

Abstract
Background
Ethanol exposure during the rodent equivalent to the 3rd trimester of human pregnancy (i.e., first 1–2 weeks of neonatal life) has been shown to produce structural and functional alterations in the CA3 hippocampal sub-region, which is involved in associative memory. Synaptic plasticity mechanisms dependent on retrograde release of brain-derived neurotrophic factor (BDNF) driven by activation of L-type voltage-gated Ca2+ channels (L-VGCCs) are thought to play a role in stabilization of both GABAergic and glutamatergic synapses in CA3 pyramidal neurons. We previously showed that ethanol exposure during the first week of life blocks BDNF/L-VGCC-dependent long-term potentiation of GABAA receptor-mediated synaptic transmission in these neurons. Here, we tested whether this effect is associated with lasting alterations in GABAergic and glutamatergic transmission.
Methods
Rats were exposed to air or ethanol for 3 h/day between postnatal days three and five in vapor inhalation chambers, a paradigm that produces peak serum ethanol levels near 0.3 g/dl. Whole-cell patch-clamp electrophysiological recordings of spontaneous inhibitory and excitatory postsynaptic currents (sIPSCs and sEPSCs, respectively) were obtained from CA3 pyramidal neurons in coronal brain slices prepared at postnatal days 13–17.
Results
Ethanol exposure did not significantly affect the frequency, amplitude, rise-time and half-width of either sIPSCs or sEPSCs.
Conclusions
We show that an ethanol exposure paradigm known to inhibit synaptic plasticity mechanisms that may participate in the stabilization of GABAergic and glutamatergic synapses in CA3 pyramidal neurons does not produce lasting functional alterations in these synapses, suggesting that compensatory mechanisms restored the balance of excitatory and inhibitory synaptic transmission.
Keywords: Ethanol, Alcohol, Development, Synaptic, Transmission, GABA, Glutamate, Electrophysiology, Channel, Receptor
Background
The hippocampus, a brain region located in the medial portion of the temporal lobe, is involved in memory formation, learning, and mood. A number of studies have documented alterations in these processes both in humans and animals exposed to ethanol during development [1]. Deficits in hippocampal function are thought to contribute to these alterations, playing a role the pathophysiology of fetal alcohol spectrum disorders [2, 3]. Although the mechanisms responsible for the hippocampal alterations associated with developmental alcohol exposure are not well understood, several studies suggest that these are, in part, a consequence of damage to neurons located in the CA3 sub-region, which is involved in associative memory [1, 4].
Prenatal exposure to ethanol in rodents (equivalent to the 1st and 2nd trimesters of human pregnancy), has been shown to induce structural and functional changes in the CA3 sub-region. Gestational exposure to ethanol triggered the formation of a hypertrophic infra-pyramidal mossy fiber bundle in the CA3 sub-region [5, 6]. A reduction in 3H-vinlidene kainate binding sites was demonstrated in the CA3 stratum lucidum of the ventral hippocampus from 45 day-old rat offspring exposed to ethanol during fetal development [7]. Using electron microscopy, Tanaka et al [8] showed that prenatal ethanol exposure decreases the number of synapses in the CA3 sub-region at gestational day 21. Studies suggest that exposure during periods equivalent to the human 3rd trimester of pregnancy can have even more significant effects on this hippocampal sub-region. West and Hamre [9] reported that exposure to ethanol between postnatal day (P) 1 and P10 was associated with the presence of aberrant intra-pyramidal and infra-pyramidal mossy fibers across the CA3 sub-region. Binge-like ethanol exposure during P4-P10 (but not gestational days 1–20) decreased the number and density of pyramidal cells in this sub-region [10, 11]. A similar finding was reported by Miki et al [12] who detected a reduction in CA3 pyramidal neuron number in rats exposed to ethanol between P10 and P15. However, it is noteworthy that studies using both guinea pigs and rats have failed to detect alterations in the number of pyramidal neurons in this hippocampal sub-region [13, 14]. Therefore, several studies have investigated the possibility that developmental ethanol exposure impairs the function of CA3 neurons rather than affecting their morphology.
An electrophysiological study with 50–70 day-old offspring from rats exposed to ethanol throughout gestation reported a reduction in the frequency of high potassium-induced epileptiform bursts in the CA3 stratum radiatum [15]. Galindo et al [16] found that acute ethanol exposure increased network-driven giant depolarizing potentials in CA3 pyramidal neurons from neonatal rats, an effect that is likely a consequence of increased GABAA receptor-mediated excitatory synaptic transmission. It was also demonstrated that these immature neurons do not develop tolerance to this effect of ethanol [17]. Acute and repeated ethanol exposure between P2 and P6 was shown to inhibit brain-derived neurotrophic factor (BDNF)- and L-type voltage-gated Ca2+ channel (L-VGCC)-dependent long-term potentiation of GABAA receptor-mediated spontaneous postsynaptic currents in CA3 pyramidal neurons [18]. BDNF/L-VGCC-dependent plasticity mechanisms are thought to play a role in the stabilization of both GABAergic and glutamatergic synapses in developing hippocampal neurons [19–21]. Based on these findings, we hypothesized that the ethanol-induced alterations of BDNF/L-VGCC-dependent synaptic plasticity result in a persistent reduction in both GABAergic and glutamatergic synaptic currents in CA3 pyramidal neurons. To test this hypothesis, we exposed neonatal rats to ethanol from P3–P5 and measured GABAA receptor- and AMPA receptor-dependent spontaneous postsynaptic currents at P13–P17 using patch-clamp slice electrophysiological techniques.
Results
Pups were exposed to high doses of ethanol in vapor chambers between P3-5, as described below. Average pup weights were: P3 (control = 7.7 ± 0.1 g, n = 13; ethanol = 7.7 ± 0.2 g, n = 13), P5 (control = 10.5 ± 0.3 g, n = 14; ethanol = 8.6 ± 0.3 g, n = 13), P10 (control = 20.3 ± 0.4 g, n = 11; ethanol = 17.6 ± 1.0 g, n = 10), P15 (control = 35.3 ± 1.0 g, n = 12; ethanol = 30.1 ± 1.5 g, n = 11) and P20 (control = 49.9 ± 2.0 g, n = 10; ethanol = 43.5 ± 2.3 g, n = 12). Two-way ANOVA revealed a significant decrease in weight in the ethanol group at P15 and P20 (age: F (4, 109) = 407.0, P <0.0001; ethanol treatment: F (1, 109) = 19.55, P <0.0001; Interaction: F (4, 109) = 2.557, P = 0.04; P = 0.05 by Bonferroni’s test at these ages). In a previous study [22], we did not find a significant effect of this ethanol exposure paradigm on pup body weight, suggesting that offspring from different batches of timed-pregnant Sprague-Dawley rats may display differential sensitivity to ethanol. This may be related to exposure of animals to different stress levels during transport or housing (e.g., exposure to new animal care personnel). The average numbers of pups/litter at the start of the exposure paradigm (P3) were 9.8 ± 0.6 (n = 14 litters) and 10.1 ± 0.4 (n = 14 litters) for the control and ethanol groups, respectively (U = 97.5; P =0.99 by Mann-Whitney test). The average concentration of ethanol in the vapor chamber was 7.9 ± 0.24 g/dl (n = 7 rounds of exposure). We have previously shown that this exposure paradigm results in peak serum ethanol levels of 0.33 g/dl (73 mM) in the pup and 0.02 g/dl (5 mM) in the dam [22].
We evaluated the impact of this ethanol exposure paradigm on GABAA receptor-mediated spontaneous inhibitory postsynaptic currents (sIPSCs). Slice electrophysiological recordings were performed at P13-17. Fig. 1a shows examples of sIPSCs recorded from CA3 pyramidal neurons in slices from control and ethanol treated rats. Neither the frequency of these events (U = 20; P = 0.39 by Mann-Whitney test) nor their amplitude (U = 26; P = 0.86 by Mann-Whitney test) were significantly different between treatment groups (Fig. 1b–c). Likewise, the half-width of the events was not significantly affected by ethanol exposure (t(12) = 0.96; P = 0.35 by t-test; Fig. 1d) nor was the rise time (control = 2.04 ± 0.42 ms, n = 8; ethanol = 1.71 ± 0.53 ms, n = 6; t(12) = 0.48; P = 0.63 by t-test).
Fig 1.
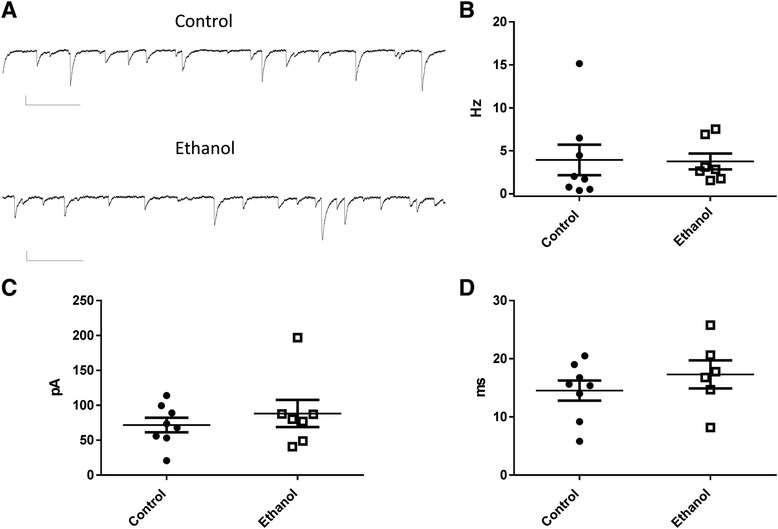
Third trimester-equivalent ethanol exposure did not significantly affect spontaneous inhibitory postsynaptic currents (sIPSCs) in CA3 pyramidal neurons. a Sample traces illustrating sIPSC recordings obtained from a postnatal day 15 control rat exposed to air and a postnatal day 16 rat exposed to ethanol. Scale bars = 66.7 pA and 256 ms. Ethanol exposure did not significantly affect sIPSC frequency (b), amplitude (c) or half-width (d)
We then assessed the impact of P3-5 ethanol exposure on AMPA receptor mediated sEPSCs. Slice electrophysiological recordings were also performed at P13-17. Sample sEPSC recordings are shown in Fig. 2a. Neither the frequency of these events (t(14) = 1.35; P = 0.19 by t-test) nor their amplitude (t(14) = 0.76; P = 0.45 by t-test) were significantly different between treatment groups (Fig. 2b-c). Moreover, neither the sEPSC half-width (t(14) = 0.78; P = 0.44 by t-test; Fig. 2d) nor the rise time (control = 1.82 ± 0.07 ms, n = 8; ethanol = 1.71 ± 0.11 ms, n = 8, t(14) = 0.77; P = 0.45 by t-test) were significantly affected by ethanol exposure. Spontaneous EPSCs triggered by glutamate release from mossy fibers have been demonstrated to have amplitudes ≥100 pA [23]. Therefore, we analyzed the effect of ethanol exposure on these large events. As shown in Table 1, ethanol exposure did not significantly affect the frequency, amplitude, rise-time or half-width of these events. Small events with an amplitude ≤100 pA were not significantly affected either. The percent of events with amplitudes ≥100 pA was 17.5 ± 4.6 % for the control group and 17.2 ± 2.5 % for the ethanol group (t(14) = 0.063; P = 0.95 by unpaired t-test; n = 8).
Fig 2.
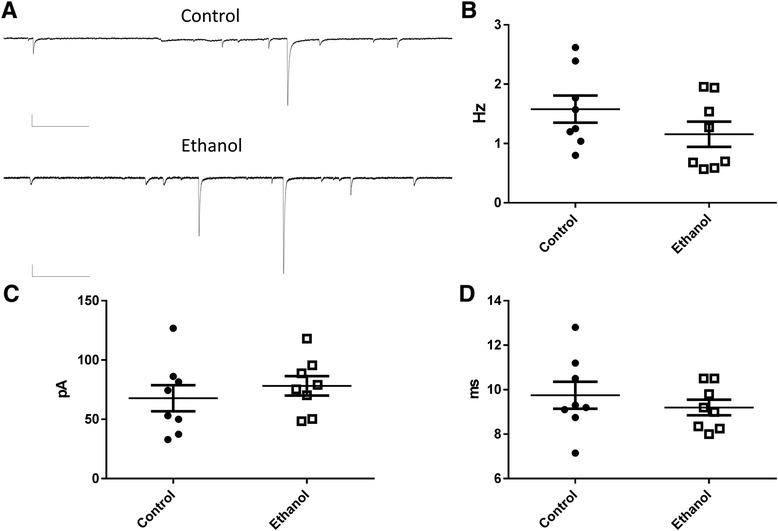
Third trimester-equivalent ethanol exposure did not significantly affect spontaneous excitatory postsynaptic currents (sEPSCs) in CA3 pyramidal neurons. a Sample traces illustrating sEPSC recordings obtained from a postnatal day 17 control rat exposed to air and a postnatal day 15 rat exposed to ethanol. Scale bars = 66.7 pA and 328 ms. Ethanol exposure did not significantly affect sEPSC frequency (b), amplitude (c) or half-width (d)
Table 1.
Summary of characteristics of large (≥100 pA) and small (≤ 100 pA) sEPSCs
| Parameter | Control | Ethanol | Statistics |
|---|---|---|---|
| Frequency (Hz) | |||
| Large events | 0.35 ± 0.04 (n = 6) | 0.24 ± 0.05 (n = 6) | U = 12; P = 0.14 |
| Small events | 1.35 ± 0.25 (n = 8) | 0.96 ± 0.16 (n = 8) | t(14) = 1.26; P = 0.22 |
| Amplitude (pA) | |||
| Large events | 232.2 ± 14 (n = 8) | 255.2 ± 26 (n = 8) | t(14) = 0.78;P = 0.44 |
| Small events | 34.6 ± 1.9 (n = 8) | 41.5 ± 3.1 (n = 8) | t(14) = 1.88; P = 0.08 |
| Rise time (ms) | |||
| Large events | 2.08 ± 0.11 (n = 8) | 1.86 ± 0.10 (n = 7) | t(13) = 1.33; P = 0.2 |
| Small events | 1.5 ± 0.12 (n = 8) | 1.47 ± 0.13 (n = 8) | t(14) = 0.16; P = 0.86 |
| Half-width (ms) | |||
| Large events | 10.08 ± 0.67 (n = 8) | 8.9 ± 0.46 (n = 7) | t(13) = 1.38; P = 0.18 |
| Small events | 8.76 ± 0.69 (n = 8) | 8.3 ± 0.86 (n = 8) | t(14) = 0.4; P = 0.69 |
Discussion
Our ethanol exposure paradigm did not significantly affect the properties of sIPSCs, a finding that does not support the hypothesis that ethanol exposure during this critical period, when CA3 pyramidal neurons express BDNF/L-VGCC-dependent synaptic plasticity, produces a lasting impairment in GABAA receptor-dependent synaptic transmission. We did not observe changes in sIPSC frequency, amplitude, rise-time or half-width, indicating that ethanol exposure neither affects spontaneous GABA release nor the function of postsynaptic GABAA receptors. These findings suggest that ethanol did not have an overall effect on the function of GABAergic interneurons that provide inhibitory inputs to the soma and proximal dendrites of CA3 pyramidal neurons. Due to space-clamp limitations, GABAergic inputs to distal dendritic sites cannot be reliably measured using somatic patch-clamp electrodes [24]. Therefore, we cannot discount the possibility that these were affected by ethanol exposure. This issue could be addressed in the future using current-clamp recordings of spontaneous inhibitory postsynaptic potentials or voltage-clamp experiments of IPSCs evoked by electrical stimulation of distal inputs. As mentioned above, BDNF/L-VGCC-dependent long-term potentiation of GABAergic transmission is thought to play a role in stabilizing GABAA receptor-containing synapses in the soma and/or proximal dendrites of CA3 pyramidal neurons [19]. Therefore, it is somewhat surprising that ethanol-induced inhibition of this plasticity mechanism did not affect the properties of GABAA-dependent sIPSCs [18]. A potential explanation for the lack of a lasting effect is that the action of ethanol is transient, occurring only while this agent is present during the 3 h/day exposure on P3-P5 (and several hours/day post-exposure while ethanol is being metabolized) and reversing at some point between this period and the time at which the electrophysiological recordings were acquired (P13-P17). We previously demonstrated that ethanol vapor exposure from P2-P16 causes a significantly delay in the acquisition of action potential-independent GABAA receptor mediated postsynaptic currents in CA3 pyramidal neurons [25], suggesting that repeated ethanol exposure encompassing a longer period of development could alter plasticity mechanisms involved in the maturation of GABAergic synapses.
Out studies also failed to demonstrate an effect of ethanol exposure on the frequency, amplitude, rise-time or half-width of AMPA receptor-mediated sEPSCs, including large events likely mediated by glutamate release from mossy fibers. These results suggest that ethanol did not have lasting effects on glutamatergic transmission at the pre- or post-synaptic levels. The lack of an effect of ethanol on large sEPSCs is unexpected given that BDNF/L-VGCC-dependent synaptic plasticity has been shown to strengthen mossy fiber-to-CA3 pyramidal neuron synapses during postnatal development [20]. Moreover, we have previously demonstrated that acute ethanol exposure inhibits glutamate release and AMPA receptor-mediated EPSCs in slices from P3-P10 rats [26] and it would have been expected for these acute effects to cause lasting compensatory pre- and/or post-synaptic alterations in glutamatergic transmission [27–29]. As was the case for GABAergic transmission, it is possible that neurons were able to counteract the effects of ethanol, leading to a normalization of glutamatergic transmission at the time of our electrophysiological experiments. For instance, neurons could have engaged alternative pathways involved in synapse stabilization, including upregulation of modulatory neurotransmitter systems (e.g., serotonin) [30, 31].
Conclusions
We demonstrate here that a 3rd trimester-equivalent ethanol exposure paradigm that significantly impairs synaptic plasticity mechanisms thought to be involved in the stabilization of GABAergic and glutamatergic synapses in CA3 pyramidal neurons does not produce lasting alterations in spontaneous synaptic transmission mediated by GABAA and AMPA receptors. These findings suggest that neonatal CA3 pyramidal neurons deploy compensatory mechanisms in response to this exposure paradigm that ultimately restore the balance of excitatory and inhibitory synaptic transmission. It should be emphasized, however, that even if the effects of our P3–P5 exposure paradigm are transient, these could still alter the trajectory of developmental processes other than the refinement of GABAergic and glutamatergic synaptic transmission [32] and this issue should be investigated in the future.
Methods
All chemicals were obtained from Sigma-Aldrich (St. Louis, MO), except when noted. The University of New Mexico Health Sciences Center Institutional Animal Care and Use Committee approved the procedures followed in this study. Timed-pregnant Sprague-Dawley rats (Harlan, Indianapolis, IN) were used in these experiments. The gestational age upon arrival to our animal facility was 12–15 days. Between P3 and P5, pups were exposed to air (control) or ethanol for 3 h/day using a previously described vapor inhalation chamber paradigm [22]. We previously showed that the paradigm minimally disrupts maternal care [22]. Coronal brain slices (300 μm) containing the dorsal hippocampal formation were prepared from ketamine-anesthetized rats at P13–17 using a vibrating slicer (Leica Microsystems, Bannockburn, IL) and sucrose-based cutting solution, as previously described [22]. Subsequently, slices were allowed to recover in oxygenated artificial cerebral spinal fluid containing (in mM): NaCl, 125; KCl, 2; NaH2PO4, 1.3; NaCO3, 26; glucose, 10, CaCl2, 2; MgSO4, 1 at 35 °C for 40 min in a water bath. The chamber containing the slices was then taken out of the water bath and stored at 21–22 °C for at least 30 min prior to the start of electrophysiological recordings. Neurons were visualized using an Olympus BX50WI upright microscope (Olympus, Center Valley, PA) equipped with infra-red Differential Interference Contrast optics and connected to a charge-coupled device camera (CCD100, DAGE-MTI, Michigan City, IN). Whole-cell patch-clamp electrophysiological recordings were obtained at -70 mV and 32 °C using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA). Spontaneous IPSCs were recorded in presence of 3 mM kynurenate using an internal solution containing (in mM): KCl, 135; HEPES, 10; EGTA, 0.5; MgCl2, 2; ATP-Mg, 5; GTP-Na, 1; QX-314-Cl, 1; pH 7.3 with KOH. These events were blocked by the GABAA receptor antagonist, gabazine (Tocris, Bristol, U.K.) (50 μM; n = 3). AMPA receptor-mediated sEPSCs were recorded in presence of 50 μM gabazine using an internal solution containing (in mM): K-Gluconate, 120; KCl, 15; EGTA, 0.1; HEPES, 10; MgCl2, 4; ATP-Mg, 4; GTP-Na, 0.3; phosphocreatine, 7; pH 7.3 with KOH. These events were blocked by the AMPA receptor antagonist, 2,3-Dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX, 10 μM; n = 3). The access resistance was between 5 and 26 MΩ and if it changed more than 30 %, the recording was rejected. Mini-Analysis program (Synaptosoft, Decatur, GA) was used to analyze the electrophysiological data. Statistical analyses were performed with Prism Version 6.05 (GraphPad Software, San Diego, CA). Data were initially analyzed with the Kolmogorov-Smirnov normality test and if they followed a normal distribution, they were subsequently analyzed using parametric tests (t-tests or two-way ANOVA). If data did not pass the normality test, they were analyzed using two-tailed Mann-Whitney tests. Data are presented as mean ± SEM. The unit of determination is the average of results obtained with slices from a single animal (1–3 cells/animal; each cell from a different slice).
Acknowledgements
This work was supported by R37-AA015614 and P50AA022534.
Abbreviations
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- ANOVA
analysis of variance
- BDNF
brain-derived neurotrophic factor
- EGTA
ethylene glycol tetra acetic acid
- GABAA
γ-amino butyric acid receptor, type-A
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- L-VGCC
L-type voltage-gated Ca2+ channel
- NBQX
2,3-Dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide
- P
postnatal day
- QX-314
N-(2,6-Dimethylphenylcarbamoylmethyl) triethylammonium chloride
- sEPSC
spontaneous excitatory postsynaptic current
- sIPSC
spontaneous inhibitory postsynaptic current
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BCB provided intellectual contribution, exposed the animals to ethanol vapor, performed the electrophysiological recordings, performed data analysis, and edited the manuscript. CFV contributed intellectually, assisted with the vapor chamber exposure paradigm, performed data analyses, interpreted the data, and wrote/edited the manuscript. Both authors read and approved the final manuscript.
Contributor Information
Brian Charles Baculis, Email: bbaculis@salud.unm.edu.
Carlos Fernando Valenzuela, Phone: (505) 272-3128, Email: fvalenzuela@salud.unm.edu.
References
- 1.Berman RF, Hannigan JH. Effects of prenatal alcohol exposure on the hippocampus: spatial behavior, electrophysiology, and neuroanatomy. Hippocampus. 2000;10(1):94–110. doi: 10.1002/(SICI)1098-1063(2000)10:1<94::AID-HIPO11>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 2.Hamilton DA, Kodituwakku P, Sutherland RJ, Savage DD. Children with Fetal Alcohol Syndrome are impaired at place learning but not cued-navigation in a virtual Morris water task. Behav Brain Res. 2003;143(1):85–94. doi: 10.1016/S0166-4328(03)00028-7. [DOI] [PubMed] [Google Scholar]
- 3.Norman AL, Crocker N, Mattson SN, Riley EP. Neuroimaging and fetal alcohol spectrum disorders. Dev Disabil Res Rev. 2009;15(3):209–17. doi: 10.1002/ddrr.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Langston RF, Stevenson CH, Wilson CL, Saunders I, Wood ER. The role of hippocampal subregions in memory for stimulus associations. Behav Brain Res. 2010;215(2):275–91. doi: 10.1016/j.bbr.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 5.West JR, Hodges CA, Black AC., Jr Prenatal exposure to ethanol alters the organization of hippocampal mossy fibers in rats. Science. 1981;211(4485):957–9. doi: 10.1126/science.7466371. [DOI] [PubMed] [Google Scholar]
- 6.West JR, Pierce DR. The effect of in utero ethanol exposure on hippocampal mossy fibers: an HRP study. Brain Res. 1984;317(2):275–9. doi: 10.1016/0165-3806(84)90104-4. [DOI] [PubMed] [Google Scholar]
- 7.Farr KL, Montano CY, Paxton LL, Savage DD. Prenatal ethanol exposure decreases hippocampal 3H-vinylidene kainic acid binding in 45-day-old rats. Neurotoxicol Teratol. 1988;10(6):563–8. doi: 10.1016/0892-0362(88)90093-1. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka H, Nasu F, Inomata K. Fetal alcohol effects: decreased synaptic formations in the field CA3 of fetal hippocampus. Int J Dev Neurosci. 1991;9(5):509–17. doi: 10.1016/0736-5748(91)90037-M. [DOI] [PubMed] [Google Scholar]
- 9.West JR, Hamre KM. Effects of alcohol exposure during different periods of development: changes in hippocampal mossy fibers. Brain Res. 1985;349(1-2):280–4. doi: 10.1016/0165-3806(85)90155-5. [DOI] [PubMed] [Google Scholar]
- 10.Livy DJ, Miller EK, Maier SE, West JR. Fetal alcohol exposure and temporal vulnerability: effects of binge-like alcohol exposure on the developing rat hippocampus. Neurotoxicol Teratol. 2003;25(4):447–58. doi: 10.1016/S0892-0362(03)00030-8. [DOI] [PubMed] [Google Scholar]
- 11.Maier SE, West JR. Regional differences in cell loss associated with binge-like alcohol exposure during the first two trimesters equivalent in the rat. Alcohol. 2001;23(1):49–57. doi: 10.1016/S0741-8329(00)00133-6. [DOI] [PubMed] [Google Scholar]
- 12.Miki T, Harris SJ, Wilce PA, Takeuchi Y, Bedi KS. Effects of age and alcohol exposure during early life on pyramidal cell numbers in the CA1-CA3 region of the rat hippocampus. Hippocampus. 2004;14(1):124–34. doi: 10.1002/hipo.10155. [DOI] [PubMed] [Google Scholar]
- 13.Byrnes ML, Reynolds JN, Brien JF. Brain growth spurt-prenatal ethanol exposure and the guinea pig hippocampal glutamate signaling system. Neurotoxicol Teratol. 2003;25(3):303–10. doi: 10.1016/S0892-0362(02)00354-9. [DOI] [PubMed] [Google Scholar]
- 14.Tran TD, Kelly SJ. Critical periods for ethanol-induced cell loss in the hippocampal formation. Neurotoxicol Teratol. 2003;25(5):519–28. doi: 10.1016/S0892-0362(03)00074-6. [DOI] [PubMed] [Google Scholar]
- 15.Swartzwelder HS, Farr KL, Wilson WA, Savage DD. Prenatal exposure to ethanol decreases physiological plasticity in the hippocampus of the adult rat. Alcohol. 1988;5(2):121–4. doi: 10.1016/0741-8329(88)90008-0. [DOI] [PubMed] [Google Scholar]
- 16.Galindo R, Zamudio PA, Valenzuela CF. Alcohol is a potent stimulant of immature neuronal networks: implications for fetal alcohol spectrum disorder. J Neurochem. 2005;94(6):1500–11. doi: 10.1111/j.1471-4159.2005.03294.x. [DOI] [PubMed] [Google Scholar]
- 17.Galindo R, Valenzuela CF. Immature hippocampal neuronal networks do not develop tolerance to the excitatory actions of ethanol. Alcohol. 2006;40(2):111–8. doi: 10.1016/j.alcohol.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zucca S, Valenzuela CF. Low concentrations of alcohol inhibit BDNF-dependent GABAergic plasticity via L-type Ca2+ channel inhibition in developing CA3 hippocampal pyramidal neurons. J Neurosci. 2010;30(19):6776–81. doi: 10.1523/JNEUROSCI.5405-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gubellini P, Ben-Ari Y, Gaiarsa JL. Endogenous neurotrophins are required for the induction of GABAergic long-term potentiation in the neonatal rat hippocampus. J Neurosci. 2005;25(24):5796–802. doi: 10.1523/JNEUROSCI.0824-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sivakumaran S, Mohajerani MH, Cherubini E. At immature mossy-fiber-CA3 synapses, correlated presynaptic and postsynaptic activity persistently enhances GABA release and network excitability via BDNF and cAMP-dependent PKA. J Neurosci. 2009;29(8):2637–47. doi: 10.1523/JNEUROSCI.5019-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohajerani MH, Sivakumaran S, Zacchi P, Aguilera P, Cherubini E. Correlated network activity enhances synaptic efficacy via BDNF and the ERK pathway at immature CA3 CA1 connections in the hippocampus. Proc Natl Acad Sci U S A. 2007;104(32):13176–81. doi: 10.1073/pnas.0704533104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baculis BC, Diaz MR, Valenzuela CF. Third trimester-equivalent ethanol exposure increases anxiety-like behavior and glutamatergic transmission in the basolateral amygdala. Pharmacol Biochem Behav. 2015;137:78–85. doi: 10.1016/j.pbb.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henze DA, Card JP, Barrionuevo G, Ben-Ari Y. Large amplitude miniature excitatory postsynaptic currents in hippocampal CA3 pyramidal neurons are of mossy fiber origin. J Neurophysiol. 1997;77(3):1075–86. doi: 10.1152/jn.1997.77.3.1075. [DOI] [PubMed] [Google Scholar]
- 24.Williams SR, Mitchell SJ. Direct measurement of somatic voltage clamp errors in central neurons. Nat Neurosci. 2008;11(7):790–8. doi: 10.1038/nn.2137. [DOI] [PubMed] [Google Scholar]
- 25.Everett JC, Licon-Munoz Y, Valenzuela CF. Effects of third trimester-equivalent ethanol exposure on Cl(-) co-transporter expression, network activity, and GABAergic transmission in the CA3 hippocampal region of neonatal rats. Alcohol. 2012;46(6):595–601. doi: 10.1016/j.alcohol.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mameli M, Zamudio PA, Carta M, Valenzuela CF. Developmentally regulated actions of alcohol on hippocampal glutamatergic transmission. J Neurosci. 2005;25(35):8027–36. doi: 10.1523/JNEUROSCI.2434-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iqbal U, Brien JF, Kapoor A, Matthews SG, Reynolds JN. Chronic prenatal ethanol exposure increases glucocorticoid-induced glutamate release in the hippocampus of the near-term foetal guinea pig. J Neuroendocrinol. 2006;18(11):826–34. doi: 10.1111/j.1365-2826.2006.01479.x. [DOI] [PubMed] [Google Scholar]
- 28.Butters NS, Reynolds JN, Brien JF. Effects of chronic prenatal ethanol exposure on cGMP content and glutamate release in the hippocampus of the neonatal guinea pig. Neurotoxicol Teratol. 2003;25(1):59–68. doi: 10.1016/S0892-0362(02)00325-2. [DOI] [PubMed] [Google Scholar]
- 29.Bellinger FP, Davidson MS, Bedi KS, Wilce PA. Neonatal ethanol exposure reduces AMPA but not NMDA receptor levels in the rat neocortex. Brain Res Dev Brain Res. 2002;136(1):77–84. doi: 10.1016/S0165-3806(02)00363-2. [DOI] [PubMed] [Google Scholar]
- 30.Choi IS, Cho JH, Kim JT, Park EJ, Lee MG, Shin HI, et al. Serotoninergic modulation of GABAergic synaptic transmission in developing rat CA3 pyramidal neurons. J Neurochem. 2007;103(6):2342–53. doi: 10.1111/j.1471-4159.2007.04945.x. [DOI] [PubMed] [Google Scholar]
- 31.Rayen I, Gemmel M, Pauley G, Steinbusch HW, Pawluski JL. Developmental exposure to SSRIs, in addition to maternal stress, has long-term sex-dependent effects on hippocampal plasticity. Psychopharmacology. 2015;232(7):1231–44. doi: 10.1007/s00213-014-3758-0. [DOI] [PubMed] [Google Scholar]
- 32.Thompson BL, Levitt P. Now you see it, now you don’t--closing in on allostasis and developmental basis of psychiatric disorders. Neuron. 2010;65(4):437–9. doi: 10.1016/j.neuron.2010.02.010. [DOI] [PubMed] [Google Scholar]