

Abstract
Study Objectives:
To test the hypothesis that greater resting sympathetic activity in obstructive sleep apnea (OSA) syndrome would not induce a lesser sympathetic neurovascular transduction.
Design:
Case-controlled cohort study.
Participants:
33 patients with newly diagnosed OSA without comorbidities and 14 healthy controls.
Interventions:
6 months of continuous positive airway pressure (CPAP) treatment for OSA patients and follow-up for 9 healthy controls.
Measurements and Results:
We assessed resting sympathetic outflow and sympathetic neurovascular transduction. Sympathetic activity was directly measured (microneurography) at rest and in response to sustained isometric handgrip exercise. Neurovascular transduction was derived from the relationship of sympathetic activity and blood pressure to leg blood flow during exercise. Despite an elevated sympathetic activity of ∼50% in OSA compared to controls, neurovascular transduction was not different (i.e., absence of tachyphylaxis). After six months of CPAP, there were significant declines in diastolic pressure, averaging ∼4 mm Hg, and in sympathetic activity, averaging ∼20% with no change in transduction.
Conclusions:
Greater sympathetic activity in obstructive sleep apnea does not appear to be associated with lesser neurovascular transduction. Hence, elevated sympathetic outflow without lesser transduction may underlie the prevalent development of hypertension in this population that is well controlled by continuous positive airway pressure treatment.
Citation:
Tamisier R, Tan CO, Pepin JL, Levy P, Taylor JA. Blood pressure increases in OSA due to maintained neurovascular sympathetic transduction: impact of CPAP. SLEEP 2015;38(12):1973–1980.
Keywords: obstructive sleep apnea, sympathetic nervous system, hypertension, continuous positive airway pressure, arterial blood pressure
INTRODUCTION
Obstructive sleep apnea syndrome (OSAS) is increasingly recognized as a significant health hazard for a substantial portion of the adult population primarily because of its association with several serious cardiovascular disorders.1 Although it remains uncertain what facilitates cardiovascular disease in the apneic patient, OSAS is recognized as a risk factor for the development of hypertension and a primary etiology for secondary resistant hypertension.2;3 Thus, the development of hypertension is the key initiating factor that results in the comorbidities of ischemic stroke, cardiac ischemia, and chronic heart failure.4–6 Moreover, elevated tonic sympathetic outflow to the vasculature elicited by sleep apnea persists into the waking daytime hours, and this, at least in part, mediates the elevated pressure seen in those with OSAS.7–9 In fact, OSAS is now presented as a model of neurogenic hypertension.10 Hence, understanding the mechanisms underlying sustained sympatho-excitation in OSAS may provide new strategies to prevent and mitigate the accompanying cardiac and vascular disease associated with neurogenic hypertension. Moreover, continuous positive airway pressure (CPAP), which is the reference treatment for the OSAS patient, has a major effect in preventing the development of hypertension,11 and it would be important to identify a mechanism for this effect.
Prior work suggests that vascular sympathetic outflow is increased even in those who are otherwise healthy but have OSAS.12;13 Hence, the increase in tonic sympathetic activity appears to be due to OSAS per se. Interestingly, the 50% to 100% greater sympathetic activity in OSAS compared to age-matched controls14 may not be accompanied by alterations in resting limb blood flow.15 Although this fits with data suggesting a functional downregulation of sympatho-adrenergic α-receptors in those with OSAS,16 it may also indicate one important mechanism for sustained sympathoexcitation: reduced sympathetic neurovascular transduction (i.e., a sustained tachyphylaxis)17 that in turn results in elevated outflow to effect appropriate hemodynamic control.18 For example, healthy older individuals demonstrate greater sympathetic activity than young healthy individuals that is proportional to their lower sympathetic neurovascular transduction.19 Hence, an appropriate response to sympatho-excitation appears to be a decrease in sympathetic neurovascular transduction, which may subserve a regulatory function to maintain blood pressure.
We sought to determine if the ability of sympathetic nervous activity to control limb blood flow is impaired by the pathophysiology of OSAS and whether this impairment may be a trigger that promotes the elevation in blood pressure. In addition, we sought to determine if CPAP effectively treats sympathetic overactivity in those with OSAS without cardiometabolic comorbidities, and if this is accompanied by an increase in sympathetic transduction and hence no change in blood pressure. We assessed resting sympathetic outflow and sympathetic neurovascular transduction in individuals newly diagnosed with OSAS but without other cardiometabolic comorbidities and in a control group of age-matched healthy individuals. In addition, we examined a subgroup of individuals with OSAS six months after treatment with CPAP. We hypothesized that the greater resting sympathetic activity in normotensive individuals with OSAS would relate to lesser transduction and that a decline in sympathetic activity after CPAP would result in increased transduction and hence maintain (normotensive) blood pressure.
METHODS
Subjects
Individuals with OSAS aged 24–76 years (n = 33; 91% male) were recruited from those reporting to a sleep clinic with the complaint of snoring and or sleepiness and with subsequent confirmation of sleep apnea after full polysomnography. Continuous recordings were taken with electrode positions C3/ A2-C4/A1-Cz/O1 of the international 10–20 electrode placement system, eye movements, chin electromyogram, and ECG with modified V2 lead. Sleep was scored manually according to standard criteria.20 Airflow was measured mainly with nasal pressure (nasal cannula) and completed by the sum of oral and nasal thermistor signals. Respiratory efforts were monitored with abdominal and thoracic bands. An additional signal of respiratory effort (i.e., pulse transit time) was recorded concurrently. Oxygen saturation was measured using a digital pulse oximeter. An apnea was defined as a complete cessation of airflow ≥ 10 s and a hypopnea as reduction ≥ 50% in the nasal pressure signal or a decrease between 30% and 50% associated with either oxygen desaturation ≥ 3% or EEG arousal defined by the Chicago report21 and the latest AASM statement,20 both lasting ≥ 10 s. Apneas were classified as obstructive, central, or mixed according to the presence or absence of respiratory efforts, on pulse transit time22 and shape of the respiratory curve of nasal pressure (flow limited aspect or not).23 AHI was calculated and defined as the number of apneas and hypopneas per hour of sleep (full polysomnography). Sleep apnea was defined as an apnea-hypopnea index (AHI) ≥ 15/h of sleep since this rule of scoring with a nasal cannula are much more sensitive. Subsequently, all OSAS patients were screened for cardiovascular and metabolic disease based on medical history, clinical exam, office blood pressure (to exclude hypertensive patients, i.e., those with systolic and diastolic blood pressure over 140 mm Hg and 90 mm Hg obtained as the average of the 3 measurements during an office session), and fasting glucose. Age-matched control subjects (n = 14) were recruited from the general population and completed a full polysomnography with an AHI < 10/h of sleep. All subjects were non-smokers, and free of elevated resting blood pressures and overt autonomic or cardiovascular diseases. All subjects provided written informed consent approved by the ethical committee at the Grenoble University Hospital Center (protocol number NCT00764218).
Protocol and Measurements
A standard 3-lead electrocardiogram was recorded continuously throughout the study. Beat-by-beat arterial blood pressure via finger photoplethysmography (CNAP 500, CN-Systems, Austria) was measured and was calibrated against oscillometric brachial pressures measured throughout the study session (automatic device calibration). Multiunit post-ganglionic muscle sympathetic activity was recorded from the peroneal nerve.24 Neural activity was amplified, and band pass was filtered, rectified, and integrated to create the sympathetic neurogram for real-time inspection. The raw, unfiltered neurogram was recorded at 10 kHz for processing using our algorithm for identification of sympathetic nerve signals.25 Subjects were instrumented for measurement of blood flow velocity in the popliteal artery (4-MHz probe; EZdop, DWL) at the popliteal fossa of the leg contralateral to the nerve recording. All signals were digitized and stored (Windaq, DATAQ Instruments, or PowerLab, ADInstruments) for subsequent analysis.
Subsequent to instrumentation, each subject's maximal voluntary handgrip force was determined from ≥ 3 maximal contractions on a handgrip dynamometer. After acquisition of a suitable sympathetic nerve recording, a 5-min resting baseline recording was obtained. Subjects then performed sustained isometric handgrip exercise with the dominant hand to fatigue. Target force was displayed on an oscilloscope and set at 35% of the mean of the 2 greatest maximal contractions for the hand. Continuous visual and auditory feedback (from the investigators) was provided to subjects to ensure maintenance of target force. Force was maintained until this level could no longer be achieved. The point of exhaustion was defined as a decrease in handgrip force ≥ 10% below target level for > 2 s despite continued verbal encouragement and the attainment of a maximal perceived exertion. During exercise, breathing was monitored to ensure that no Valsalva maneuvers (breath-holding strains) were performed.
CPAP Intervention and Follow-Up
Patients with diagnosed OSAS were offered the opportunity to receive CPAP treatment for 6 months as part of the research protocol. Of the 33 OSAS patients, 4 refused treatment, 4 received CPAP but did not complete the follow-up visit, and 2 preferred treatment via a mandibular advancement device. Those who did and did not receive CPAP did not differ in age (50.7 ± 2.2 and 52.7 ± 3.0), body mass index (BMI; 25.7 ± 0.6 vs. 25.7 ± 0.7), sleep AHI (33.3 ± 3.6 vs. 35.2 ± 6.1), or oxygen saturation (94.5% ± 0.3% vs. 94.0% ± 0.5%). To assess stability of our measures over time, 9 of the age-matched healthy controls were also studied again after 6 months. Of 14 healthy controls, 5 declined to participate in the follow-up study (see Figure 1).
Figure 1.
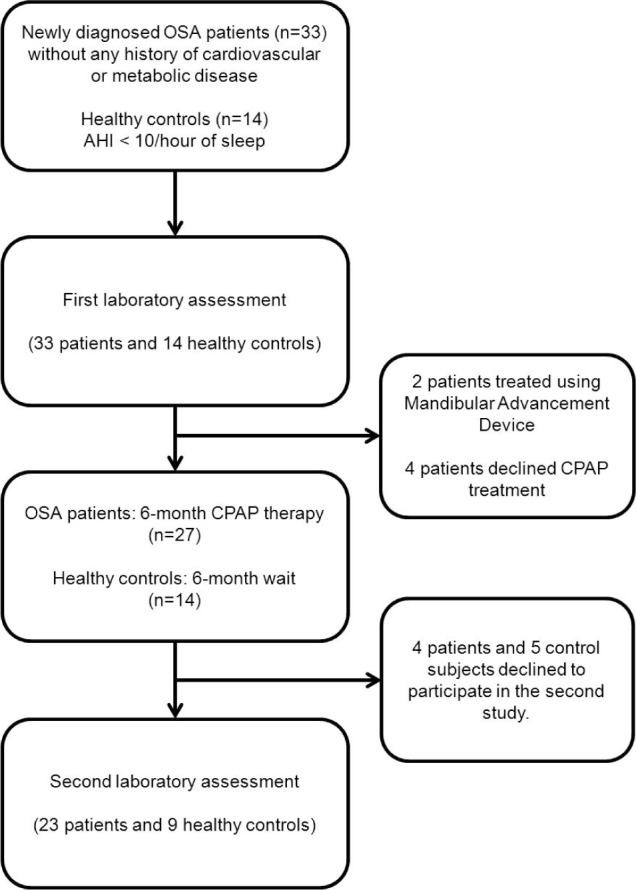
Subject enrollment and study design.
Data Analysis
Sympathetic activity was analyzed as described previously25 to obtain a clean representation of nerve activity in the form of a spike train that would provide necessary time resolution for accurate assessment of the relation between sympathetic activity and blood flow. We derived sympathetic neurovascular transduction for each subject from the sympathetic, arterial pressure, and popliteal flow signals sampled at 4 Hz, as previously described.19 This approach entails a linear transfer function model analogous to Poiselle's Law and produces reliable and interpretable parameters. The time-dependent transfer function between sympathetic nerve activity and blood flow was estimated for each individual, and the strength of the relation (i.e., gain of the transfer function) between sympathetic activity and blood flow was then quantified as the total area under the transfer function. It should be noted that simple derivations of either vascular resistance or vascular conductance provided markedly lower explained variance than the above approach.19 Since basal blood flow to the muscle is generally proportional to the tissue volume, inter-individual differences in body size and composition may confound assessment of sympathetic neurovascular transduction. To account for differences in tissue volume when assessing sympathetic neurovascular transduction, we normalized blood flow velocity for each individual to the flow velocity during supine rest, as described previously.19 All data were analyzed using custom-written software in Matlab (Mathworks, Natick, MA).
Statistics
Group comparisons between healthy individuals and OSAS patients were performed via one-way analysis of variance (ANOVA). Comparison within healthy individuals (0 vs 6 months) and within OSAS patients (before vs after 6 months of CPAP treatment) were performed via one-way repeated measures ANOVA. Conformity of the data to statistical assumptions that underlie ANOVA was verified via standard statistical tests. Correlations of sympathetic transduction gain to mean arterial pressure and to resting sympathetic activity were explored and when a significant relation was observed, linear and nonlinear regressions were explored. A P value < 0.05 was considered significant and all data are presented as means ± standard error of the mean (SEM).
RESULTS
Subject characteristics are shown in Table 1. BMI ranged from 19.2 to 30.9 kg/m2 in the OSAS patients and from 20.3 to 28.4 kg/m2 in the healthy controls. The distribution of values was similar in the 2 groups and average BMI did not differ. Only resting sympathetic activity was different between those with and without OSAS; sympathetic activity was elevated by ∼50% in OSAS compared to the age-matched control group (P < 0.05; Figure 2).
Table 1.
Subject characteristics.
Figure 2.
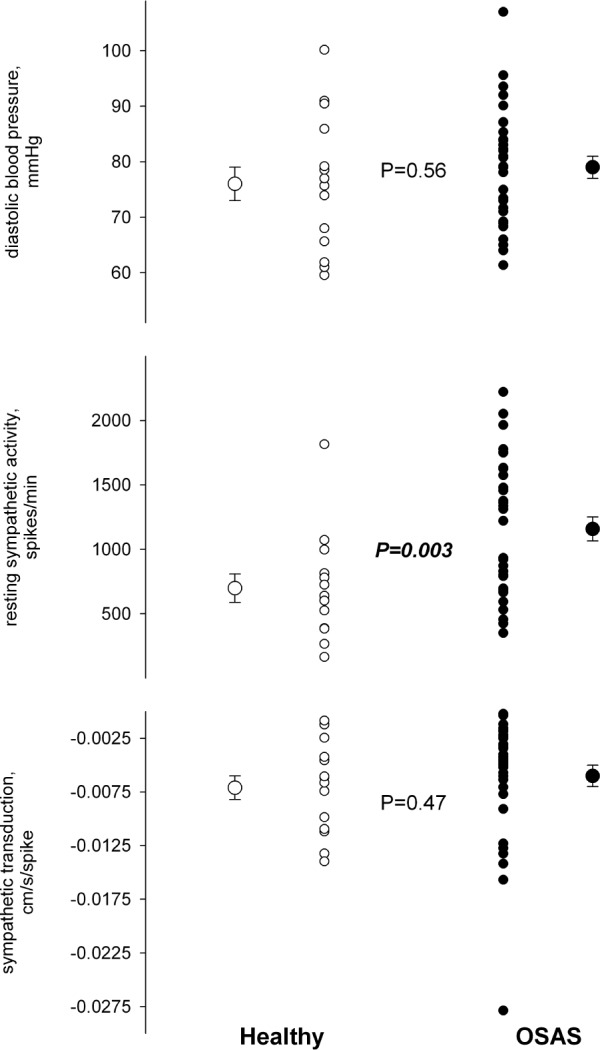
Diastolic blood pressure, resting sympathetic nerve activity, and sympathetic neurovascular transduction in patients with OSAS and age-matched controls.
The relationship of blood flow to both blood pressure and sympathetic activity during the isometric handgrip exercise was well-described by the sympathetic neurovascular transduction assessment. The explained variance averaged 67% in the OSAS and 68% for the age-matched healthy controls. As shown in Figure 2, despite the markedly higher sympathetic activity, sympathetic transduction was similar between the OSAS and healthy controls. Moreover, despite both higher sympathetic activity and similar sympathetic transduction, diastolic pressure did not differ between OSAS and healthy control groups (Figure 2). Within the OSAS group, sympathetic activity did not relate to AHI severity and had only a tendency to relate to sympathetic transduction (r = 0.33, P = 0.12), whereas diastolic pressure was significantly related to leg vascular resistance (r = 0.55, P = 0.002).
In the 23 individuals who agreed to 6 months of CPAP treatment, compliance averaged 4.5 h/night, but ranged widely from noncompliance (0 h/night, n = 4) to essentially full compliance (≥ 6 h/night, n = 11). Nonetheless, AHI declined on average by 73%, from 32.3 ± 3.6 to 7.1 ± 1.3. After 6 months of CPAP, there were significant declines in diastolic pressure, averaging ∼4 mm Hg, and in sympathetic activity, averaging ∼20% (Table 2; Figure 3). There was no significant effect of CPAP on sympathetic neurovascular transduction (Table 2). There was no relation between the change in sympathetic nerve activity and CPAP compliance and either absolute or relative change in AHI, suggesting that the effect of CPAP on sympathetic activity was not dose dependent. This is consistent with the fact that the level of sympathetic activity prior to treatment was not related to AHI severity. There was essentially no change in any other variable over 6 months in the age-matched healthy controls. As in the larger group of OSAS patients, the subgroup receiving CPAP demonstrated only a tendency for a relationship between sympathetic activity and sympathetic transduction prior to treatment; however, the decline in sympathetic activity after treatment resulted in a closer relation between the level of sympathetic activity and sympathetic transduction (Figure 4). In addition, there was a relation between diastolic pressure and leg vascular resistance prior to CPAP treatment, but the strength of this relation was roughly halved after treatment (Figure 5). Given these relations and their changes, we performed a multiple linear regression on the pretreatment and posttreatment data to explore potential alterations in the determinants of diastolic pressure due to CPAP treatment. Diastolic pressure was regressed on AHI, leg vascular resistance, sympathetic activity, sympathetic transduction, and the interaction between sympathetic activity and transduction. Prior to CPAP treatment, all variables provided significant explanatory power for the level of diastolic pressure with an overall r-square of 0.60 at a P value of 0.007. However, after CPAP treatment, AHI no longer provided explanatory power for the level of diastolic pressure, and although the other variables still had predictive value, the overall r-square was lesser (0.37) with a P value of only 0.08.
Table 2.
Hemodynamic variables measured 6-months apart (healthy volunteers) or before and after 6 months of CPAP therapy (OSAS patients).
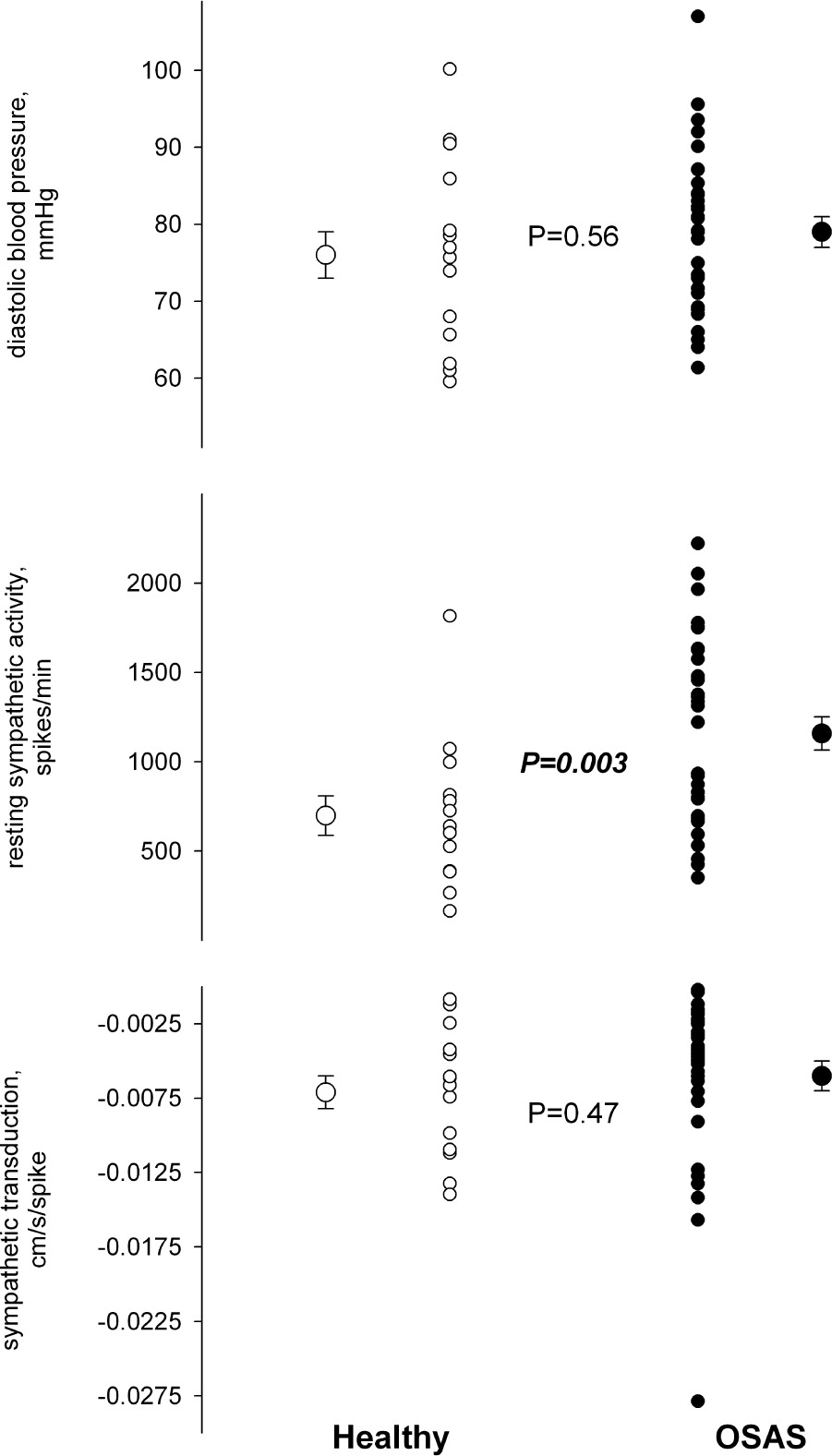
Figure 3.

Diastolic blood pressure and sympathetic nerve activity before and after 6 months of CPAP therapy.
Figure 4.
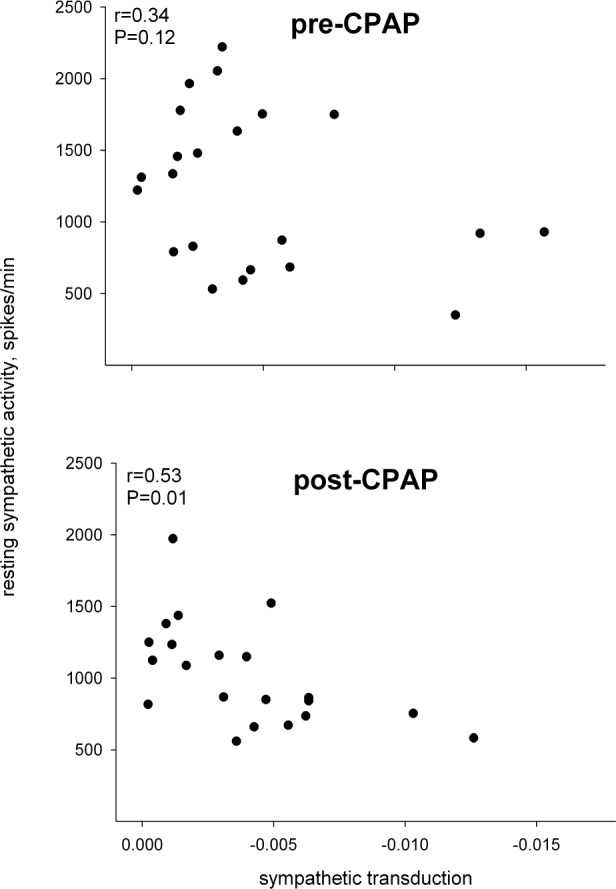
The relation between sympathetic neurovascular transduction and resting sympathetic activity before and after 6 months of CPAP therapy.
Figure 5.
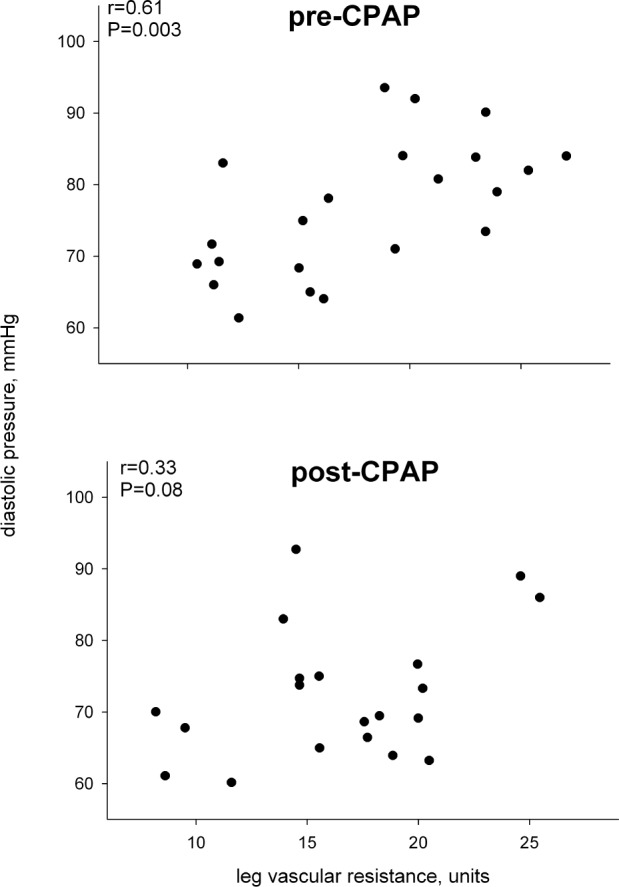
The relation between diastolic blood pressure and leg vascular resistance before and after 6 months of CPAP therapy.
DISCUSSION
Our results suggest that the elevated sympathetic activity associated with OSAS does not result from lesser sympathetic neurovascular transduction. Although our OSAS patients were not hypertensive according to either laboratory measurements or ambulatory monitoring, they demonstrated sympathetic activity that was significantly higher than their healthy counterparts. A comparably normotensive pressure with elevated sympathetic activity in OSAS would seem incompatible with a sympathetic transduction similar to healthy subjects. However, the data from CPAP treatment provided insight to this apparent incongruency. After six months of treatment, reductions in nighttime apneic events not only resulted in the expected decline in sympathetic activity, but also a decrease in arterial pressure. These data indicate that despite pressures similar to healthy controls at baseline, the elevated sympathetic activity coupled with similar neurovascular transduction in the OSAS group did drive pressure to higher levels, though it remained within the normotensive range. Hence, contrary to our hypothesis, the decline in sympathetic activity after CPAP did not alter transduction but did reduce (the already normotensive) blood pressure.
Our findings are particularly intriguing, given that we studied individuals who were newly diagnosed with OSAS and had no other comorbidities that might elevate sympathetic activity or increase risk for hypertension. Indeed, individuals with existing elevated pressure were intentionally excluded from the study. Although several prior studies have demonstrated an elevated muscle sympathetic nervous activity in those with OSAS, most included individuals who were obese, smokers, had metabolic syndrome, diabetes, heart failure, or established hypertension.15;26–35 Each of these comorbidities can elevate sympathetic activity and it is unclear how they might interact with OSAS to alter the hemodynamic profile. There is only one prior published study with a lean OSAS population comparable to our own. Grassi et al.12 found that lean individuals without elevated blood pressures but with OSAS demonstrate sympathetic activity ∼50% higher than those without OSAS. However, this prior work did not explore the impact of treatment and so, unlike the current findings, did not provide any perspective on how elevated sympathetic activity might relate to the hemodynamic profile.
There may be several potential mechanisms for the sustained sympatho-excitation in OSAS, among them augmented peripheral chemoreceptor stimulation36 and impaired baroreflex restraint31 of sympathetic tone. Indeed, prior work has demonstrated that two weeks of intermittent hypoxic exposure impairs baroreflex restraint and increases both peripheral chemoreceptor sensitivity and sympathetic activity.37 However, our data do not lend themselves to exploration of either baroreflex or chemoreflex control of sympathetic outflow. We reasoned that in response to a sustained sympathoexcitation in the absence of overt increases in blood pressure, those with OSAS would demonstrate lower sympathetic neurovascular transduction. This would fit with prior work suggesting an attenuated vasoconstriction with extrinsically applied α-adrenergic receptor stimulation in normotensive patients with OSAS.16 However, we found that neurovascular transduction was no different between those with OSAS and their matched healthy controls. Hence, the increased sympathetic activity in those with OSAS but without elevated pressure does not subserve a regulatory function to maintain blood pressure by countering lower sympathetic neurovascular transduction.
In fact, CPAP treatment revealed that the higher sympathetic activity coupled with unaltered transduction resulted in greater resting blood pressure at baseline, despite no obvious hypertension in those with OSAS. These findings contrast with Narkiewicz et al.,38 who found reduced sympathetic activity without reduced pressure after six months of CPAP treatment in normotensive OSAS patients. However, it should be noted that these individuals were obese (BMI 33 ± 5 kg/m2) and gained weight over the 6-month treatment period. The OSAS patients we studied were all within normal weight (no BMI above 30 kg/m2) and remained within that weight over the 6 months of treatment. In fact, these are the first data on the effect of CPAP on sympathetic activity in those with newly diagnosed OSAS without any other comorbidity. Our results suggest that the hyper-sympathetic state resulting from OSAS results in an unrecognized form of “pre-hypertension.” Though these individuals did not demonstrate a blood pressure clinically defined as pre-hypertension, CPAP treatment revealed that their hyper-sympathetic state at baseline was driving blood pressure to higher levels within the normotensive range.
After receiving CPAP, the relationship between sympathetic activity and sympathetic transduction became closer in those with OSAS. This suggests that, prior to treatment, the higher tonic sympathetic outflow was inappropriate to or actually exceeded the level of neurovascular transduction. Hence, we found that the level of leg vascular resistance, which is sympathetically driven, directly related to greater diastolic pressure prior to CPAP. After CPAP, with a decrease in sympathetic outflow that is in closer alignment to neurovascular transduction, leg vascular resistance was a lesser determinant of diastolic pressure. In addition, as might be expected after CPAP, apnea-hypopnea index no longer provided explanatory power for the level of diastolic pressure. Thus, our data suggest that CPAP-mediated reduction in sympathetic activity without concomitant reduction in sympathetic neurovascular transduction underlies the reduction in diastolic pressure.
Intermittent hypoxia, both in animal models8;39 and in humans37;40 leads to a sustained sympathetic activation. This sympathetic hyperactivity due to chronic intermittent hypoxia is thought to be a main mechanism leading to hyper-tension OSAS.41 Hence, OSAS may be thought of as a model for neurogenic hypertension wherein different mechanisms for sustained sympathetic hyperactivity are implicated, for example, increased peripheral chemoreception36 and decrease in baroreflex control,42 as well as the potential of primary aldosteronism.43 We explored the potential of an attendant tachyphylaxis that could also underlie sustained sympathetic hyperactivity. However, we found that despite greater sympathetic activity and similar blood pressure in those with OSAS, their sympathetic neurovascular transduction was similar to matched healthy controls. Surprisingly, the explanation for these seemingly contradictory findings was found in the reduction in both sympathetic activity and blood pressure with CPAP treatment. Hence, the absence of a lower sympathetic transduction in OSAS results in a relatively elevated blood pressure due to the higher tonic sympathetic outflow. It is possible that the high sympathetic activity with unaltered transduction results in vascular remodeling and sustained hypertension over time, in the absence of treatment. Moreover, it may be prudent to target therapeutic strategies to counteract this deleterious effect and prevent cardiovascular morbidity in patients who refuse or are not able to use CPAP.
DISCLOSURE STATEMENT
This work was supported by the National Institutes of Health, National Heart Lung and Blood Institute (grant HL81693 in the U.S. [JAT]) and by grants from the Regional Research Council (PHRC 2006) to the Grenoble Hospital in France (RT, JLP, and PL), and an unrestricted grant from Agiradom who partly supports the cost. Dr. Tamisier has received research support from ResMed. The other authors have indicated no financial conflicts of interest. The work was performed at CHU de Grenoble, Clinique Physiologie, Sommeil et Exercice, Grenoble, France.
ACKNOWLEDGMENTS
The authors thank the volunteers for their participation in these studies.
Author contributions: Conception and design of the paper: Drs. Tamisier, Tan, Pepin, Levy and Taylor. Analysis and interpretation: Drs. Tamisier, Tan, and Taylor. Drafting the manuscript for important intellectual content: Drs. Tamisier, Tan, Pepin, Levy and Taylor.
REFERENCES
- 1.Somers VK, White DP, Amin R, et al. Sleep apnea and cardiovascular disease: an American Heart Association/American College of Cardiology Foundation Scientific Statement from the American Heart Association Council for High Blood Pressure Research Professional Education Committee, Council on Clinical Cardiology, Stroke Council, and Council on Cardiovascular Nursing. In collaboration with the National Heart, Lung, and Blood Institute National Center on Sleep Disorders Research (National Institutes of Health) Circulation. 2008;118:1080–111. doi: 10.1161/CIRCULATIONAHA.107.189375. [DOI] [PubMed] [Google Scholar]
- 2.Mancia G, Fagard R, Narkiewicz K, et al. 2013 ESH/ESC guidelines for the management of arterial hypertension: the Task Force for the Management of Arterial Hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC) Eur Heart J. 2013;34:2159–219. doi: 10.1093/eurheartj/eht151. [DOI] [PubMed] [Google Scholar]
- 3.Parati G, Lombardi C, Hedner J, et al. Position paper on the management of patients with obstructive sleep apnea and hypertension: joint recommendations by the European Society of Hypertension, by the European Respiratory Society and by the members of European COST (COoperation in Scientific and Technological research) ACTION B26 on obstructive sleep apnea. J Hypertens. 2012;30:633–46. doi: 10.1097/HJH.0b013e328350e53b. [DOI] [PubMed] [Google Scholar]
- 4.Drager LF, Bortolotto LA, Krieger EM, Lorenzi-Filho G. Additive effects of obstructive sleep apnea and hypertension on early markers of carotid atherosclerosis. Hypertension. 2009;53:64–9. doi: 10.1161/HYPERTENSIONAHA.108.119420. [DOI] [PubMed] [Google Scholar]
- 5.Levy D, Larson MG, Vasan RS, Kannel WB, Ho KK. The progression from hypertension to congestive heart failure. JAMA. 1996;275:1557–62. [PubMed] [Google Scholar]
- 6.Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med. 2005;353:2034–41. doi: 10.1056/NEJMoa043104. [DOI] [PubMed] [Google Scholar]
- 7.Fletcher EC, Lesske J, Culman J, Miller CC, Unger T. Sympathetic denervation blocks blood pressure elevation in episodic hypoxia. Hypertension. 1992;20:612–9. doi: 10.1161/01.hyp.20.5.612. [DOI] [PubMed] [Google Scholar]
- 8.Lesske J, Fletcher EC, Bao G, Unger T. Hypertension caused by chronic intermittent hypoxia--influence of chemoreceptors and sympathetic nervous system. J Hypertens. 1997;15:1593–603. doi: 10.1097/00004872-199715120-00060. [DOI] [PubMed] [Google Scholar]
- 9.Somers VK, Dyken ME, Clary MP, Abboud FM. Sympathetic neural mechanisms in obstructive sleep apnea. J Clin Invest. 1995;96:1897–904. doi: 10.1172/JCI118235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abboud F, Kumar R. Obstructive sleep apnea and insight into mechanisms of sympathetic overactivity. J Clin Invest. 2014;124:1454–7. doi: 10.1172/JCI70420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marin JM, Agusti A, Villar I, et al. Association between treated and untreated obstructive sleep apnea and risk of hypertension. JAMA. 2012;307:2169–76. doi: 10.1001/jama.2012.3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grassi G, Facchini A, Trevano FQ, et al. Obstructive sleep apnea-dependent and -independent adrenergic activation in obesity. Hypertension. 2005;46:321–5. doi: 10.1161/01.HYP.0000174243.39897.6c. [DOI] [PubMed] [Google Scholar]
- 13.Grassi G, Seravalle G, Brambilla G, et al. Regional differences in sympathetic activation in lean and obese normotensive individuals with obstructive sleep apnoea. J Hypertens. 2014;32:383–8. doi: 10.1097/HJH.0000000000000034. [DOI] [PubMed] [Google Scholar]
- 14.Carlson JT, Hedner J, Elam M, Ejnell H, Sellgren J, Wallin BG. Augmented resting sympathetic activity in awake patients with obstructive sleep apnea. Chest. 1993;103:1763–8. doi: 10.1378/chest.103.6.1763. [DOI] [PubMed] [Google Scholar]
- 15.Moradkhan R, Spitnale B, McQuillan P, Hogeman C, Gray KS, Leuenberger UA. Hypoxia-induced vasodilation and effects of regional phentolamine in awake patients with sleep apnea. J Appl Physiol. 2010;108:1234–40. doi: 10.1152/japplphysiol.90855.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grote L, Kraiczi H, Hedner J. Reduced alpha- and beta(2)-adrenergic vascular response in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 2000;162:1480–7. doi: 10.1164/ajrccm.162.4.9912028. [DOI] [PubMed] [Google Scholar]
- 17.D'Almeida MS, Lautt WW. Expression of vascular escape: conductance or resistance? Am J Physiol. 1992;262:H1196. doi: 10.1152/ajpheart.1992.262.4.H1191. [DOI] [PubMed] [Google Scholar]
- 18.Scherrer U, Pryor SL, Bertocci LA, Victor RG. Arterial baroreflex buffering of sympathetic activation during exercise-induced elevations in arterial pressure. J Clin Invest. 1990;86:1855–61. doi: 10.1172/JCI114916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan CO, Tamisier R, Hamner JW, Taylor JA. Characterizing sympathetic neurovascular transduction in humans. PLoS One. 2013;8:e53769. doi: 10.1371/journal.pone.0053769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berry RB, Budhiraja R, Gottlieb DJ, et al. Rules for scoring respiratory events in sleep: update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events. Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med. 2012;8:597–619. doi: 10.5664/jcsm.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sleep-related breathing disorders in adults: recommendations for syndrome definition and measurement techniques in clinical research. The Report of an American Academy of Sleep Medicine Task Force. Sleep. 1999;22:667–89. [PubMed] [Google Scholar]
- 22.Argod J, Pepin JL, Levy P. Differentiating obstructive and central sleep respiratory events through pulse transit time. Am J Respir Crit Care Med. 1998;158:1778–83. doi: 10.1164/ajrccm.158.6.9804157. [DOI] [PubMed] [Google Scholar]
- 23.Hosselet JJ, Norman RG, Ayappa I, Rapoport DM. Detection of flow limitation with a nasal cannula/pressure transducer system. Am J Respir Crit Care Med. 1998;157:1461–7. doi: 10.1164/ajrccm.157.5.9708008. [DOI] [PubMed] [Google Scholar]
- 24.Hagbarth KE, Vallbo AB. Pulse and respiratory grouping of sympathetic impulses in human muscle-nerves. Acta Physiol Scand. 1968;74:96–108. doi: 10.1111/j.1748-1716.1968.tb04218.x. [DOI] [PubMed] [Google Scholar]
- 25.Tan CO, Taylor JA, Ler AS, Cohen MA. Detection of multifiber neuronal firings: a mixture separation model applied to sympathetic recordings. IEEE Trans Biomed Eng. 2009;56:147–58. doi: 10.1109/TBME.2008.2002138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donadio V, Liguori R, Vetrugno R, et al. Daytime sympathetic hyperactivity in OSAS is related to excessive daytime sleepiness. J Sleep Res. 2007;16:327–32. doi: 10.1111/j.1365-2869.2007.00602.x. [DOI] [PubMed] [Google Scholar]
- 27.Grassi G, Seravalle G, Quarti-Trevano F, et al. Reinforcement of the adrenergic overdrive in the metabolic syndrome complicated by obstructive sleep apnea. J Hypertens. 2010;28:1313–20. doi: 10.1097/HJH.0b013e328337a9fd. [DOI] [PubMed] [Google Scholar]
- 28.Imadojemu VA, Gleeson K, Quraishi SA, Kunselman AR, Sinoway LI, Leuenberger UA. Impaired vasodilator responses in obstructive sleep apnea are improved with continuous positive airway pressure therapy. Am J Respir Crit Care Med. 2002;165:950–3. doi: 10.1164/ajrccm.165.7.2102003. [DOI] [PubMed] [Google Scholar]
- 29.Imadojemu VA, Mawji Z, Kunselman A, Gray KS, Hogeman CS, Leuenberger UA. Sympathetic chemoreflex responses in obstructive sleep apnea and effects of continuous positive airway pressure therapy. Chest. 2007;131:1406–13. doi: 10.1378/chest.06-2580. [DOI] [PubMed] [Google Scholar]
- 30.Leuenberger U, Jacob E, Sweer L, Waravdekar N, Zwillich C, Sinoway L. Surges of muscle sympathetic nerve activity during obstructive apnea are linked to hypoxemia. J Appl Physiol. 1995;79:581–8. doi: 10.1152/jappl.1995.79.2.581. [DOI] [PubMed] [Google Scholar]
- 31.Toschi-Dias E, Trombetta IC, Dias dS, V, et al. Time delay of baroreflex control and oscillatory pattern of sympathetic activity in patients with metabolic syndrome and obstructive sleep apnea. Am J Physiol Heart Circ Physiol. 2013;304:H1038–44. doi: 10.1152/ajpheart.00848.2012. [DOI] [PubMed] [Google Scholar]
- 32.Trombetta IC, Somers VK, Maki-Nunes C, et al. Consequences of comorbid sleep apnea in the metabolic syndrome--implications for cardiovascular risk. Sleep. 2010;33:1193–9. doi: 10.1093/sleep/33.9.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trombetta IC, Maki-Nunes C, Toschi-Dias E, et al. Obstructive sleep apnea is associated with increased chemoreflex sensitivity in patients with metabolic syndrome. Sleep. 2013;36:41–9. doi: 10.5665/sleep.2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ueno LM, Drager LF, Rodrigues AC, et al. Effects of exercise training in patients with chronic heart failure and sleep apnea. Sleep. 2009;32:637–47. doi: 10.1093/sleep/32.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Waradekar NV, Sinoway LI, Zwillich CW, Leuenberger UA. Influence of treatment on muscle sympathetic nerve activity in sleep apnea. Am J Respir Crit Care Med. 1996;153:1333–8. doi: 10.1164/ajrccm.153.4.8616563. [DOI] [PubMed] [Google Scholar]
- 36.Narkiewicz K, van de Borne PJ, Pesek CA, Dyken ME, Montano N, Somers VK. Selective potentiation of peripheral chemoreflex sensitivity in obstructive sleep apnea. Circulation. 1999;99:1183–9. doi: 10.1161/01.cir.99.9.1183. [DOI] [PubMed] [Google Scholar]
- 37.Tamisier R, Pepin JL, Remy J, et al. 14 nights of intermittent hypoxia elevate daytime blood pressure and sympathetic activity in healthy humans. Eur Respir J. 2011;37:119–28. doi: 10.1183/09031936.00204209. [DOI] [PubMed] [Google Scholar]
- 38.Narkiewicz K, Kato M, Phillips BG, Pesek CA, Davison DE, Somers VK. Nocturnal continuous positive airway pressure decreases daytime sympathetic traffic in obstructive sleep apnea. Circulation. 1999;100:2332–5. doi: 10.1161/01.cir.100.23.2332. [DOI] [PubMed] [Google Scholar]
- 39.Zoccal DB, Bonagamba LG, Oliveira FR, ntunes-Rodrigues J, Machado BH. Increased sympathetic activity in rats submitted to chronic intermittent hypoxia. Exp Physiol. 2007;92:79–85. doi: 10.1113/expphysiol.2006.035501. [DOI] [PubMed] [Google Scholar]
- 40.Gilmartin GS, Lynch M, Tamisier R, Weiss JW. Chronic intermittent hypoxia in humans during 28 nights results in blood pressure elevation and increased muscle sympathetic nerve activity. Am J Physiol Heart Circ Physiol. 2010;299:H925–31. doi: 10.1152/ajpheart.00253.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moraes DJ, Zoccal DB, Machado BH. Medullary respiratory network drives sympathetic overactivity and hypertension in rats submitted to chronic intermittent hypoxia. Hypertension. 2012;60:1374–80. doi: 10.1161/HYPERTENSIONAHA.111.189332. [DOI] [PubMed] [Google Scholar]
- 42.Carlson JT, Hedner JA, Sellgren J, Elam M, Wallin BG. Depressed baroreflex sensitivity in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 1996;154:1490–6. doi: 10.1164/ajrccm.154.5.8912770. [DOI] [PubMed] [Google Scholar]
- 43.Di Murro A, Petramala L, Cotesta D, et al. Renin-angiotensinaldosterone system in patients with sleep apnoea: prevalence of primary aldosteronism. J Renin Angiotensin Aldosterone Syst. 2010;11:165–72. doi: 10.1177/1470320310366581. [DOI] [PubMed] [Google Scholar]