

Abstract
Key points
RGK (Rad, Rem, Rem2, Gem/Kir) proteins are small monomeric G‐proteins implicated in various cellular functions and disease states. All RGK proteins potently inhibit voltage‐gated calcium (CaV) channels.
It is unclear whether RGK proteins are regulated by guanine nucleotide binding in a manner that conforms to a canonical G‐protein regulation paradigm.
We utilized strategic Rad and Rem mutants together with a range of functional readouts (CaV1.2 currents, Ca2+ transients, CaVβ binding) in heterologous cells and cardiomyocytes to determine whether the function of these RGK proteins is regulated in a canonical manner.
Our results demonstrate that Rad and Rem are non‐canonical G‐proteins with respect to the regulatory role of their guanine nucleotide binding domain in CaV1.2 channel regulation.
Our findings offer deepened insights into cellular mechanisms governing RGK regulation and function, and contribute towards our understanding of their pathophysiological roles.
Abstract
Rad and Rem are Ras‐like G‐proteins linked to diverse cardiovascular functions and pathophysiology. Understanding how Rad and Rem are regulated is important for deepened insights into their pathophysiological roles. As in other Ras‐like G‐proteins, Rad and Rem contain a conserved guanine‐nucleotide binding domain (G‐domain). Canonically, G‐domains are key control modules, functioning as nucleotide‐regulated switches of G‐protein activity. Whether Rad and Rem G‐domains conform to this canonical paradigm is ambiguous. Here, we used multiple functional measurements in HEK293 cells and cardiomyocytes (CaV1.2 currents, Ca2+ transients, CaVβ binding) as biosensors to probe the role of the G‐domain in regulation of Rad and Rem function. We utilized RadS105N and RemT94N, which are the cognate mutants to RasS17N, a dominant‐negative variant of Ras that displays decreased nucleotide binding affinity. In HEK293 cells, over‐expression of either RadS105N or RemT94N strongly inhibited reconstituted CaV1.2 currents to the same extent as their wild‐type (wt) counterparts, contrasting with reports that RadS105N is functionally inert in HEK293 cells. Adenovirus‐mediated expression of either wt Rad or RadS105N in cardiomyocytes dramatically blocked L‐type calcium current (I Ca,L) and inhibited Ca2+‐induced Ca2+ release, contradicting reports that RadS105N acts as a dominant negative in heart. By contrast, RemT94N was significantly less effective than wt Rem at inhibiting I Ca,L and Ca2+ transients in cardiomyocytes. FRET analyses in cardiomyocytes revealed that both RadS105N and RemT94N had moderately reduced binding affinity for CaVβs relative to their wt counterparts. The results indicate Rad and Rem are non‐canonical G‐proteins with respect to the regulatory role of their G‐domain in CaV1.2 regulation.
Abbreviations
- AFU
acceptor fluorescence units
- CaV
voltage‐dependent calcium channel
- CaV1.2
voltage‐gated L‐type calcium channel
- CaVβ
voltage‐dependent calcium channel auxiliary β subunit
- CFP
cyan fluorescent protein
- FRET
fluorescent resonance energy transfer
- GAP
GTPase‐activating protein
- GEF
guanine nucleotide exchange factor
- GNBP
guanine nucleotide binding pocket
- IBa,L
barium currents through L‐type calcium channel
- ICa
calcium current
- ICa,L
L‐type calcium current
- Ipeak
peak current density
- RGK
Rad, Rem, Rem2, Gem/Kir
- wt
wild‐type
- YFP
yellow fluorescent protein
Introduction
RGK (Rad, Rem, Rem2, Gem/Kir) proteins are a four‐member sub‐group of the superfamily of Ras‐like monomeric G‐proteins (Colicelli, 2004; Flynn & Zamponi, 2010; Yang & Colecraft, 2013). RGKs have distinctive tissue distributions, with Rad and Rem expressed in cardiac, skeletal and smooth muscle (Reynet & Kahn, 1993; Finlin & Andres, 1997; Finlin et al. 2003; Chang et al. 2007; Wang et al. 2010). Several studies suggest Rad and Rem play important roles in the cardiovascular system: Rad expression is decreased in failing human heart, and Rad knockout mice display increased susceptibility to transverse aortic constriction‐induced cardiac hypertrophy (Chang et al. 2007); Rad expression is elevated in injured blood vessels where it suppresses neointimal formation by preventing vascular smooth muscle cell migration (Fu et al. 2005); Rem expression in ventricular myocytes is down‐regulated during conditions mimicking inflammation (Finlin & Andres, 1997); and Rem knockout mice display a moderate increase in L‐type (CaV1.2) Ca2+ currents (I Ca,L) in cardiomyocytes (Magyar et al. 2012). RGKs have been linked to regulation of cell cytoskeleton dynamics, and interact with important kinases including Ca2+ and calmodulin‐dependent protein kinase II (CaMKII) and Rho‐dependent kinase (ROK) (Moyers et al. 1997; Ward et al. 2002; Chang et al. 2007; Correll et al. 2008). Finally, RGKs are the most potent known intracellular inhibitors of high‐voltage‐activated CaV1 and CaV2 family calcium channels (Beguin et al. 2001; Finlin et al. 2003; Chen et al. 2005; Flynn & Zamponi, 2010; Yang et al. 2010; Yang & Colecraft, 2013). Despite their important biological functions, it is unclear how RGKs are regulated in cells. Understanding their regulatory mechanism(s) is critical for deepened insights into their physiological roles and how their dysfunction leads to disease.
All small G‐proteins contain a conserved guanine nucleotide binding domain (G‐domain) which canonically acts as a switch to facilitate cycling between an active GTP‐bound or inactive GDP‐bound state (Sprang, 1997; Colicelli, 2004). Transitions between active and inactive states are catalysed by guanine nucleotide exchange factors (GEFs) and GTPase‐activating proteins (GAPs), respectively (Sprang, 1997; Colicelli, 2004). In the prototypical small G‐protein, Ras, GTP binding to the guanine nucleotide binding pocket (GNBP) stabilizes two disordered regions in the G‐domain referred to as switch I and switch II, respectively. Distinct mutations that impair various facets of the canonical regulatory paradigm in Ras and other small G‐proteins are leading causes of cancer and heart disease, emphasizing the importance of this regulatory mechanism (Sprang, 1997; Colicelli, 2004; Loirand et al. 2013). In Ras, a serine to asparagine mutation at residue 17 disrupts the GNBP. Consequently, RasS17N displays a markedly reduced affinity for GTP (Feig & Cooper, 1988 a), loses the capacity to bind downstream effectors, and acts as a dominant negative in situ due to increased avidity for GEF (Feig, 1999). Introducing mutations cognate to RasS17N is a widely used method to inactivate guanine nucleotide regulation of G‐domains, and to generate putative dominant negative variants of diverse small G‐proteins (Feig, 1999).
Similar to Ras, RGKs possess a G‐domain that binds GTP and GDP (Reynet & Kahn, 1993; Maguire et al. 1994; Finlin et al. 2000; Opatowsky et al. 2006; Yanuar et al. 2006; Splingard et al. 2007) and displays intrinsic GTPase activity (Zhu et al. 1995; Finlin et al. 2000; Splingard et al. 2007; Sasson et al. 2011). Furthermore, biochemical studies confirm that point mutations cognate to RasS17N in the GNBP of the RGK proteins Rad and Gem significantly disrupt guanine nucleotide binding (Sasson et al. 2011). Despite these manifestations of classical small G‐proteins, RGKs display certain divergent features that raise questions as to whether they conform to a canonical guanine nucleotide‐regulated switch paradigm. Chief among these are: (1) structural comparisons between GTP‐ and GDP‐bound RGKs do not reveal the classical conformational change that occurs in the switch I and II regions of Ras (Vetter & Wittinghofer, 2001; Sasson et al. 2011); and (2) the switch I and II regions are divergent among RGKs, suggesting they do not participate in functions that are conserved among all RGKs, such as inhibition of CaV1/CaV2 channels (Beguin et al. 2001; Finlin et al. 2003; Chen et al. 2005; Yang & Colecraft, 2013). Given these reasons, it is quite surprising then that some studies which utilized the RGK cognate mutants to RasS17N (RadS105N, RemT94N, Rem2S129N, GemS89N) suggested that the functional states of RGKs are regulated by guanine nucleotides in a manner analogous to Ras (Ward et al. 2004; Beguin et al. 2005 a,b; Yada et al. 2007; Xu et al. 2010). This has been most definitively demonstrated for Rad where RadS105N was found ineffective at blocking recombinant CaV1.2 channels reconstituted in human embryonic kidney (HEK293) cells, in contrast to the strong inhibition observed with wild‐type (wt) Rad in this system (Yada et al. 2007). Moreover, whereas over‐expression of wt Rad in cardiomyocytes strongly inhibited endogenous CaV1.2, RadS105N displayed a dominant negative phenotype and significantly increased I Ca,L (Yada et al. 2007). These results suggest that Rad, and perhaps other RGKs, may have co‐opted the guanine nucleotide‐regulated switch mechanism and customized it for their functional regulation since their switch I and II regions appear uninvolved. If confirmed, this would be an important phenomenon to understand as both a new mechanism for guanine nucleotide regulation of small G‐proteins as well as for deepened insights into pathophysiological roles of RGK proteins. By contrast with Rad, similar studies with other RGKs suggest that they do not conform to a canonical guanine nucleotide‐regulated switch mechanism. For example, in sympathetic neurons, Rem2S129N blocked endogenous N‐type (CaV2.2) channels just as effectively as wt Rem2 (Chen et al. 2005). It is unclear whether these variances reflect genuine distinctions among RGK proteins or are related to the different cellular contexts.
Overall, there remain serious questions about the putative role of guanine nucleotide binding in the functional regulation of RGK proteins. Resolving current ambiguities requires comparative functional studies among distinct RGKs across different cellular contexts. Here, we investigated Rad and Rem because of precedent studies and because they are both basally expressed in heart cells where they are believed to exert a tonic inhibitory effect on CaV1.2 channels (Wang et al. 2010; Magyar et al. 2012; Manning et al. 2013). We used RadS105N and RemT94N as tools to evaluate the necessity of an intact GNBP in the regulation of Rad and Rem function, respectively. We first compare functional effects of wt and mutant RGKs on I Ca across two cellular contexts: recombinant CaV1.2 channels reconstituted in HEK293 cells, and endogenous CaV1.2 in adult rat ventricular myocytes. In cardiomyocytes, we further analyse the impact on Ca2+ transients and differences in binding affinity to CaVβ to gain new insights into whether an intact GNBP is necessary for Rad and Rem function in the cardiac environment. The results reveal that Rad and Rem are non‐canonical G‐proteins because their inhibition of CaV1.2 channels does not require an intact GNBP. Nevertheless, we observed that disrupting the GNBP led to cell‐context‐dependent reductions in Rad and Rem protein stability and diminished potency in inhibiting CaV1.2 channels.
Methods
Molecular biology
To generate bicistronic constructs, an internal ribosomal entry site (ires)‐mCherry cassette was generated by overlap extension PCR and cloned into pcDNA3 vector using EcoRI and HindIII sites. Mouse Rad (NM_019662) or Rem (NM_009047) were ligated upstream of the ires sequence using AflII and EcoRI sites to generate Rad‐ires‐mCherry or Rem‐ires‐mCherry, respectively. Cyan fluorescent protein (CFP)‐tagged Rad and Rem constructs were generated by PCR amplification and cloned into pcDNA4.1 mammalian expression vectors as described previously (Yang et al. 2010). Point mutations in Rad and Rem were generated using a QuikChange Site‐Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA). All PCR products were verified by sequencing. The linker residues for CFP‐128‐YFP and CFP‐50‐YFP plasmids were derived from the unstructured C‐terminus of the auxiliary CaVβ3 subunit (NM_012828.2), where YFP is yellow fluorescent protein.
Generation of adenoviruses
CFP‐tagged RGK adenoviruses (Ad) were generated using the AdEasy XL system (Stratagene) as described previously (Xu et al. 2010). Ad RGK‐ires‐mCherry adenoviral vectors were generated using the Adeno‐X CMV vector kit (Clontech, Mountain View, CA, USA) according to the manufacturer's instructions. Adenoviruses were purified using a caesium chloride discontinuous gradient as described previously (Colecraft et al. 2002).
Cell culture and transfection
Low‐passage‐number HEK293 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 μg ml−1 penicillin–streptomycin. HEK293 cells cultured in 35 mm tissue culture dishes were transiently transfected with CaV1.2 α1C (6 μg), CaV β2a (4 μg), T antigen (2 μg), and the appropriate RGK construct (4 μg), using the calcium‐phosphate precipitation method. Cells were washed with serum‐free DMEM after 6–8 h and maintained in supplemented DMEM. Proteasome inhibitor lactacystin (Sigma‐Aldrich, St. Louis, MO, USA) was added to HEK293 cells at a final concentration of 10 μm. Cells remained at 37°C in 5% CO2 humidified incubators for 24–48 h before patch experiments.
Myocyte isolation and culture
Primary cultures of adult rat heart ventricular cells were prepared as previously described (Xu & Colecraft, 2009; Subramanyam et al. 2013). Procedures were in accordance with the guidelines of the Columbia University Animal Care and Use Committee. Adult male Sprague–Dawley rats (Harlan) were killed with halothane, and ventricular myocytes isolated by enzymatic digestion with 1.7 mg Liberase enzyme mix (Roche) using a Langendorff perfusion apparatus. Myocytes were cultured on laminin‐coated glass coverslips or MatTek dishes and maintained in supplemented Medium 199. Cells were infected with 10–20 μl of viral stock in a final volume of 1–2 ml.
Electrophysiology
Whole‐cell recordings were carried out at room temperature on HEK293 cells 48–72 h after transfection using an EPC‐8 patch clamp amplifier controlled by PULSE software (HEKA Elektronik, Lambrecht/Pfalz, Germany) as previously described (Yang et al. 2010, 2012). Transfected cells were split and cultured on fibronectin‐coated 9 mm × 9 mm glass coverslips (Bellco Glass, Inc., Vineland, NJ, USA) 24 h prior to patching. Micropipettes were fashioned from 1.5 mm thin‐walled glass (World Precision Instruments, Sarasota, FL, USA) using a P97 microelectrode puller (Sutter Instruments, Novato, CA, USA). Pipette resistance was typically between 1.5 and 2.5 MΩ when filled with internal solution containing (in mM): 135 caesium methanesulfonate, 5 caesium chloride, 5 EGTA, 1 MgCl2, 4 MgATP added fresh, 10 Hepes (pH 7.5). External solution contained (mm): 140 tetraethylammonium‐methanesulfonate, 5 BaCl2, and 10 Hepes (pH 7.4). Leak and capacitive currents were subtracted using a P/8 protocol. For experiments using non‐hydrolysable analogues of guanine nucleotides, 2 mm of either GDP‐β‐S or GTP‐γ‐S were included in the patch pipette.
Whole‐cell recordings of cultured rat ventricular myocytes were conducted as previously described (Colecraft et al. 2002; Subramanyam et al. 2013). Patch pipettes typically had a resistance between 1 and 2 MΩ when filled with internal solution containing (in mm): 150 caesium methanesulfonate, 10 EGTA, 5 CsCl, 1 MgCl2, 4 MgATP added fresh, 10 Hepes (pH 7.3). The larger pipette tips minimized series resistance errors and enhanced voltage and space clamp of cardiomyocytes. Nevertheless, the ample transverse tubules present in adult rat cardiomyocytes had a filtering effect on tail currents which were consequently not analysed in this study. Cells were perfused with normal Tyrode external solution containing (in mm): 138 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 0.33 NaH2PO4, 10 Hepes (pH 7.4) during gigaseal formation. Following the initial break‐in to the whole‐cell configuration, external recording solution containing (in mm): 155 N‐methy‐d‐glucamine aspartate, 10 4‐aminopyridine, 1 MgCl2, 5 BaCl2, 10 Hepes (pH 7.4) was perfused on the cells during current recordings unless otherwise specified. Leak and capacitive currents were subtracted using a P/8 protocol.
Western blotting
Western blots were performed as previously described (Subramanyam et al. 2013). Briefly, proteins from whole‐cell lysates were resolved on a 4–12% Bis·Tris gel and transferred onto a nitrocellulose membrane. Membranes were blocked with 5% milk for 1 h and then incubated with primary antibody at 4°C overnight. Membranes were washed and incubated with horseradish peroxidase‐conjugated secondary antibody for 1 h at room temperature. Protein bands were detected by chemiluminescence on a gel imager. Primary antibodies used (dilution, vendor): Rad goat polyclonal (1:200; Santa Cruz Biotechnology, Santa Cruz, CA, USA), Rem mouse polyclonal (1:200; Santa Cruz Biotechnology), glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) rabbit polyclonal (1:200; Santa Cruz Biotechnology), mCherry rabbit polyclonal (1:400; Biovision), GFP rabbit polyclonal (1:5000, Invitrogen, Carlsbad, CA, USA).
Confocal imaging
Confocal images were obtained with a Leica SP2 confocal microscope using a ×63 oil immersion objective (NA 1.4). Laser lines and detector settings used were: CFP, 405 nm diode laser, 465–502 nm detection; mCherry, 543 nm HeNe, 550–600 nm detection.
Calcium transient imaging
Primary cardiomyocytes cultured on MatTek dishes were loaded with the acetoxymethylester form of rhod‐2, rhod‐2 AM (5 μm with 0.05% Pluronic F127 detergent) in Tyrode solution containing (in mm): 138 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 0.33 NaH2PO4, 10 Hepes for 15 min in a 37°C incubator. Cells were then washed and maintained in Tyrode solution. Confocal line scan imaging was conducted using a Leica SP2 microscope equipped with a ×63 oil immersion, NA 1.4, objective. Rhod‐2 was excited at 543 nm and emission detected between 550 and 600 nm. Cells were paced at 0.7 Hz using a Myopacer EP (IonOptix, Westwood, MA, USA). Confocal line scan frequency was set at 400 Hz and cells were scanned for 20 s. For experiments involving caffeine, Tyrode solution containing 10 mm caffeine was perfused onto the cells during the line scan. Confocal line scan signals were analysed using ImageJ (NIH, Bethesda, MD, USA) and MATLAB (MathWorks, Natick, MA, USA).
Fluorescent resonance energy transfer (FRET) imaging
We adapted a previously described 3‐cube FRET approach based on CFP‐ (donor) and YFP‐tagged (acceptor) molecules to probe specific protein–protein interactions in live cells (Erickson et al. 2003; Chen et al. 2006, 2007). Cells were imaged using a ×40 oil objective (NA 1.3) on a Nikon Eclipse Ti‐U inverted microscope and fluorescence images acquired using an electron‐multiplying gain CCD camera (QuantEM:512SC, Photometrics). Excitation wavelengths of 440 nm (CFP and FRET cubes) and 500 nm (YFP cube) were applied using a random access monochromator with a 75 watt xenon arc lamp housing (PTI DeltaRam X, Photon Technology International). Filter cubes used were (dichroic, emission): DD (455DCLP, D480/30M); AA (525DRLP, 530EFLP); DA (455DRLP, 535DF25). Cross‐talk parameters were determined by imaging cells expressing either donor (CFP) or acceptor (YFP) fluorescent proteins alone. FRET efficiency (E) and relative donor (D) and acceptor (A) concentrations were calculated as described previously (Chen et al. 2007). Relative K d and E max values were calculated assuming a bi‐molecular interaction and performing a least‐squares fit of the data as described before (Chen et al. 2007).
Structural representation
Ribbon and surface representations of crystal structures were generated and rendered using a PyMOL Molecular Graphics System (Schrödinger, LLC, New York, NY, USA). Structures used were RasS17N (PDB: 3LO5), Rem (PDB: 2NZJ) and Rad (PDB: 2DPX).
Data and statistical analyses
Data were analysed off‐line using PulseFit (HEKA), Microsoft Excel, MATLAB, and Origin software. Statistical analyses were performed in Origin and Microsoft Excel using built‐in functions. Pooled data are presented as means ± SEM, and P values were calculated using Student's two‐tailed unpaired t test. Comparisons involving more than two groups were analysed using one‐way ANOVA with Bonferroni post hoc analyses. P < 0.05 was considered significant.
Results
Justification for using mutations cognate to RasS17N to study the regulatory role of RGK G‐domains
Random mutagenesis studies identified RasS17N as a mutant Ras protein that could no longer interact with downstream effector proteins, and exerted a dominant negative effect in situ (Feig & Cooper, 1988 a). Crystal structures revealed that Ser17 is important for Mg2+ binding to the GNBP of Ras (Vetter & Wittinghofer, 2001; Nassar et al. 2010). Accordingly, RasS17N displays a reduced affinity for guanine nucleotides, a factor that contributes prominently to both the loss of interaction with effector proteins and the dominant negative properties (Feig & Cooper, 1988 a,b). The use of mutations cognate to RasS17N has been a popular and effective tool to generate loss‐of‐function and/or dominant negative variants of several Ras‐like G‐proteins (Feig, 1999). The effectiveness of such mutations can provide useful insights into whether G‐domains of other monomeric G‐proteins function in a manner analogous to Ras.
Crystal structures of RGK proteins indicate that residues corresponding to Ser17 in Ras play analogous roles in coordinating Mg2+ binding in the GNBP (Opatowsky et al. 2006; Yanuar et al. 2006; Splingard et al. 2007; Sasson et al. 2011; Reymond et al. 2012) (Fig. 1). Accordingly, biochemical studies confirm that mutating these residues to Asn in RGKs leads to markedly reduced affinity for guanine nucleotides (Sasson et al. 2011), indicating a compromised GNBP. In particular, RadS105N displays over 200‐fold reduced affinity for GDP compared to wt, with no measurable binding to GTP (Sasson et al. 2011). If RGKs with mutations cognate to RasS17N display loss‐of‐function characteristics reminiscent of Ras, this can be a powerful demonstration of canonical G‐protein regulation properties. Currently, there is only one reported GAP for an RGK protein, nm23 (also referred to as nucleoside diphosphate kinase), which was identified as a GAP for Rad (Moyers et al. 1998; Zhu et al. 1999). Thus far, no traditional GEF has been discovered for any RGK protein. If RGKs with mutations cognate to RasS17N display dominant negative properties in situ, this would suggest the presence of a conventional GEF in the particular cellular context. These considerations justify and motivate the use of RadS105N and RemT94N to determine whether the functional properties of these RGK proteins are regulated in a manner analogous to Ras.
Figure 1. Structural representations of RasS17N, Rad and Rem .
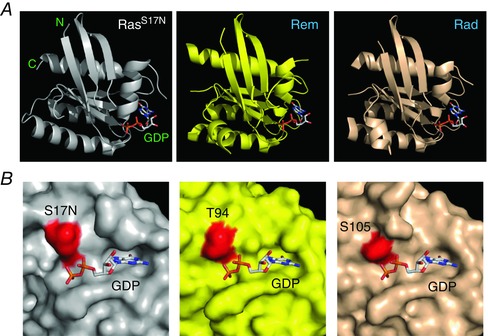
A, from left to right, ribbon structures of RasS17N (PDB: 3LO5), Rem (PDB: 2NZJ) and Rad (PDB: 2DPX), all bound to GDP. B, close‐up surface representation of RasS17N (left) with the S17N residue coloured in red. Rem (middle) and Rad (right) are shown in the same orientation with the homologous residues T94 and S105 shown in red.
Impact of RadS105N and RemT94N on CaV1.2 reconstituted in HEK293 cells
We first determined the impact of RadS105N and RemT94N on recombinant CaV1.2 (α1C + β2a) channels reconstituted in HEK293 cells in comparison to their wt counterparts (Fig. 2). To ensure that cells selected for whole‐cell recordings expressed RGK proteins, we used bicistronic internal ribosomal entry site (ires) vectors that separately expressed RGK and mCherry proteins from the same mRNA (Fig. 2 A). Hence, mCherry fluorescence was used to select transfected cells for electrophysiological analyses. Cells expressing α1C + β2a gave rise to large whole‐cell Ba2+ currents (I Ba,L) that peaked with a 0 mV test pulse (Fig. 2 B and C; I peak,0mV = −52.6 ± 5.3 pA pF−1, n = 36). As expected, co‐expressing wt Rad with CaV1.2 led to a dramatically inhibited I Ba,L (Fig. 2 D and E; I peak,0mV = −6.8 ± 1.7 pA pF−1, n = 14). Cells expressing RadS105N also displayed a depressed I Ba,L, although the magnitude of inhibition was significantly less than observed with wt Rad (Fig. 2 D and E; I peak,0mV = −16.5 ± 2.3 pA pF−1, n = 18; P < 0.05 compared to wt Rad). We observed a similar trend with wt Rem and RemT94N: cells co‐expressing Rem displayed a deeply inhibited I Ba,L (Fig. 2 F and G; I peak,0mV = −1.4 ± 0.5 pA pF−1, n = 7), whereas RemT94N caused a significantly less potent, but still strong, inhibition (Fig. 2 F and G; I peak,0mV = −11.7 ± 1.7 pA pF−1, n = 9, P < 0.05 compared to wt Rem). The robust inhibition of I Ba,L by wt Rad and Rem persisted when either 2 mm GDP‐β‐S or 2 mm GTP‐γ‐S was included in the patch pipette (not shown). To accurately interpret the weaker effects of RadS105N and RemT94N on I Ba,L, we determined whether these mutations had any effect on protein expression. Western blot analyses demonstrated a ∼70% decreased protein expression levels for RadS105N and RemT94N compared to wt Rad and Rem, respectively (Fig. 2 H and I). This result suggested that the weaker inhibition of I Ba,L seen with RadS105N and RemT94N could be due to reduced protein expression, given that inhibitory effects of RGKs on CaV channels are dose dependent (Seu & Pitt, 2006).
Figure 2. Diminished impact of RadS105N/RemT94N compared to wt Rad/Rem on reconstituted CaV1.2 channels .
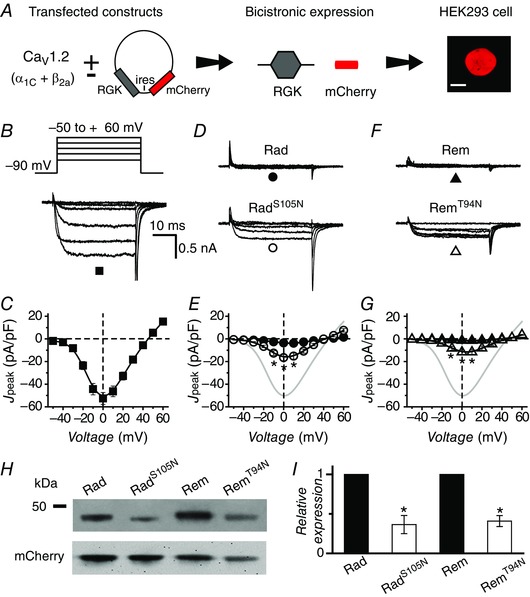
A, schematic diagram of experimental protocol: CaV1.2 subunits are transiently transfected into HEK293 cells with or without bicistronic RGK plasmid vectors. Scale bar is 5 μm. B, representative traces from control cells expressing α1C + β2a. C, population peak current density vs. test pulse voltage (J peak−V) relationship for control cells (▪, n = 36). D, representative traces for CaV1.2 channels co‐expressed with either wt Rad (top) or RadS105N (bottom). E, population J peak−V for channels co‐expressed with wt Rad (●, n = 14) or RadS105N (○, n = 18). Data for control cells (grey trace) are reproduced for visual comparison. F and G, data for cells co‐expressing CaV1.2 and either wt Rem (▲, n = 7) or RemT94N (△, n = 9), same format as D and E. H, representative Western blots. I, normalized RGK protein expression obtained by densitometric analyses. RGK protein bands were normalized to mCherry as an internal control; n = 7 for Rad, n = 9 for Rem. *P < 0.005, Student's unpaired t test. Data are presented as means ± SEM.
We took two approaches to determine whether the weaker effects of RadS105N and RemT94N on I Ba,L in HEK293 cells could be entirely explained by the observed decrease in protein expression. First, we hypothesized that RadS105N and RemT94N would be stabilized by fusing them to the highly stable cyan fluorescent protein (CFP) (Fig. 3 A). Indeed, Western blots indicated CFP‐RadS105N had similar expression levels to CFP‐Rad (Fig. 3 B and C). The expression level of CFP‐RemT94N was also significantly boosted although it still showed a 30% reduction in expression compared to CFP‐Rem (Fig. 3 B and C). With the increased protein expression, both CFP‐RadS105N and CFP‐RemT94N blocked I Ba,L to the same extent as wt CFP‐Rad and CFP‐Rem, respectively (Fig. 3 D–G).
Figure 3. Fusing RadS105N and RemT94N to CFP rescues protein expression and restores wt inhibition of reconstituted CaV1.2A .
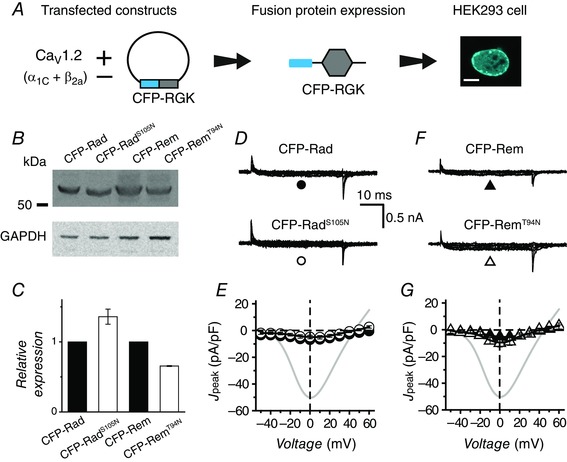
A, schematic diagram of experimental protocol: CaV1.2 subunits are transiently transfected into HEK293 cells with or without CFP‐fused RGK proteins. Scale bar is 5 μm. B, representative Western blots. C, normalized CFP‐tagged RGK protein expression obtained from densitometric analyses. RGK protein bands were normalized to GAPDH, n = 3 for each. D, representative traces for CaV1.2 channels co‐expressed with either wt CFP‐Rad (top) or CFP‐RadS105N (bottom). E, population J peak−V for channels co‐expressed with wt CFP‐Rad (●, n = 12) or CFP‐RadS105N (○, n = 9). Data for control cells (grey trace) are reproduced for visual comparison. F and G, data for cells co‐expressing CaV1.2 and either wt CFP‐Rem (▲, n = 8) or CFP‐RemT94N (△, n = 7), same format as D and E. Data are presented as means ± SEM.
In the second approach, we used the proteasomal inhibitor, lactacystin, to slow the rate of degradation of untagged RadS105N and RemT94N (Fenteany et al. 1995). Treatment with 10 μm lactacystin elevated expression of all RGK proteins in HEK293 cells, and increased RadS105N/RemT94N expression levels similar to their wt counterparts (Fig. 4 A and B). Most importantly, in cells treated with lactacystin, both RadS105N and RemT94N caused a deeper inhibition of I Ba,L (I peak,0mV = −8.3 ± 1.4 pA pF−1, n = 5 for RadS105N, and I peak,0mV = −5.3 ± 1.3 pA pF−1, n = 5 for RemT94N) (Fig. 4 C and D). Lactacystin had no effect on I Ba,L in control cells expressing α1C + β2a alone (Fig. 4 E).
Figure 4. Rescue of RadS105N and RemT94N expression and inhibition of ICa,L with lactacystin .
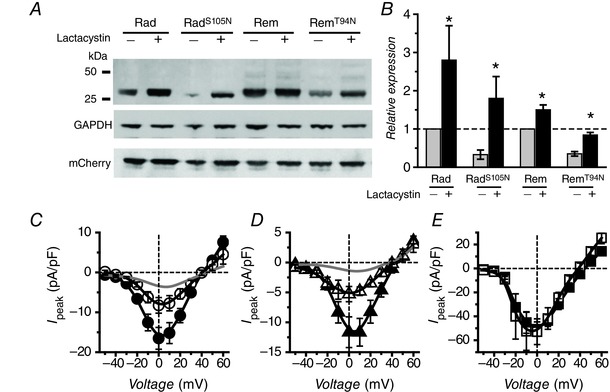
A, representative Western blots. B, normalized RGK protein expression in the absence and presence of 10 μm lactacystin. C, population J peak–V relationships for α1C + β2a + RadS105N in the absence (●, n = 18) or presence (○, n = 7) of lactacystin. D, population J peak–V relationships for α1C + β2a + RemT94N in the absence (▲, n = 9) or presence (△, n = 5) of lactacystin. Grey lines in C and D represent data obtained with co‐expression of wt Rad and Rem, respectively. E, population J peak–V relationships for α1C + β2a channels without (▪, n = 5) or with (□, n = 7) lactacystin.
Overall, these results indicate that in HEK293 cells, RadS105N and RemT94N show a weakened ability to block CaV1.2 channels solely due to a decrease in protein expression. Once expression of these proteins is normalized, they block reconstituted CaV1.2 just as effectively as wt Rad and Rem, respectively. These data disagree with a previous report that RadS105N is ineffective at blocking CaV1.2 reconstituted in HEK293 cells (Yada et al. 2007).
Impact of RadS105N and RemT94N on endogenous CaV1.2 in adult cardiomyocytes
It was possible that the cellular context could significantly impact the prevalence of a canonical switch mechanism in RGKs owing to potential differential expression of GEFs and GAPs in distinct cell types. Both Rad and Rem are expressed in heart cells where knockdown/knockout experiments suggest they exert a tonic inhibitory effect on I Ca,L (Reynet & Kahn, 1993; Finlin & Andres, 1997; Wang et al. 2010; Magyar et al. 2012; Manning et al. 2013). Accordingly, we next examined whether an intact GNBP is essential for Rad and Rem function in adult rat ventricular cardiomyocytes. We generated bicistronic adenoviruses encoding Rad or Rem and mCherry (Fig. 5 A). Unlike our observations in HEK293 cells, Western blots indicated protein levels of RemT94N and wt Rem were equivalent (Fig. 5 B). Control cardiomyocytes infected with GFP adenovirus (Ad‐GFP) yielded robust I Ba,L that peaked with a −10 mV test pulse (Fig. 5 C and D; I peak,‐10mV = −19.9 ± 1.7 pA pF−1, n = 18). Both wt Rad and RadS105N produced a deep inhibition of I Ba,L (I peak,‐10mV = −1.9 ± 1.4 pA pF−1, n = 10 for wt Rad; and I peak,‐10mV = −3.2 ± 0.4 pA pF−1, n = 15 for RadS105N) (Fig. 5 E and F). By contrast, RemT94N was clearly less effective in blocking I Ba,L in cardiomyocytes compared to wt Rem (Fig. 5 G and H; I peak,‐10mV = −8.6 ± 0.9 pA pF−1, n = 13 for RemT94N and I peak,‐10mV = −1.6 ± 0.3 pA pF−1, n = 9 for wt Rem). Given the similar expression levels of RemT94N and wt Rem (Fig. 5 B) the weaker effect of RemT94N on I Ba,L compared to wt Rem in heart cells was not due to reduced protein expression. In agreement with this, we observed overall similar responses when cardiomyocytes were infected with CFP‐tagged RGK proteins (Fig. 6 A and B). Both wt CFP‐Rad and CFP‐RadS105N strongly inhibited I Ba,L, although the effect of CFP‐RadS105N was slightly weaker (I peak,‐10mV = −5.2 ± 0.6 pA pF−1, n = 18 for CFP‐RadS105N compared to I peak,‐10mV = −2.2 ± 0.2 pA pF−1, n = 14 for wt CFP‐Rad) (Fig. 6 C and D). While both CFP‐Rem and CFP‐RemT94N inhibited I Ba,L, the effect of the mutant (I peak,‐10mV = −9.8 ± 0.6 pA pF−1, n = 6) was markedly weaker compared to wt (I peak,‐10mV = −2.2 ± 0.3 pA pF−1, n = 6) (Fig. 6 E and F).
Figure 5. Impact of RadS105N/RemT94N on endogenous CaV1.2 in cardiomyocytes compared to wt Rad/Rem .
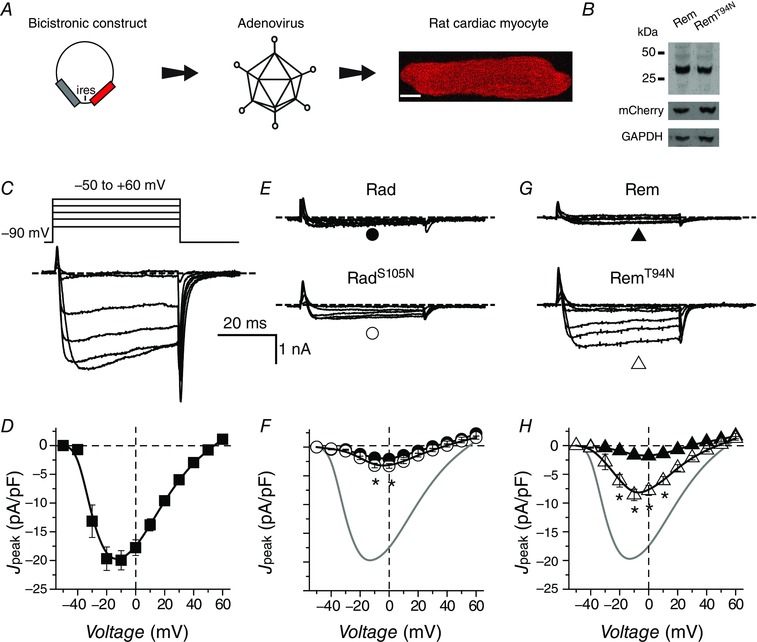
A, experimental protocol: adult rat cardiomyocytes are infected with adenoviruses encoding RGK and mCherry in a bicistronic vector. Scale bar is 10 μm. B, representative Western blot showing Rem and RemT94N expression in adenovirus‐infected adult rat cardiac myocytes. Experiment was reproduced 4 times. C, representative traces from adult rat cardiomyocyte expressing GFP. D, population J peak−V for control cardiomyocytes expressing GFP (▪, n = 18). E, representative I Ba traces for cardiomyocytes expressing either wt Rad (top) or RadS105N (bottom). F, population J peak−V for cardiomyocytes expressing either wt Rad (●, n = 10) or RadS105N (○, n = 15). G and H, data for cardiomyocytes expressing either CFP‐Rem (▲, n = 9) or CFP‐RemT94N (△, n = 14), same format as E and F. Data are presented as means ± SEM. *P < 0.05, Student's unpaired t test.
Figure 6. Impact of CFP‐RadS105N/CFP‐RemT94N on IBa,L in cardiomyocytes compared to wt CFP‐Rad/CFP‐Rem .
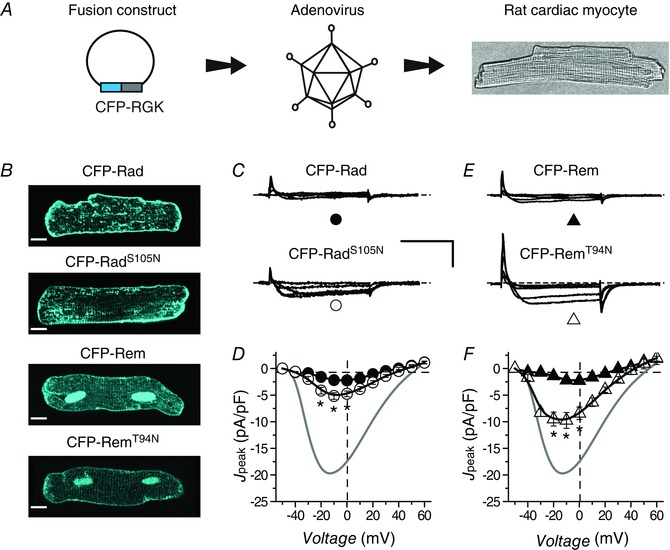
A, experimental protocol: adult rat cardiomyocytes are infected with adenoviruses encoding CFP‐tagged RGK proteins. Scale bar is 10 μm. B, exemplar confocal images showing sub‐cellular localization of CFP‐tagged RGK proteins. C, representative traces for cardiomyocytes expressing either wt CFP‐Rad (top) or CFP‐RadS105N (bottom). D, population J peak−V for cardiomyocytes expressing either wt CFP‐Rad (●, n = 14) or CFP‐RadS105N (○, n = 18). Data for control cardiomyocytes (grey trace) are reproduced for visual comparison. E and F, data for cardiomyocytes expressing either CFP‐Rem (▲, n = 6) or CFP‐RemT94N (△, n = 6), same format as C and D. Data are presented as means ± SEM. P < 0.05, Student's unpaired t test.
The finding that both wt Rad and RadS105N markedly inhibit CaV1.2 channels in cardiomyocytes contradicts reports that RadS105N displays a dominant negative effect in heart cells as reported by an increase in I Ca,L (Yada et al. 2007). The discrepant results were not due to differences in charge carrier as we found that RadS105N still markedly inhibited I Ca,L in adult rat ventricular cardiomyocytes when we used 2 mm Ca2+ as the charge carrier (not shown). With respect to Rem, the data indicate that the guanine nucleotide binding status of its G‐domain may be important for its ability to fully block CaV1.2 in cardiomyocytes, consistent with our previous observation in guinea pig ventricular myocytes (Xu et al. 2010).
Impact of RadS105N and RemT94N on Ca2+‐induced Ca2+ release
To obtain a broader perspective on the requirement for an intact GNBP on Rad/Rem function in heart cells, we assessed the impact of RadS105N and RemT94N on Ca2+‐induced Ca2+ release in adult rat cardiomyocytes. Control cardiomyocytes loaded with rhod‐2 AM responded to 0.7 Hz field stimulation with robust Ca2+ transients (Fig. 7 A and B; ΔF/F 0 = 8.3 ± 1.3, n = 12, where ΔF = F − F 0). Over‐expression of all the RGKs except CFP‐RemT94N led to significant decreases in Ca2+ transient amplitude, albeit with quantitative differences in the extent of inhibition (Fig. 7 A and B). The decreased Ca2+ transient amplitude was not due to insufficient Ca2+ in the sarcoplasmic reticulum Ca2+ stores, as indicated by caffeine release experiments (not shown). CFP‐Rem produced the deepest inhibition (ΔF/F 0 = 2.0 ± 0.5, n = 22), with a large fraction (43%) of cells non‐responsive to field stimulation (Fig. 7 C). Further insights were obtained by plotting relative Ca2+ transient amplitudes as a function of CFP fluorescence (which provides an index of RGK protein expression levels). CFP‐Rad and CFP‐RadS105N displayed a similar protein concentration dependence of Ca2+ transient inhibition (Fig. 7 D). By contrast, CFP‐Rem and CFP‐RemT94N yielded clearly divergent profiles of inhibition (Fig. 7 E). At moderate levels of expression, CFP‐RemT94N had no impact on Ca2+ transients whereas CFP‐Rem produced a near maximal inhibition (Fig. 7 E). At higher expression levels, CFP‐RemT94N is able to inhibit Ca2+ transient amplitude, but the extent of inhibition plateaus at a level higher than observed with wt CFP‐Rem (Fig. 7 E).
Figure 7. Relative impact of CFP‐RadS105N and CFP‐RemT94N on Ca2+‐induced Ca2+ release .
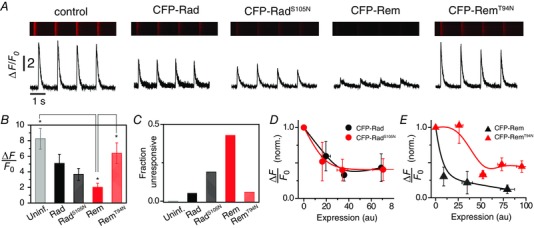
A, exemplar recordings showing effect of CFP‐tagged RGK proteins on field stimulation‐evoked rhod‐2‐reported Ca2+ transients in adult rat cardiomyocytes. B, relative impact of distinct RGK proteins on Ca2+ transient amplitude for uninfected (light grey, n = 12), CFP‐Rad (black, n = 19), CFP‐RadS105N (checkered black, n = 15), CFP‐Rem (red, n = 21), CFP‐RemT94N (checkered red, n = 17). C, fraction of cells unresponsive to field‐stimulation for distinct RGKs. D, dependence of relative Ca2+ transient amplitude on expression levels of CFP‐Rad (black circles, n = 19) and CFP‐RadS105N (red circles, n = 15). E, dependence of relative Ca2+ transient amplitude on expression levels of CFP‐Rem (black triangles, n = 21) and CFP‐RemT94N (red triangles, n = 17). P < 0.05, one‐way ANOVA with Bonferroni post hoc analyses.
Impact of G‐domain mutations on Rad and Rem interaction with auxiliary CaVβ
All RGKs bind auxiliary CaVβ subunits (Beguin et al. 2001; Correll et al. 2008; Yang & Colecraft, 2013), and for Rad and Rem, this interaction is at least partially responsible for their potent inhibition of reconstituted CaV1.2 channels (Yang et al. 2012). Hence, not only could assessing the impact of RadS105N and RemT94N on CaVβ binding in cardiomyocytes yield further perspectives on the necessity of an intact GNBP for this interaction, but it could also potentially provide an explanation for why Rem and RemT94N differ in their degree of I Ca,L block in heart. We adapted a previously described three‐cube FRET approach (Chen et al. 2006, 2007) to probe RadS105N and RemT94N binding to CaVβ (compared to their wt counterparts) in live cardiomyocytes. Prior to imaging cardiomyocytes, we calibrated our three‐cube FRET set‐up in HEK293 cells using CFP–YFP fusion constructs with varying linker lengths as described before (Chen et al. 2006, 2007). We then verified our set‐up by using the rapamycin‐induced dimerization system of FKBP: rapamycin binding domain (FRB) and FK506 binding protein (FKBP) (Banaszynski et al. 2005). In the absence of 1 μm rapamycin, FRB and FKBP displayed low FRET efficiency (E = 0.026 ± 0.004, n = 78) whereas the addition of rapamycin elevated FRET efficiency (E = 0.167 ± 0.008, n = 105), faithfully representing the non‐interacting and interacting conditions, respectively.
We then applied three‐cube FRET to investigate RadS105N/RemT94N interaction with CaVβ in live cardiomyocytes. As a positive control, we assessed the binding of CaV1.2 α1C I–II loop (I–II) to CaVβ, a well‐known and characterized high‐affinity protein interaction (Chen et al. 2004; Opatowsky et al. 2004; Van Petegem et al. 2004; 2008). We used adenovirus to co‐express CFP‐I–II and YFP‐β3 in adult rat cardiomyocytes. This pair resulted in a significantly elevated FRET efficiency (E = 0.15 ± 0.01, n = 44) compared to negative controls expressing non‐interacting CFP‐tagged loops of α1C and YFP‐β3 (E = 0.04 ± 0.01, n = 27) (Fig. 8 A). A useful extension of the three‐cube FRET method permits estimates of relative protein interaction affinities by fitting scatter plots of E versus free acceptor concentration (A free) from different experiments to a 1:1 binding model (Erickson et al. 2003; Chen et al. 2007). Applying this analysis to CFP‐I–II + YFP‐β3 FRET data yielded values for the maximal intrinsic FRET efficiency (E max = 0.25) and K d (1840 acceptor fluorescence units, AFU) (Fig. 8 B). A scatter plot of CFP‐tagged negative control loops + YFP‐β3 data on the same graph reports on the trajectory attributable to spurious FRET (Fig. 8 B).
Figure 8. FRET evaluation of wt and mutant Rad/Rem interactions with CaVβ3 in cardiomyocytes .
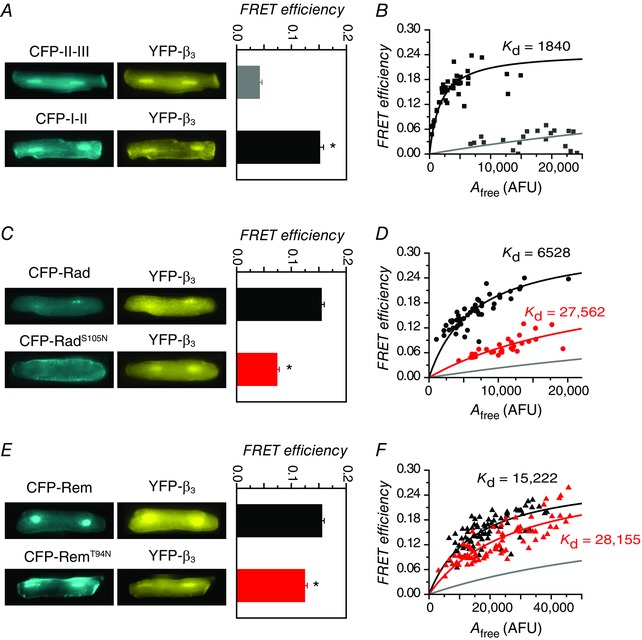
A, left, epifluorescence images of cardiomyocytes co‐expressing YFP‐β3 and either CFP‐I–II (bottom) or negative controls (CFP‐tagged N‐terminus, II–III, or III–IV loops of α1C) (top). Right, FRET efficiency. B, binding analyses for CFP‐I–II (black squares, n = 44) or negative controls (grey squares, n = 27) co‐expressed with YFP‐β3 in cardiomyocytes. C and D, data for CFP‐Rad (black circles, n = 52) and CFP‐RadS105N (red circles, n = 29) co‐expressed with YFP‐β3. Same format as A and B. E and F, data for CFP‐Rem (black triangles, n = 81) and CFP‐RemT94N (red triangles, n = 82) co‐expressed with YFP‐β3. Same format as A and B.
When co‐expressed with YFP‐β3, CFP‐Rad yielded a robust average FRET efficiency (0.156 ± 0.005, n = 52) that was significantly higher than obtained with CFP‐RadS105N (0.074 ± 0.004, n = 29) (Fig. 8 C). Binding analyses indicated that while both CFP‐Rad and CFP‐RadS105N bound YFP‐β3, the S105N mutation resulted in a 4‐fold decrease in binding affinity (K d = 6528 AFU for Rad; K d = 27,562 AFU for RadS105N) (Fig. 8 D). Similarly, we detected FRET when YFP‐β3 was co‐expressed with either CFP‐Rem (0.155 ± 0.004, n = 82) or CFP‐RemT94N (0.124 ± 0.005, n = 81) (Fig. 8 E). Binding analysis suggested that CFP‐Rem has a lower affinity for YFP‐β3 (K d = 15,222 AFU) compared to CFP‐Rad, and the T94N mutation results in a further ∼2‐fold decrease in binding affinity (K d = 28,155 AFU for RemT94N) (Fig. 8 F). Overall, these data demonstrate that mutations cognate to RasS17N in the G‐domains of Rad and Rem decrease, but do not abolish, their interaction with CaVβ in cardiac myocytes. Moreover, because RadS105N and RemT94N display comparable affinities for CaVβ in cardiomyocytes, the functional differences between them with respect to inhibition of cardiac CaV1.2 cannot be explained by variations in their binding to this auxiliary subunit. A caveat here is that splice variants of CaVβ2 rather than CaVβ3 are probably the dominant CaVβ isoforms in rat ventricular myocytes (Colecraft et al. 2002; Takahashi et al. 2003). Our interpretation of the data assumes there are no substantive differences in how Rad and Rem bind to distinct CaVβ isoforms.
Discussion
In this work, we have used CaV1.2 channel currents, intracellular Ca2+ transients, and binding to auxiliary CaVβ as biosensors to determine whether an intact GNBP is necessary for the function of the RGK proteins Rad and Rem. The approach relied on assessing the functional impact of RadS105N and RemT94N, which are mutations analogous to RasS17N. In Ras, the S17N mutation destabilizes guanine nucleotide binding and functionally locks the protein in an inactive position where it does not interact with effectors (Feig & Cooper, 1988 a). Moreover, RasS17N also displays a high affinity for Ras‐GEF, leading to dominant negative effects in situ (Feig, 1999). If RGK proteins with cognate mutations to RasS17N also displayed similar functional outcomes (i.e. loss‐of‐function on effectors and dominant negative properties in situ) this would be a strong indication that they are regulated by guanine nucleotides in a manner similar to Ras. Indeed, a previous study that used CaV1.2 inhibition as a functional outcome indicated a Ras‐like regulatory phenotype for Rad: RadS105N lost the ability to block recombinant CaV1.2 reconstituted in HEK293 cells, and over‐expressing RadS105N in guinea pig ventricular myocytes resulted in a markedly elevated I Ca,L, consistent with a dominant negative effect (Yada et al. 2007). Our results contrast starkly with this previous report. We found that when protein expression is normalized, RadS105N and RemT94N effectively inhibit recombinant CaV1.2 channels reconstituted in HEK293 cells to the same extent as wt Rad and Rem, respectively (Figs 3 and 4). Hence, a destabilized GNBP does not lead to a loss‐of‐function on effector CaV1.2 channels. Moreover, over‐expressing RadS105N in rat cardiomyocytes deeply inhibited both I Ca,L and Ca2+ transients to nearly the same extent as wt Rad, providing no indication of a dominant negative effect (Fig. 5 F and Fig. 7 D). There are several possible reasons for the discrepant results. In HEK293 cells, we found RadS105N has reduced protein stability compared to wt Rad (Fig. 2 H). Combined with the stochastic nature of transient transfections, it would appear necessary to carefully select cells for whole‐cell recordings that have an adequate expression of RadS105N. We achieved this here by using bicistronic vectors expressing mCherry as well as CFP‐tagged RadS105N, enabling fluorescent protein selection of cells that expressed adequate levels of RadS105N with near certainty. A failure to use such precautionary measures could potentially lead to selection of cells with little to no expression of RadS105N possibly leading to an erroneous conclusion that RadS105N is inert on reconstituted CaV1.2 in HEK293 cells. Over‐expression of RadS105N in transgenic mice led to prolonged action potentials and cardiac arrhythmias that was interpreted as a dominant negative effect, though no direct measurement of I Ca,L was done using cardiomyocytes from these transgenic mice (Yada et al. 2007). The variance with our results could potentially be due to compensatory mechanisms in the transgenic mouse model, differences in expression levels, or possibly, species differences. Overall, our results suggest that, with respect to inhibition of CaV1.2 in HEK293 cells and cardiomyocytes, the functional activity of Rad is not regulated by a canonical guanine nucleotide‐regulated switch mechanism. This conclusion more closely aligns with the available structural data that Rad does not undergo a GTP‐regulated conformational change similar to Ras (Sasson et al. 2011).
Despite our overall conclusion that RGK regulation of CaV1.2 channels does not conform to a canonical small G‐protein regulatory paradigm, we did observe some notable functional distinctions between RadS105N and RemT94N compared to their wt counterparts. First, in HEK293 cells, both RadS105N and RemT94N showed diminished protein expression levels compared to wt Rad and Rem, respectively, suggesting a potential role for guanine nucleotide binding in regulating RGK protein stability in this cellular context (Fig. 2). Similar findings have been made for other small G‐proteins (Cherfils & Chardin, 1999; Sasson et al. 2011). Second, both RadS105N and RemT94N displayed a reduced binding affinity for CaVβ compared to wt Rem and Rad in cardiomyocytes (Fig. 8). This result largely agrees with previous reports that mutations which disrupt the GNBP of RGKs disrupt their binding to CaVβ, though there is a noteworthy difference in the extent of abrogation of the interaction. Whereas previous studies using pull‐down assays indicate RGK mutants with a disrupted GNBP display a dramatically diminished or completely ablated binding to CaVβ (Beguin et al. 2005 a,b, 2006), we show a more moderate effect in live cardiomyocytes using FRET. This intermediate outcome is more easily reconciled with findings that RadS105N and RemT94N are still capable of inhibiting CaV1.2 channels, given that this effect is at least partially mediated through RGK binding to CaVβ (Yang et al. 2012). Overall, the impact of RadS105N and RemT94N mutations compared to wt can be classified as having either no effect or an intermediate effect, rather than a complete elimination of function as would be expected for a canonical Ras‐like G‐protein with an impaired GNBP. Finally, we observed a fundamental qualitative difference in the ability of RemT94N to inhibit CaV1.2 channels in HEK293 cells (strong inhibition with normalized expression) compared to cardiomyocytes (weak inhibition). The weaker effect of RemT94N in rat cardiomyocytes was not due to decreased protein expression, and is in accord with our previous observations in guinea pig ventricular myocytes (Xu et al. 2010). Because RemT94N in HEK293 cells can fully block CaV1.2 when protein expression is normalized, the most parsimonious interpretation of the data is that in heart, an as‐yet‐unknown cardiomyocyte‐specific factor neutralizes the effectiveness of RemT94N to inhibit I Ca,L. Potential candidate mechanisms include a protein that binds strongly to and effectively sequesters RemT94N or a post‐translational modification that affects the intrinsic ability of the RGK to inhibit CaV1.2. A caveat is that to date there is no direct biochemical confirmation that RemT94N displays a compromised GNBP with reduced affinity for guanine nucleotides. We assume this feature based on the similar roles of RasS17 and RemT94 in co‐ordinating Mg2+ in the GNBP, as revealed by crystal structures (Fig. 1). The functional deficits we observe with RemT94N in this study suggest this assumption is justified. Nevertheless, confirmation of the role of guanine nucleotides in Rem (and Rad) signalling requires more detailed information on the nucleotide binding status of wt and mutant versions of these proteins both in vitro and in situ.
Rad and Rem are endogenously expressed in cardiac myocytes (Reynet & Kahn, 1993; Finlin & Andres, 1997; Chang et al. 2007). Knockdown of Rad using shRNA (70% knockdown) in rat cardiomyocytes led to increases in I Ca,L (50%), Ca2+ transient amplitude (52%), and contractility (58%) (Wang et al. 2010). Rem knockout mice displayed a moderately increased I Ca,L (Magyar et al. 2012). These previous results suggest RGKs have a physiological role in maintaining cardiac Ca2+ homeostasis, making the question of how they themselves are regulated highly relevant. Both Rad and RadS105N yielded a similar concentration‐dependent profile of inhibition of Ca2+ transients (Fig. 7 D), suggesting this response is largely independent of the guanine‐nucleotide binding status of the Rad G‐domain. These results suggest that for Rad, controlling protein expression levels may be the dominant mechanism by which the influence of this RGK on cardiac Ca2+ homeostasis is regulated. A decrease in Rad protein expression occurs in the failing human heart and is probably an important contributor to the complex remodelling of cardiac Ca2+ homeostasis that occurs in this condition (Chang et al. 2007). Nucleotide diphosphate kinase (NDPK; also known as nm23) has been identified as a GAP for Rad (Zhu et al. 1999). NDPK is present in cardiomyocytes (Hippe et al. 2007) and its activity would be expected to regulate the GTPase activity of Rad in heart cells. As our results suggest that calcium handling by Rad is independent of its G‐domain nucleotide binding cycle, further work will be needed to elucidate a potential physiological role for putative nm23 regulation of Rad in heart cells. In contrast to Rad, our results predict that the nucleotide binding status of Rem G‐domain may have a large influence on cardiac excitation–contraction coupling since at moderate levels of expression, wt Rem strongly inhibited Ca2+‐induced Ca2+ release whereas RemT94N was without effect.
Additional information
Competing interests
None declared.
Author contributions
D.D.C. designed and performed experiments, analysed data, and wrote the paper. H.M.C. designed experiments, analysed data, obtained funding and wrote the paper. Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
D.D.C. was supported by a Multidisciplinary Training Grant in Translational Cardiovascular Research (T32‐HL087745). This work was supported by grants RO1 HL 084332 and 1RO1‐GM107585 from the National Institutes of Health to H.M.C., and an Established Investigator Award from the American Heart Association to H.M.C.
Acknowledgements
We thank Ming Chen for technical support and Dr Prakash Subramanyam for adenovirus encoding CFP‐tagged α1C N‐terminus and I–II loops. We thank Drs Akil Puckerin and Prakash Subramanyam for comments on the manuscript, Dr Wenjun Xie for advice on Ca2+ transient measurements, Dr Brent Osbourne for guidance with PyMOL rendering, and Robert Vogel (Weill Cornell Medical College) for MATLAB consultation and advice.
References
- Banaszynski LA, Liu CW & Wandless TJ (2005). Characterization of the FKBP.rapamycin.FRB ternary complex. J Am Chem Soc 127, 4715–4721. [DOI] [PubMed] [Google Scholar]
- Beguin P, Mahalakshmi RN, Nagashima K, Cher DH, Ikeda H, Yamada Y, Seino Y & Hunziker W (2006). Nuclear sequestration of β‐subunits by Rad and Rem is controlled by 14‐3‐3 and calmodulin and reveals a novel mechanism for Ca2+ channel regulation. J Mol Biol 355, 34–46. [DOI] [PubMed] [Google Scholar]
- Beguin P, Mahalakshmi RN, Nagashima K, Cher DH, Kuwamura N, Yamada Y, Seino Y & Hunziker W (2005. a). Roles of 14‐3‐3 and calmodulin binding in subcellular localization and function of the small G‐protein Rem2. Biochem J 390, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beguin P, Mahalakshmi RN, Nagashima K, Cher DH, Takahashi A, Yamada Y, Seino Y & Hunziker W (2005. b). 14‐3‐3 and calmodulin control subcellular distribution of Kir/Gem and its regulation of cell shape and calcium channel activity. J Cell Sci 118, 1923–1934. [DOI] [PubMed] [Google Scholar]
- Beguin P, Nagashima K, Gonoi T, Shibasaki T, Takahashi K, Kashima Y, Ozaki N, Geering K, Iwanaga T & Seino S (2001). Regulation of Ca2+ channel expression at the cell surface by the small G‐protein kir/Gem. Nature 411, 701–706. [DOI] [PubMed] [Google Scholar]
- Chang L, Zhang J, Tseng YH, Xie CQ, Ilany J, Bruning JC, Sun Z, Zhu X, Cui T, Youker KA, Yang Q, Day SM, Kahn CR & Chen YE (2007). Rad GTPase deficiency leads to cardiac hypertrophy. Circulation 116, 2976–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Puhl HL 3rd & Ikeda SR (2007). Estimating protein‐protein interaction affinity in living cells using quantitative Förster resonance energy transfer measurements. J Biomed Opt 12, 054011. [DOI] [PubMed] [Google Scholar]
- Chen H, Puhl HL 3rd, Koushik SV, Vogel SS & Ikeda SR (2006). Measurement of FRET efficiency and ratio of donor to acceptor concentration in living cells. Biophys J 91, L39–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Puhl HL 3rd, Niu SL, Mitchell DC & Ikeda SR (2005). Expression of Rem2, an RGK family small GTPase, reduces N‐type calcium current without affecting channel surface density. J Neurosci 25, 9762–9772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Li MH, Zhang Y, He LL, Yamada Y, Fitzmaurice A, Shen Y, Zhang H, Tong L & Yang J (2004). Structural basis of the α1–β subunit interaction of voltage‐gated Ca2+ channels. Nature 429, 675–680. [DOI] [PubMed] [Google Scholar]
- Cherfils J & Chardin P (1999). GEFs: structural basis for their activation of small GTP‐binding proteins. Trends Biochem Sci 24, 306–311. [DOI] [PubMed] [Google Scholar]
- Colecraft HM, Alseikhan B, Takahashi SX, Chaudhuri D, Mittman S, Yegnasubramanian V, Alvania RS, Johns DC, Marban E & Yue DT (2002). Novel functional properties of Ca2+ channel β subunits revealed by their expression in adult rat heart cells. J Physiol 541, 435–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colicelli J (2004). Human RAS superfamily proteins and related GTPases. Sci STKE 2004, RE13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correll RN, Pang C, Niedowicz DM, Finlin BS & Andres DA (2008). The RGK family of GTP‐binding proteins: regulators of voltage‐dependent calcium channels and cytoskeleton remodeling. Cell Signal 20, 292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson MG, Liang H, Mori MX & Yue DT (2003). FRET two‐hybrid mapping reveals function and location of L‐type Ca2+ channel CaM preassociation. Neuron 39, 97–107. [DOI] [PubMed] [Google Scholar]
- Feig LA (1999). Tools of the trade: use of dominant‐inhibitory mutants of Ras‐family GTPases. Nat Cell Biol 1, E25–27. [DOI] [PubMed] [Google Scholar]
- Feig LA & Cooper GM (1988. a). Inhibition of NIH 3T3 cell proliferation by a mutant ras protein with preferential affinity for GDP. Mol Cell Biol 8, 3235–3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig LA & Cooper GM (1988. b). Relationship among guanine nucleotide exchange, GTP hydrolysis, and transforming potential of mutated ras proteins. Mol Cell Biol 8, 2472–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ & Schreiber SL (1995). Inhibition of proteasome activities and subunit‐specific amino‐terminal threonine modification by lactacystin. Science 268, 726–731. [DOI] [PubMed] [Google Scholar]
- Finlin BS & Andres DA (1997). Rem is a new member of the Rad‐ and Gem/Kir Ras‐related GTP‐binding protein family repressed by lipopolysaccharide stimulation. J Biol Chem 272, 21982–21988. [DOI] [PubMed] [Google Scholar]
- Finlin BS, Crump SM, Satin J & Andres DA (2003). Regulation of voltage‐gated calcium channel activity by the Rem and Rad GTPases. Proc Natl Acad Sci USA 100, 14469–14474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlin BS, Shao H, Kadono‐Okuda K, Guo N & Andres DA (2000). Rem2, a new member of the Rem/Rad/Gem/Kir family of Ras‐related GTPases. Biochem J 347, 223–231. [PMC free article] [PubMed] [Google Scholar]
- Flynn R & Zamponi GW (2010). Regulation of calcium channels by RGK proteins. Channels (Austin) 4, 434–439. [DOI] [PubMed] [Google Scholar]
- Fu M, Zhang J, Tseng YH, Cui T, Zhu X, Xiao Y, Mou Y, De Leon H, Chang MM, Hamamori Y, Kahn CR & Chen YE (2005). Rad GTPase attenuates vascular lesion formation by inhibition of vascular smooth muscle cell migration. Circulation 111, 1071–1077. [DOI] [PubMed] [Google Scholar]
- Hippe HJ, Luedde M, Lutz S, Koehler H, Eschenhagen T, Frey N, Katus HA, Wieland T & Niroomand F (2007). Regulation of cardiac cAMP synthesis and contractility by nucleoside diphosphate kinase B/G protein βγ dimer complexes. Circ Res 100, 1191–1199. [DOI] [PubMed] [Google Scholar]
- Loirand G, Sauzeau V & Pacaud P (2013). Small G proteins in the cardiovascular system: physiological and pathological aspects. Physiol Rev 93, 1659–1720. [DOI] [PubMed] [Google Scholar]
- Maguire J, Santoro T, Jensen P, Siebenlist U, Yewdell J & Kelly K (1994). Gem: an induced, immediate early protein belonging to the Ras family. Science 265, 241–244. [DOI] [PubMed] [Google Scholar]
- Magyar J, Kiper CE, Sievert G, Cai W, Shi GX, Crump SM, Li L, Niederer S, Smith N, Andres DA & Satin J (2012). Rem‐GTPase regulates cardiac myocyte L‐type calcium current. Channels (Austin) 6, 166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning JR, Yin G, Kaminski CN, Magyar J, Feng HZ, Penn J, Sievert G, Thompson K, Jin JP, Andres DA & Satin J (2013). Rad GTPase deletion increases L‐type calcium channel current leading to increased cardiac contraction. J Am Heart Assoc 2, e000459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyers JS, Bilan PJ, Zhu J & Kahn CR (1997). Rad and Rad‐related GTPases interact with calmodulin and calmodulin‐dependent protein kinase II. J Biol Chem 272, 11832–11839. [DOI] [PubMed] [Google Scholar]
- Moyers JS, Zhu J & Kahn CR (1998). Effects of phosphorylation on function of the Rad GTPase. Biochem J 333, 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar N, Singh K & Garcia‐Diaz M (2010). Structure of the dominant negative S17N mutant of Ras. Biochemistry 49, 1970–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opatowsky Y, Chen CC, Campbell KP & Hirsch JA (2004). Structural analysis of the voltage‐dependent calcium channel β subunit functional core and its complex with the α1 interaction domain. Neuron 42, 387–399. [DOI] [PubMed] [Google Scholar]
- Opatowsky Y, Sasson Y, Shaked I, Ward Y, Chomsky‐Hecht O, Litvak Y, Selinger Z, Kelly K & Hirsch JA (2006). Structure‐function studies of the G‐domain from human gem, a novel small G‐protein. FEBS Lett 580, 5959–5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond P, Coquard A, Chenon M, Zeghouf M, El Marjou A, Thompson A & Menetrey J (2012). Structure of the GDP‐bound G domain of the RGK protein Rem2. Acta Crystallogr Sect F Struct Biol Cryst Commun 68, 626–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynet C & Kahn CR (1993). Rad: a member of the Ras family overexpressed in muscle of type II diabetic humans. Science 262, 1441–1444. [DOI] [PubMed] [Google Scholar]
- Sasson Y, Navon‐Perry L, Huppert D & Hirsch JA (2011). RGK family G‐domain:GTP analog complex structures and nucleotide‐binding properties. J Mol Biol 413, 372–389. [DOI] [PubMed] [Google Scholar]
- Seu L & Pitt GS (2006). Dose‐dependent and isoform‐specific modulation of Ca2+ channels by RGK GTPases. J Gen Physiol 128, 605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splingard A, Menetrey J, Perderiset M, Cicolari J, Regazzoni K, Hamoudi F, Cabanie L, El Marjou A, Wells A, Houdusse A & de Gunzburg J (2007). Biochemical and structural characterization of the gem GTPase. J Biol Chem 282, 1905–1915. [DOI] [PubMed] [Google Scholar]
- Sprang SR (1997). G protein mechanisms: insights from structural analysis. Annu Rev Biochem 66, 639–678. [DOI] [PubMed] [Google Scholar]
- Subramanyam P, Chang DD, Fang K, Xie W, Marks AR & Colecraft HM (2013). Manipulating L‐type calcium channels in cardiomyocytes using split‐intein protein transsplicing. Proc Natl Acad Sci USA 110, 15461–15466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi SX, Mittman S & Colecraft HM (2003). Distinctive modulatory effects of five human auxiliary β2 subunit splice variants on L‐type calcium channel gating. Biophys J 84, 3007–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Petegem F, Clark KA, Chatelain FC & Minor DL Jr (2004). Structure of a complex between a voltage‐gated calcium channel β‐subunit and an α‐subunit domain. Nature 429, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Petegem F, Duderstadt KE, Clark KA, Wang M & Minor DL Jr (2008). Alanine‐scanning mutagenesis defines a conserved energetic hotspot in the CaVα1 AID‐CaVβ interaction site that is critical for channel modulation. Structure 16, 280–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter IR & Wittinghofer A (2001). The guanine nucleotide‐binding switch in three dimensions. Science 294, 1299–1304. [DOI] [PubMed] [Google Scholar]
- Wang G, Zhu X, Xie W, Han P, Li K, Sun Z, Wang Y, Chen C, Song R, Cao C, Zhang J, Wu C, Liu J & Cheng H (2010). Rad as a novel regulator of excitation‐contraction coupling and β‐adrenergic signaling in heart. Circ Res 106, 317–327. [DOI] [PubMed] [Google Scholar]
- Ward Y, Spinelli B, Quon MJ, Chen H, Ikeda SR & Kelly K (2004). Phosphorylation of critical serine residues in Gem separates cytoskeletal reorganization from down‐regulation of calcium channel activity. Mol Cell Biol 24, 651–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward Y, Yap SF, Ravichandran V, Matsumura F, Ito M, Spinelli B & Kelly K (2002). The GTP binding proteins Gem and Rad are negative regulators of the Rho‐Rho kinase pathway. J Cell Biol 157, 291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X & Colecraft HM (2009). Primary culture of adult rat heart myocytes. J Vis Exp; doi:10.3791/1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Marx SO & Colecraft HM (2010). Molecular mechanisms, and selective pharmacological rescue, of Rem‐inhibited CaV1.2 channels in heart. Circ Res 107, 620–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yada H, Murata M, Shimoda K, Yuasa S, Kawaguchi H, Ieda M, Adachi T, Murata M, Ogawa S & Fukuda K (2007). Dominant negative suppression of Rad leads to QT prolongation and causes ventricular arrhythmias via modulation of L‐type Ca2+ channels in the heart. Circ Res 101, 69–77. [DOI] [PubMed] [Google Scholar]
- Yang T & Colecraft HM (2013). Regulation of voltage‐dependent calcium channels by RGK proteins. Biochim Biophys Acta 1828, 1644–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Puckerin A & Colecraft HM (2012). Distinct RGK GTPases differentially use α1‐ and auxiliary β‐binding‐dependent mechanisms to inhibit CaV1.2/CaV2.2 channels. PLoS One 7, e37079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Xu X, Kernan T, Wu V & Colecraft HM (2010). Rem, a member of the RGK GTPases, inhibits recombinant CaV1.2 channels using multiple mechanisms that require distinct conformations of the GTPase. J Physiol 588, 1665–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanuar A, Sakurai S, Kitano K & Hakoshima T (2006). Crystal structure of human Rad GTPase of the RGK‐family. Genes Cells 11, 961–968. [DOI] [PubMed] [Google Scholar]
- Zhu J, Reynet C, Caldwell JS & Kahn CR (1995). Characterization of Rad, a new member of Ras/GTPase superfamily, and its regulation by a unique GTPase‐activating protein (GAP)‐like activity. J Biol Chem 270, 4805–4812. [DOI] [PubMed] [Google Scholar]
- Zhu J, Tseng YH, Kantor JD, Rhodes CJ, Zetter BR, Moyers JS & Kahn CR (1999). Interaction of the Ras‐related protein associated with diabetes Rad and the putative tumor metastasis suppressor NM23 provides a novel mechanism of GTPase regulation. Proc Natl Acad Sci USA 96, 14911–14918. [DOI] [PMC free article] [PubMed] [Google Scholar]