engineering. PLoS One 9, e105171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghodsizadeh A, Taei A, Totonchi M, Seifinejad A, Gourabi H, Pournasr B, Aghdami N, Malekzadeh R, Almadani N, Salekdeh GH, et al (2010). Generation of liver disease‐specific induced pluripotent stem cells along with efficient differentiation to functional hepatocyte‐like cells. Stem Cell Rev 6, 622–632. [DOI] [PubMed] [Google Scholar]
- Godoy P, Hewitt NJ, Albrecht U, Andersen ME, Ansari N, Bhattacharya S, Bode JG, Bolleyn J, Borner C, Bottger J, et al (2013). Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non‐parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch Toxicol 87, 1315–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Lechon MJ, Tolosa L, Conde I & Donato MT (2014). Competency of different cell models to predict human hepatotoxic drugs. Expert Opin Drug Metab Toxicol 10, 1553–1568. [DOI] [PubMed] [Google Scholar]
- Goodwin B, Watson MA, Kim H, Miao J, Kemper JK & Kliewer SA (2003). Differential regulation of rat and human CYP7A1 by the nuclear oxysterol receptor liver X receptor‐α. Mol Endocrinol 17, 386–394. [DOI] [PubMed] [Google Scholar]
- Guillouzo A, Corlu A, Aninat C, Glaise D, Morel F & Guguen‐Guillouzo C (2007). The human hepatoma HepaRG cells: a highly differentiated model for studies of liver metabolism and toxicity of xenobiotics. Chem Biol Interact 168, 66–73. [DOI] [PubMed] [Google Scholar]
- Gunness P, Mueller D, Shevchenko V, Heinzle E, Ingelman‐Sundberg M & Noor F (2013). 3D organotypic cultures of human HepaRG cells: a tool for in vitro toxicity studies. Toxicol Sci 133, 67–78. [DOI] [PubMed] [Google Scholar]
- Halilbasic E, Claudel T & Trauner M (2013). Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J Hepatol 58, 155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuman DM (1989). Quantitative estimation of the hydrophilic‐hydrophobic balance of mixed bile salt solutions. J Lipid Res 30, 719–730. [PubMed] [Google Scholar]
- Hofmann AF & Hagey LR (2008). Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell Mol Life Sci 65, 2461–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houten SM, Watanabe M & Auwerx J (2006). Endocrine functions of bile acids. EMBO J 25, 1419–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizawa M, Matsunawa M, Adachi R, Uno S, Ikeda K, Masuno H, Shimizu M, Iwasaki K, Yamada S & Makishima M (2008). Lithocholic acid derivatives act as selective vitamin D receptor modulators without inducing hypercalcemia. J Lipid Res 49, 763–772. [DOI] [PubMed] [Google Scholar]
- Jacquemin E & Hadchouel M (1999). Genetic basis of progressive familial intrahepatic cholestasis. J Hepatol 31, 377–381. [DOI] [PubMed] [Google Scholar]
- Jemnitz K, Veres Z & Vereczkey L (2010). Contribution of high basolateral bile salt efflux to the lack of hepatotoxicity in rat in response to drugs inducing cholestasis in human. Toxicol Sci 115, 80–88. [DOI] [PubMed] [Google Scholar]
- Jones ML, Martoni CJ, Ganopolsky JG, Labbe A & Prakash S (2014). The human microbiome and bile acid metabolism: dysbiosis, dysmetabolism, disease and intervention. Expert Opin Biol Ther 14, 467–482. [DOI] [PubMed] [Google Scholar]
- Kanebratt KP & Andersson TB (2008. a). Evaluation of HepaRG cells as an in vitro model for human drug metabolism studies. Drug Metab Dispos 36, 1444–1452. [DOI] [PubMed] [Google Scholar]
- Kanebratt KP & Andersson TB (2008. b). HepaRG cells as an in vitro model for evaluation of cytochrome P450 induction in humans. Drug Metab Dispos 36, 137–145. [DOI] [PubMed] [Google Scholar]
- Kaplan MM & Gershwin ME (2005). Primary biliary cirrhosis. N Engl J Med 353, 1261–1273. [DOI] [PubMed] [Google Scholar]
- Karasov WH, Martinez del Rio C & Caviedes‐Vidal E (2011). Ecological physiology of diet and digestive systems. Annu Rev Physiol 73, 69–93. [DOI] [PubMed] [Google Scholar]
- Keitel V, Burdelski M, Warskulat U, Kuhlkamp T, Keppler D, Haussinger D & Kubitz R (2005). Expression and localization of hepatobiliary transport proteins in progressive familial intrahepatic cholestasis. Hepatology 41, 1160–1172. [DOI] [PubMed] [Google Scholar]
- Keitel V, Kubitz R & Haussinger D (2008). Endocrine and paracrine role of bile acids. World J Gastroenterol 14, 5620–5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna R & Burrows SR (2011). Human immunology: a case for the ascent of non‐furry immunology. Immunol Cell Biol 89, 330–331. [DOI] [PubMed] [Google Scholar]
- Khetani SR & Bhatia SN (2008). Microscale culture of human liver cells for drug development. Nat Biotechnol 26, 120–126. [DOI] [PubMed] [Google Scholar]
- Kleensang A, Maertens A, Rosenberg M, Fitzpatrick S, Lamb J, Auerbach S, Brennan R, Crofton KM, Gordon B, Fornace AJ Jr, et al (2014). t4 Workshop Report: Pathways of toxicity. ALTEX 31, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein S & Heinzle E (2012). Isotope labeling experiments in metabolomics and fluxomics. Wiley Interdiscip Rev Syst Biol Med 4, 261–272. [DOI] [PubMed] [Google Scholar]
- Klein S, Maggioni S, Bucher J, Mueller D, Niklas J, Shevchenko V, Mauch K, Heinzle E & Noor F (2015). In silico modeling for the prediction of dose and pathway related adverse effects in humans from in vitro repeated‐dose studies. Tox Sci DOI: 10.1093/toxsci/kfv218. [DOI] [PubMed] [Google Scholar]
- Krawczyk M, Mullenbach R, Weber SN, Zimmer V & Lammert F (2010). Genome‐wide association studies and genetic risk assessment of liver diseases. Nat Rev Gastroenterol Hepatol 7, 669–681. [DOI] [PubMed] [Google Scholar]
- Kurdi P, Kawanishi K, Mizutani K & Yokota A (2006). Mechanism of growth inhibition by free bile acids in lactobacilli and bifidobacteria. J Bacteriol 188, 1979–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam P, Wang R & Ling V (2005). Bile acid transport in sister of P‐glycoprotein (ABCB11) knockout mice. Biochemistry 44, 12598–12605. [DOI] [PubMed] [Google Scholar]
- Landesmann B, Mennecozzi M, Berggren E & Whelan M (2013). Adverse outcome pathway‐based screening strategies for an animal‐free safety assessment of chemicals. Altern Lab Anim 41, 461–471. [DOI] [PubMed] [Google Scholar]
- Lankisch TO, Metzger J, Negm AA, Vosskuhl K, Schiffer E, Siwy J, Weismuller TJ, Schneider AS, Thedieck K, Baumeister R, et al (2011). Bile proteomic profiles differentiate cholangiocarcinoma from primary sclerosing cholangitis and choledocholithiasis. Hepatology 53, 875–884. [DOI] [PubMed] [Google Scholar]
- Le Beyec J, Xu R, Lee SY, Nelson CM, Rizki A, Alcaraz J & Bissell MJ (2007). Cell shape regulates global histone acetylation in human mammary epithelial cells. Exp Cell Res 313, 3066–3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre P, Cariou B, Lien F, Kuipers F & Staels B (2009). Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev 89, 147–191. [DOI] [PubMed] [Google Scholar]
- Leslie EM, Watkins PB, Kim RB & Brouwer KL (2007). Differential inhibition of rat and human Na+‐dependent taurocholate cotransporting polypeptide (NTCP/SLC10A1) by bosentan: a mechanism for species differences in hepatotoxicity. J Pharmacol Exp Ther 321, 1170–1178. [DOI] [PubMed] [Google Scholar]
- Li T & Chiang JY (2015). Bile acids as metabolic regulators. Curr Opin Gastroenterol 31, 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Meyer C, Xu C, Weng H, Hellerbrand C, ten Dijke P & Dooley S (2013). Animal models of chronic liver diseases. Am J Physiol Gastrointest Liver Physiol 304, G449–G468. [DOI] [PubMed] [Google Scholar]
- Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, Haussler MR & Mangelsdorf DJ (2002). Vitamin D receptor as an intestinal bile acid sensor. Science 296, 1313–1316. [DOI] [PubMed] [Google Scholar]
- Mann ER, Landy JD, Bernardo D, Peake ST, Hart AL, Al‐Hassi HO & Knight SC (2013). Intestinal dendritic cells: their role in intestinal inflammation, manipulation by the gut microbiota and differences between mice and men. Immunol Lett 150, 30–40. [DOI] [PubMed] [Google Scholar]
- Mantis NJ & Forbes SJ (2010). Secretory IgA: arresting microbial pathogens at epithelial borders. Immunol Invest 39, 383–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martignoni M, Groothuis GM & de Kanter R (2006). Species differences between mouse, rat, dog, monkey and human CYP‐mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol 2, 875–894. [DOI] [PubMed] [Google Scholar]
- Martinez‐Becerra P, Briz O, Romero MR, Macias RI, Perez MJ, Sancho‐Mateo C, Lostao MP, Fernandez‐Abalos JM & Marin JJ (2011). Further characterization of the electrogenicity and pH sensitivity of the human organic anion‐transporting polypeptides OATP1B1 and OATP1B3. Mol Pharmacol 79, 596–607. [DOI] [PubMed] [Google Scholar]
- McCracken KW, Cata EM, Crawford CM, Sinagoga KL, Schumacher M, Rockich BE, Tsai YH, Mayhew CN, Spence JR, Zavros Y, et al (2014). Modelling human development and disease in pluripotent stem‐cell‐derived gastric organoids. Nature 516, 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mells GF, Kaser A & Karlsen TH (2013). Novel insights into autoimmune liver diseases provided by genome‐wide association studies. J Autoimmun 46, 41–54. [DOI] [PubMed] [Google Scholar]
- Mueller D, Kramer L, Hoffmann E, Klein S & Noor F (2014). 3D organotypic HepaRG cultures as in vitro model for acute and repeated dose toxicity studies. Toxicol In Vitro 28, 104–112. [DOI] [PubMed] [Google Scholar]
- Mueller D, Muller‐Vieira U, Biemel KM, Tascher G, Nussler AK & Noor F (2012). Biotransformation of diclofenac and effects on the metabolome of primary human hepatocytes upon repeated dose exposure. Eur J Pharm Sci 45, 716–724. [DOI] [PubMed] [Google Scholar]
- Mullenbach R & Lammert F (2011). An update on genetic analysis of cholestatic liver diseases: digging deeper. Dig Dis 29, 72–77. [DOI] [PubMed] [Google Scholar]
- Navaneethan U, Lourdusamy V, Gk Venkatesh P, Willard B, Sanaka MR & Parsi MA (2015). Bile proteomics for differentiation of malignant from benign biliary strictures: a pilot study. Gastroenterol Rep (Oxf) 3, 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwdorp M, Gilijamse PW, Pai N & Kaplan LM (2014). Role of the microbiome in energy regulation and metabolism. Gastroenterology 146, 1525–1533. [DOI] [PubMed] [Google Scholar]
- OECD (2012). The Adverse Outcome Pathway for Skin Sensitization Initiated by Covalent Binding to Proteins. Part I: Scientific Evidence. Series on Testing and Assessment No. 168. OECD, Paris. [Google Scholar]
- Oertelt S, Lian ZX, Cheng CM, Chuang YH, Padgett KA, He XS, Ridgway WM, Ansari AA, Coppel RL, Li MO, et al (2006). Anti‐mitochondrial antibodies and primary biliary cirrhosis in TGF‐β receptor II dominant‐negative mice. J Immunol 177, 1655–1660. [DOI] [PubMed] [Google Scholar]
- Osterreicher CH & Trauner M (2012). Animal models of biliary tract injury. Curr Opin Gastroenterol 28, 239–243. [DOI] [PubMed] [Google Scholar]
- Paine AJ & Andreakos E (2004). Activation of signalling pathways during hepatocyte isolation: relevance to toxicology in vitro. Toxicol In Vitro 18, 187–193. [DOI] [PubMed] [Google Scholar]
- Pang X, Hua X, Yang Q, Ding D, Che C, Cui L, Jia W, Bucheli P & Zhao L (2007). Inter‐species transplantation of gut microbiota from human to pigs. ISME J 1, 156–162. [DOI] [PubMed] [Google Scholar]
- Patlewicz G, Simon T, Goyak K, Phillips RD, Rowlands JC, Seidel SD & Becker RA (2013). Use and validation of HT/HC assays to support 21st century toxicity evaluations. Regul Toxicol Pharmacol 65, 259–268. [DOI] [PubMed] [Google Scholar]
- Pollheimer MJ, Fickert P & Stieger B (2014). Chronic cholestatic liver diseases: clues from histopathology for pathogenesis. Mol Aspects Med 37, 35–56. [DOI] [PubMed] [Google Scholar]
- Pound P & Bracken MB (2014). Is animal research sufficiently evidence based to be a cornerstone of biomedical research? BMJ 348, g3387. [DOI] [PubMed] [Google Scholar]
- Poupon R, Chazouilleres O & Poupon RE (2000). Chronic cholestatic diseases. J Hepatol 32, 129–140. [DOI] [PubMed] [Google Scholar]
- Ramaiahgari SC, den Braver MW, Herpers B, Terpstra V, Commandeur JN, van de Water B & Price LS (2014). A 3D in vitro model of differentiated HepG2 cell spheroids with improved liver‐like properties for repeated dose high‐throughput toxicity studies. Arch Toxicol 88, 1083–1095. [DOI] [PubMed] [Google Scholar]
- Ramirez T, Daneshian M, Kamp H, Bois FY, Clench MR, Coen M, Donley B, Fischer SM, Ekman DR, Fabian E, et al (2013). Metabolomics in toxicology and preclinical research. ALTEX 30, 209–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell DW (2009). Fifty years of advances in bile acid synthesis and metabolism. J Lipid Res 50(Suppl), S120–S125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz RE, Fleming HE, Khetani SR & Bhatia SN (2014). Pluripotent stem cell‐derived hepatocyte‐like cells. Biotechnol Adv 32, 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmi C, Mayo MJ, Bach N, Ishibashi H, Invernizzi P, Gish RG, Gordon SC, Wright HI, Zweiban B, Podda M, et al (2004). Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics, and environment. Gastroenterology 127, 485–492. [DOI] [PubMed] [Google Scholar]
- Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald‐Smith GP, Gao H, Hennessy L, et al (2013). Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA 110, 3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen NJ, Hansen RA, Morgan DR, Gangarosa LM, Ringel Y, Thiny MT, Russo MW & Sandler RS (2006). The burden of gastrointestinal and liver diseases, 2006. Am J Gastroenterol 101, 2128–2138. [DOI] [PubMed] [Google Scholar]
- Siller R, Greenhough S, Park IH & Sullivan GJ (2013). Modelling human disease with pluripotent stem cells. Curr Gene Ther 13, 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirenko O, Hesley J, Rusyn I & Cromwell EF (2014). High‐content assays for hepatotoxicity using induced pluripotent stem cell‐derived cells. Assay Drug Dev Technol 12, 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirica AE, Nathanson MH, Gores GJ & Larusso NF (2008). Pathobiology of biliary epithelia and cholangiocarcinoma: proceedings of the Henry M. and Lillian Stratton Basic Research Single‐Topic Conference. Hepatology 48, 2040–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Z, Ryerson D & Kemper JK (2013). Epigenomic regulation of bile acid metabolism: emerging role of transcriptional cofactors. Mol Cell Endocrinol 368, 59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer VA, Xu R & Bissell MJ (2007). Extracellular matrix, nuclear and chromatin structure, and gene expression in normal tissues and malignant tumors: a work in progress. Adv Cancer Res 97, 275–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter CM, Martin DI & Ward RL (2004). Germline epimutation of MLH1 in individuals with multiple cancers. Nat Genet 36, 497–501. [DOI] [PubMed] [Google Scholar]
- Thomas C, Pellicciari R, Pruzanski M, Auwerx J & Schoonjans K (2008). Targeting bile‐acid signalling for metabolic diseases. Nat Rev Drug Discov 7, 678–693. [DOI] [PubMed] [Google Scholar]
- Tolosa L, Carmona A, Castell JV, Gomez‐Lechon MJ & Donato MT (2014). High‐content screening of drug‐induced mitochondrial impairment in hepatic cells: effects of statins. Arch Toxicol 89, 1847–1860. [DOI] [PubMed] [Google Scholar]
- Torres DM, Williams CD & Harrison SA (2012). Features, diagnosis, and treatment of nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 10, 837–858. [DOI] [PubMed] [Google Scholar]
- Trottier J, Bialek A, Caron P, Straka RJ, Heathcote J, Milkiewicz P & Barbier O (2012). Metabolomic profiling of 17 bile acids in serum from patients with primary biliary cirrhosis and primary sclerosing cholangitis: a pilot study. Dig Liver Dis 44, 303–310. [DOI] [PubMed] [Google Scholar]
- Tsuneyama K, Moritoki Y, Kikuchi K & Nakanuma Y (2012). Pathological features of new animal models for primary biliary cirrhosis. Int J Hepatol 2012, 403954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno Y, Ambrosini YM, Moritoki Y, Ridgway WM & Gershwin ME (2010). Murine models of autoimmune cholangitis. Curr Opin Gastroenterol 26, 274–279. [DOI] [PubMed] [Google Scholar]
- Villeneuve DL, Crump D, Garcia‐Reyero N, Hecker M, Hutchinson TH, LaLone CA, Landesmann B, Lettieri T, Munn S, Nepelska M, et al (2014. a). Adverse outcome pathway (AOP) development I: strategies and principles. Toxicol Sci 142, 312–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeneuve DL, Crump D, Garcia‐Reyero N, Hecker M, Hutchinson TH, LaLone CA, Landesmann B, Lettieri T, Munn S, Nepelska M, et al (2014. b). Adverse outcome pathway development II: best practices. Toxicol Sci 142, 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinken M, Landesmann B, Goumenou M, Vinken S, Shah I, Jaeschke H, Willett C, Whelan M & Rogiers V (2013). Development of an adverse outcome pathway from drug‐mediated bile salt export pump inhibition to cholestatic liver injury. Toxicol Sci 136, 97–106. [DOI] [PubMed] [Google Scholar]
- Wang L, Wang J, Shi Y, Zhou X, Wang X, Li Z, Huang X, Han Z, Li T, Wang M, et al (2013). Identification of a primary biliary cirrhosis associated protein as lysosome‐associated membrane protein‐2. J Proteomics 91, 569–579. [DOI] [PubMed] [Google Scholar]
- Wang R, Salem M, Yousef IM, Tuchweber B, Lam P, Childs SJ, Helgason CD, Ackerley C, Phillips MJ & Ling V (2001). Targeted inactivation of sister of P‐glycoprotein gene (spgp) in mice results in nonprogressive but persistent intrahepatic cholestasis. Proc Natl Acad Sci USA 98, 2011–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T, et al (2006). Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439, 484–489. [DOI] [PubMed] [Google Scholar]
- Wei J, Qiu de K & Ma X (2009). Bile acids and insulin resistance: implications for treating nonalcoholic fatty liver disease. J Dig Dis 10, 85–90. [DOI] [PubMed] [Google Scholar]
- Willett C, Caverly Rae J, Goyak KO, Landesmann B, Minsavage G & Westmoreland C (2014). Pathway‐based toxicity: history, current approaches and liver fibrosis and steatosis as prototypes. ALTEX 31, 407–421. [DOI] [PubMed] [Google Scholar]
- Williams R (2006). Global challenges in liver disease. Hepatology 44, 521–526. [DOI] [PubMed] [Google Scholar]
- Woolbright BL, Dorko K, Antoine DJ, Clarke JI, Gholami P, Li F, Kumer SC, Schmitt TM, Forster J, Fan F, et al (2015). Bile acid‐induced necrosis in primary human hepatocytes and in patients with obstructive cholestasis. Toxicol Appl Pharmacol 283, 168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Spencer VA & Bissell MJ (2007). Extracellular matrix‐regulated gene expression requires cooperation of SWI/SNF and transcription factors. J Biol Chem 282, 14992–14999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Hong JY, Rockwell CE, Copple BL, Jaeschke H & Klaassen CD (2012). Effect of bile duct ligation on bile acid composition in mouse serum and liver. Liver Int 32, 58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zollner G & Trauner M (2009). Nuclear receptors as therapeutic targets in cholestatic liver diseases. Br J Pharmacol 156, 7–27. [DOI] [PMC free article] [PubMed] [Google Scholar]