

Abstract
Context:
TAK-385 is a highly selective, oral, nonpeptide GnRH antagonist being investigated as a possible prostate cancer treatment.
Objective:
The objectives were to evaluate safety, tolerability, pharmacokinetics, and pharmacodynamics of TAK-385 on LH and testosterone.
Design, Setting, and Participants:
This was a three-part, randomized, double-blind, placebo-controlled, phase 1 dose-escalation study in 176 healthy male UK volunteers.
Interventions:
Part 1, single doses of TAK-385 (0 [placebo], 80, 120, 180, or 360 mg). Part 2, 14-day TAK-385 (0, 20, 40, 80, or 180 mg) daily. Part 3, 28-day TAK-385 (40 [with loading dose], 60, 80, or 160 mg) or placebo daily. Parts 2 and 3 included men aged 40–75 years.
Main Outcome Measures:
Main outcome measures included plasma concentrations of TAK-385, LH, and testosterone.
Results:
Oral TAK-385 was readily absorbed, and steady state was reached in ≤14 days. Food reduced TAK-385 systemic exposure by 47–52%. Mean serum testosterone levels declined ≤6 hours after TAK-385 administration. Loading doses up to 360 mg on day 1 or 360 mg on day 1 followed by 240 mg on day 2 reduced the time to achieve castrate testosterone levels from ≥7 to <3 days. TAK-385 doses ≥80 mg/d achieved sustained medical castration and trough TAK-385 concentrations >4 ng/mL. After discontinuation of TAK-385 on day 28, testosterone levels normalized in most subjects in ≤ 28 days. Common adverse events included bradycardia, headache, and hot flush (all grade ≤2).
Conclusions:
Oral TAK-385 (40–180 mg/d) was well tolerated and effectively lowered testosterone in healthy men. Planned phase 2 doses in men with hormone-sensitive prostate cancer are 80 and 120 mg/d.
Androgen deprivation therapy (ADT) is a mainstay of first-line treatment for advanced prostate cancer (1). Modes of ADT currently in clinical use include bilateral orchiectomy to eliminate testicular testosterone production or chemical castration with depot injections of GnRH agonists (1- to 6-mo depots) or the 1-month depot peptide analog antagonist, degarelix (1). Continuous administration of GnRH agonists down-regulates and desensitizes GnRH receptors in the pituitary, leading to reduced secretion of LH and downstream testosterone, whereas antagonists of GnRH suppress testosterone by direct blockade of pituitary GnRH receptors (1). The androgen-suppressive effects of GnRH agonists are preceded by an initial stimulatory phase known as testosterone surge. This surge, along with the time required for full receptor desensitization, prolongs the time to achieve castration to 2–4 weeks (2). Testosterone surge may also lead to prostate cancer flare, which may include skeletal pain, spinal cord compression, and, rarely, death, although flare may be prevented with coadministration of antiandrogens such as bicalutamide (3). The rapid onset of effect with GnRH antagonists prevents testosterone flare and makes conventional early antiandrogen therapy unnecessary (4, 5).
Presently, the only GnRH antagonist approved for the treatment of prostate cancer is degarelix. Phase 3 trial results demonstrated that monthly sc administration of degarelix was noninferior to leuprolide at maintaining serum testosterone for 1 year below the current conventional threshold of medical castration, 1.73 nmol/L (50 ng/dL), and was also similar for attaining the lower threshold of 0.69 nmol/L (20 ng/dL), which is the usual level attained by GnRH agonists when assessed using modern immunoassay or liquid chromatography/tandem mass spectroscopy (LC/MS/MS) techniques (5, 6). Although the upper threshold of 1.73 nmol/L is the standard accepted by regulatory bodies, the lower threshold is considered the desired therapeutic goal for ADT (6). Degarelix was associated with rapid testosterone reduction, without flare, and prostate-specific antigen reduction during the first month was comparable to that observed in patients in the study receiving leuprolide and bicalutamide (5). A retrospective follow-up analysis from that study suggested that high-risk patients who received degarelix had a lower incidence of biochemical disease progression or death (P = .05) vs patients who received leuprolide (4). However, additional prospective studies are needed to confirm the potential benefits of GnRH antagonism vs agonism. In the phase 3 trial of degarelix, 40% of patients experienced adverse injection-site reactions such as pain, erythema, and swelling (5).
TAK-385 is an investigational, highly selective, orally active, nonpeptide GnRH antagonist that binds to human GnRH receptors with subnanomolar affinity (IC50 = 0.33 nm in the presence of 40% fetal bovine serum) (7, 8). Although binding studies and comparisons among species demonstrate that TAK-385 acts as a classic competitive antagonist of GnRH binding, the precise molecular mechanism of that antagonism has not been elucidated. Initial phase 1 studies of TAK-385 in healthy premenopausal women showed that TAK-385 was generally safe and well tolerated after single oral doses up to 80 mg and multiple daily doses up to 40 mg for 14 days (unpublished data). The current phase 1 study was designed to evaluate the safety and pharmacokinetics (PK)/pharmacodynamics (PD) of TAK-385 after single and multiple doses in healthy men, including those in the age range (40–75 y) most commonly associated with prostate cancer. The key objective was to identify a safe and effective TAK-385 dose range that would achieve sustained castrate testosterone levels and be suitable for long-term phase 2 evaluation in patients with prostate cancer.
Subjects and Methods
Study design
This was a randomized, placebo-controlled, single- and multiple-dose, dose-escalation, parallel-group study conducted in three parts (Figure 1). The study was designed and overseen by the authors. Subjects were recruited by and studied under the supervision of the contract research organization Hammersmith Medicines Research Ltd (London, UK). A portion of subjects in Part 3a were recruited by Simbec Research Ltd (Merthyr Tydril, South Wales, UK). In Parts 1 and 2, sequential dose cohorts were studied in the inpatient setting; in Part 3, parallel dose cohorts were studied predominantly in the outpatient setting. Part 1 included ascending single doses of TAK-385 via 20-mg tablets (0 [placebo], 80, 120, 180, or 360 mg); Part 2 included 14-day multiple daily doses of TAK-385 via 20-mg tablets (20, 40, 80, or 180 mg) or placebo; and Part 3 included 28-day daily doses of TAK-385 via 20- or 80-mg tablets (40 [with loading dose], 60, 80, or 160 mg) or placebo. A randomization schedule generated by the Sponsor was provided to the site research pharmacy, which in turn dispensed blinded active or placebo tablets. Thus, within (but not between) Parts 1 and 3, site personnel with direct contact with subjects as well as medical monitor(s) were blinded to treatment assignment. Contract research organization and Sponsor personnel participated in between-part decisions regarding subject safety, dose level, and loading dose regimens.
Figure 1.

Study design.
Subjects
The study protocol and all procedures were approved by local Ethics Committees and the UK central healthy authority (Medicines and Healthcare Products Regulatory Agency). The study was conducted in compliance with good clinical practice and the Declaration of Helsinki. All subjects provided written informed consent before initiation of any study procedures.
Healthy adult men not receiving medications known to affect the pituitary-gonadal axis were enrolled. The age ranges were 18–50 years for Part 1 and 40–70 years for Part 2. In Part 3, in keeping with patients treated for prostate cancer, older men aged 45–75 with stable chronic medical conditions (eg, diabetes, hypertension, hyperlipidemia, or cardiovascular disease) (full details are in the Supplemental Data) were included. No subject in Part 3 was allowed to be on more than two prescription medicines. Hormonal preparations, anticonvulsants, antipsychotics, anticoagulants (except aspirin), calcium channel or beta blockers, diabetes treatment other than metformin, and thyroid supplements were not allowed. To allow for increased age and background medical conditions, testosterone entry criteria were relaxed for each successive study part: >8.67 nmol/L for Part 1, >6.94 nmol/L for Part 2, and >5.21 nmol/L for Parts 3 and 3a. In all cases, screening LH was to be above the lower limits of detection. Additional subject eligibility criteria are detailed in the Supplemental Data.
Study drug administration
In Part 1, single doses of TAK-385 (80, 120, 180, and 360 mg in cohorts 1–4, respectively) were administered after an 8-hour overnight fast. After complete washout, subjects in cohort 3 of Part 1 (180-mg single dose) received a second dose of TAK-385 30 minutes after a standard US Food and Drug Administration-recommended, high-fat, high-calorie breakfast (800–1000 cal, 50% from fat).
In Part 2, TAK-385 was administered at daily doses of 20, 40, 80, or 180 mg in sequential cohorts (each cohort active, n = 6; placebo, n = 2) for 14 consecutive days. The 20- and 40-mg dose cohorts were administered various loading dose regimens for 1–3 days to further refine dose selection for Part 3.
Subjects in Part 3 were included if they were generally healthy or had well-controlled chronic medical conditions. Based on Part 2 results, two dose cohorts received either 40 or 160 mg/d, studied in parallel with a placebo cohort (n = 22 per cohort). The 40-mg cohort received a loading dose of 320 mg on day 1 and a loading dose of 160 mg on day 2. Based on Part 3 results from the 40- and 160-mg/d cohorts, two additional cohorts received 60 mg/d (n = 16) and 80 mg/d (n = 14) with no loading doses, with an additional placebo cohort (n = 8). All subjects were instructed to take the study drug upon waking in the morning, approximately 30 minutes before breakfast. Brief inpatient stays occurred on days 1–2 and days 27–29 to obtain more frequent PK and PD sampling.
Assessments and data analysis
At all clinic visits, the investigator recorded all observed or volunteered adverse events (AEs). Detailed PK and hormone sampling schedules, as well as testosterone assays and statistical analyses, are reported in the Supplemental Data. For each part of the study, blood was collected at prespecified time points to determine the plasma concentrations of TAK-385 using LC/MS/MS methodology, validated according to Good Laboratory Practice (9). LH was measured using a standard immunoassay via chemiluminescence (range, 0.2–250 mIU/mL; hLH, Beckman-Coulter). Serum testosterone (PD) was measured by immunoassay at screening (range, 20–1100 ng/dL) and by Good Laboratory Practice-validated LC/MS/MS at baseline and postdose (assay sensitive to 0.173 nmol/L [approximately 50 pg/mL]; intra- and interassay coefficients of variation [CVs] <10%). The upper limit of quantification for the LC/MS/MS assay was 3000 pg/mL (10.4 nmol/L); all values above this were imputed to 3000 pg/mL. Postbaseline sampling was more frequent during Parts 1 and 2 than Part 3.
Results
Demographics
In all, 176 male volunteers 19–75 years of age were enrolled; the majority were white (Table 1). In Parts 1, 2, and 3 of the study, age ranges were 19–50, 41–69, and 45–75 years, respectively. Weight and body mass index were similar across all cohorts. Screening testosterone levels were slightly but clinically insignificantly lower in the progressive study parts; baseline LH values were similar across study parts.
Table 1.
Demographic and Baseline Characteristics
| Part 1 | Part 2 | Part 3 | Part 3a | |
|---|---|---|---|---|
| n | 32 | 40 | 66 | 38 |
| Age, median (range), y | 28 (19–50) | 50 (41–69) | 53 (45–75) | 53 (45–71) |
| Race, n (%) | ||||
| White | 24 (75) | 37 (93) | 59 (89) | 35 (92) |
| Black/African American | 6 (19) | 2 (5) | 2 (3) | 1 (3) |
| Asian | 2 (6) | 1 (3) | 2 (3) | 1 (3) |
| Other | 0 | 0 | 3 (5) | 1 (3) |
| Weight, median (range), kg | 82.1 (60.1–101.4) | 78.7 (61.6–108.4) | 80.1 (60.6–107.2) | 78.8 (66.4–96.6) |
| BMI, median (range), kg/m2 | 25.1 (19.2–30.3) | 25.8 (20.4–31.6) | 25.8 (20.9–32.0) | 26.3 (20.7–31.5) |
| Screening testosterone, mean (SD), nmol/L | 18.2 (4.7) | 15.2 (4.4) | 13.8a (4.6) | |
Abbreviation: BMI, body mass index.
Pooled value as patients were recruited as one group for each part, with similar demographics and baseline characteristics.
Pharmacokinetics
Part 1 single-dose PK are summarized in the Supplemental Table 1. The multidose PK of TAK-385 (Part 2) showed that exposure was generally dose proportional (Table 2). Across dose levels, TAK-385 was readily absorbed after oral administration, with a median time to maximum plasma concentration (Tmax) of 1.01–1.59 hours postdose (Table 2). Based on area under the curve (AUC)0-τ, there was an approximate 2-fold accumulation over the 14-day dosing period. The Tmax was similar on days 1 and 14, and on day 14 it occurred within 1–2 hours after dosing at all dose levels (Table 2 and Figure 2A).
Table 2.
Mean (%CV) Plasma and Urine PK Parameters of TAK-385 (Day 14, Multiple Oral Dosing [Part 2])
| Parameter | 20 mgb (Cohort 4) | 40 mgc (Cohort 3) | 80 mg (Cohort 1) | 180 mg (Cohort 2) |
|---|---|---|---|---|
| n | 6 | 6 | 6 | 6 |
| AUC0–24, ng · h/mL | 42.3 (24.9) | 121 (55.5) | 203 (26.9) | 704 (50.8) |
| Cmax, ng/mL | 3.91 (35.8) | 20.8 (64.0) | 35.6 (54.0) | 168 (97.4) |
| Cmin, ng/mL | 1.27 (25.4) | 2.74 (46.0) | 3.48 (31.2) | 11.4 (42.8) |
| Tmax, ha | 1.01 (0.50–1.50) | 1.59 (0.50–2.00) | 1.26 (0.50–4.00) | 1.50 (1.00–4.00) |
| t1/2, h | 64.5 (52.6)d | 49.4 (30.6) | 36.5 (17.4) | 35.8 (23.0) |
| R(AUC) | — | — | 1.92 (23.5) | 2.16 (43.0) |
| R(Cmax) | — | — | 1.52 (62.7) | 1.80 (37.7) |
| Fe, % | 1.44 (31.8) | 1.76 (38.6) | 1.89 (20.2) | 2.88 (45.6) |
| CLr (L/h) | 6.92 (29.5) | 6.42 (25.0) | 7.66 (17.4) | 7.43 (16.2) |
Abbreviations: AUC0–24, area under the plasma concentration time curve from time 0 to 24 h; CLr, renal clearance; Cmin, minimum observed plasma concentration over the 24-hour dosing interval (day 14); Fe, fraction of the dose excreted unchanged in urine; R(AUC), accumulation factor based on AUC; R(Cmax), accumulation factor based on Cmax; —, not applicable.
Median (range).
Loading doses of 320 mg on day 1, 240 mg on day 2, and 160 mg on day 3.
Loading dose of 360 mg on day 1.
n = 4.
Figure 2.
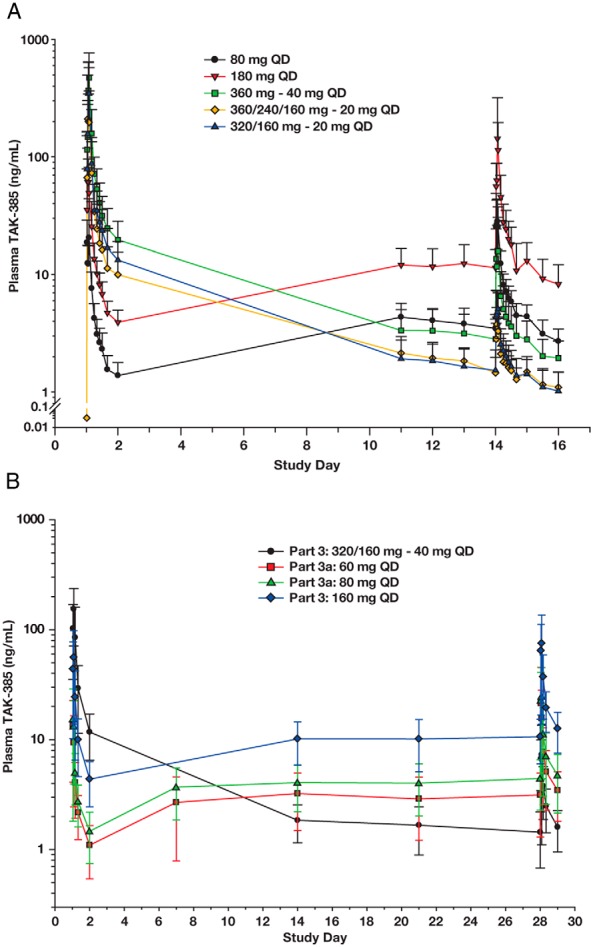
Mean plasma concentration-time profiles of TAK-385. A, Semilogarithmic plot of the mean (+SD) concentration-time profiles from subjects in Part 2 who received daily doses of TAK-385 for 14 days. B, Semilogarithmic plot of the mean (+ SD) concentration-time profiles from subjects in Parts 3 and 3a who received daily doses of TAK-385 for 28 days.
Plasma concentrations of TAK-385 decreased in a multiexponential manner with a mean disposition phase elimination half-life (t1/2) of approximately 36–65 hours across the 20- to 180-mg/d dose range. Systemic exposure to TAK-385 exhibited large to moderate variability after repeat dosing: mean %CV for maximum observed plasma concentration (Cmax) and AUC ranged from 24.9–64.0% between 20 and 80 mg/d and were 50.8 and 97.4%, respectively, at 180 mg/d (Table 2). Only a small percentage of TAK-385 was excreted unchanged in urine (Table 2).
After oral administration of a single 180-mg dose, TAK-385 absorption in plasma was decreased and delayed 30 minutes after ingestion of a high-fat, high-calorie meal vs under fasted conditions. Cmax and AUC0-∞ decreased by approximately 52 and 47%, respectively, indicating that food had a clinically meaningful effect on TAK-385 oral bioavailability.
In Part 3, mean PK profiles were consistent with those observed in Parts 1 and 2. There was no evidence of time- or concentration-dependent change in exposure upon repeat dosing for 28 days (Figure 2B).
Pharmacodynamics (hormone assessments)
After a single dose of TAK-385 in Part 1, serum LH levels decreased within 6 hours of administration and remained suppressed for up to 48 hours thereafter (Supplemental Figure 1A). Similar results were observed after repeated dosing in Part 2, with all doses 40 mg/d or higher effectively reducing LH by day 14; LH suppression was partial in subjects receiving TAK-385 20 mg/d (Supplemental Figure 1B). FSH levels were also reduced in a dose-proportional manner, and with 14- or 28-day dosing (Parts 2 and 3), levels were reduced by >90% at daily doses of 80 mg or greater (Supplemental Table 2).
In Part 1, mean serum testosterone levels also began to decrease 4–6 hours after a single dose of TAK-385 and reached a nadir at 36 hours postdose (Figure 3A). The time course of testosterone reduction was qualitatively similar for all doses but appeared most rapid and profound in the 360-mg cohort. In Part 2, after multiple doses of TAK-385, mean testosterone levels below the threshold for chemical castration (≤50 ng/dL or 1.73 nmol/L) (6) were achieved within 48 hours postdose (d 3) in the 180-mg cohort and in the cohorts receiving at least one loading dose (Figure 3B). Despite effective loading dose regimens, 20 mg was ineffective at maintaining effective castration, and the dose range of 40–160 mg daily was thus selected for Part 3.
Figure 3.

Mean (+ SD) serum concentration-time profiles of testosterone. A and B, Data from subjects who received single (A) or multiple (B) doses of TAK-385 or placebo in Parts 1 and 2, respectively. C and D, Data from subjects who received multiple doses of TAK-385 or placebo for 28 days in Parts 3 and 3a, respectively. Dashed and solid lines represent medical castration levels of 1.73 and 0.69 nmol/L, respectively. Note: All testosterone values for Figure 3 were measured by LC/MS/MS, with the upper range of the standard assay curve (ULQ) of 3000 pg/mL (10.4 nmol/L). All baseline values above 10.4 nmol/L (3000 pg/ml) were thus set to the ULQ.
In Part 3, reduction in serum testosterone levels was most rapid in subjects who received a loading dose on days 1 and 2 (40-mg cohort), and in the 160-mg cohort. In the 160-mg cohort, mean serum testosterone concentrations fell below 1.73 nmol/L by day 7 and below 0.69 nmol/L by day 28. However, in the 40-mg cohort (with a loading dose), unlike the result observed in Part 2, mean serum testosterone levels did not fall below 1.73 nmol/L at any time point after day 4 (Figure 3C).
In the 160-mg cohort, a serum testosterone level <1.73 nmol/L was achieved in 100% of subjects by day 28, and a serum testosterone level <0.69 nmol/L was achieved in 75% of subjects (Supplemental Table 3). To further identify an effective daily dose below 160 mg/d, two additional smaller cohorts were subsequently enrolled in Part 3 and received either 60 or 80 mg/d (or placebo; Figure 3D) (termed Part 3a in tables and figures because the data were evaluated sequentially and thus separately from the 40-mg and 160-mg cohorts). Mean serum testosterone levels <0.69 nmol/L were achieved by day 28 in >70% of subjects in the 60- and 80-mg cohorts (Supplemental Table 3).
In Part 3, testosterone recovery was monitored after completion of dosing on day 28 (Supplemental Table 4). In the 160-mg cohort, levels recovered to 8.67 nmol/L in most subjects (68%) by day 56 and returned to the subject's baseline level in almost 60% of subjects. Recovery was more rapid in the 60- and 80-mg cohorts; testosterone levels were ≥8.67 nmol/L (250 ng/dL) by day 56 in all but one subject and recovered to baseline levels or above in almost 80% of subjects.
Safety
Most subjects experienced at least one AE, all mild to moderate in severity (grade ≤2). Bradycardia (<60 beats/min) was the most common AE, but the incidence was similar in subjects receiving TAK-385 or placebo, and it was considered by the investigator to be clinically insignificant in this healthy male population (Table 3). Headache and hot flush were also common, and several other AEs appeared to be related to the effects of acute medical castration (Table 3). Seven subjects in Part 3 experienced prolongation of the QTc interval to >450 milliseconds on day 14 or day 28. The mean increases in QTcF (QTc interval calculated per Fridericia's formula) (10) on day 28 vs baseline in the 60-, 80-, and 160-mg cohorts were 14.2, 14.3, and 19.1 milliseconds, respectively. No subject experienced a QTc greater than 480 milliseconds. The observed changes were similar before and after dosing (ie, were not related to drug concentration) and were considered related to medical castration and not clinically significant (see Discussion). Per phase 1 protocol subject discontinuation rules, three subjects were taken off study because of elevations of alanine and aspartate aminotransferase: two subjects in the 160-mg cohort and one subject in the 60-mg cohort. These liver function abnormalities were generally mild in severity (<3× upper limit of normal) and resolved by the end-of-study follow-up visit.
Table 3.
Most Common (>10% of Subjects) AEs, Safety Population
| Part 1 |
Part 2 |
Part 3 |
Part 3a |
|||||
|---|---|---|---|---|---|---|---|---|
| TAK-385 (80–360 mg) | Placebo | TAK-385 (20–180 mg) | Placebo | TAK-385 (40–160 mg) | Placebo | TAK-385 (60–80 mg) | Placebo | |
| n | 24 | 8 | 30 | 10 | 44 | 22 | 30 | 8 |
| Bradycardia | 19 (79) | 7 (88) | 27 (90) | 9 (90) | 33 (75) | 17 (77) | — | — |
| Headache | 1 (4) | — | 7 (23) | 2 (20) | 8 (18) | 4 (18) | 11 (37) | 4 (50) |
| Hot flush | 1 (4) | — | 7 (23) | — | 19 (43) | 2 (9) | 11 (37) | — |
| AV block first degree | 1 (4) | — | 6 (20) | — | — | — | — | — |
| Decreased libido | — | — | 5 (17) | — | — | — | — | — |
| Prolonged QTc | 1 (4) | — | 4 (13) | — | 7 (16) | — | — | — |
| Nasopharyngitis | — | — | 4 (13) | — | — | — | 6 (20) | 2 (25) |
| Hyperhidrosis | — | — | — | — | 8 (18) | — | — | — |
| Loss of libido | — | — | — | — | — | — | 5 (17) | — |
Abbreviation: AV, atrioventricular. Data are expressed as number (percentage); —, none reported.
Discussion
The goal of this study was to establish the clinical PK, endocrine PD, and safety profiles of TAK-385 in healthy, predominantly older men and to determine whether TAK-385 is a suitable drug candidate for phase 2 studies in men requiring medical castration therapy for hormone-sensitive prostate cancer. TAK-385 appeared well tolerated over dosing periods up to 28 days, and the minimum effective dose for sustained castration was determined to be ≥80 mg/d. A loading dose regimen shortened the time interval required to achieve castrate testosterone levels but, contrary to previously reported findings with a peptide antagonist (11), did not reduce the daily dose of TAK-385 required to achieve sustained medical castration.
TAK-385 was readily absorbed in plasma after single and multiple oral dosing, although absorption was variable. Importantly, a high-fat meal had a significant impact in reducing the exposure of TAK-385, suggesting that the drug would optimally be administered without food. After repeated daily dosing, generally dose-proportional steady-state plasma concentrations of TAK-385 were reached within 11–14 days with an approximate 2-fold accumulation over that time period.
As expected, the PK profile of TAK-385 differs significantly from that of an injectable depot peptide analog, such as degarelix, with a prolonged time to steady state of 5–6 months (12) and a reported mean terminal t1/2 of 23–61 days (11, 12). As a nonpeptidic small molecule, TAK-385 has the advantages of oral bioavailability and longer intrinsic t1/2 vs a depot peptide analog. Dosing flexibility and potentially more rapid withdrawal of effect must be weighed against relative advantages of a long-acting depot, which may include the simplicity of peptide metabolism and long-term patient compliance.
Parts 1 and 2 of the study clearly demonstrated the kinetics of testosterone lowering, with the fall in testosterone uniformly associated with prior and/or concurrent reduced LH, consistent with the GnRH antagonist mechanism of action. FSH levels were also profoundly reduced, a finding consistent with GnRH antagonism but different from the reported effects of LHRH agonists (13). No such changes occurred in subjects randomized to placebo, effectively ruling out a significant contribution to lowering of the normal diurnal variation of the pituitary-gonadal axis. In Parts 2 and 3, sustained, medical castration was consistently achieved at TAK-385 daily doses of 80, 160, and 180 mg. In the controlled dosing conditions of Part 2, a 40-mg daily dose appeared effective to achieve medical castration, whereas with less-controlled outpatient dosing in Part 3, 40 mg daily was relatively ineffective, and 20-mg daily doses were uniformly ineffective, regardless of various loading dose regimens. However, to achieve castration within 2–3 days, loading doses are required at all dose levels below 180 mg daily. The dose-dependent effect on medical castration generally correlated with steady-state PK as reflected by trough concentrations (24 h after last dose and just prior to the next) of TAK-385. Thus, sustained castrate levels of serum testosterone (Supplemental Figure 2) were more likely associated with trough TAK-385 concentrations ≥4 ng/mL, achieved more reliably with daily doses ≥80 mg.
Transient increases in serum testosterone above the castrate level (testosterone breakthrough) have been observed in subjects treated with ADT and may be associated with poorer clinical outcomes (14). In Part 3 of this study, the 60-mg/d dosing regimen resulted in serum testosterone levels of 0.69 nmol/L by day 21 in 43% of subjects, but testosterone breakthrough on day 28 was observed in some subjects at this dose level. No breakthrough was observed on day 28 in subjects receiving 80 or 160 mg/d dosing (Supplemental Figure 3). Therefore, daily doses of TAK-385 ≥80 mg, with single loading dose on day 1, are planned for further phase 2 clinical testing in men with prostate cancer.
There were no deaths, serious AEs, or severe AEs in this study. The most common AE was bradycardia, which was similar in incidence across all dose cohorts and between subjects receiving active or placebo doses. The second most common cardiovascular AE was QTc prolongation. However, changes in QTc were observed predose when TAK-385 plasma concentrations were low (≤12.8 ng/mL), and the QTc interval remained generally unchanged 4 hours after dosing. Moreover, no changes in QTc intervals were observed on day 1 when single doses up to 360 mg were administered. There is evidence that the male sex hormone dihydrotestosterone influences QT by affecting ventricular repolarization (15), and epidemiological data have shown that low levels of testosterone are associated with longer QT intervals (16). Increases in QTcF of 10–20 milliseconds have also been reported with medical castration agents such as degarelix and leuprolide (17–19). Vasomotor symptoms and other AEs were consistent with ADT and were observed in studies of degarelix (5, 12). A similar proportion of subjects experienced AEs across all dosing cohorts, and in Part 3 (daily dosing for 28 d), safety profiles in the 40-mg and 160-mg cohorts were similar. Mild to moderate transaminase non-dose-related elevations were observed, which were asymptomatic and either transient or rapidly reversible. Transaminase abnormalities have also been reported in studies of peptide GnRH analogs including degarelix (5), but prospective comparison is required.
Another oral nonpeptide GnRH antagonist, elagolix (Neurocrine Biosciences, AbbVie) is currently in development as a treatment for women with endometriosis or uterine fibroids (20); studies in men have not been reported. Based on results of TAK-385 studied in women (Takeda; unpublished data), a postmenopausal castrate state can be achieved with lower doses vs those required in this study to achieve castration in men. This may be due to the tonic and relatively more resistant negative feedback system in men.
In summary, in healthy volunteers evaluated for up to 28 days of treatment, administration of TAK-385 at doses ≥80 mg/d produced sustained reductions in serum testosterone levels to below castration levels. Loading doses shortened the time to medical castration but did not affect the required maintenance dose. Testosterone flare did not occur. Data from the phase 3 trial of degarelix suggest that GnRH antagonists are an attractive option for testosterone suppression in prostate cancer. The use of degarelix has been relatively limited because of the requirement for large volume loading doses and monthly injections. A once-daily oral GnRH antagonist might retain many of the potential benefits of an antagonist while avoiding injection site reactions associated with peptide antagonists. Although long-term medication compliance is a potential issue with daily oral medications, other oral therapies in prostate cancer, such as androgen receptor antagonists or the more recently approved therapies for castration-resistant disease (21–23), are effective treatment options. Based on these promising phase 1 results in healthy older men, a phase 2 program has been initiated to evaluate the long-term safety and efficacy of TAK-385 in patients with prostate cancer.
Acknowledgments
The authors thank all of the volunteers for their participation in the study and acknowledge Steve Warrington, Hammersmith Medicines Research Ltd medical director and lead pharmacologist, and all staff members of Hammersmith Medicines Research Ltd and Simbec Research Ltd. The authors also acknowledge the editorial assistance of Dawn L. Lee of FireKite (an Ashfield company, part of UDG Healthcare plc) in the development of this manuscript.
This work was supported by Millennium Pharmaceuticals, Inc, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
European Clinical Trials Database (EudraCT) no. 2011-002868-24.
Disclosure Summary: D.B.M. and H.M.F. are employees of Millennium Pharmaceuticals, Inc. H.S. was an employee of Millennium Pharmaceuticals, Inc. at the time the work was done but is no longer employed by the company. F.S. is a consultant and an investigator for Millennium Pharmaceuticals, Inc.
Footnotes
- ADT
- androgen deprivation therapy
- AE
- adverse event
- AUC
- area under the curve
- Cmax
- maximum observed plasma concentration
- CV
- coefficient of variation
- LC/MS/MS
- liquid chromatography/tandem mass spectroscopy
- PD
- pharmacodynamics
- PK
- pharmacokinetics
- t1/2
- half-life
- Tmax
- time to maximum plasma concentration.
References
- 1. Shore ND, Abrahamsson PA, Anderson J, Crawford ED, Lange P. New considerations for ADT in advanced prostate cancer and the emerging role of GnRH antagonists. Prostate Cancer Prostatic Dis. 2013;16(1):7–15. [DOI] [PubMed] [Google Scholar]
- 2. Rick FG, Block NL, Schally AV. An update on the use of degarelix in the treatment of advanced hormone-dependent prostate cancer. Onco Targets Ther. 2013;6:391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thompson IM. Flare associated with LHRH-agonist therapy. Rev Urol. 2001;3(suppl 3):S10–S14. [PMC free article] [PubMed] [Google Scholar]
- 4. Tombal B, Miller K, Boccon-Gibod L, et al. Additional analysis of the secondary end point of biochemical recurrence rate in a phase 3 trial (CS21) comparing degarelix 80 mg versus leuprolide in prostate cancer patients segmented by baseline characteristics. Eur Urol. 2010;57(5):836–842. [DOI] [PubMed] [Google Scholar]
- 5. Klotz L, Boccon-Gibod L, Shore ND, et al. The efficacy and safety of degarelix: a 12-month, comparative, randomized, open-label, parallel-group phase III study in patients with prostate cancer. BJU Int. 2008;102(11):1531–1538. [DOI] [PubMed] [Google Scholar]
- 6. Mottet N, Bastian PJ, Bellmunt J, et al. Guidelines on Prostate Cancer. European Association of Urology. Version 2.2014. http://uroweb.org/wp-content/uploads/1607-Prostate-Cancer_LRV3.pdf. Updated April 2014; accessed September 28, 2015.
- 7. Nakata D, Masaki T, Tanaka A, et al. Suppression of the hypothalamic-pituitary-gonadal axis by TAK-385 (relugolix), a novel, investigational, orally active, small molecule gonadotropin-releasing hormone (GnRH) antagonist: studies in human GnRH receptor knock-in mice. Eur J Pharmacol. 2014;723:167–174. [DOI] [PubMed] [Google Scholar]
- 8. Miwa K, Hitaka T, Imada T, et al. Discovery of 1-{4-[1-(2,6-difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (TAK-385) as a potent, orally active, non-peptide antagonist of the human gonadotropin-releasing hormone receptor. J Med Chem. 2011;54(14):4998–5012. [DOI] [PubMed] [Google Scholar]
- 9. World Health Organization. Handbook: Good Laboratory Practice (GLP): Quality Practices for Regulated Non-clinical Research and Development 2009. 2nd ed http://www.who.int/tdr/publications/documents/glp-handbook.pdf. Published January 2009; accessed August 29, 2015.
- 10. Fridericia LS. The duration of systole in an electrocardiogram in normal humans and in patients with heart disease. 1920. Ann Noninvasive Electrocardiol. 2003;8(4):343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Behre HM, Kliesch S, Pühse G, Reissmann T, Nieschlag E. High loading and low maintenance doses of a gonadotropin-releasing hormone antagonist effectively suppress serum luteinizing hormone, follicle-stimulating hormone, and testosterone in normal men. J Clin Endocrinol Metab. 1997;82(5):1403–1408. [DOI] [PubMed] [Google Scholar]
- 12. Van Poppel H, Tombal B, de la Rosette JJ, Persson BE, Jensen JK, Kold Olesen T. Degarelix: a novel gonadotropin-releasing hormone (GnRH) receptor blocker–results from a 1-yr, multicentre, randomised, phase 2 dosage-finding study in the treatment of prostate cancer. Eur Urol. 2008;54(4):805–813. [DOI] [PubMed] [Google Scholar]
- 13. Beer TM. Experimental use of GnRH antagonists as second-line hormonal therapy. Rev Urol. 2004;6(suppl 7):S33–S38. [PMC free article] [PubMed] [Google Scholar]
- 14. Morote J, Orsola A, Planas J, et al. Redefining clinically significant castration levels in patients with prostate cancer receiving continuous androgen deprivation therapy. J Urol. 2007;178:1290–1295. [DOI] [PubMed] [Google Scholar]
- 15. Liu XK, Katchman A, Whitfield BH, et al. In vivo androgen treatment shortens the QT interval and increases the densities of inward and delayed rectifier potassium currents in orchiectomized male rabbits. Cardiovasc Res. 2003;57(1):28–36. [DOI] [PubMed] [Google Scholar]
- 16. Zhang Y, Ouyang P, Post WS, et al. Sex-steroid hormones and electrocardiographic QT-interval duration: findings from the third National Health and Nutrition Examination Survey and the Multi-Ethnic Study of Atherosclerosis. Am J Epidemiol. 2011;174(4):403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smith MR, Klotz L, Persson BE, Olesen TK, Wilde AA. Cardiovascular safety of degarelix: results from a 12-month, comparative, randomized, open label, parallel group phase III trial in patients with prostate cancer. J Urol. 2010;184(6):2313–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garnick MB, Pratt CM, Campion M, Shipley J. The effect of hormonal therapy for prostate cancer on electrocardiographic QT interval: phase 3 results following treatment with leuprolide and goserelin, alone or with bicalutamide, and the GnRH antagonist abarelix. J Clin Oncol. 2004;22:4578. [Google Scholar]
- 19. Saglam H, Çakar A, Köse O, et al. Changes in electrocardiogram findings during treatment with gonadotropin-releasing hormone agonist and surgical castration for prostate carcinoma. Open J Urol. 2012;2(3A):153–156. [Google Scholar]
- 20. Carr B, Giudice L, Dmowski WP, et al. Elagolix, an oral GnRH antagonist for endometriosis-associated pain: a randomized controlled study. J Endometr. 2013;5(3):105–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Beer TM, Armstrong AJ, Rathkopf DE, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014;371(5):424–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cannata DH, Kirschenbaum A, Levine AC. Androgen deprivation therapy as primary treatment for prostate cancer. J Clin Endocrinol Metab. 2012;97(2):360–365. [DOI] [PubMed] [Google Scholar]
- 23. de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364(21):1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]