

Abstract
Key points
Cardiomyocytes withdraw from the cell cycle late in prenatal and early in postnatal life and subsequent heart growth occurs via cellular hypertrophy. The signals regulating this transition remain unknown.
Lesion of the neonatal sympathetic nervous system results in decreased heart size.
In vitro investigations show that sympathetic neurons delay cardiomyocyte maturation and cell cycle withdrawal.
Early sympathetic innervation may contribute to the determination of adult heart size by regulating the total number of cardiomyocytes via regulation of the proliferative/hypertrophic transition.
Early perturbations of sympathetic structure or function could have long‐term effects on adult heart function.
Abstract
Sympathetic drive to the heart is a key modulator of cardiac function and interactions between heart tissue and innervating sympathetic fibres are established early in development. Significant innervation takes place during postnatal heart development, a period when cardiomyocytes undergo a rapid transition from proliferative to hypertrophic growth. The question of whether these innervating sympathetic fibres play a role in regulating the modes of cardiomyocyte growth was investigated using 6‐hydroxydopamine (6‐OHDA) to abolish early sympathetic innervation of the heart. Postnatal chemical sympathectomy resulted in rats with smaller hearts, indicating that heart growth is regulated by innervating sympathetic fibres during the postnatal period. In vitro experiments showed that sympathetic interactions resulted in delays in markers of cardiomyocyte maturation, suggesting that changes in the timing of the transition from hyperplastic to hypertrophic growth of cardiomyocytes could underlie changes in heart size in the sympathectomized animals. There was also an increase in the expression of Meis1, which has been linked to cardiomyocyte cell cycle withdrawal, suggesting that sympathetic signalling suppresses cell cycle withdrawal. This signalling involves β‐adrenergic activation, which was necessary for sympathetic regulation of cardiomyocyte proliferation and hypertrophy. The effect of β‐adrenergic signalling on cardiomyocyte hypertrophy underwent a developmental transition. While young postnatal cardiomyocytes responded to isoproterenol (isoprenaline) with a decrease in cell size, mature cardiomyocytes showed an increase in cell size in response to the drug. Together, these results suggest that early sympathetic effects on proliferation modulate a key transition between proliferative and hypertrophic growth of the heart and contribute to the sympathetic regulation of adult heart size.
Abbreviations
- DIV
days in vitro
- 6‐OHDA
6‐hydroxydopamine
- P
postnatal day
- PH3
phospho‐histone H3
- WGA
wheat germ agglutinin
Introduction
The embryonic heart grows primarily through hyperplasia, or cellular proliferation. However, the proliferative period of cardiomyocytes is limited and rat cardiomyocytes withdraw from the cell cycle shortly after birth (Li et al. 1996). Following cell cycle withdrawal, heart size increases predominantly via cellular hypertrophy, or the enlargement of individual cardiomyocytes (Ahuja et al. 2007). While many organs retain the ability to replenish cell number throughout the lifespan, the regenerative capacity of cardiomyocytes is, at best, limited (Zak, 1973). A population of cardiac stem cells has been putatively identified; however, the ability of these cells to reliably regenerate cardiac tissue following injury is still an area of debate and active investigation (Murry et al. 2004; Bergmann et al. 2009). Thus, the number of postnatal cardiomyocyte divisions, and the timing of the developmental transition between proliferative and hypertrophic growth in the young animal are significant factors for determining the total number of cardiomyocytes in the adult heart. In turn, the proper number of cardiomyocytes is critical for normal heart function, with either an increase (Chang et al. 2010) or decrease (Levkau et al. 2008) in cardiomyocyte proliferation resulting in heart failure. Thus, early determination of cardiomyocyte cell number regulates the ability of the heart to maintain its output in a physiologically relevant range.
Several regulators of cardiomyocyte proliferation have been identified, including ALMS1 (Zulato et al. 2011), CASZ1 (Dorr et al. 2015) and the homeodomain transcription factor, Meis1 (Mahmoud et al. 2013). Knockout of Meis1 leads to an extended period of cardiomyocyte proliferation, while overexpression leads to a decrease in cardiomyocyte number. These findings position Meis1 as a potential master regulator of postnatal cardiomyocyte cell cycle arrest, and one that has the potential to interact with signals that coordinate cardiomyocyte development. However, the factors that drive the signalling pathway that result in transcriptional regulation by Meis1 are not known.
The sympathetic nervous system is one system that provides an important pathway for developmental signalling in the heart. The innervation of the heart by sympathetic neurons takes place late in the prenatal and early postnatal period, a period marked by the functional maturation of sympathetic neurons, and the establishment of sympathetic drive to the heart (Dowell, 1985). The role of sympathetic signalling in heart development is well established. Mice lacking the ability to synthesize noradrenaline (norepinephrine) die in utero, probably as a result of massive cardiac defects (Thomas et al. 1995). Additionally, noradrenergic signalling in other systems is a known regulator of gene regulation (Briest et al. 2003; Yang et al. 2006) and cellular proliferation (Bevan, 1975). Cardiomyocytes undergo the transition from hyperplastic to hypertrophic growth during the late prenatal and early postnatal period, as innervating fibres from the sympathetic nervous system reach the heart, raising the possibility of a regulatory role of these fibres in cardiomyocyte cell cycle arrest. While a role for sympathetic signalling in the cardiac hyperplastic–hypertrophic transition has not been established, the developmental actions of the sympathetic system suggest a potential role in the regulation of this important developmental transition.
We investigated whether sympathetic innervation of the heart regulates the developmental transition between proliferation and hypertrophy in neonatal cardiomyoyctes. We found that an in vivo developmental lesion of the sympathetic nervous system resulted in a decrease in heart size that could potentially be accounted for by a delay in cardiomyocyte cell cycle arrest via a β‐adrenergic signalling pathway. Sympathetic innervation also reduced expression of cardiac Meis1, identifying a potential genetic mechanism for sympathetic regulation of cardiomyocyte proliferation. We also found that β‐adrenergic signalling influenced cardiomyocyte hypertrophy in a developmentally dependent manner, consistent with adult pathological responses. These data implicate the sympathetic nervous system as a regulator in the developmental transition from hyperplastic to hypertrophic growth and for setting the total number of cardiomyocytes in the adult heart. The finding of a developmentally changing role for β‐adrenergic signalling in cardiomyocyte proliferation and hypertrophy suggests that state‐specific effects of sympathetic drive could contribute to pathological cardiac hypertrophy in the adult.
Methods
Ethical approval
All tissue was obtained from male and female Sprague–Dawley rats aged from postnatal day (P) 1 to 8 weeks in accordance with the Brandeis University Institutional Animal Care and Use Committee.
Animals
Chemical sympathectomy
Chemical sympathectomies were performed on neonatal rats (P0) with a single intraperitoneal injection of 100 mg kg−1 6‐hydroxydopamine (6‐OHDA; Sigma, St Louis, MO, USA) in 0.9% saline solution with 1 mg ml−1 ascorbic acid. Control animals were given a sham injection of the same saline solution without the 6‐OHDA. This treatment protocol has previously (Angeletti & Levi‐Montalcini, 1970; Kostrzewa & Jacobowitz, 1974) been shown to result in extensive fibre loss across the entire sympathetic nervous system of neonatal rat pups. Denervation can be variable across different organ systems; in this work we analysed cardiac innervation following lesion. Pups were killed 48 h after birth/injection (P2), 1 week after birth/injection (P7), or 8 weeks after birth/injection. Body weight and heart weight were assayed for each animal at the time they were killed.
Isolation and culture of sympathetic neurons and cardiomyocytes
Sympathetic neurons were isolated from the superior cervical ganglia and cardiomyocytes were isolated from the ventricles of P1–P3 rats and were cultured as previously described (Lockhart et al. 1997; Luther & Birren, 2006).
Pharmacological treatments
β‐Adrenergic signalling was manipulated using the β‐adrenergic agonist isoproterenol (isoprenaline; 10 μm) and the β‐adrenergic antagonist propranolol (2 μm). α‐Adrenergic signalling was manipulated with the α‐adrenergic agonist phenylephrine (50 μm) and the α‐adrenergic antagonist prazosin (20 μm). β1‐Adrenergic signalling was blocked with the β1‐specific antagonist bisoprolol (1 mm) and β2‐adrenergic signalling was antagonized with ICI‐118,551 (0.1 μm). Cholinergic signalling was manipulated with the cholinergic agonist muscarine (2.5 μm) and the cholinergic antagonist scopolamine (10 μm, all from Sigma). Concentrations of pharmacological agents were set at pharmacologically relevant doses, as shown in previously published cardiomyocyte in vitro work, both from our lab and others (Bristow et al. 1986; Iwaki et al. 1990; Zimmer et al. 1995; Yang et al. 2002; Luther et al. 2013).
Immunochemistry
Immunohistochemistry
Whole hearts were fixed in 4% paraformaldehyde overnight. The hearts were submerged in a solution of 30% sucrose overnight and flash frozen in Tissue‐Tek OCT media (VWR, Radnor, PA, USA). Sections, 15 μm thick, were cut on a cryostat (Leica CM3050, Buffalo Grove, IL, USA). Sections were matched using anatomical landmarks. All immunohistochemistry was performed at room temperature. Sections were rehydrated in PBS for 30 min and treated for 30 min with 10 mg ml−1 sodium borohydride to reduce autofluorescence.
Sympathetic innervation of heart tissue
To assess the extent of sympathetic innervation in 6‐OHDA and sham‐treated hearts, sections were permeabilized with 0.1% Triton X‐100 in 10% donkey serum for 30 min, followed by 30 min of blocking in 10% donkey serum. Sections were incubated with primary antibody (anti‐tyrosine hydroxylase; EMD Millipore, Darmstadt, Germany; 1:500) overnight at room temperature. Following a 30 min wash with PBS, sections were incubated for 45 min in the dark with secondary antibodies (donkey anti‐rabbit Cy5; Jackson Immunoresearch, West Grove, PA, USA; 1:500). Slides were rinsed for 30 min in PBS and allowed to dry before affixing a coverslip with Fluromount G (Southern Biotech, Birmingham, AL, USA). Stained sections were kept at −20°C and imaged within 48 h. At least five images were taken from at least two different sections from each of three different hearts at each condition and age. Exposure time was kept constant between hearts that were processed together at each age. Images were analysed and innervation density was calculated using the NeuronJ plug‐in for ImageJ (NIH, Bethesda, MD, USA).
Cell size
Cardiomyocyte cell size in vivo was determined in matched sections. Sections were rinsed with Hanks’ balanced salt solution (HBSS; Sigma, St. Louis, MO, USA) for 30 min and then incubated with 10 μg ml−1 wheat germ agglutinin (WGA; Life Technologies, Carlsbad, CA, USA) for 10 min at room temperature in the dark. Slides were rinsed for 30 min with PBS and allowed to dry before affixing a coverslip, as above. At least five images from at least three different sections from each of three different hearts in each condition and at each age were captured and at least 10 cardiomyocytes from each image were analysed using ImageJ software. Cardiomyocytes were defined as large cells that had a clearly defined cross section, an identification that was confirmed by double‐labelling some sections with WGA and α‐actinin, which is specific for the developing cardiomyocytes in our cultures as assessed by cardiomyocyte morphology and the presence of sarcomeric structure.
Immunocytochemistry
Cultures were fixed for 10 min in 4% paraformaldehyde. Cells were permeablized for 10 min in a solution of 1% normal donkey serum and 0.1% NP‐40 in PBS. Ten per cent normal donkey serum was used to block non‐specific antibody binding. Primary antibody was applied in 1% normal donkey serum either overnight at 4°C or between 1 and 3 h at room temperature. No detectable difference was observed in the outcomes of the different staining protocols. Antibodies were used at the following concentrations: α‐actinin (Sigma;1:800); peripherin (EMD Millipore, Darmstadt, Germany; 1:1000); phospho‐histone H3 (Cell Signaling Technology, Danvers, MA, USA; 1:1500). Following 3 × 10 min PBS washes, secondary antibodies, also in 1% donkey serum, were applied in the dark for 45 min at room temperature (donkey anti‐mouse Rhodamine, donkey anti‐rabbit FITC; Jackson Immunoresearch) and used at a 1:600 dilution. Cultures were rinsed with 3 × 10 min PBS and 4′,6‐diamidino‐2‐phenylindole (DAPI, Life Technologies; 1 μg ml−1) was added to one of the final 10 min PBS rinses to visualize nuclei.
Cellular hypertrophy
Cardiomyocytes, as identified by α‐actinin staining, were imaged using Volocity software (PerkinElmer, Waltham, MA, USA) on an Olympus IX‐81 microscope. Cell size was measured using ImageJ software. For each experiment, 20 cardiomyocytes per dish were analysed from two culture dishes and counted as a single independent preparation. This was repeated at each time point for at least three independent preparations.
Cellular proliferation
Random fields containing cardiomyocytes were captured using Volocity. Rates of cellular proliferation were calculated as the percentage of cardiomyocyte nuclei double‐labelled for both DAPI and phospho‐histone H3. For each experiment, 60–70 random fields were imaged from two dishes and the images containing cardiomyocytes (approximately 200–300 cardiomyocytes per independent preparation) were analysed and counted as a single independent preparation. This was repeated at each time point for at least three independent preparations.
Binucleation
Prior to immunostaining, cellular membranes were visualized with Alexa Fluor 680‐conjugated wheat germ agglutinin (WGA). Cells were washed with HBSS 3 × 10 min, incubated with WGA (10 μg ml−1) for 10 min and then washed with HBSS for another 3 × 10 min. They were then stained as above for α‐actinin. Random fields containing cardiomyocytes were analysed for the percentage of binucleated cardiomyocytes per dish as described for cellular proliferation experiments (above). For each experiment, 60–70 random fields were imaged from two dishes and the images containing cardiomyocytes were analysed and counted as a single independent preparation. This was repeated at each time point at least 3 times from separate preparations.
Gene expression
Meis1 expression was assayed using real time RT–PCR. Portions of P2, P7 and 8 week ventricles were flash frozen in liquid nitrogen and stored at −80°C until used. RNA was extracted using a Direct‐zol RNA MiniPrep Kit (Zymo Research, Irvine, CA, USA). Reverse transcriptase PCR was performed as previously described (Moon & Birren, 2008). Pre‐validated TaqMan primers were used to assay levels of Meis1, using glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) as a reference gene (Life Technologies). We ran real‐time PCR assays on a Rotogene6000 real‐time PCR machine (Corbett Life Sciences, Qiagen, Valencia, CA, USA) and quantified data as previously described (Ramakers et al. 2003).
Statistical analysis
All data are presented as the mean of at least three independent experiments (n ≥ 3) with the standard error of the mean (SEM) indicated by error bars. Statistical significance was determined using Student's t test (for two groups) or ANOVA with the appropriate post hoc test (for more than two groups). Data were compiled using Excel 2013 software and analysed using Excel (version 2013 for Windows; Microsoft, Seattle, WA, USA) or the Statistical Package for Social Sciences (version 13.0 for Windows; SPSS, Chicago, IL, USA).
Results
Early sympathetic innervation coordinates cardiac development in vivo
Postnatal heart development takes place during a period of active innervation by the sympathetic nervous system. We investigated the role of sympathetic innervation in the postnatal development of the heart by inducing selective neonatal lesions of the developing sympathetic nervous system. Neonatal rat pups were injected with 6‐hydroxydopamine (6‐OHDA) or with saline, and heart weight and body weight were analysed 2 days, 7 days and 8 weeks following the lesion. Hearts from lesioned animals were significantly smaller than hearts from sham‐injected animals as early as P2 (Fig. 1 A and B; (5.8 ± 0.2) × 10−2 g vs. (6.6 ± 0.2) × 10−2 g, mean ± SEM, n = 13, P = 0.01) and this difference persisted at both 7 days and 8 weeks post‐injection (Fig. 1 A and B; P7: (11.5 ± 0.5) × 10−2 g vs. (13.7 ± 0.6) × 10−2 g, n = 14, P = 0.006; 8 weeks: 1.2 ± 0.1 g vs. 1.5 ± 0.3 g, n = 7, P = 0.05). This difference cannot be attributed to a lesion‐induced decrease in overall body size, as there was no significant difference in body weight between lesioned and control animals (Fig. 1 C; P2: 10.00 ± 0.19 g vs. 10.05 ± 0.19 g, P7: 20.34 ± 0.78 g vs. 19.96 ± 0.50 g, 8 weeks: 322.61 ± 51.91 g vs. 312.47 ± 27.79 g). When normalized to body weight, we found that while, as expected, the total heart weight:body weight ratio decreased over the 8 week period in both the lesioned and control animals, a significant decrease was seen in the lesioned animals compared to the sham‐treated controls at all ages tested (Fig. 1 D; P2: (5.9 ± 0.2) × 10−3 vs. (6.5 ± 0.2) × 10−3, n = 14, P = 0.04; P7: (5.6 ± 0.2) × 10−3 vs. (6.3 ± 0.3) × 10−3, n = 14, P = 0.03; 8 weeks: (3.7 ± 0.2) × 10−3 vs. (4.5 ± 0.3) × 10−3, n = 7, P = 0.01).
Figure 1. Decreased heart size following 6‐OHDA‐induced neonatal chemical sympathectomy .
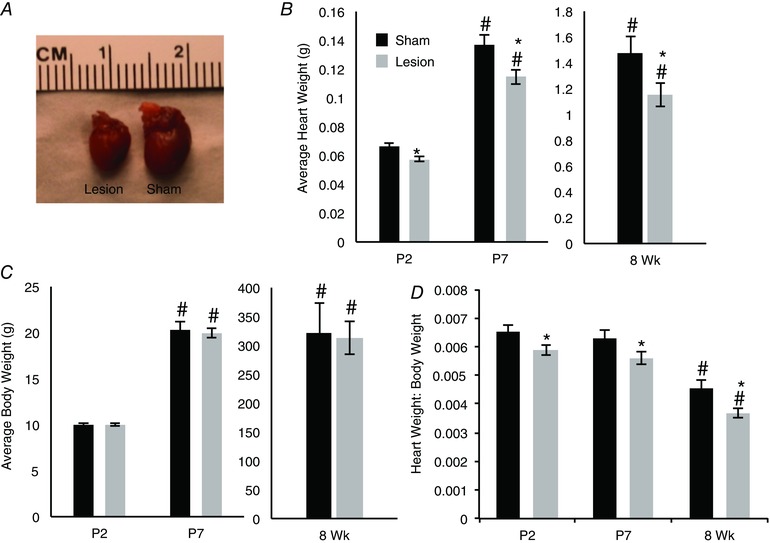
A, hearts from 1‐week‐old rats injected with 6‐OHDA at P0 were visibly smaller than hearts from sham‐injected animals. B, a time course of heart weight in animals injected with a saline vehicle control (black) or 6‐OHDA (grey) shows a reduction in heart weight in chemically sympathectomized animals compared to age‐matched vehicle controls. C, there was no difference in average body weight of rat pups that received either an injection of saline (black) or 6‐OHDA (grey). D, the difference in heart weight alone resulted in a significant decrease in the heart weight to body weight ratio (heart weight:body weight) in the lesioned animals. (P2 n = 13; P7 n = 14; 8 week (Wk) n = 7; *P < 0.05 vs. age‐matched control; # P < 0.05 vs.P2 within condition.)
We confirmed that the decreased heart size in 6‐OHDA‐treated animals was associated with an effective lesion of the sympathetic system by examining the density of sympathetic innervation of the heart at P2, P7 and 8 weeks following lesion. Sympathetic nerve fibre density was determined by staining heart sections for tyrosine hydroxylase (TH) and analysing fibre length in matched sections of left ventricles for 6‐OHDA‐treated animals and sham‐injected controls (Fig. 2 A and B). We found that a single injection of 6‐OHDA at P0 (see Methods) was sufficient to disrupt left ventricular sympathetic innervation to a level below our ability to detect it at P2 and P7. Modest, but measurable, sympathetic reinnervation was observed by 8 weeks post‐injection (Fig. 2 C; P2: (9.9 ± 0.5) × 10−3 μm μm−2 vs. 0 ± 0 μm μm−2, n = 3, P = 9.2 × 10−5; P7: (4.2 ± 0.2) × 10−3 μm μm−2 vs. 0 ± 0 μm μm−2, n = 3, P = 0.0001; 8 weeks: 2.7 × 10−3 ± 8.4 × 10−5 μm μm−2 vs. (0.6 ± 0.5) × 10−3 μm μm−2, n = 3, P = 0.02). We also observed a significant decrease in overall sympathetic innervation density through development, which can be attributed to the size of the heart increasing disproportionately with respect to increases in sympathetic innervation. These data demonstrate that loss of sympathetic innervation is associated with the development of smaller hearts.
Figure 2. Neonatal injection with 6‐OHDA depletes sympathetic innervation of the heart .
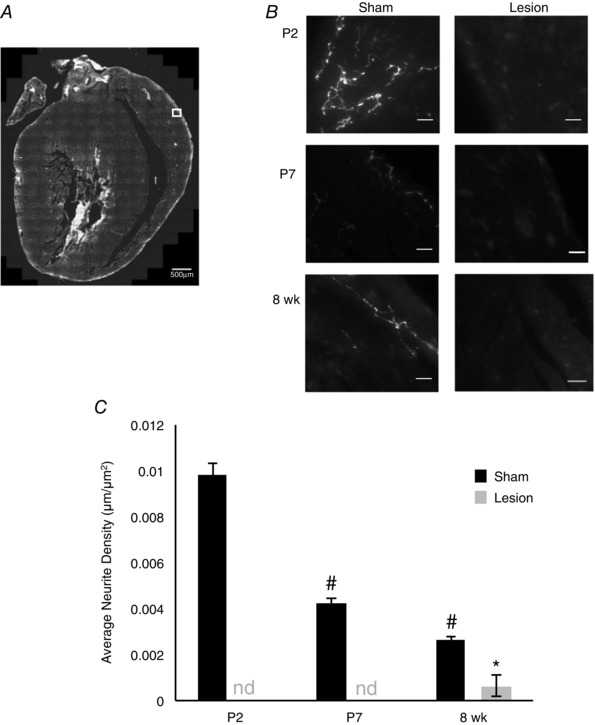
A, representative composite image of a section from a P2 heart. Hearts were sectioned longitudinally and innervation within matched images of the anterior epicardium of the left ventricle was analysed (box) using ImageJ (scale bar = 500 μm). B, representative images of matched sections stained with tyrosine hydroxylase (TH). TH staining was quantified to determine the innervation density in sham (left) and lesioned (right) hearts at P2 (top), P7 (middle) and 8 weeks (bottom). Sympathetic innervation was not detectable in lesioned animals at P2 and P7 and significantly reduced at 8 weeks. (Scale bar = 10 μm.) C, bar plot showing quantification of left ventricular sympathetic nerve density for sham (black) or 6‐OHDA (grey) injected animals demonstrating that injection with 6‐OHDA significantly decreases sympathetic innervation in the heart. (n = 3; nd, not detectable; *P < 0.05 vs. age‐matched sham; # P < 0.05 vs. P2 and P7 within condition.)
Sympathetic innervation delays cardiomyocyte maturation in culture
The proliferative window for developing rat cardiomyocytes closes during the postnatal period of sympathetic innervation. During the final division, karyokinesis becomes decoupled from cytokinesis, resulting in binucleated cardiomyocytes that grow via cellular hypertrophy (Thornburg et al. 2011). This transition provides a possible site of regulation of heart size by the sympathetic nervous system. We assessed three markers of postnatal cardiomyocyte maturation: cell cycle withdrawal, binucleation and size, for cardiomyocytes grown in the presence or absence of co‐cultured postnatal sympathetic neurons. For each analysis, cardiomyocytes and sympathetic neurons were co‐cultured for 4 or 5 days and the cultures were fixed and stained for α‐actinin to identify the cardiomyocytes (Fig. 3 A, C and E). Proliferating cardiomyocytes were identified via double‐labelling for phospho‐histone H3 (PH3) staining (Fig. 3 A) and the percentage of PH3‐positive (PH3+) cardiomyocyte nuclei were compared between conditions. We found a greater percentage of proliferating cardiomyocytes in dishes co‐cultured with sympathetic neurons than for cardiomyocytes grown in the absence of neurons (Fig. 3 B; 10.4 ± 0.012% vs. 4.6 ± 0.01%, n = 4, P = 0.01). This suggests that sympathetic innervation acts to maintain cardiomyocytes in a proliferative state and is consistent with a role for innervating sympathetic fibres in delaying cardiomyocyte maturation.
Figure 3. Sympathetic innervation of cardiomyocytes in culture inhibits cardiomyocyte maturation .
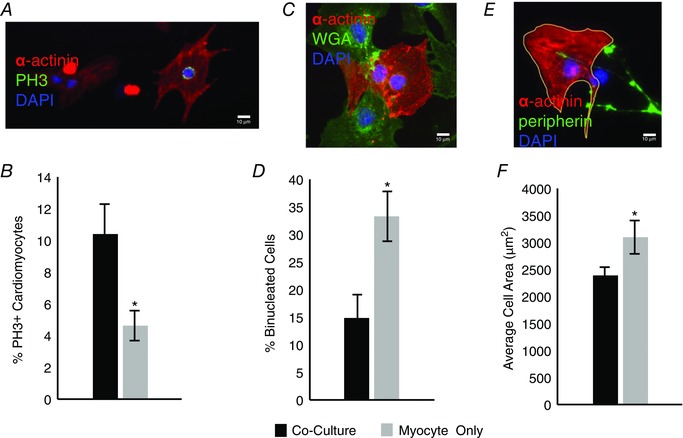
A, representative image showing a proliferating cardiomyocyte double‐labelled with α‐actinin and phospho‐histone H3 (PH3; scale bar = 10 μm). B, bar plot showing a higher rate of proliferation for cardiomyoctes co‐cultured with sympathetic neurons for 5 days (black) than for cardiomyocytes grown in isolation (grey; n = 4). C, representative image showing a binucleated cardiomyocyte identified by α‐actinin, staining of cell membranes with wheat germ agglutinin (WGA), and DAPI nuclear staining (scale bar = 10 μm). D, bar plot showing co‐cultures (black) had a decreased number of binucleated cardiomyocytes compared to myocyte‐only cultures (grey; n = 3). E, representative image showing a cardiomyocyte, visualized with α‐actinin staining, outlined with ImageJ, neurons visualized with peripherin, and DAPI nuclear staining (scale bar = 10 μm). F, cardiomyocytes co‐cultured with sympathetic neurons for 4 days (black) were smaller than cardiomyocytes that were cultured for the same period in isolation (grey; n = 3; *P < 0.05 vs. co‐culture.)
We also examined the appearance of binucleated cells as an additional marker of cardiomyocyte maturation. Cells were triple‐labelled with α‐actinin to identify cardiomyocytes, fluorescently conjugated wheat germ agglutinin (WGA) to delineate the membranes of individual cells, and DAPI to label nuclei (Fig. 3 C). We counted the percentage of individual cardiomyocytes containing two DAPI‐labelled nuclei and found significantly more binucleated cardiomyocytes in myocyte‐only cultures than in co‐cultures (Fig. 3 D; 33.3 ± 0.04% vs. 14.7 ± 0.04%, n = 3, P = 0.02), consistent with a role for the innervating neurons in delaying the final round of cell division.
If cardiomyocytes mature later in the presence of innervating neurons, we would expect the cells to show reduced hypertrophic growth in the presence of co‐cultured sympathetic neurons. We tested this by culturing cardiomyocytes in the presence and absence of sympathetic neurons for 4 days, staining for α‐actinin, and using ImageJ software to measure the area of individual cardiomyocytes independent of their proliferative state. We found that cardiomyocytes grown in the presence of sympathetic neurons were significantly smaller than those grown in isolation (Fig. 3 F; 2380.9 ± 153.0 μm2 vs. 3093.0 ± 69.5 μm2, n = 3, P = 0.01), indicating that sympathetic innervation limits hypertrophic growth while maintaining the proliferative capacity of cardiomyocytes.
Sympathetic neurons act via β‐adrenergic signalling to regulate the cardiomyocyte transition from hyperplasia to hypertrophy
We asked whether noradrenergic sympathetic neurotransmission underlies the sympathetic regulation of cardiomyocyte proliferation and hypertrophy. Cardiomyocytes grown in the presence of sympathetic neurons were treated with propranolol, a β‐adrenergic antagonist, to determine if blocking β‐adrenergic transmission occluded the effects of sympathetic fibres on cardiomyocyte proliferation. In addition, cardiomyocytes grown in the absence of neurons were treated with isoproterenol, a β‐adrenergic agonist, to determine the effects of pharmacologically triggering the signalling pathway. We found that treatment of co‐cultures with 2 μm propranolol for 5 days decreased proliferation of the co‐cultured cardiomyocytes, as measured by the percentage of cardiomyocytes staining for PH3 (Fig. 4 A; 3.49 ± 0.95% vs. 6.61 ± 0.75%, n = 5, P = 0.03), consistent with a role of β‐adrenergic signalling in maintaining cardiomyocyte proliferation. We did not observe any independent effect of the propranolol on myocyte‐only cultures (data not shown). We also observed increased proliferation of cardiomyocytes cultured in the presence of sympathetic neurons compared to cardiomyocytes grown alone, consistent with the results shown in Fig. 3. However, we observed no significant effect on proliferation when myocyte‐only cultures were treated with 10 μm isoproterenol for the same period (3.24 ± 0.006% vs. 2.05 ± 0.003%, n = 5, P = 0.93), suggesting that co‐cultured sympathetic neurons may provide multiple signals that act together to regulate the proliferation of the cardiomyocytes. We also used an alternative proliferative marker, Ki67, as a second approach to test the effects of these pharmacological manipulations. These experiments confirmed that inhibition of β‐adrenergic signalling with propranolol blocked the ability of co‐cultured sympathetic neurons to increase proliferation, while treatment of isolated cardiomyocytes with isoproterenol had no effect on proliferation (data not shown). Together, these data suggest that noradrenergic signalling is necessary, but not sufficient for the maintenance of cardiomyocyte proliferation in culture.
Figure 4. Sympathetic neurons promote cardiomyocyte proliferation via β‐adrenergic signalling .
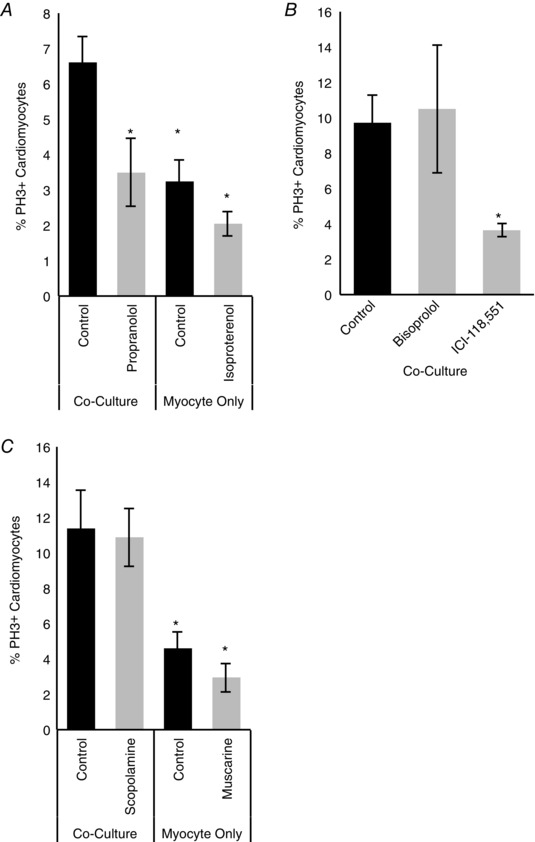
A, when β‐adrenergic signalling was blocked in co‐cultures with propranolol (2 μm) during the entire 5 day culture period there was a decrease in cardiomyocyte proliferation, as measured by the number of PH3+ cardiomyocytes. There was no significant effect of treatment with the β‐adrenergic agonist isoproterenol (10 μm) in myocyte‐only cultures, although proliferation was decreased compared to co‐culture conditions (n = 5). B, β1 and β2 signalling was blocked in 5 day co‐cultures with bisoprolol (left) and ICI‐118,551 (right), respectively. When β2‐adrenergic receptors were blocked with ICI‐118,551, there was a significant decrease in cardiomyocyte proliferation (n = 3). C, when cholinergic signalling was blocked with the cholinergic antagonist scopolamine (10 μm) in co‐cultures, we could not detect an effect on the number of PH3+ cells. Stimulating cholinergic signalling in myocyte‐only cultures with the cholinergic agonist muscarine (2.5 μm) also had no effect. (n = 3; *P < 0.05 vs. co‐culture.)
There are two isoforms of β‐adrenergic receptors found in cardiac tissue: β1‐ and β2‐receptors. We characterized the contribution of these receptors to sympathetic regulation of cardiomyocyte proliferation by co‐culturing sympathetic neurons and cardiomyocytes for 5 days and blocking β1‐adrenergic receptors with bisoprolol (1 mm) and β2‐adrenergic receptors with ICI‐118,551 (0.1 μm). We were unable to detect any changes in cardiomyocyte proliferation when β1‐adrenergic receptors were blocked. However, there was a decrease in the percentage of PH3+ cardiomyocytes in co‐cultures when β2‐adrenergic receptors were blocked with ICI‐118,551 (Fig. 4 B; 10.4 ± 2% vs. 3.6 ± 0.4%, n = 3, P = 0.04), suggesting that sympathetic regulation of cardiomyocyte proliferation is regulated via β2 receptors.
We also assessed the potential contributions of α‐adrenergic signalling to sympathetic regulation of cardiomyocyte proliferation by triggering α‐adrenergic signalling with the α‐adrenergic agonist phenylephrine (50 μm) in myocyte‐only cultures and blocking α‐adrenergic signalling with the α‐adrenergic antagonist prazosin in co‐cultures (20 μm). We were unable to detect an effect of either manipulation on levels of cardiomyocyte proliferation (data not shown), suggesting that noradrenergic control of cardiomyocyte proliferation is mediated primarily via the β2‐receptors.
Given that sympathetic neurons are known to release acetylcholine both in vitro (Furshpan et al. 1976; Yang et al. 2002) and in vivo (Landis & Keefe, 1983; Kimura et al. 2012), we also investigated whether cholinergic transmission could account for effects of sympathetic neurons on cardiomyocyte proliferation. We treated myocyte‐only and co‐cultures for 5 days in vitro and used the cholinergic agonist muscarine (2.5 μm) or the antagonist scopolamine (10 μm) to perturb cholinergic signalling. Co‐culture with sympathetic neurons, again, resulted in an increase in cardiomyocyte proliferation (Fig. 4 C; 11.4 ± 0.02% vs. 4.60 ± 0.01%, n = 5, P = 0.01). Although there was a trend towards a decrease in proliferation following muscarine treatment, we did not find a significant difference for either muscarine or scopolamine, consistent with the previous experiments suggesting that β2‐receptors mediate the effects of sympathetic actions on cardiomyocyte cell cycle withdrawal. These experiments focused on the role of muscarinic receptors, the primary pathway for cholinergic regulation of cardiomyocyte function, but we cannot rule out a possible action of nicotinic signalling in this system.
We also asked if sympathetic innervation limits cardiomyocyte hypertrophy via a β‐adrenergic signalling pathway by treating co‐cultures with propranolol and cardiomyocyte cultures with isoproterenol and measuring cardiomyocyte size. Treatment of co‐cultures with 2 μm propranolol for 4 days resulted in an increase in cardiomyocyte cell size (Fig. 5 A; 3489.0 ± 216.4 μm2 vs. 2558.7 ± 86.5 μm2, n = 3, P = 0.03). In addition, triggering β‐adrenergic signalling in myocyte‐only cultures by treatment with 10 μm isoproterenol for the same period resulted in a reduction of cell size (Fig. 5 A; 2217.5 ± 223.5 μm2 vs. 3159.2 ± 126.9 μm2, n = 3, P = 0.02). There was not a significant additive effect of isoproterenol treatment on the size of co‐cultured cardiomyocytes compared to cardiomyocytes grown alone, nor was there an independent effect of propranolol on myocyte‐only cultures. These data suggest that sympathetic β‐adrenergic signalling is both necessary and sufficient to decrease the hypertrophic response of cardiomyocytes and that noradrenaline release from sympathetic neurons regulates the developmental switch from hyperplasia to hypertrophy in the heart.
Figure 5. Sympathetic neurons decrease cardiomyocyte size via β‐adrenergic signalling .
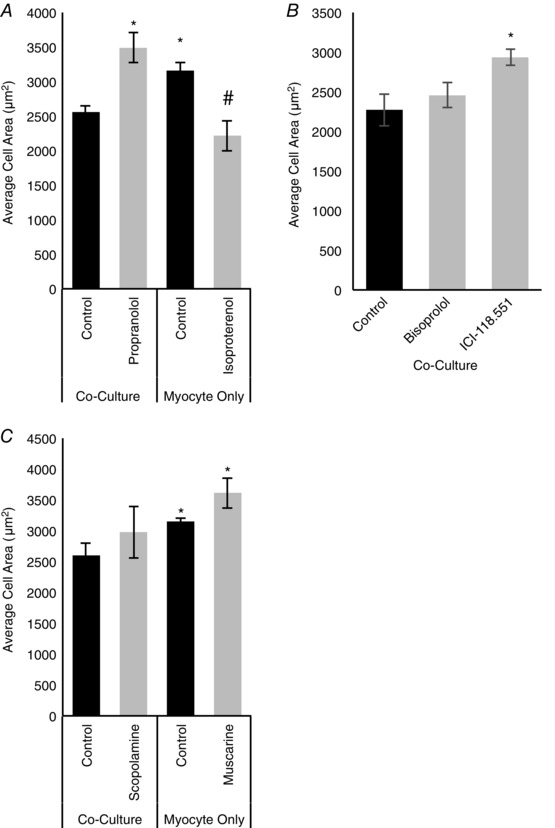
A, treatment of co‐cultured cardiomyocytes with propranolol (2 μm) resulted in an increase in average cardiomyocyte cell size, while treatment of myocte‐only cultures with isoproterenol (10 μm) resulted in a decrease in cell size (n = 3). B, β1‐ and β2‐receptors in co‐cultures were blocked with bisoprolol (left) and ICI‐118,551 (right), respectively. There was no effect of bisoprolol on cardiomyocyte size, but when β2‐receptors were blocked with ICI‐118,551 there was a significant increase in cell size (n = 3). C, when cholinergic signalling was blocked with the cholinergic antagonist scopolamine (10 μm) in co‐cultures, there was no effect on size of cardiomyocytes. Stimulating cholinergic signalling in myocyte‐only cultures with the cholinergic agonist muscarine (2.5 μm) also had no statistically significant effect. (n = 3; *P < 0.05 vs. co‐culture control; # P < 0.05 vs. myocyte‐only control.)
We next asked whether, as for cell cycle regulation, the effects of sympathetic neurons on cardiomyocyte cell size were mediated via β2‐receptors. β1‐ and β2‐receptors were blocked with bisoprolol or ICI‐118,551, respectively, for 4 days. We did not detect an effect of bisoprolol on cardiomyocyte cell size, but treatment with ICI‐118,551 increased cardiomyocyte size for cardiomyocytes co‐cultured with sympathetic neurons (Fig. 5 B; 2934.77 ± 100.34 μm2 vs. 2272.30 ± 205.66 μm2, n = 3, P = 0.03). Again, we were unable to detect an effect of either the manipulation of α‐adrenergic receptors or cholinergic receptors on cardiomyocyte size, although there was a non‐significant trend towards an increase in size following muscarine treatment (Fig. 5 C; data not shown.) These results are consistent with a model in which β‐adrenergic stimulation is a predominant pathway for sympathetic regulation of cardiomyocyte maturation with a specific role for β2‐adrenergic receptors.
Sympathetic regulation of cardiomyocyte growth is transient
Sympathetic innervation of cultured neonatal cardiomyocytes affects both their proliferative capacity and their hypertrophic growth, yet the transition from hyperplasia to hypertrophy continues to take place, suggesting that sympathetic signalling affects the timing of the transition. We therefore investigated the effect of sympathetic co‐culture on cardiomyocyte size and proliferation across the first postnatal week. We cultured cardiomyocytes in the presence or absence of sympathetic neurons for 1–7 days in vitro (DIV) and assayed average cell size, as described above. We found that cardiomyocytes undergo significant hypertrophy over the 7 days in the presence or absence of neurons, increasing their size more than 5‐fold (Fig. 6 A). During this period, cardiomyctes co‐cultured with sympathetic neurons showed a transient delay in growth at days 3 and 4 compared to the myocyte cultures (Fig. 6 A; 3 DIV: 1812.11 ± 174.94 μm2 vs. 2403.55 ± 128.07 μm2, n = 3, P = 0.05; 4 DIV: 2380.9 ± 146.7 μm2 vs. 3092.99 ± 69.52 μm2, n = 3, P = 0.01). This demonstrates an effect of sympathetic signalling on cardiomyocyte hypertrophy. While caution is warranted in relating the results of in vitro experiments to the effects of sympathectomy in vivo, the transient nature of the sympathetic effect on cardiomyocyte growth may suggest that sympathetic regulation of cardiomyocyte proliferation plays a greater role than regulation of cell size in setting adult heart size.
Figure 6. Sympathetic regulation of cardiomyocyte proliferation and cell growth is transient .
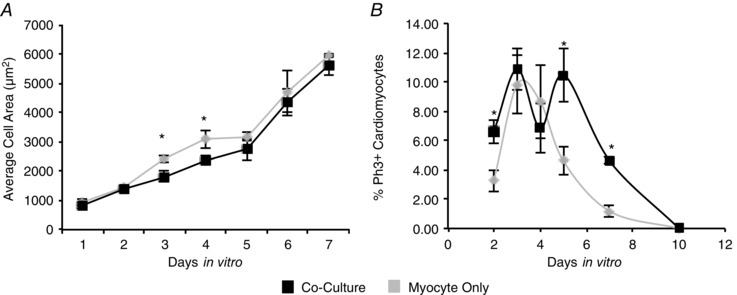
A, the area of individual cardiomyocytes was measured for cells cultured in the presence or absence of co‐cultured sympathetic neurons. Cultures were fixed and stained for α‐actinin each day for a 7 day period and cardiomyocyte area was measured. A plot of average cell size over days in vitro shows that cardiomyocytes are significantly smaller at 3 and 4 days when grown with sympathetic neurons. B, a plot of percentage of proliferating cardiomyocytes for cultures grown for 2, 3, 4, 5, 7 and 10 days in vitro shows that neuronal co‐culture delayed cell cycle withdrawal. (n ≥ 3; *P < 0.05 vs. age‐matched co‐culture.)
We also examined cardiomyocyte proliferation during the postnatal period by quantifying the percentage of PH3‐positive cells in cultures grown in the presence or absence of co‐cultured neurons. All cardiomyocytes showed complete withdrawal from the cell cycle by 10 days in vitro, but cardiomyocytes in co‐cultures showed higher rates of proliferation at 2, 5 and 7 days compared to the myocyte‐only cultures (Fig. 6 B; 2 DIV: 7 ± 0.008% vs. 3.25 ± 0.007%, n = 6, P = 0.02; 5 DIV: 10 ± 0.02% vs. 4.60 ± 0.009%, n = 4, P = 0.01; 7 DIV: 5 ± 0.002% vs. 1.18 ± 0.004%, n = 3, P = 0.002). These data show that in the absence of sympathetic innervation, cardiomyocytes exit the cell cycle earlier, suggesting a model in which the reduced heart size of animals with neonatal sympathectomies results from a long‐term reduction in the number of cardiomyocytes.
Developmental changes in β‐adrenergic regulation of cardiomyocyte size
The transient effect of sympathetic co‐culture on cardiomyocyte size suggests that β‐adrenergic control of cellular hypertrophy is developmentally regulated. We investigated this possibility by examining the effect of β‐adrenergic signalling on the cell size of more mature cardiomyocytes. Cardiomyocytes were grown alone or in co‐culture for 3 weeks to allow for cardiomyocyte maturation (Lockhart et al. 2000) and cultures were treated with the β‐adrenergic agonist isoproterenol for the last 4 days of the growth period. In contrast to our results for young cardiomyocytes, we observed a significant increase in cardiomyocyte size for isoproterenol‐treated cultures, both for cardiomyocytes grown alone and in co‐culture (Fig. 7 A; myocyte‐only isoproterenol‐treated: 116.06 ± 2.65% of control, n = 4, P = 0.003; co‐culture isoproterenol‐treated: 128.07 ± 7.01% of control, n = 3, P = 0.04). These results suggest that the response to sympathetic transmission undergoes a postnatal developmental switch.
Figure 7. β‐Adrenergic signalling undergoes a developmental shift from a negative to a positive regulator of cardiomyocyte hypertrophy .
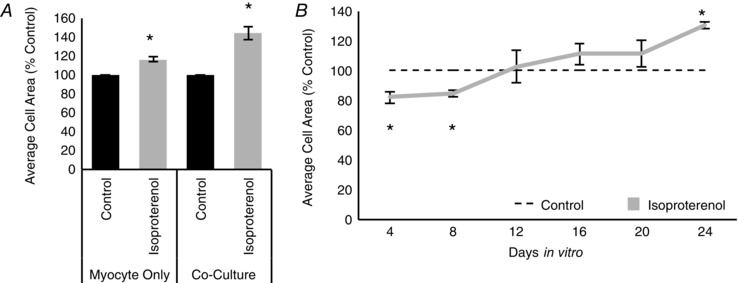
A, bar plot comparing the average cell size of 3‐week‐old cardiomyocytes cultured without (left) or with (right) sympathetic neurons. Cultures were either treated with isoproterenol (10 μm; grey) for the last 4 days in vitro or not treated (black). Four days of isoproterenol treatment was sufficient to induce an increase in cardiomyocyte size. B, a plot of average cardiomyocyte size over days in vitro shows that, at 4 and 8 DIV, treatment with isoproterenol (grey line) decreases cardiomyocyte size, compared to the control. As the cardiomyocytes mature in culture, the effect of isoproterenol reverses, with a significant increase in cardiomyocyte size seen at 24 DIV. For each time point, cultures were treated with isoproterenol for the last 4 days of the culture period. (n = 3; *P < 0.05 vs. age‐matched control.)
We directly tested whether there was a developmentally coordinated change in the effects of β‐adrenergic signalling by carrying out a time course in which cardiomyocytes were cultured in isolation for 4–24 days and treated with isoproterenol for the last 4 days of the culture period. We found that until 8 DIV, isoproterenol‐treated cardiomyocytes were smaller than untreated control cells. However, we observed a trend towards an increase in cardiomyocyte size in response to isoproterenol after 12 days DIV, which reached significance by 24 DIV (Fig. 7 B; 4 DIV isoproterenol‐treated: 82.19 ± 0.04% of control, n = 3, P = 0.04; 8 DIV = 84.74 ± 0.02% of control, n = 3, P = 0.003; 24 DIV = 130.38 ± 0.02% of control, n = 3, P = 0.001). These data show that sympathetic innervation plays an ongoing, but changing role in regulating the size of cardiomyocytes.
Sympathetic innervation in vivo regulates expression of the cell cycle arrest‐associated transcription factor Meis1
Recently, Meis1 was identified as a master regulator of cardiomyocyte proliferation, with increased expression of Meis1 driving cell cycle withdrawal (Mahmoud et al. 2013). We therefore asked whether Meis1 expression was regulated during the period of sympathetic innervation in vivo. We extracted RNA from sham‐injected and 6‐OHDA‐lesioned hearts at P2, P7 and 8 weeks and assayed levels of Meis1 expression. We found that expression levels of Meis1 mRNA increased with age between P2 and P7, with a return to a lower level of expression in 8‐week‐old animals (Fig. 8 A). We went on to ask whether this expression pattern was regulated by the presence of innervating sympathetic neurons in vivo. We found that neonatal sympathetic lesion resulted in an increase in Meis1 mRNA over the sham for animals analysed at P2 and P7 (Fig. 8 B), with a greater increase at P7. In contrast, there was no significant difference in levels of Meis1 expression between sham and lesioned animals at 8 weeks. The increase in Meis1 expression in animals lacking sympathetic innervation suggests a genetic mechanism through which the sympathetic innervation may act to regulate cardiomyocyte proliferation.
Figure 8. Meis1, a cell cycle arrest‐associated transcription factor, is developmentally regulated in vivo, and is upregulated following sympathetic lesion .
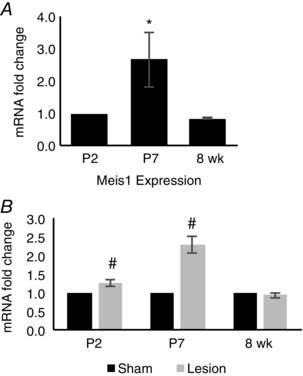
A, bar plot showing that Meis1 mRNA expression transiently increases between P2 and P7 in control animals, returning to P2 levels by 8 weeks. B, bar plot demonstrating that lesion of the sympathetic nervous system at P0 leads to an increase in Meis1 expression at P2 and P7. By 8 weeks, there was no difference in Meis1 expression compared to the sham control. (*P < 0.05 vs. P2; # P < 0.05 vs. sham.)
Sympathetic innervation regulates heart cell density and number in vivo
We tested the hypothesis that sympathetic regulation of cardiomyocyte proliferation could account for the smaller hearts observed in animals with sympathetic lesions by assessing the density of cardiomyocytes in sections of intact heart tissue from lesioned and sham control animals. We visualized the outlines of cardiomyocytes via fluorescent WGA staining and confirmed that large cells identified by WGA staining were cardiomyocytes by double‐labelling some sections with α‐actinin (Fig. 9 A). WGA also marked a population of previously identified non‐myocyte cells that were much smaller than the neighbouring cardiomyocytes (Gelpi et al. 2011). We used the WGA staining to identify these two populations in adolescent (8 weeks) animals that had received sham and 6‐OHDA injections at P0 (Fig. 9 B, asterisks and arrowheads, respectively). Analysis of cardiomyocyte density showed that density was significantly lower in the lesioned animals, consistent with a role for sympathetic innervation in increasing cardiomyocyte cell number in the normal heart (Fig. 9 C; 1.45 ± 0.04 cells mm−2 vs. 2.14 ± 0.2 cells mm−2, n = 3, P = 0.03). While lesioned hearts showed a decrease in the number of cardiomyocytes per section, we found an increase in the density of non‐myocyte cells in the lesioned animals (Fig. 9 D; 1.9 ± 0.08 cells mm−2 vs. 3.04 ± 0.14 cells mm−2, n = 4, P = 7.5 × 10−5).
Figure 9. Sympathetic innervation is required for the regulation of cardiomyocyte density in vivo .
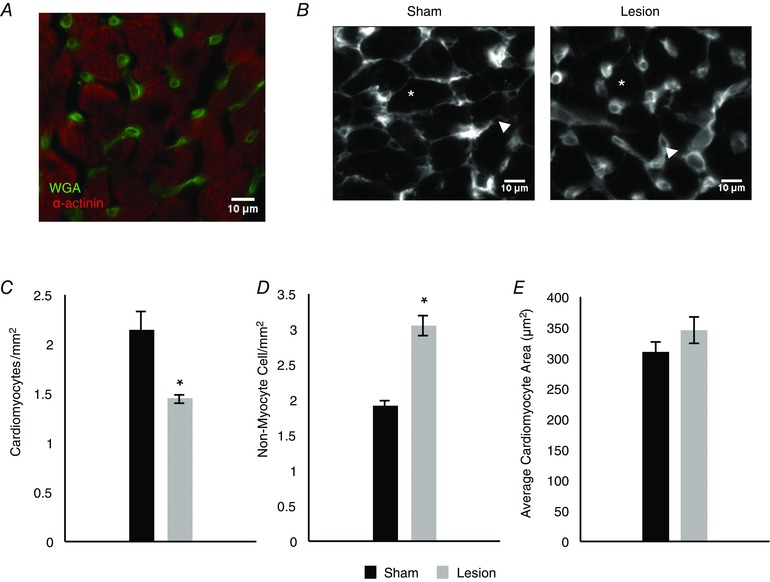
A, representative image of wheat germ agglutinin (WGA) and α‐actinin staining in ventricular sections of 8‐week‐old rats. WGA delineates the cellular membrane of cardiomyocytes as well as staining non‐myocyte cells in the slice. B, representative images of WGA staining showing cardiomyocytes (asterisk) and non‐cardiomyocyte cells (arrowhead) in sham (left) and lesioned (right) animals. C, bar plot showing that, compared to sham (black), neonatal sympathetic lesions (grey) result in a decrease in cardiomyocyte density in 8‐week‐old animals (n = 3). D, bar plot comparing the density of non‐myocyte cells in sham (black) and lesioned (grey) animals, showing an increase in the density of non‐myocyte cells in lesioned animals (n = 4). E, bar plot showing that the average cardiomyocyte size in sham (black) and lesioned (grey) animals is not significantly different, suggesting that the difference in heart size in lesioned animals cannot be accounted for by decreased cell size. (n = 3; *P < 0.05 vs. sham.)
We also analysed cardiomyocyte size in heart sections from the same 8‐week‐old lesioned and sham‐injected animals by measuring the perimeter of WGA‐labelled cardiomyocytes and calculating cell area in matched sections. We found that average cardiomyocyte size was not significantly different in the different conditions (Fig. 9 E). Our in vitro findings showed an early, transient effect of sympathetic fibres on cardiomyocyte size; we therefore also looked at cell size in vivo at P2, P4 and P7 following a neonatal sympathectomy or sham injection. We were unable, however, to detect a change in cardiomyocyte size at these early time points (data not shown). We therefore cannot account for smaller heart size following sympathetic lesion by a change in cardiomyocyte cell size. In contrast, the decreased density of cardiomyocytes in lesioned hearts, along with the increase in non‐myocyte cells in the sections, is consistent with a smaller total number of cardiomyocytes as a result of premature withdrawal from the cell cycle following sympathetic lesion.
Discussion
Cardiomyocytes withdraw from the cell cycle shortly after birth and, for the most part, remain quiescent throughout an organism's life. This work identifies the sympathetic nervous system as a regulator of this developmental transition, after which cardiac tissue primarily grows via hypertrophy. Following a neonatal lesion of the sympathetic nervous system, rat hearts are smaller, with fewer cardiomyocytes. The presence of sympathetic neurons in cardiomyocyte cultures in vitro delays cardiomyocyte cell cycle withdrawal and transiently limits hypertrophy via a β‐adrenergic signalling pathway, suggesting that sympathetic innervation regulates cardiomyocyte number during the postnatal period. Sympathetic signalling is mediated via β‐adrenergic receptors and is likely to involve the Meis1 protein, which is regulated by sympathetic innervation and has been linked to cardiomyocyte cell cycle withdrawal (Mahmoud et al. 2013). Overall, this study supports a role for the sympathetic nervous system in regulating adult cardiomyocyte number and demonstrates that developmentally dynamic signalling contributes to adult heart structure.
Normal cardiac function is dependent upon the total number of cardiomyocytes in the heart (Levkau et al. 2008; Porrello et al. 2008; Chang et al. 2010) and thus, early regulation of cell number has significant implications for understanding adult cardiac properties and the development of cardiac pathologies. Studies examining mouse models containing either a greater or lesser number of cardiomyocytes support the idea that cell number must be set within a critical range for normal cardiac function (Levkau et al. 2008; Chang et al. 2010; Shenje et al. 2014). Our work demonstrates that early sympathetic interactions both prolong the period of postnatal cardiomyocyte proliferation in culture and result in the development of larger hearts with more cardiomyocytes in vivo. This suggests that sympathetic signalling is part of a homeostatic system that acts to set the final number of cardiomyocytes in the adult heart.
A role for sympathetic fibres in controlling developmental properties of innervated tissue was recently demonstrated in a study examining the role of sympathetic signalling in the development of the pancreas. Borden et al. (2013) found that sympathetic innervation was necessary for proper pancreatic islet cell development. Ablation of sympathetic signalling, either in vitro or in vivo, led to impairment of islet cell migration and aberrant cell patterning, as well as impaired glucose tolerance (Borden et al. 2013). The fact that our observations in the heart have defined a proliferative effect without obvious cellular changes in heart morphology indicates that, while sympathetic signalling may play a general role in the development of target organs, the specific effects will differ depending upon the developmental properties of each system.
While the specific cellular effects of sympathetic innervation differ between the heart and the pancreas, at least some aspects of the signalling mechanism are shared. Similar to our findings in the heart, Borden et al. (2013) identified a role for β‐adrenergic signalling in pancreatic islet cell development. However, additional pathways may also contribute to cell cycle control in the heart. Interestingly, parasympathetic cholinergic signalling has recently been reported to be required for injury‐induced cardiomyocyte regeneration (Mahmoud et al. 2015). Yet we were unable to identify an effect of sympathetic cholinergic signalling on developmentally regulated cardiomyocye proliferation and hypertrophy. Rather, we found that β‐adrenergic signalling, and specifically β2‐adrenergic signalling, was necessary for sympathetic regulation of cardiomyocyte proliferation and cardiomyocyte size, suggesting that regeneration following cardiac damage may engage discrete pathways.
Our finding that β‐adrenergic signalling delays hypertrophic growth in young cardiomyocytes contrasts with reported effects of this signalling as a positive regulator of hypertrophy, specifically pathological hypertrophy, in adult animals (Hunter & Chien, 1999; Heineke & Molkentin, 2006). These findings are consistent with our observation of a developmental shift in cardiomyocyte growth responses to β‐adrenergic signalling in which isoproterenol limited the growth of individual cardiomyocytes in the first few days after birth, but promoted hypertrophy in more mature cardiomyocytes. Such a developmental switch may reflect a shift in the distribution of β‐adrenergic receptor subtypes. Here we demonstrate that β2 receptors are the primary subtype contributing to the sympathetic regulation of early postnatal cardiomyocyte proliferation and hypertrophy. Interestingly, the ratio of β1‐ to β2‐receptors changes developmentally and alterations in β1:β2 receptor ratios have been reported to underlie pathological changes in cardiac function (Wadhawan et al. 2003). Taken together with our work, this may suggest that differential expression of β‐adrenoreceptors may underlie the changes in response to β‐adrenergic signalling as cardiomyocytes develop.
An interesting possible framework for thinking about developmental changes in β‐adrenergic receptors and responses is as a shift between a physiological and a pathological growth state of the cardiomyocytes. The idea that the identical signal could trigger alternative responses depending upon the developmental state of the tissue is consistent with work demonstrating that proteins associated with cellular division are re‐expressed during pathological hypertrophy, even though cardiomyocytes are unable to undergo complete divisions (Ahuja et al. 2007). Evidence of a bifurcated signalling pathway coupled to β‐adrenergic receptors (Xiao, 2001) provides a possible mechanistic substrate by which the same signal, in this case, noradrenaline, may trigger divergent effects. In this context, it is interesting to speculate that the early transient effect of β‐adrenergic signalling on inhibiting cardiomyocyte hypertrophy may be related to the expanded period of proliferation for these cells.
One way to begin to understand the underlying pathways is to identify candidate proteins that may contribute to the sympathetic regulation of cardiomyocyte maturation. Meis1, which is upregulated as the heart develops and cardiomyocytes withdraw from the cell cycle (Mahmoud et al. 2013), is a potential downstream effector of β‐adrenergic signalling in this system. Interestingly, we found that denervated hearts showed increased levels of Meis1 expression during the early postnatal period, within the same period that innervation extends the period of cardiomyocyte proliferation. We also observed that, in adult cardiac tissue, Meis1 expression levels are similar to that seen in early postnatal life. This would suggest that increased levels of Meis1 are not necessary to maintain cardiomyocytes in a post‐mitotic state and that the increase observed during this developmental transition is the salient signal that causes cardiomyocyte cell cycle arrest. Further work is required to place Meis1 within a pathway, and to understand how additional proteins, including ALMS1, which has been implicated in both in cell cycle control (Zulato et al. 2011) and cardiomyopathies (Chang et al. 2010), and CASZ1, which is essential for cardiomyocyte proliferation in early prenatal development (Dorr et al. 2015), contribute to sympathetic regulation of cardiomyocyte number in the heart.
Our work suggests that early perturbations of sympathetic structure or function could have long‐term effects on the function of the adult heart. This has implications for human health, as there are several clinical situations where sympathetic innervation is perturbed early in development. Surgical repair of congenital heart defects are among the most common surgeries performed on infants in the United States. These surgeries damage the sympathetic nerves innervating the heart, resulting in disordered sympathetic innervation following the regrowth of these nerves to cardiac tissue (Falkenberg et al. 2010; Kuehn et al. 2014). Children who undergo these surgeries tend to recover well and develop normally. However, adults who had congenital heart surgery are at greater risk for sudden cardiac death later in life (Silka et al. 1998). This suggests that a fruitful avenue of investigation will be to characterize how the sympathetic‐regulated shift from hyperplasia to hypertrophy affects the function of the adult heart under conditions of disordered sympathetic innervation.
Our work adds the sympathetic nervous system to the growing list of factors and genes (Machida et al. 1997; Ahuja et al. 2007 b; Shenje et al. 2014) involved in cell cycle control in the cardiac system and provides a new system for understanding cardiac disease and dysfunction.
Additional information
Competing interests
The authors have no competing interests to declare.
Author contributions
Conception and design of the experiments: R.E.K. and S.J.B. Collection, analysis, and interpretation of data: R.E.K. Drafting/revising the article: R.E.K. and S.J.B. Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by NIH R01NS057305 and NIH P30NS45713 to S.J.B.
Acknowledgements
The authors are grateful for the assistance of Sagarika Utture and Hina Bhat, as well as Dr Beth Habecker for many useful discussions.
References
- Ahuja P, Perriard E, Pedrazzini T, Satoh S, Perriard JC & Ehler E (2007. a). Re‐expression of proteins involved in cytokinesis during cardiac hypertrophy. Exp Cell Res 313, 1270–1283. [DOI] [PubMed] [Google Scholar]
- Ahuja P, Sdek P & MacLellan WR (2007. b). Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol Rev 87, 521–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeletti PU & Levi‐Montalcini R (1970). Sympathetic nerve cell destruction in newborn mammals by 6‐hydroxydopamine. Proc Natl Acad Sci USA 65, 114–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé‐Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S & Frisén J (2009). Evidence for cardiomyocyte renewal in humans. Science 324, 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan RD (1975). Effect of sympathetic denervation on smooth muscle cell proliferation in the growing rabbit ear artery. Circ Res 37, 14–19. [DOI] [PubMed] [Google Scholar]
- Borden P, Houtz J, Leach SD & Kuruvilla R (2013). Sympathetic innervation during development is necessary for pancreatic islet architecture and functional maturation. Cell Rep 4, 287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briest W, Hölzl A, Rassler B, Deten A, Baba HA & Zimmer H‐G (2003). Significance of matrix metalloproteinases in norepinephrine‐induced remodelling of rat hearts. Cardiovasc Res 57, 379–387. [DOI] [PubMed] [Google Scholar]
- Bristow MR, Ginsburg R, Umans V, Fowler M, Minobe W, Rasmussen R, Zera P, Menlove R, Shah P & Jamieson S (1986). β 1‐ and β2‐adrenergic‐receptor subpopulations in nonfailing and failing human ventricular myocardium: coupling of both receptor subtypes to muscle contraction and selective β1‐receptor down‐regulation in heart failure. Circ Res 59, 297–309. [DOI] [PubMed] [Google Scholar]
- Chang KTE, Taylor GP, Meschino WS, Kantor PF & Cutz E (2010). Mitogenic cardiomyopathy: a lethal neonatal familial dilated cardiomyopathy characterized by myocyte hyperplasia and proliferation. Hum Pathol 41, 1002–1008. [DOI] [PubMed] [Google Scholar]
- Dorr KM, Amin NM, Kuchenbrod LM, Labiner H, Charpentier MS, Pevny LH, Wessels A & Conlon FL (2015). Casz1 is required for cardiomyocyte G1‐to‐S phase progression during mammalian cardiac development. Development 142, 2037–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowell RT (1985). Postnatal development of rat heart during 6‐hydroxydopamine or propranolol treatment. Proc Soc Exp Biol Med 178, 565–574. [DOI] [PubMed] [Google Scholar]
- Falkenberg C, Hallhagen S, Nilsson K, Nilsson B & Östman‐Smith I (2010). A study of the physiological consequences of sympathetic denervation of the heart caused by the arterial switch procedure. Cardiol Young 20, 150–158. [DOI] [PubMed] [Google Scholar]
- Furshpan EJ, MacLeish PR, O'Lague PH & Potter DD (1976). Chemical transmission between rat sympathetic neurons and cardiac myocytes developing in microcultures: evidence for cholinergic, adrenergic, and dual‐function neurons. Proc Natl Acad Sci USA 73, 4225–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelpi RJ, Park M, Gao S, Dhar S, Vatner DE & Vatner SF (2011). Apoptosis in severe, compensated pressure overload predominates in nonmyocytes and is related to the hypertrophy but not function. Am J Physiol Heart Circ Physiol 300, H1062–H1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heineke J & Molkentin JD (2006). Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol 7, 589–600. [DOI] [PubMed] [Google Scholar]
- Hunter JJ & Chien KR (1999). Signaling pathways for cardiac hypertrophy and failure. N Engl J Med 341, 1276–1283. [DOI] [PubMed] [Google Scholar]
- Iwaki K, Sukhatme VP, Shubeita HE & Chien KR (1990). α‐ and β‐adrenergic stimulation induces distinct patterns of immediate early gene expression in neonatal rat myocardial cells. J Biol Chem 265, 13809–13817. [PubMed] [Google Scholar]
- Kimura K, Ieda M & Fukuda K (2012). Development, maturation, and transdifferentiation of cardiac sympathetic nerves. Circ Res 110, 325–336. [DOI] [PubMed] [Google Scholar]
- Kostrzewa RM & Jacobowitz DM (1974). Pharmacological actions of 6‐hydroxydopamine. Pharmacol Rev 26, 199–288. [PubMed] [Google Scholar]
- Kuehn A, Vogt M, Schwaiger M, Ewert P & Hauser M (2014). Ventricular sympathetic innervation in patients with transposition of the great arteries after arterial switch operation and Rastelli procedure : impact of arterial dissection and coronary reimplantation. Circ J 78, 1717–1722. [DOI] [PubMed] [Google Scholar]
- Landis SC & Keefe D (1983). Evidence for neurotransmitter plasticity in vivo: Developmental changes in properties of cholinergic sympathetic neurons. Dev Biol 98, 349–372. [DOI] [PubMed] [Google Scholar]
- Levkau B, Schäfers M, Wohlschlaeger J, von Wnuck Lipinski K, Keul P, Hermann S, Kawaguchi N, Kirchhof P, Fabritz L, Stypmann J, Stegger L, Flögel U, Schrader J, Fischer JW, Hsieh P, Ou YL, Mehrhof F, Tiemann K, Ghanem A, Matus M, Neumann J, Heusch G, Schmid KW, Conway EM & Baba HA (2008). Survivin determines cardiac function by controlling total cardiomyocyte number. Circulation 117, 1583–1593. [DOI] [PubMed] [Google Scholar]
- Li F, Wang X, Capasso JM & Gerdes AM (1996). Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol 28, 1737–1746. [DOI] [PubMed] [Google Scholar]
- Lockhart ST, Mead JN, Pisano JM, Slonimsky JD & Birren SJ (2000). Nerve growth factor collaborates with myocyte‐derived factors to promote development of presynaptic sites in cultured sympathetic neurons. J Neurobiol 42, 460–476. [PubMed] [Google Scholar]
- Lockhart ST, Turrigiano GG & Birren SJ (1997). Nerve growth factor modulates synaptic transmission between sympathetic neurons and cardiac myocytes. J Neurosci 17, 9573–9582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther JA & Birren SJ (2006). Nerve growth factor decreases potassium currents and alters repetitive firing in rat sympathetic neurons. J Neurophysiol 96, 946–958. [DOI] [PubMed] [Google Scholar]
- Luther JA, Enes J & Birren SJ (2013). Neurotrophins regulate cholinergic synaptic transmission in cultured rat sympathetic neurons through a p75‐dependent mechanism. J Neurophysiol 109, 485–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida N, Brissie N, Sreenan C & Bishop SP (1997). Inhibition of cardiac myocyte division in c‐myc transgenic mice. J Mol Cell Cardiol 29, 1895–1902. [DOI] [PubMed] [Google Scholar]
- Mahmoud AI, Kocabas F, Muralidhar SA, Kimura W, Koura AS, Thet S, Porrello ER & Sadek HA (2013). Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 497, 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud AI, O'Meara CC, Gemberling M, Zhao L, Bryant DM, Zheng R, Gannon JB, Cai L, Choi W‐Y, Egnaczyk GF, Burns CE, Burns CG, MacRae CA, Poss KD & Lee RT (2015). Nerves regulate cardiomyocyte proliferation and heart regeneration. Dev Cell 34, 387–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon J‐I & Birren SJ (2008). Target‐dependent inhibition of sympathetic neuron growth via modulation of a BMP signaling pathway. Dev Biol 315, 404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murry CE, Soonpaa MH, Reinecke H, Nakajima H, Nakajima HO, Rubart M, Pasumarthi KBS, Ismail Virag J, Bartelmez SH, Poppa V, Bradford G, Dowell JD, Williams DA & Field LJ (2004). Haematopoietic stem cells do not transdifferentiate into cardiac myocytes in myocardial infarcts. Nature 428, 664–668. [DOI] [PubMed] [Google Scholar]
- Porrello ER, Widdop RE & Delbridge LMD (2008). Early origins of cardiac hypertrophy: does cardiomyocyte attrition programme for pathological ‘catch‐up’ growth of the heart? Clin Exp Pharmacol Physiol 35, 1358–1364. [DOI] [PubMed] [Google Scholar]
- Ramakers C, Ruijter JM, Deprez RH & Moorman AF (2003). Assumption‐free analysis of quantitative real‐time polymerase chain reaction (PCR) data. Neurosci Lett 339, 62–66. [DOI] [PubMed] [Google Scholar]
- Shenje LT, Andersen P, Halushka MK, Lui C, Fernandez L, Collin GB, Amat‐Alarcon N, Meschino W, Cutz E, Chang K, Yonescu R, Batista DA, Chen Y, Chelko S, Crosson JE, Scheel J, Vricella L, Craig BD, Marosy BA, Mohr DW, Hetrick KN, Romm JM, Scott AF, Valle D, Naggert JK, Kwon C, Doheny KF & Judge DP (2014). Mutations in Alström protein impair terminal differentiation of cardiomyocytes. Nat Commun 5, 3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silka MJ, Hardy BG, Menashe VD & Morris CD (1998). A population‐based prospective evaluation of risk of sudden cardiac death after operation for common congenital heart defects. J Am Coll Cardiol 32, 245–251. [DOI] [PubMed] [Google Scholar]
- Thomas SA, Matsumoto AM & Palmiter RD (1995). Noradrenaline is essential for mouse fetal development. Nature 374, 643–646. [DOI] [PubMed] [Google Scholar]
- Thornburg K, Jonker S, O'Tierney P, Chattergoon N, Louey S, Faber J & Giraud G (2011). Regulation of the cardiomyocyte population in the developing heart. Prog Biophys Mol Biol 106, 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadhawan R, Tseng Y‐T, Stabila J, McGonnigal B, Sarkar S & Padbury J (2003). Regulation of cardiac β1‐adrenergic receptor transcription during the developmental transition. Am J Physiol Heart Circ Physiol 284, H2146–H2152. [DOI] [PubMed] [Google Scholar]
- Xiao R‐P (2001). β‐Adrenergic signaling in the heart: dual coupling of the β2‐adrenergic receptor to Gs and Gi proteins. Sci STKE 2001, re15. [DOI] [PubMed] [Google Scholar]
- Yang B, Slonimsky JD & Birren SJ (2002). A rapid switch in sympathetic neurotransmitter release properties mediated by the p75 receptor. Nat Neurosci 5, 539–545. [DOI] [PubMed] [Google Scholar]
- Yang EV, Sood AK, Chen M, Li Y, Eubank TD, Marsh CB, Jewell S, Flavahan NA, Morrison C, Yeh PE, Lemeshow S & Glaser R (2006). Norepinephrine up‐regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)‐2, and MMP‐9 in nasopharyngeal carcinoma tumor cells. Cancer Res 66, 10357–10364. [DOI] [PubMed] [Google Scholar]
- Zak R (1973). Cell proliferation during cardiac growth. Am J Cardiol 31, 211–219. [DOI] [PubMed] [Google Scholar]
- Zimmer HG, Kolbeck‐Ruhmkorff C & Zierhut W (1995). Cardiac hypertrophy induced by alpha‐and beta‐adrenergic receptor stimulation. Cardioscience 6, 47. [PubMed] [Google Scholar]
- Zulato E, Favaretto F, Veronese C, Campanaro S, Marshall JD, Romano S, Cabrelle A, Collin GB, Zavan B & Belloni AS (2011). ALMS1‐deficient fibroblasts over‐express extra‐cellular matrix components, display cell cycle delay and are resistant to apoptosis. PLoS One 6, e19081. [DOI] [PMC free article] [PubMed] [Google Scholar]