

Abstract
Key points
In cystinosis, a lysosomal storage disorder, an altered redox state has been suggested as contributing to cellular dysfunction.
Ctns gene knockdown in a pancreatic β‐cell line caused increased cystine levels.
Attenuated nutrient stimulated insulin secretion was observed after Ctns knockdown which may have been caused by an increase in oxidative stress.
Oxidative stress may reduce ATP production in pancreatic β‐cells resulting in attenuated insulin release.
The redox‐sensitive transcription factor NF‐κB was activated after Ctns knockdown which may contribute to the increased incidence of apoptosis.
Abstract
The pancreatic β‐cell has reduced antioxidant defences making it more susceptible to oxidative stress. In cystinosis, a lysosomal storage disorder, an altered redox state may contribute to cellular dysfunction. This rare disease is caused by an abnormal lysosomal cystine transporter, cystinosin, which causes excessive accumulation of cystine in the lysosome. Cystinosis associated kidney damage and dysfunction leads to the Fanconi syndrome and ultimately end‐stage renal disease. Following kidney transplant, cystine accumulation in other organs including the pancreas leads to multi‐organ dysfunction. In this study, a Ctns gene knockdown model of cystinosis was developed in the BRIN‐BD11 rat clonal pancreatic β‐cell line using Ctns‐targeting siRNA. Additionally there was reduced cystinosin expression, while cell cystine levels were similarly elevated to the cystinotic state. Decreased levels of chronic (24 h) and acute (20 min) nutrient‐stimulated insulin secretion were observed. This decrease may be due to depressed ATP generation particularly from glycolysis. Increased ATP production and the ATP/ADP ratio are essential for insulin secretion. Oxidised glutathione levels were augmented, resulting in a lower [glutathione/oxidised glutathione] redox potential. Additionally, the mitochondrial membrane potential was reduced, apoptosis levels were elevated, as were markers of oxidative stress, including reactive oxygen species, superoxide and hydrogen peroxide. Furthermore, the basal and activated phosphorylated forms of the redox‐sensitive transcription factor NF‐κB were increased in cells with silenced Ctns. From this study, the cystinotic‐like pancreatic β‐cell model demonstrated that the altered oxidative status of the cell, resulted in depressed mitochondrial function and pathways of ATP production, causing reduced nutrient‐stimulated insulin secretion.
Abbreviations
- ADP
adenosine diphosphate
- ATP
adenosine triphosphate
- Carboxy‐H2DFFDA
5‐(and‐6)‐carboxy‐2’,7’‐difluorodihydro fluorescein diacetate
- CT80
Ctns targeting siRNA pool
- CTNS
Cystinosin gene
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- GSH
glutathione
- GSSG
oxidised glutathione
- MnSOD
mitochondrial superoxide dismutase
- NF‐κB
nuclear factor kappa‐B
- NT80
non‐targeting siRNA pool
- NTP
nucleoside triphosphate
- PBS
phosphate‐buffered saline
- PCR
polymerase chain reaction
- PI
propidium iodide
- SDS‐PAGE
sodium dodecyl sulphate–polyacrylamide gel electrophoresis
- SEM
standard error mean
- SOD
superoxide dismutase
- TBST
Tris‐buffered saline with Tween‐20
Introduction
Cystinosis is an autosomal recessive, lysosomal storage disorder where the cell becomes dysfunctional due to the accumulation of cystine (Gahl et al. 2002). The cause of cystinosis is a defect in the CTNS gene that causes partial or complete functional failure of the cystinosin protein, which transports cysteine out of the lysosome. Renal dysfunction due to the progression of Fanconi syndrome, results in the loss of small molecules and nutrients in the urine (Gahl et al. 2002). The low‐molecular weight proteinuria associated with Fanconi syndrome in cystinosis has been attributed to dysfunctional endocytosis in the proximal tubule (Wilmer et al. 2010), although the expression of important receptors necessary for endocytosis, megalin and cubulin, are normal in individuals with this disease (Wilmer et al. 2008). Furthermore, the cellular dysfunction has been suggested to occur due to abnormal ATP and mitochondrial activity as well as altered antioxidant capabilities of cystinotic kidney proximal tubule cells, which have a high metabolic demand (Wilmer et al. 2010).
In the pancreas, CTNS is expressed at similar levels to those observed in the kidney and the Cystinosin LKG isoform is expressed slightly higher in the pancreas than in the kidney (Taranta et al. 2012). This suggests that the pancreas requires cystinosin at a similar level to the kidney therefore predicting likely functional failure when cystinosin is defective in metabolically active cells, such as pancreatic islet cells. Lysosomal cystine accumulation leading to cellular dysfunction in the insulin secreting pancreatic β‐cells can result in diabetes mellitus. Post‐transplant diabetes mellitus develops in less than 25% of adults between 18 and 40 years (Ammenti et al. 1986; Theodoropoulos et al. 1993; Filler et al. 1998; Gahl et al. 2007; Prokai et al. 2008). However, it develops more frequently following transplant in individuals with cystinosis compared to those without cystinosis (Gahl et al. 1986) and this is not fully associated with corticosteroid therapy (Fivush et al. 1987). With the introduction of cysteamine therapy the age of onset of diabetes is delayed and can sometimes be prevented (Brodin‐Sartorius et al. 2011).
In histological studies of individuals with cystinosis, pancreatic β‐cell hyperplasia and increased β‐cell number have been noted (Milner & Wirdnam, 1982; Gahl et al. 2001) as well as high concentrations of cystine in the β‐cells (Fivush et al. 1987). It has been suggested that in cystinosis the altered ability of cells to counter stress, including metabolic stress, is altered very slowly over the patient's lifetime eventually resulting in loss of blood glucose control and therefore diabetes which may be a consequence of altered glucose uptake and metabolism in insulin sensitive tissues and/or cystine may partially attenuate the pancreatic β‐cell insulin secretion pathway (Robert et al. 1999; Nesterova & Gahl, 2008). While insulin resistance is not evident, diabetes associated with cystinosis appears to be due to a deficient insulin release mechanism in response to elevated glucose (Robert et al. 1999). Notably, both the first and second phases of insulin secretion appear to be reduced in individuals with cystinosis and the decrease in insulin release and related C‐peptide production is slow and progressive (Filler et al. 1998). β‐Cell failure in cystinosis is not associated with pancreatic islet inflammation as known to occur in type I diabetes (Filler et al. 1998).
In this study, the effect of Ctns gene silencing on cellular function was assessed in insulin secreting rat clonal pancreatic β‐cells. The effects of cystinosin knockdown on insulin secretion, intracellular thiol levels, ATP levels and ATP production capacity, as well as levels of apoptosis and changes in mitochondrial membrane potential, were investigated.
Methods
Reagents
RPMI 1640 medium, penicillin–streptomycin, fetal bovine serum (FBS) and l‐glutamine were obtained from Gibco (Life Technologies, Carlsbad, CA, USA). Ctns‐targeting and non‐targeting siRNA and DharmaFECT 1 transfection reagent were obtained from GE Dharmacon (Lafayette, CO, USA). Fluorescent probes including Annexin‐V‐Fluorescein, DFF‐DA, hydroethidine and JC‐1 are from Molecular Probes (Life Technologies). All other reagents were obtained from Sigma‐Aldrich (St Louis, MO, USA) unless stated otherwise.
Cell culture
RPMI 1640 medium supplemented with 10% (v/v) FBS, 100units ml–1 penicillin, 0.1 mg ml−1 streptomycin and 2 mm glutamine was used to culture the clonal rat insulin‐secreting β‐cell line, BRIN‐BD11. These cells have been extensively characterised with respect to metabolic stimulus–secretion coupling, intracellular signalling, redox regulation, apoptosis and proliferation in response to various stimuli (McClenaghan et al. 1996; McClenaghan & Flatt, 1999; Brennan et al. 2003; Krause et al. 2011; Mullooly et al. 2014). Cells were seeded at 1.5 × 105cells ml–1 in 24‐well or 6‐well plates. Cells were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air using a Forma Scientific incubator.
Ctns gene knockdown
Ctns‐targeting siRNA were selected and produced based on the rat Ctns gene (homologous to the human CTNS gene), using ON‐TARGETplus SMARTpool technology by GE Dharmacon. BRIN‐BD11 cells were seeded and allowed to adhere overnight. Ctns‐targeting siRNA pool (80 pmol μl−1; CT80) was transiently transfected into the cells for 18 h using DharmaFECT 1, according to the instructions of the manufacturer. Mock transfection involved the transfection in the absence of siRNA, while the negative control was the non‐targeting siRNA pool (NT80). Real‐time PCR was performed at 24 h post‐transfection and subsequent experiments were performed at 72 h post‐transfection.
Real‐time PCR
TRIzol reagent (Life Technologies) was used to extract total RNA from BRIN‐BD11 cells, before mRNA was reverse‐transcribed into cDNA using SuperScript II Reverse Transcriptase (Life Technologies), according to the instructions of the manufacturer. The cDNA was combined with Taqman primers for rat Ctns and β‐actin and PCR master mix, which contained AmpliTaq Gold DNA polymerase, dNTP mix, uracil‐DNA glycosylase and ROX passive reference dye. The multiplex reaction mixture for duplicate reactions was created as follows: 1 μl target and control probes, 10 μl Taqman master mix, 1 μl cDNA template, brought up to 20 μl with RNase‐free water. Results are presented as relative quantity based on delta Ct values relative to the Mock TF.
Western blot
Cell lysates containing 20 μg protein were prepared and subjected to 7.5% SDS‐PAGE before being electrophoretically transferred to a nitrocellulose membrane. Blocking buffer and antibodies were prepared in 5% non‐fat milk in Tris‐base saline buffer (pH 7.4) containing 0.1% (v/v) Tween‐20 (TBST). The membranes were incubated for 60 min at room temperature (RT) with blocking buffer, followed by overnight incubation with primary antibody (dilutions Cystinosin monoclonal antibody 1:5,000 (Abnova, Taipei, Taiwan), NF‐κB 1:5000 (Cell Signalling Technology, Danvers, MA, USA) or β‐actin 1:10,000) at 4°C. Three TBST washes were then performed before incubation with secondary antibody conjugated to horseradish peroxidase followed by three TBST washes. Enhanced Chemiluminescence Kit (Thermo Fisher Scientific, Rockford, IL, USA) was used to detect the bound secondary antibody following the instructions of the manufacturer. Densitometry analysis was performed on the bands developed on autoradiographic film using ImageJ software.
Insulin secretion
At 72 h post‐transfection, the cell supernatant was removed and used to determine chronic insulin release (over the previous 24 h). The cells were then washed with PBS and acute insulin secretion was stimulated after cells were starved for 40 min with Krebs Ringer buffer (KRB), pH 7.4, containing 1.1 mm d‐glucose. The cells were then stimulated with KRB containing 16.7 mm glucose plus 10 mm alanine for 20 min at 37 °C (a standard potent stimulus for insulin secretion). The KRB was collected and insulin release was determined using the Mercodia ultra‐sensitive rat insulin ELISA kit (Uppsala, Sweden), according to the instructions of the manufacturer.
Glucose consumption
The cell supernatant was removed at 72 h post transfection and used to determine the concentration of glucose consumed (over the previous 24 h) using the glucose liquicolour enzymatic colorimetric determination assay (HUMAN, Wiesbaden, Germany), according to manufacturer's instructions.
Intracellular cysteine and glutathione levels
Total and free intracellular cysteine concentrations were determined using a method developed by Gaitonde (1967) with the modifications described by Dominy et al (2007). In brief, cell lysates were acidified with acetic acid before reaction with acid ninhydrin reagent at 100°C for 10 min and rapidly cooled on ice. The acid ninhydrin reagent forms a pink colour in an acidic solution following reaction with cysteine that can be detected at 560 nm. Dithiothreitol was used to reduce cystine to cysteine for detection by disrupting the disulphide bonds. The difference between intracellular total and free cysteine (Cys) concentrations were used to determine cystine content. All concentrations were normalised for protein content.
Using the enzyme‐recycling method developed by Baker et al (1990) with slight modifications, total glutathione (GSH) and oxidised glutathione (GSSG) concentrations were determined and levels were normalised for protein content.
The Nernst equation (E h = E 0 + RT/nFln([disulphide]/[thiol]2) was used to calculate the theoretical redox potential (E h) of the GSH/GSSG couple. (E 0, standard potential for the redox couple (−264 mV for GSSG/GSH2); R, gas constant; T, absolute temperature; n, number of transferred electrons (e.g. 2); F, Faraday's constant; Jones et al. 2000). A dilution volume of 5 μl (mg protein)–1 was assumed when calculating the redox couple. The value of E h was expressed in millivolts.
Intracellular ATP content and production
Cells were detached using 0.05% trypsin–EDTA and washed with ice‐cold PBS. The cell pellets were resuspended in 100 μl ice‐cold PBS. The cell suspension was then diluted 25‐fold and the ATP content was assessed using the ATP Bioluminescence Assay Kit HSII (Roche Diagnostics, Mannheim, Germany), according to the instructions of the manufacturer. Protein concentration of the cell lysates was used to normalise the ATP content.
ATP production was assessed by inhibiting glycolysis and mitochondrial ATP production. Cells were incubated for 1 h at 37°C with sodium iodoacetate (300 μm SIA), which inhibits GAPDH and therefore glycolysis, or 1.8 μg ml−1 oligomycin (Brennan et al. 2002), which inhibits mitochondrial complex V (ATP synthase). Following incubation with the inhibitors the intracellular ATP levels were determined.
Apoptosis and necrosis
Apoptotic and necrotic cell death were determined using Annexin V‐FITC and propidium iodide (PI), respectively. Cell pellets were resuspended in 1× Annexin‐V binding buffer (BD Biosciences, San Jose, CA, USA) containing 5 μl of both Annexin V‐FITC and 1 μg ml−1 PI and incubated for 5 min at RT before being analysed using the BD Accuri C6 flow cytometer (BD Biosciences).
Mitochondrial membrane potential
JC‐1 lipophilic cationic dye enters the mitochondria and reversibly emits red fluorescence in healthy cells with high mitochondrial transmembrane potential, while JC‐1 emits green fluorescence in cells with low mitochondrial transmembrane potential. Cell pellets were resuspended in media containing 2 μm JC‐1 dye and incubated at 37°C for 30 min. Cells were pelleted by centrifugation and resuspended in PBS before being analysed using the BD Accuri C6 flow cytometer. The ratio of green to red fluorescence represented the mitochondrial transmembrane potential.
Reactive oxygen species (ROS), superoxide and hydrogen peroxide
Generation of ROS and superoxide were detected using the ROS‐sensitive probe, 5‐(and‐6)‐carboxy‐2’,7’‐difluorodihydro fluorescein diacetate (carboxy‐H2DFFDA) or the fluorogenic probe hydroethidine, respectively. Cells were incubated with 20 μm carboxy‐H2DFFDA for 45 min or 5 μm hydroethidine for 30 min. Cells were detached using trypsin–EDTA and the cell suspension was analysed immediately using the BD Accuri C6 flow cytometer.
The intracellular hydrogen peroxide concentration was determined using the Amplex Red hydrogen peroxide and peroxidase assay kit (Life Technologies) according to the instructions of the manufacturer.
Protein concentration
Protein concentrations were determined using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific) according to the instructions of the manufacturer.
Statistical analysis
Data are presented as the means of three independent experiments ± SEM and were analysed with the GraphPad Prism 5.0 software (GraphPad Software Inc., San Diego, CA, USA). Data were analysed by Student's t test and differences at P value of < 0.05 were considered significant.
Results
Effect of Ctns targeting siRNA on Ctns gene expression and cystinosin protein expression
Ctns mRNA levels were potently and significantly decreased by 85% compared to Mock transfection (MT) (P < 0.001) and non‐targeting siRNA control (NT80 P < 0.001) as determined by real‐time quantitative PCR following 24 h transfection with Ctns targeting siRNA (Fig. 1 A). This was associated with a 50% decrease in cystinosin expression compared to both MT (P < 0.001) and NT80 (P < 0.001) as determined by Western blot at 72 h post‐transfection (Fig. 1 B).
Figure 1. Assessment of Ctns gene knockdown by real‐time PCR and Western blot analysis .
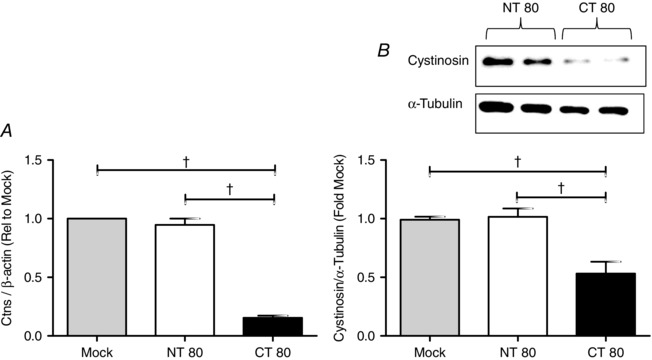
BRIN‐BD11 cells were transiently transfected with a Ctns targeting siRNA pool (80 pmol μl−1, CT80) for 24 h using DharmaFECT 1 transfection reagent, according to manufacturer's instructions. Mock transfection (Mock) was performed in the absence of siRNA, while the negative control contained scrambled non‐targeting siRNA pool (NT80). Ctns mRNA and cystinosin levels were assessed by real‐time PCR (A) and Western blot (B) at 24 and 72 h post transfection, respectively. Lanes 1 and 2 of the representative Western blot analysis correspond to NT 80, and lanes 3 and 4 correspond to CT 80. Densitometry analysis of Western blots are expressed as ratio of cystinosin to α‐tubulin expression and are presented as mean fold change relative to Mock TF ± SEM of three or more independent experiments. †statistically significant difference from Mock TF or NT80, as indicated, at P < 0.05 and P < 0.001, respectively. Mock TF, mock transfection; NT, non‐targeting siRNA; CT, Ctns targeting siRNA.
Effect of Ctns gene knockdown on insulin release over 24 h and following nutrient stimulation
Ctns gene knockdown caused a 50% decrease in chronic insulin release over 24 h compared to NT80 (21.53 ± 3.642 to 46.55 ± 6.386 ng (mg protein)−1, respectively; P < 0.01; Fig. 2 A). Acute insulin release following nutrient stimulation over 20 min was also significantly reduced from 2.238 ± 0.415 ng mg−1 protein in NT80 cells to 0.989 ± 0.187 nmol (mg protein)−1 in cells treated with CT80 (P < 0.01) (Fig. 2 B).
Figure 2. Effect of Ctns knockdown on chronic and acute nutrient stimulated insulin secretion and glucose consumption .
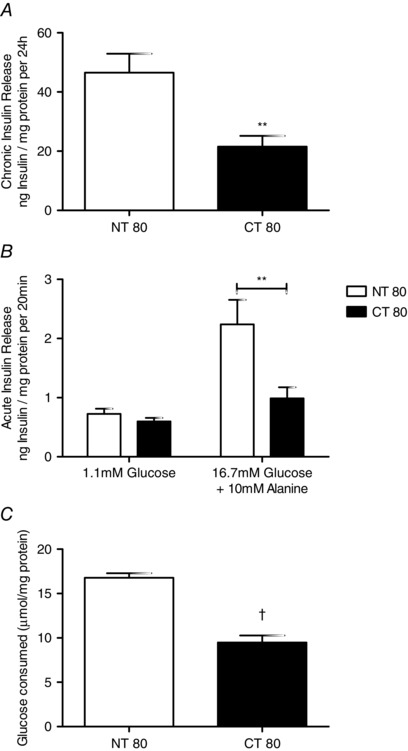
A, the chronic release of insulin was assessed in BRIN‐BD11 cells between 48–72 h following transfection. Cells were initially incubated with KRB containing 1.1 mm glucose for 40 min at 37°C before insulin release was stimulated for 20 min using KRB containing 1.1 mm glucose (basal) or 16.7 mm glucose and 10 mm alanine. B, acute stimulated insulin secretion was determined in BRIN‐BD11 cells at 72 h post‐transfection. C, glucose consumed was determined between 48–72 h post transfection. Results are expressed as mean ± SEM of at least four independent experiments. ** and †, statistically significant difference from NT80 at P < 0.01 and P < 0.001, respectively. NT, non‐targeting siRNA; CT, Ctns targeting siRNA.
Effect of Ctns gene knockdown on glucose consumption over 24 h
Cells transfected with CT80 utilised only half of the glucose consumed by NT80 (9.47 ± 0.81% versus 16.14 ± 0.85%, respectively). This decrease in glucose consumption was significantly different compared to control (P < 0.001).
Effect of Ctns gene knockdown on intracellular thiol/disulphide systems
The hallmark of cystinosis is the accumulation of cystine in the lysosome. The accumulation of cystine in the cells with decreased Ctns expression was assessed by finding the difference between the total and free cysteine levels (Fig. 3). Cystine concentration was increased over 3‐fold in cells treated with CT80 compared to the non‐targeting control, NT80 (1.826 ± 0.177 vs. 0.623 ± 0.161 nmol (mg protein)−1, respectively; P < 0.01) (Fig. 3 A). A smaller but still significant 2‐fold increase was also observed in total cysteine levels between NT80‐ and CT80‐treated cells from 5.574 ± 0.293 to 10.64 ± 0.880 nmol (mg protein)−1 (P < 0.001) (Fig. 3 B).
Figure 3. Effect of Ctns knockdown on thiol and disulphide levels .
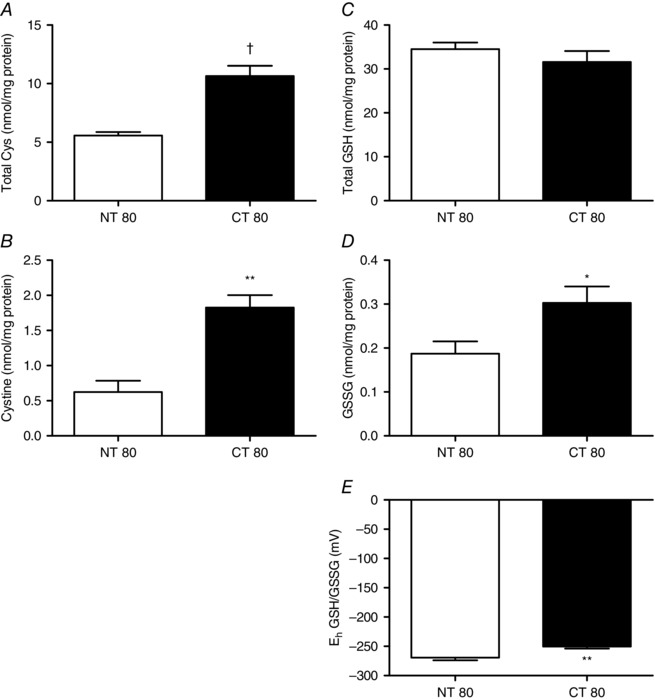
Following transfection with Ctns siRNA, intracellular total cysteine (A), cystine (B), total glutathione (C) and oxidised glutathione (GSSG) (D) levels in BRIN‐BD11 cells were determined. Total cysteine was determined using acid ninhydrin reagent developed by Gaitonde (1967), while total GSH and GSSG levels were determined using an enzyme recycling assay. E, using the Nernst equation, theoretical redox potential (E h) of GSH/GSSG couple was assessed. Results are expressed as the means ± SEM of at least three independent experiments. *, ** and †, statistically significant difference from NT80 control at P < 0.05, P < 0.01 and P < 0.001, respectively. NT, non‐targeting siRNA; CT, Ctns targeting siRNA.
There was no significant change in total glutathione levels as NT80‐treated cells at 34.50 ± 1.503 nmol (mg protein)−1 were comparable to CT80‐treated cells 31.58 ± 2.492 nmol (mg protein)−1 (Fig. 3 C). In contrast, oxidised glutathione was increased in CT80 cells compared to NT80 (0.303 ± 0.037 vs. 0.187 ± 0.028 nmol (mg protein)−1, respectively; P < 0.05) (Fig. 3 D). This alteration in the oxidised glutathione was associated with a slight but significant, less negative glutathione redox potential compared to NT80 (−250.4 ± 3.437 vs. −269.4 ± 4.59 mV, respectively; P < 0.01) (Fig. 3 E).
Effect of Ctns gene knockdown on ATP concentrations and production
Total intracellular ATP levels were significantly decreased in cells with decreased cystinosin expression compared to NT80 (3.167 ± 0.248 vs. 5.764 ± 0.529 nmol (mg protein)−1, respectively; P < 0.01) (Fig. 4).
Figure 4. Effect of Ctns gene knockdown on intracellular ATP levels, ATP production and mitochondrial membrane potential .
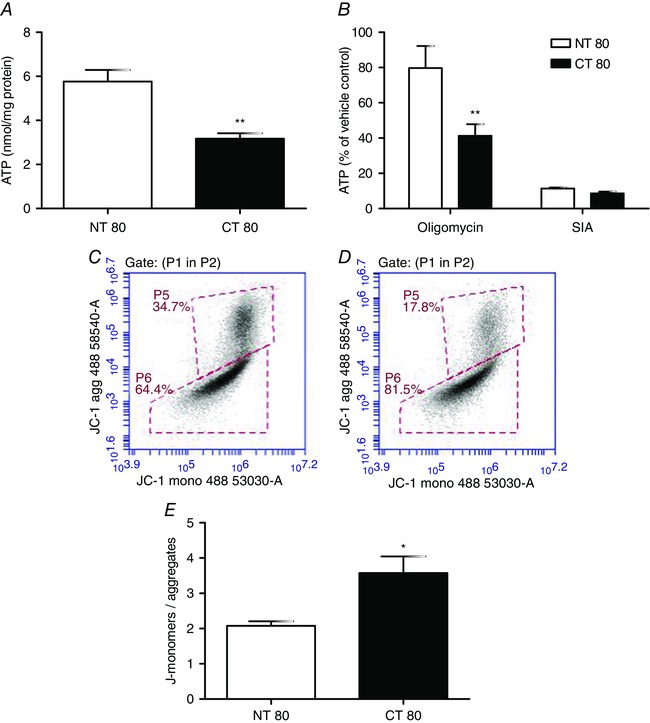
A, total ATP concentration was determined in BRIN‐BD11 cells at 72 h post‐transfection with Ctns targeting siRNA. B, ATP production was assessed following inhibition of mitochondrial ATP production with oligomycin or inhibition of glycolysis using sodium iodoacetate. E, at 72 h post‐transfection, the mitochondrial transmembrane potential was determined by flow cytometry using the JC‐1 dye. Data are presented as means ± SEM of at least three independent experiments. * and **, statistically significant difference from NT80 control at P < 0.05 and P < 0.01, respectively. C and D, representative 2D density plots of green (x‐axis: FL‐1; ex: 488 nm, em: 530 ± 30 nm) vs. red fluoresence (y‐axis: FL‐2; excitation: 488 nm, emission: 585 ± 40 nm) are shown for NT80 (C) and CT80 (D). NT, non‐targeting siRNA; CT, Ctns targeting siRNA.
The source of ATP was further investigated by inhibiting glycolysis or mitochondrial ATP production. Glycolytic capacity as indicated by ATP concentration was assessed by inhibiting mitochondrial ATP production using oligomycin. ATP derived from glycolysis was 50% lower in CT80‐treated cells than NT80 control (41.316 ± 6.538% vs. 79.686 ± 12.482% of control) (P < 0.01) (Fig. 4). In contrast, ATP derived from mitochondrial oxidative phosphorylation was slightly but not significantly decreased following Ctns knockdown compared to NT80 (8.6 ± 0.991% vs. 11.334 ± 0.628% of control), as assessed after addition of the glycolytic inhibitor, sodium iodoacetate.
In cells with high mitochondrial membrane potential (ΔΨm), JC‐1 spontaneously forms complexes (J‐aggregates) with intense red fluorescence. In cells with compromised mitochondrial function, with low ΔΨm, JC‐1 remains in the monomeric form, as indicated by green fluorescence. Using JC‐1 to assess BRIN‐BD11 mitochondrial membrane potential changes, the ratio of green to red fluorescence was significantly higher in CT80‐treated cells compared to NT80 (3.571 ± 0.472 vs. 2.075 ± 0.133, respectively) (P < 0.05) (Fig. 4 E). This indicated a decrease in the mitochondrial membrane potential, thus impaired ability to generate ATP.
Effect of Ctns gene knockdown on cell viability
The effect of cystinosin knockdown on cell death and apoptosis was assessed by flow cytometry using propidium iodide and Annexin‐V‐FITC, respectively. Propidium iodide measures cells in late apoptosis or necrosis when the cell plasma membrane becomes permeable. This is not a measure of total cell death. Annexin‐V on the other hand binds to the phosphatidylserine that has translocated from the inner leaflet in healthy cells to the outer leaflet in cells undergoing apoptosis. An increase in apoptotic cells was observed in CT80‐treated cells, which was indicated by an increase in Annexin‐V positive fluorescence, compared to NT80 (12.05 ± 2.515% vs. 7.260 ± 0.527%, respectively) (P < 0.01) (Fig. 5). There was no change in the percentage of dead cells which were identified as those that stained positive for PI between the two treatments.
Figure 5. Effect of Ctns knockdown on necrosis and apoptosis levels .
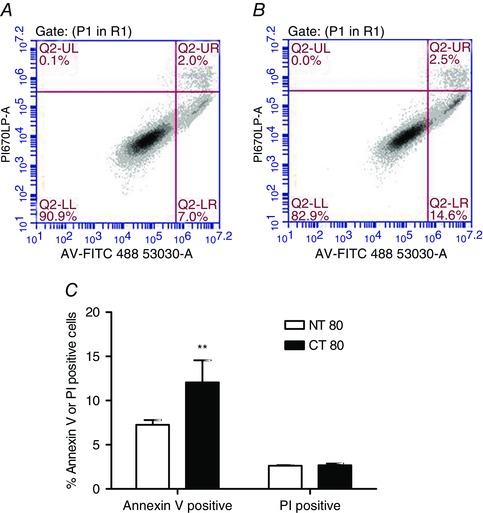
Following transfection of BRIN‐BD11 cells with Ctns targeting siRNA, apoptosis levels and late apoptosis or necrosis were determined at 72 h post transfection by flow cytometry using the Annexin‐V‐FITC and propidium iodide (PI), respectively. A and B, representative 2D density plots of Annexin‐V‐FITC (x‐axis: FL‐1 488 excitation, 530 ± 30 nm emission) versus PI (y‐axis: FL‐3 488 excitation, 675LPnm emission) for NT80, (A) and CT80 (B). C, results are presented as the mean of the percentage of apoptotic cells or dead cells ± SEM of three independent experiments. **, statistically significant difference from NT80 control at P < 0.01. NT, non‐targeting siRNA; CT, Ctns targeting siRNA.
Effect of Ctns gene knockdown on the intracellular oxidative stress levels
Three markers of oxidative stress including reactive oxygen species, superoxide and hydrogen peroxide were found to be significantly increased in the cells where cystinosin was knocked‐down. ROS was increased from 2875 ± 116.5 RFU in control NT80 cells to 3997 ± 249.5 RFU in CT80 (P < 0.01) (Fig. 6). Superoxide was elevated from 21710 ± 568.1 RFU in NT80 cells to 27201 ± 1542 RFU in CT80 cells (P < 0.01). Similarly, hydrogen peroxide was increased from 3.792 ± 0.109 nmol (mg protein)−1 in NT80 cells to 5.314 ± 0.344 nmol (mg protein)−1 in CT80 (P < 0.01).
Figure 6. Effect of Ctns knockdown on intracellular reactive oxygen species and oxidative stress .
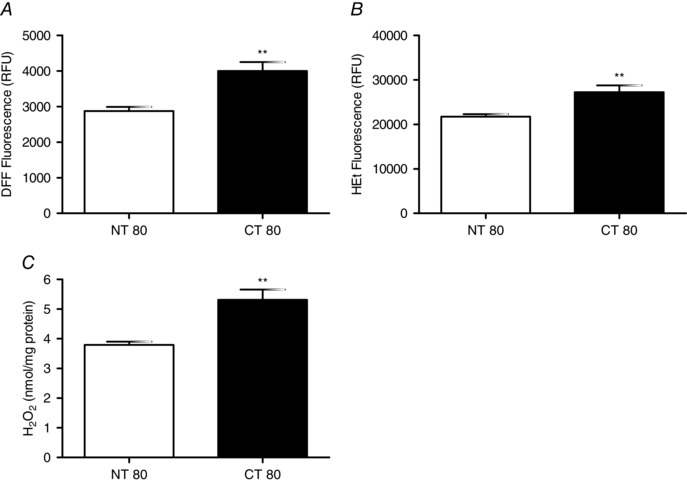
A and B, intracellular reactive oxygen species were determined in BRIN‐BD11 cells at 72 h post‐transfection with Ctns targeting siRNA by flow cytometry using DFF‐DA, a redox sensitive probe (A), while superoxide levels were measured using hydroethidine (HEt), a superoxide‐sensitive probe (B). C, intracellular hydrogen peroxide levels were assessed by amplex red assay. Flow cytometry data are presented as mean fluorescence (RFU) ± SEM of at least three independent experiments. **, statistically significant difference from NT80 at P < 0.01. NT, non‐targeting siRNA; CT, Ctns targeting siRNA.
Effect of Ctns gene knockdown on the expression of NF‐κB
One of the transcription factors affected by altered oxidative stress is NF‐κB. Interestingly, this protein was potently and significantly increased in both its basal and phosphorylated activated forms. Following transfection with Ctns targeting siRNA, basal NF‐κB was increased from 1.008 ± 0.107 in NT80 cells to 1.921 ± 0.259 in CT80 (P < 0.01) (Fig. 7). This increase was even more pronounced for the phosphorylated form, increasing from 1.176 ± 0.167 to 2.995 ± 0.264 in NT80 and CT80 cells, respectively (P < 0.001). The more pronounced increase in phosphorylation of NF‐κB (compared to the level of NF‐κB) indicates an increase in the phosphorylation level (per unit of NF‐κB), and is not simply a secondary effect of the increase in NF‐κB.
Figure 7. Effect of Ctns knockdown on NF‐κB protein expression .

At 72 h post‐transfection with Ctns targeting siRNA in BRIN‐BD11 cells, NF‐κB p65 and phosphorylated NF‐κB p65 protein levels were determined by Western blot. A and B, densitometry analysis of blots are expressed as ratio of NF‐κB/α‐tubulin (A) or phosphorylated‐NF‐κB/α‐tubulin (C) expression and are presented as mean fold change relative to Mock TF ± SEM of three independent experiments. ** and †, statistically significant difference from NT80 at P < 0.01 and P < 0.001, respectively. Mock, mock transfection containing no siRNA; NT, non‐targeting siRNA; CT, Ctns targeting siRNA.
Discussion
The response of the pancreatic β‐cell to glucose is critical to its function as an insulin secreting cell and it is known that appropriate levels of intracellular reactive oxygen and nitrogen species regulate this process. High levels of oxidative stress may adversely affect insulin secretion (Newsholme et al. 2012). Pancreatic β‐cells are particularly susceptible to oxidative stress due to decreased levels of antioxidant defence. In the genetic lysosomal storage disorder cystinosis, oxidative stress, as a consequence of lysosomal cystine accumulation, has been reported and may contribute to the high (5–25%) incidence of diabetes in individuals with this disease (Gahl et al. 2007; Wilmer et al. 2010). The occurrence of diabetes is reduced with long‐term therapy with the cystine depleting aminothiol cysteamine (Cystagon) (Gahl et al. 2007), which may enhance antioxidant defences. The effect of cystine accumulation on pancreatic β‐cell antioxidant defence and metabolic stimulus–secretion coupling has not been previously investigated.
In the present study, clonal insulin‐secreting BRIN‐BD11 pancreatic β‐cells were utilised, based on their well characterised responses to glucose and amino acids, as well as their reproducible secretory responses, consistent receptor/signalling protein levels, and cell survival/death responses (Corless et al. 2006; Kiely et al. 2007; Krause et al. 2011). Due to the complexity of the pancreas and limited access to viable human islets, the development of a stable human β‐cell line has been difficult (Scharfmann et al. 2013). However, the development of rodent cell lines, such as the BRIN‐BD11 cell line, has provided a valuable resource in investigating the pancreatic β‐cell and in advancing the understanding of diabetes and β‐cell dysfunction (Steiner et al. 2010; Newsholme et al. 2012). In the present study, this rat cell line was used to determine the potential impact of substantial loss of the cystinosin transporter on the function of the β‐cell including nutrient stimulated insulin secretion.
Alteration to the CTNS gene sequence results in the cystinosis phenotype due to the inability of the lysosome to export cystine following protein degradation (Gahl et al. 2002). In the model of cystinosis used in this study, the rat Ctns gene (homologous to human CTNS) mRNA transcripts were targeted for degradation by sequence specific siRNA. This model resulted in a biochemical and molecular model similar to cellular dysfunction in cystinotic disease. The decrease in Ctns mRNA was associated with a reduction in the expression of the cystine lysosomal transporter, cystinosin, in pancreatic β‐cells, which was similar to levels detected in individuals heterozygous for cystinosis and in the milder variants of the disease, late‐onset and ocular (Gahl et al. 1982 a,b). Further confirmation of the model used in this study was achieved by demonstrating a 3‐fold increase in intracellular cystine levels. Similar levels of cystinosin knockdown and cystine accumulation were also observed in human kidney cells (HK‐2) using a similar model (Bellomo et al. 2010; Sumayao et al. 2013) and in rabbit primary renal proximal tubule cells (Taub et al. 2011; Taub & Cutuli, 2012).
In the present study, the cystinosin knockdown caused attenuation of both chronic and acute nutrient stimulated insulin release to be reduced, while glucose consumption was also diminished. The redox balance of the cells was also adversely altered as oxidised glutathione (GSSG) was increased. Interestingly, the ATP levels in the cells were also decreased which appeared to be due to decreased capacity to generate ATP from glycolysis. While the mitochondrial ATP generating capacity was intact following inhibition of glycolysis, the mitochondrial membrane potential was adversely altered. The rate of apoptosis also appeared to be elevated in these cells in the absence of an inducer of apoptosis. More pronounced rates of apoptosis have been observed following the addition of inducers of apoptosis, such as TNF‐α which has been shown to increase apoptosis in normal and cystinotic renal proximal tubule epithelial cells (Park et al. 2006). Finally, the oxidative stress levels of the pancreatic β‐cells following cystinosin knockdown were greater than control levels as indicated by elevated intracellular ROS, superoxide and hydrogen peroxide. Remarkably, the transcription factor NF‐κB was elevated in both its basal and phosphorylated active form.
Pancreatic β‐cells produce and release insulin in response to circulating nutrient concentration in a biphasic manner and decreased insulin secretion is a hallmark of diabetes mellitus (Straub & Sharp, 2002). Chronic insulin secretion (24 h) in cystinosin depleted BRIN‐BD11 cells was partially attenuated, as was acutely stimulated insulin secretion (20 min). In individuals with cystinosis, insulin secretion and C‐peptide production declines slowly and both phases of insulin secretion are reduced (Filler et al. 1998). It is likely, based on the results presented herein, that a deficiency of insulin secretion arises in individuals with cystinosis due to cell dysfunction rather than either β‐cell destruction, such as occurs in type I diabetes, or insulin resistance as can occur in type II diabetes (Filler et al. 1998). Notably, the consumption of glucose was also reduced following Ctns knockdown, in agreement with the result reported above that maximal glycolytic ATP generating capacity was reduced. Mathematical models of metabolism in the β‐cell have also highlighted the importance of glycolysis for ATP production (Salvucci et al. 2013). It is possible that a defect in the ‘nutrient sensing’ mechanism of the cell, which links nutrient availability with insulin secretion has occurred in the Ctns knockdown β‐cells. This defect may result from inhibition of glycolytic enzymes, or glucose transporters due to altered redox balance, oxidative stress or mitochondrial dysfunction (as pyruvate metabolism could be impaired). It has been reported that inhibition of GAPDH activity by hyperglycaemia does not occur when mitochondrial overproduction of superoxide is prevented by either UCP‐1 or MnSOD (Giacco & Brownlee, 2010). When GAPDH activity is inhibited, the levels of all the glycolytic intermediates that are upstream of GAPDH increase, resulting in a reduction in glucose consumption. Previously, treatment with cysteine caused reduced insulin secretion in a study by Kaneko et al., which was suggested to be caused by inhibition of glucose metabolism and/or alterations to the secretory machinery (Kaneko et al. 2006). A study by Kiely et al. reported that pro‐inflammatory cytokines caused a 50% decrease in chronic insulin secretion (Kiely et al. 2007), which could be rescued by treatment with l‐arginine (Krause et al. 2011), which bypassed metabolic stimulus–secretion coupling due to direct membrane depolarisation. It is possible that cystine associated cell stress could be bypassed by application of agents that directly lead to membrane depolarisation and stimulation of insulin secretion (e.g. use of sulphonylurea drugs).
While we have reported herein that CTNS knockdown (similar to the CTNS expression levels of individuals heterozygous for cystinosis) resulted in a reduced level of nutrient stimulated insulin secretion, cystinotic patients are usually asymptomatic and do not develop diabetes. This apparent discrepancy may be explained by two factors: (i) there is large variability in regulation of β‐cell mass in the population. For normal glycaemic control only 20–30% of β‐cells need to be fully functional (Matveyenko & Butler, 2008); (ii) rates of β‐cell regeneration may be higher in some individuals than others, leading to a continuous supply of functionally intact new β‐cells (Bouwens & Rooman, 2005). Interestingly, individuals with cystinosis display increased β‐cell number (Milner & Wirdnam, 1982) but their level of functionality is not known. If full functionality is not achieved, then the number of pancreatic β‐cells may be increased to provide sufficient insulin to maintain the glucose homeostasis in the body.
Alterations in ATP metabolism was originally speculated as causing the cellular dysfunction in cystinotic cells based on experiments using the CDME model of cystinosis (Foreman & Benson, 1990; Coor et al. 1991). Interestingly in a study by Laube et al. (2006 ), cystinotic kidney proximal tubular cells under hypoxic conditions were not able to increase mitochondrial complex activity above basal levels in response to a low oxygen environment. In the present study, intracellular ATP levels were decreased in BRIN‐BD11 cells with silenced Ctns, while those treated with oligomycin, a mitochondrial ATP synthase inhibitor, resulted in a decrease in ATP production. This indicated that the ability of the cells to produce ATP from glycolytic sources was greatly impacted. Notably, the inhibition of glycolysis by SIA had no significant effect on already depressed ATP levels (due to decreased glycolytic flux) in cystinotic β‐cells compared to control. Similar reduced ATP levels were observed in rabbit proximal tubular cells and HK‐2 cells with silenced CTNS (Taub et al. 2011; Sumayao et al. 2013) as well as in fibroblasts and renal proximal tubular cells from individuals with cystinosis (Levtchenko et al. 2006; Sansanwal et al. 2010 c). The production of ATP from HK‐2 cells with silenced CTNS demonstrated a slight but not significant decrease in ATP following oligomycin and SIA treatment (Sumayao et al. 2013). In addition, altered mitochondrial structures have been observed in cystinotic kidney cells as well as increased ROS levels (Jackson et al. 1962; Sansanwal et al. 2010 c). Although mitochondrial derived ATP did not appear to be affected in BRIN‐BD11 cells with decreased cystinosin expression, mitochondrial DNA damage may still have been caused by the observed increase in ROS. The ability to generate ATP and alter the ATP/ADP ratio is an essential step for pancreatic β‐cells to secrete insulin in response to a nutrient stimulus (Straub & Sharp, 2002). Furthermore, the mitochondrial membrane potential was adversely affected in pancreatic β‐cells with decreased cystinosin expression. This suggests altered kinetics of electron transport and/or uncoupling of the mitochondrial proton gradient as well as the associated depolarisation of the mitochondrial inner membrane may adversely affect cells with decreased cystinosin expression. A loss in mitochondrial integrity has been observed in cystinotic renal proximal tubular epithelial cells, assessed by cytochrome C release (Park et al. 2006). In BRIN‐BD11 cells and primary mouse islets, diminished ATP levels and chronic insulin secretion were also observed following treatment with sub‐lethal concentrations of hydrogen peroxide (Michalska et al. 2010) suggesting that elevated oxidative stress can impact on mitochondrial function and the insulin secreting ability of these cells.
The pancreatic β‐cell is very sensitive to oxidative stress and the expression of catalase and peroxidase enzymes are relatively low in these cells compared to other tissues (Lenzen et al. 1996; Sakai et al. 2003). Glutathione levels are also much lower in pancreatic β‐cells compared to other cell types, suggesting they may be more at risk from oxidative stress than other cells (Numazawa et al. 2008). In BRIN‐BD11 cells with Ctns knockdown, there was a 2‐fold increase in GSSG, while total and reduced GSH remained close to control levels. Overall there was a more oxidised GSH/GSSG redox potential. These observations are similar to the increase in GSSG exhibited by cystinotic fibroblasts (Levtchenko et al. 2005) and a cystinotic proximal tubular epithelial cell line (Wilmer et al. 2005). Although, HK‐2 kidney cells with a similar level of cystinosin knockdown display decreased total GSH and GSSG (Bellomo et al. 2010; Sumayao et al. 2013), a similar change in the GSH/GSSG redox potential was observed in HK‐2 cells following CTNS knockdown (Sumayao et al. 2013). The levels of intracellular cysteine have been shown to regulate CTNS expression in response to more oxidised environments (Bellomo et al. 2010). In pancreatic β‐cells, GSH antioxidant levels can be enhanced by the availability of extracellular cysteine and cystine and it is suggested that the cystine/glutamate exchanger controls GSH levels (Numazawa et al. 2008). However, higher concentrations of cysteine can suppress insulin secretion possibly as a consequence of increased hydrogen sulphide (Kaneko et al. 2006). Although intracellular cysteine levels may be elevated in the BRIN‐BD11 cells with decreased cystinosin expression, alterations in the redox status are evident. The elevated GSSG levels in the BRIN‐BD11 cells may contribute to the reduced insulin secretion as the cells may be focusing resources on responding to an oxidative insult, which itself may also prevent insulin secretion.
The mitochondria are a major source of reactive oxygen species, through oxidative phosphorylation and energy production, and so can be more disposed to oxidative stress and damage caused by ROS. Increases in ROS and oxidative stress negatively affect pancreatic β‐cell mitochondrial function contributing to pancreatic β‐cell dysfunction in diabetes (Sakai et al. 2003). Intracellular reactive oxygen species, hydrogen peroxide and superoxide levels were all elevated in BRIN‐BD11 cells with silenced Ctns. Although HK‐2 cells with decreased cystinosin expression had a comparable increase in superoxide concentration, hydrogen peroxide levels and catalase activity were not affected, while a slight but not significant increase in ROS was detected (Sumayao et al. 2013). Increased SOD was also observed in a cystinotic fibroblast cell line (Chol et al. 2004). Intracellular hydrogen peroxide has been implicated in β‐cell dysfunction associated with diabetes (Krippeit‐Drews et al. 1999; Maechler et al. 1999), and exposure of BRIN‐BD11 cells to the saturated fatty acid palmitic acid caused elevated superoxide production and depressed insulin secretion (Keane et al. 2011). Together the observed increases in hydrogen peroxide, ROS and superoxide could be detrimental for β‐cell function and could contribute to the dysfunction observed with increased cystine accumulation leading to depressed cell function and increased cell death.
Altered apoptosis has been suggested as a potential mechanism of pathophysiology associated with cystinosis (Thoene, 2007). In BRIN‐BD11 cells with Ctns‐knockdown there was an increase in the percentage of apoptotic cells but not in dead cells. As apoptosis is a time‐dependent phenomenon, sampling at later time points may demonstrate a shift from apoptotic cells to dead cells. Elevated apoptosis levels have been detected in cystinotic proximal tubular cells and in rabbit proximal tubular cells with cystinosin knockdown (Laube et al. 2006; Sansanwal et al. 2010 c; Taub & Cutuli, 2012). Not only are basal apoptosis levels increased in cystinosis, but response to stress factors and apoptosis inducers causes a more significant response in cystinotic fibroblasts and kidney proximal tubular cells than healthy cells (Park et al. 2002; Laube et al. 2006). AMPK signalling, the energy metabolic regulator, has also been suggested as contributing to apoptosis in the cystinotic cell (Taub & Cutuli, 2012), while caspase‐4 has been implicated in the loss of proximal tubules (Sansanwal et al. 2010 a). Interestingly, when ATP levels were reduced in nephropathic cystinotic fibroblasts, cell growth was adversely affected but there was no observable increase in cell death (Mannucci et al. 2006). The observed increase in apoptosis in BRIN‐BD11 cells following cystinosin knockdown may be due to the increased levels of reactive oxygen species and an altered mitochondrial membrane potential.
Uncontrolled alterations in redox homeostasis can activate transcription factors such as NF‐κB, resulting in an increase in transcription of certain genes. NF‐κB is part of an important signalling cascade and has been implicated in the progression of diabetes (Salem et al. 2014), inflammation (Baker et al. 2011) and cancer (Tornatore et al. 2012). In BRIN‐BD11 cells with silenced Ctns, NF‐κB is activated and increased in these cells. Previously, NF‐κB gene expression was reported to be upregulated following gene expression profiling from the peripheral blood from individuals with cystinosis (Sansanwal et al. 2010 b). In pancreatic β‐cells, NF‐κB activation, which is stimulated by the pro‐inflammatory cytokines, interleukin‐1β (IL‐1β) and tumour necrosis factor‐α (TNF‐α), has been linked to apoptosis and cell death (Ortis et al. 2012). Interestingly, IL‐1β is secreted in response to cystine crystals (perhaps due to macrophage inflammasome activation) and is found to be increased in individuals with cystinosis (Prencipe et al. 2014). The altered redox state with increased oxidative species observed in the BRIN‐BD11 cells with decreased cystinosin expression may activate the NF‐κB pathway. The redox status of the cell has been suggested to activate upstream kinases that signal through the NF‐κB pathway (Pantano et al. 2006). The downstream genes activated by this pathway have not yet been investigated, but may be pro‐apoptotic. In relation to cystinosis, the impact of NF‐κB activation on genes involved in stress response is of interest (Pantano et al. 2006), including superoxide dismutase, NADPH oxidase and those involved in glutathione synthesis. The activity of superoxide dismutase has previously been identified as increased in cystinotic cell lines (Chol et al. 2004) and proximal tubular cells with decreased cystinosin expression (Sumayao et al. 2013). Alterations in the glutathione and redox status of cystinotic cells have been identified in many models of cystinosis (Chol et al. 2004; Wilmer et al. 2005; Sumayao et al. 2013). NF‐κB has also been identified as a transcription factor regulator of apoptosis, affecting genes such as CASP4 and BCL2L1 (Nakanishi & Toi, 2005), which have been found to be up‐ or down‐regulated, respectively, in the gene expression profile of individuals with cystinosis (Sansanwal et al. 2010 b).
Recent studies have described additional functions for cystinosin, over and above its role in cystine transport. The absence of cystinosin resulted in an acute disorganisation of endo‐lysosomal compartments in kidney proximal tubule cells, with clustering of endocytotic vesicles in the perinuclear region, decreased expression of multiligand receptors on the cell surface and delayed processing of the ligands. Abnormal co‐localization of late endosomal and lysosomal marker LAMP‐1 with the motor protein kinesin‐1 in cystinosin‐deficient cells was also reported, suggesting that cystinosin functions as an important regulator of endo‐lysosomal dynamics (Ivanova et al. 2015). The absence of cystinosin may lead to a defective movement of endosomes to the cell surface. In β‐cells this would severely impact on receptor‐mediated action of endocrine and neuroendocrine regulators of insulin secretion including GLP‐1 (Kuna et al. 2013).
In conclusion, a cystinosin knockdown and the associated cystine accumulation detrimentally affects the function of the pancreatic β‐cell (overview Fig. 8). The increased dysfunction and apoptosis in these cells may be a consequence of alterations in glucose sensing mechanisms (including glycolysis), or mitochondrial dysfunction caused by increased oxidative stress, and results in decreased ATP production. Insulin secretion may be affected by mitochondrial deficiencies or decreased glucose sensing and metabolism. In cystinosis, depressed pancreatic β‐cell metabolism and decreased ability to handle oxidative insults, due to an altered redox status within the cell, may result in insufficient insulin release for normal metabolic requirements, leading to hyperglycaemia and eventually diabetes. These findings suggest that even a small increase in intracellular cystine may cause stress to the pancreatic β‐cell which would make all individuals with cystinosis more susceptible to developing diabetes and insulin insufficiency, which would cause further distress to already laboured cells of the patient.
Figure 8. Suggested mechanism of pathogenesis in cystinosis .
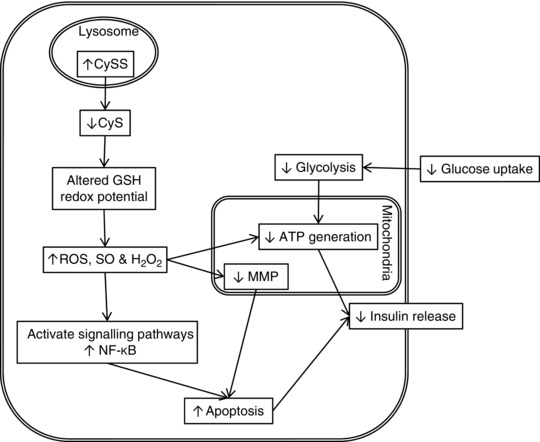
Decreased expression of the cystinosin transporter cause increased cystine (CySS) levels. This results in increased ROS, superoxide (SO) and hydrogen peroxide (H2O2) which can cause detrimental damage to the cell. In the β‐cell uncontrolled ROS/RNS could quickly overwhelm antioxidant defences and act on the mitochondria causing a decrease in ATP. This results in a decrease in insulin secretion. Increased ROS/RNS may lead to apoptosis through multiple mechanisms. Decreases in glutathione (GSH) through depletion or decreased synthesis may cause alterations in the status of GSH/GSSG couple and can activate signalling pathways, leading to augmented apoptosis. Insufficient cellular defence against ROS due to GSH depletion can lead to loss of mitochondrial membrane potential (MMP), causing apoptotic effectors to be released and activate apoptosis.
Additional information
Competing interests
None declared.
Author contributions
B.M., with contribution from R.S. and C.S., performed all experiments. B.M., T.M. and P.N. designed the study and all authors participated in the interpretation of the results. All authors contributed to writing the manuscript and to subsequent revisions. All authors approved the submission of this version to The Journal of Physiology.
Funding
This study was supported by a joint grand from Cystinosis Foundation Ireland, the Irish Health Research Board (MRCG/2008/11), Cystinosis Research Foundation USA (CRF 2012‐McMorrow‐Newsholme) and the EU SysKid project (HEALTH‐F2‐2009‐241544). P.N. is grateful to the School of Biomedical Sciences, Curtin University for research support.
Acknowledgements
We gratefully acknowledge the UCD renal disease research group for their technical assistance and scientific discussion. We also thank the School of Biomedical Sciences and Faculty of Health Sciences, Curtin University for research support.
References
- Ammenti A, Grossi A & Bernasconi S (1986). Infantile cystinosis and insulin‐dependent diabetes mellitus. Eur J Pediatr 145, 548–549. [DOI] [PubMed] [Google Scholar]
- Baker MA, Cerniglia GJ & Zaman A (1990). Microtiter plate assay for the measurement of glutathione and glutathione disulfide in large numbers of biological samples. Anal Biochem 190, 360–365. [DOI] [PubMed] [Google Scholar]
- Baker RG, Hayden MS & Ghosh S (2011). NF‐κB, inflammation, and metabolic disease. Cell Metab 13, 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellomo F, Corallini S, Pastore A, Palma A, Laurenzi C, Emma F & Taranta A (2010). Modulation of CTNS gene expression by intracellular thiols. Free Radic Biol Med 48, 865–872. [DOI] [PubMed] [Google Scholar]
- Bouwens L & Rooman I (2005). Regulation of pancreatic beta‐cell mass. Physiol Rev 85, 1255–1270. [DOI] [PubMed] [Google Scholar]
- Brennan L, Corless M, Hewage C, Malthouse JP, McClenaghan NH, Flatt PR & Newsholme P (2003). 13C NMR analysis reveals a link between l‐glutamine metabolism, d‐glucose metabolism and γ‐glutamyl cycle activity in a clonal pancreatic β‐cell line. Diabetologia 46, 1512–1521. [DOI] [PubMed] [Google Scholar]
- Brennan L, Shine A, Hewage C, Malthouse JP, Brindle KM, McClenaghan N, Flatt PR & Newsholme P (2002). A nuclear magnetic resonance‐based demonstration of substantial oxidative l‐alanine metabolism and l‐alanine‐enhanced glucose metabolism in a clonal pancreatic β‐cell line: metabolism of l‐alanine is important to the regulation of insulin secretion. Diabetes 51, 1714–1721. [DOI] [PubMed] [Google Scholar]
- Brodin‐Sartorius A, Tete MJ, Niaudet P, Antignac C, Guest G, Ottolenghi C, Charbit M, Moyse D, Legendre C, Lesavre P, Cochat P & Servais A (2011). Cysteamine therapy delays the progression of nephropathic cystinosis in late adolescents and adults. Kidney Int 81, 179–189. [DOI] [PubMed] [Google Scholar]
- Chol M, Nevo N, Cherqui S, Antignac C & Rustin P (2004). Glutathione precursors replenish decreased glutathione pool in cystinotic cell lines. Biochem Biophys Res Commun 324, 231–235. [DOI] [PubMed] [Google Scholar]
- Coor C, Salmon RF, Quigley R, Marver D & Baum M (1991). Role of adenosine triphosphate (ATP) and NaK ATPase in the inhibition of proximal tubule transport with intracellular cystine loading. J Clin Invest 87, 955–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corless M, Kiely A, McClenaghan NH, Flatt PR & Newsholme P (2006). Glutamine regulates expression of key transcription factor, signal transduction, metabolic gene, and protein expression in a clonal pancreatic beta‐cell line. J Endocrinol 190, 719–727. [DOI] [PubMed] [Google Scholar]
- Dominy JE, Hwang J & Stipanuk MH (2007). Overexpression of cysteine dioxygenase reduces intracellular cysteine and glutathione pools in HepG2/C3A cells. Am J Physiol Endocrinol Metab 293, E62–E69. [DOI] [PubMed] [Google Scholar]
- Filler G, Amendt P, von Bredow MA, Rohde W & Ehrich JH (1998). Slowly deteriorating insulin secretion and C‐peptide production characterizes diabetes mellitus in infantile cystinosis. Eur J Pediatr 157, 738–742. [DOI] [PubMed] [Google Scholar]
- Fivush B, Green OC, Porter CC, Balfe JW, O'Regan S & Gahl WA (1987). Pancreatic endocrine insufficiency in posttransplant cystinosis. Am J Dis Child 141, 1087–1089. [DOI] [PubMed] [Google Scholar]
- Foreman JW & Benson LL (1990). Effect of cystine loading on substrate oxidation by rat renal tubules. Pediatr Nephrol 4, 236–239. [DOI] [PubMed] [Google Scholar]
- Gahl WA, Balog JZ & Kleta R (2007). Nephropathic cystinosis in adults: natural history and effects of oral cysteamine therapy. Ann Intern Med 147, 242–250. [DOI] [PubMed] [Google Scholar]
- Gahl WA, Bashan N, Tietze F, Bernardini I & Schulman JD (1982. a). Cystine transport is defective in isolated leukocyte lysosomes from patients with cystinosis. Science 217, 1263–1265. [DOI] [PubMed] [Google Scholar]
- Gahl WA, Schneider JA, Thoene JG & Chesney R (1986). Course of nephropathic cystinosis after age 10 years. J Pediatr 109, 605–608. [DOI] [PubMed] [Google Scholar]
- Gahl WA, Thoene JG & Schneider JA (2001). Cystinosis: A disorder of lysosomal membrane transport In The Metabolic & Molecular Bases of Inherited Disease, 8th edn, eds Scriver CR, Beaudet AL, Sly WS. & Valle D, pp. 5085–5108. McGraw‐Hill, New York. [Google Scholar]
- Gahl WA, Thoene JG & Schneider JA (2002). Cystinosis. N Engl J Med 347, 111–121. [DOI] [PubMed] [Google Scholar]
- Gahl WA, Tietze F, Bashan N, Steinherz R & Schulman JD (1982. b). Defective cystine exodus from isolated lysosome‐rich fractions of cystinotic leucocytes. J Biol Chem 257, 9570–9575. [PubMed] [Google Scholar]
- Gaitonde MK (1967). A spectrophotometric method for the direct determination of cysteine in the presence of other naturally occurring amino acids. Biochem J 104, 627–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacco F & Brownlee M (2010). Oxidative stress and diabetic complications. Circ Res 107, 1058–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova EA, De Leo MG, Van Den Heuvel L, Pastore A, Dijkman H, De Matteis MA & Levtchenko EN (2015). Endo‐lysosomal dysfunction in human proximal tubular epithelial cells deficient for lysosomal cystine transporter cystinosin. PLoS One 10, e0120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson JD, Smith FG, Litman NN, Yuile CL & Latta H (1962). The Fanconi syndrome with cystinosis: Electron microscopy of renal biopsy specimens from five patients. Am J Med 33, 893–910. [DOI] [PubMed] [Google Scholar]
- Jones DP, Carlson JL, Mody VC, Cai J, Lynn MJ & Sternberg P (2000). Redox state of glutathione in human plasma. Free Radic Biol Med 28, 625–635. [DOI] [PubMed] [Google Scholar]
- Kaneko Y, Kimura Y, Kimura H & Niki I (2006). L‐cysteine inhibits insulin release from the pancreatic beta‐cell: possible involvement of metabolic production of hydrogen sulfide, a novel gasotransmitter. Diabetes 55, 1391–1397. [DOI] [PubMed] [Google Scholar]
- Keane DC, Takahashi HK, Dhayal S, Morgan NG, Curi R & Newsholme P (2011). Arachidonic acid actions on functional integrity and attenuation of the negative effects of palmitic acid in a clonal pancreatic beta‐cell line. Clin Sci (Lond) 120, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiely A, McClenaghan NH, Flatt PR & Newsholme P (2007). Pro‐inflammatory cytokines increase glucose, alanine and triacylglycerol utilization but inhibit insulin secretion in a clonal pancreatic beta‐cell line. J Endocrinol 195, 113–123. [DOI] [PubMed] [Google Scholar]
- Krause MS, McClenaghan NH, Flatt PR, de Bittencourt PI, Murphy C & Newsholme P (2011). L‐arginine is essential for pancreatic β‐cell functional integrity, metabolism and defense from inflammatory challenge. J Endocrinol 211, 87–97. [DOI] [PubMed] [Google Scholar]
- Krippeit‐Drews P, Kramer C, Welker S, Lang F, Ammon HP & Drews G (1999). Interference of H2O2 with stimulus–secretion coupling in mouse pancreatic β‐cells. J Physiol 514, 471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuna RS, Girada SB, Asalla S, Vallentyne J, Maddika S, Patterson JT, Smiley DL, DiMarchi RD & Mitra P (2013). Glucagon‐like peptide‐1 receptor‐mediated endosomal cAMP generation promotes glucose‐stimulated insulin secretion in pancreatic β‐cells. Am J Physiol Endocrinol Metab 305, E161–E170. [DOI] [PubMed] [Google Scholar]
- Laube GF, Shah V, Stewart VC, Hargreaves IP, Haq MR, Heales SJ & van't Hoff WG (2006). Glutathione depletion and increased apoptosis rate in human cystinotic proximal tubular cells. Pediatr Nephrol 21, 503–509. [DOI] [PubMed] [Google Scholar]
- Lenzen S, Drinkgern J & Tiedge M (1996). Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med 20, 463–466. [DOI] [PubMed] [Google Scholar]
- Levtchenko E, de Graaf‐Hess A, Wilmer M, van den Heuvel L, Monnens L & Blom H (2005). Altered status of glutathione and its metabolites in cystinotic cells. Nephrol Dial Transplant 20, 1828–1832. [DOI] [PubMed] [Google Scholar]
- Levtchenko EN, Wilmer MJ, Janssen AJ, Koenderink JB, Visch HJ, Willems PH, de Graaf‐Hess A, Blom HJ, van den Heuvel LP & Monnens LA (2006). Decreased intracellular ATP content and intact mitochondrial energy generating capacity in human cystinotic fibroblasts. Pediatr Res 59, 287–292. [DOI] [PubMed] [Google Scholar]
- McClenaghan NH, Barnett CR, Ah‐Sing E, Abdel‐Wahab YH, O'Harte FP, Yoon TW, Swanston‐Flatt SK & Flatt PR (1996). Characterization of a novel glucose‐responsive insulin‐secreting cell line, BRIN‐BD11, produced by electrofusion. Diabetes 45, 1132–1140. [DOI] [PubMed] [Google Scholar]
- McClenaghan NH & Flatt PR (1999). Engineering cultured insulin‐secreting pancreatic B‐cell lines. J Mol Med (Berl) 77, 235–243. [DOI] [PubMed] [Google Scholar]
- Maechler P, Jornot L & Wollheim CB (1999). Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic beta cells. J Biol Chem 274, 27905–27913. [DOI] [PubMed] [Google Scholar]
- Mannucci L, Pastore A, Rizzo C, Piemonte F, Rizzoni G & Emma F (2006). Impaired activity of the gamma‐glutamyl cycle in nephropathic cystinosis fibroblasts. Pediatr Res 59, 332–335. [DOI] [PubMed] [Google Scholar]
- Matveyenko AV & Butler PC (2008). Relationship between beta‐cell mass and diabetes onset. Diabetes Obes Metab 10 (Suppl. 4), 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalska M, Wolf G, Walther R & Newsholme P (2010). Effects of pharmacological inhibition of NADPH oxidase or iNOS on pro‐inflammatory cytokine, palmitic acid or H2O2‐induced mouse islet or clonal pancreatic β‐cell dysfunction. Biosci Rep 30, 445–453. [DOI] [PubMed] [Google Scholar]
- Milner RD & Wirdnam PK (1982). The pancreatic beta cell fraction in children with errors of amino acid metabolism. Pediatr Res 16, 213–216. [DOI] [PubMed] [Google Scholar]
- Mullooly N, Vernon W, Smith DM & Newsholme P (2014). Elevated levels of branched‐chain amino acids have little effect on pancreatic islet cells, but l‐arginine impairs function through activation of the endoplasmic reticulum stress response. Exp Physiol 99, 538–551. [DOI] [PubMed] [Google Scholar]
- Nakanishi C & Toi M (2005). Nuclear factor‐κB inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer 5, 297–309. [DOI] [PubMed] [Google Scholar]
- Nesterova G & Gahl W (2008). Nephropathic cystinosis: late complications of a multisystemic disease. Pediatr Nephrol 23, 863–878. [DOI] [PubMed] [Google Scholar]
- Newsholme P, Rebelato E, Abdulkader F, Krause M, Carpinelli A & Curi R (2012). Reactive oxygen and nitrogen species generation, antioxidant defenses, and β‐cell function: a critical role for amino acids. J Endocrinol 214, 11–20. [DOI] [PubMed] [Google Scholar]
- Numazawa S, Sakaguchi H, Aoki R, Taira T & Yoshida T (2008). Regulation of the susceptibility to oxidative stress by cysteine availability in pancreatic β‐cells. Am J Physiol Cell Physiol 295, C468–C474. [DOI] [PubMed] [Google Scholar]
- Ortis F, Miani M, Colli ML, Cunha DA, Gurzov EN, Allagnat F, Chariot A & Eizirik DL (2012). Differential usage of NF‐κB activating signals by IL‐1β and TNF‐α in pancreatic beta cells. FEBS Lett 586, 984–989. [DOI] [PubMed] [Google Scholar]
- Pantano C, Reynaert NL, van der Vliet A & Janssen‐Heininger YM (2006). Redox‐sensitive kinases of the nuclear factor‐κB signaling pathway. Antioxid Redox Signal 8, 1791–1806. [DOI] [PubMed] [Google Scholar]
- Park M, Helip‐Wooley A & Thoene J (2002). Lysosomal cystine storage augments apoptosis in cultured human fibroblasts and renal tubular epithelial cells. J Am Soc Nephrol 13, 2878–2887. [DOI] [PubMed] [Google Scholar]
- Park MA, Pejovic V, Kerisit KG, Junius S & Thoene JG (2006). Increased apoptosis in cystinotic fibroblasts and renal proximal tubule epithelial cells results from cysteinylation of protein kinase Cδ. J Am Soc Nephrol 17, 3167–3175. [DOI] [PubMed] [Google Scholar]
- Prencipe G, Caiello I, Cherqui S, Whisenant T, Petrini S, Emma F & De Benedetti F (2014). Inflammasome activation by cystine crystals: implications for the pathogenesis of cystinosis. J Am Soc Nephrol 25, 1163–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokai A, Fekete A, Kis E, Reusz GS, Sallay P, Korner A, Wagner L, Tulassay T & Szabo AJ (2008). Post‐transplant diabetes mellitus in children following renal transplantation. Pediatr Transplant 12, 643–649. [DOI] [PubMed] [Google Scholar]
- Robert JJ, Tete MJ, Guest G, Gagnadoux MF, Niaudet P & Broyer M (1999). Diabetes mellitus in patients with infantile cystinosis after renal transplantation. Pediatr Nephrol 13, 524–529. [DOI] [PubMed] [Google Scholar]
- Sakai K, Matsumoto K, Nishikawa T, Suefuji M, Nakamaru K, Hirashima Y, Kawashima J, Shirotani T, Ichinose K, Brownlee M & Araki E (2003). Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic beta‐cells. Biochem Biophys Res Commun 300, 216–222. [DOI] [PubMed] [Google Scholar]
- Salem HH, Trojanowski B, Fiedler K, Maier HJ, Schirmbeck R, Wagner M, Boehm BO, Wirth T & Baumann B (2014). Long‐term IKK2/NF‐κB signaling in pancreatic β‐cells induces immune‐mediated diabetes. Diabetes 63, 960–975. [DOI] [PubMed] [Google Scholar]
- Salvucci M, Neufeld Z & Newsholme P (2013). Mathematical model of metabolism and electrophysiology of amino acid and glucose stimulated insulin secretion: in vitro validation using a β‐cell line. PLoS One 8, e52611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansanwal P, Kambham N & Sarwal MM (2010. a). Caspase‐4 may play a role in loss of proximal tubules and renal injury in nephropathic cystinosis. Pediatr Nephrol 25, 105–109. [DOI] [PubMed] [Google Scholar]
- Sansanwal P, Li L, Hsieh SC & Sarwal MM (2010. b). Insights into novel cellular injury mechanisms by gene expression profiling in nephropathic cystinosis. J Inherit Metab Dis 33, 775–786. [DOI] [PubMed] [Google Scholar]
- Sansanwal P, Yen B, Gahl WA, Ma Y, Ying L, Wong LJ & Sarwal MM (2010. c). Mitochondrial autophagy promotes cellular injury in nephropathic cystinosis. J Am Soc Nephrol 21, 272–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfmann R, Rachdi L & Ravassard P (2013). Concise review: in search of unlimited sources of functional human pancreatic beta cells. Stem Cells Transl Med 2, 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner DJ, Kim A, Miller K & Hara M (2010). Pancreatic islet plasticity: interspecies comparison of islet architecture and composition. Islets 2, 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub SG & Sharp GW (2002). Glucose‐stimulated signaling pathways in biphasic insulin secretion. Diabetes Metab Res Rev 18, 451–463. [DOI] [PubMed] [Google Scholar]
- Sumayao R, McEvoy B, Martin‐Martin N, McMorrow T & Newsholme P (2013). Cystine dimethylester loading promotes oxidative stress and a reduction in ATP independent of lysosomal cystine accumulation in a human proximal tubular epithelial cell line. Exp Physiol 98, 1505–1517. [DOI] [PubMed] [Google Scholar]
- Taranta A, Petrini S, Citti A, Boldrini R, Corallini S, Bellomo F, Levtchenko E & Emma F (2012). Distribution of cystinosin‐LKG in human tissues. Histochem Cell Biol 138, 351–363. [DOI] [PubMed] [Google Scholar]
- Taub M & Cutuli F (2012). Activation of AMP kinase plays a role in the increased apoptosis in the renal proximal tubule in cystinosis. Biochem Biophys Res Commun 426, 516–521. [DOI] [PubMed] [Google Scholar]
- Taub ML, Springate JE & Cutuli F (2011). Reduced phosphate transport in the renal proximal tubule cells in cystinosis is due to decreased expression of transporters rather than an energy defect. Biochem Biophys Res Commun 407, 355–359. [DOI] [PubMed] [Google Scholar]
- Theodoropoulos DS, Krasnewich D, Kaiser‐Kupfer MI & Gahl WA (1993). Classic nephropathic cystinosis as an adult disease. JAMA 270, 2200–2204. [PubMed] [Google Scholar]
- Thoene JG (2007). A review of the role of enhanced apoptosis in the pathophysiology of cystinosis. Mol Genet Metab 92, 292–298. [DOI] [PubMed] [Google Scholar]
- Tornatore L, Thotakura AK, Bennett J, Moretti M & Franzoso G (2012). The nuclear factor kappa B signaling pathway: integrating metabolism with inflammation. Trends Cell Biol 22, 557–566. [DOI] [PubMed] [Google Scholar]
- Wilmer MJ, Christensen EI, van den Heuvel LP, Monnens LA & Levtchenko EN (2008). Urinary protein excretion pattern and renal expression of megalin and cubilin in nephropathic cystinosis. Am J Kidney Dis 51, 893–903. [DOI] [PubMed] [Google Scholar]
- Wilmer MJ, de Graaf‐Hess A, Blom HJ, Dijkman HB, Monnens LA, van den Heuvel LP & Levtchenko EN (2005). Elevated oxidized glutathione in cystinotic proximal tubular epithelial cells. Biochem Biophys Res Commun 337, 610–614. [DOI] [PubMed] [Google Scholar]
- Wilmer MJ, Emma F & Levtchenko EN (2010). The pathogenesis of cystinosis: mechanisms beyond cystine accumulation. Am J Physiol Renal Physiol 299, F905–F916. [DOI] [PubMed] [Google Scholar]