

Abstract
Background
Algae accumulate lipids to endure different kinds of environmental stresses including macronutrient starvation. Although this response has been extensively studied, an in depth understanding of the transcriptional regulatory network (TRN) that controls the transition into lipid accumulation remains elusive. In this study, we used a systems biology approach to elucidate the transcriptional program that coordinates the nitrogen starvation-induced metabolic readjustments that drive lipid accumulation in Chlamydomonas reinhardtii.
Results
We demonstrate that nitrogen starvation triggered differential regulation of 2147 transcripts, which were co-regulated in 215 distinct modules and temporally ordered as 31 transcriptional waves. An early-stage response was triggered within 12 min that initiated growth arrest through activation of key signaling pathways, while simultaneously preparing the intracellular environment for later stages by modulating transport processes and ubiquitin-mediated protein degradation. Subsequently, central metabolism and carbon fixation were remodeled to trigger the accumulation of triacylglycerols. Further analysis revealed that these waves of genome-wide transcriptional events were coordinated by a regulatory program orchestrated by at least 17 transcriptional regulators, many of which had not been previously implicated in this process. We demonstrate that the TRN coordinates transcriptional downregulation of 57 metabolic enzymes across a period of nearly 4 h to drive an increase in lipid content per unit biomass. Notably, this TRN appears to also drive lipid accumulation during sulfur starvation, while phosphorus starvation induces a different regulatory program. The TRN model described here is available as a community-wide web-resource at http://networks.systemsbiology.net/chlamy-portal.
Conclusions
In this work, we have uncovered a comprehensive mechanistic model of the TRN controlling the transition from N starvation to lipid accumulation. The program coordinates sequentially ordered transcriptional waves that simultaneously arrest growth and lead to lipid accumulation. This study has generated predictive tools that will aid in devising strategies for the rational manipulation of regulatory and metabolic networks for better biofuel and biomass production.
Electronic supplementary material
The online version of this article (doi:10.1186/s13068-015-0391-z) contains supplementary material, which is available to authorized users.
Keywords: Network modeling, Phenotypic transition, Transcriptional regulatory network, Metabolic network, Lipid accumulation, Chlamydomonas reinhardtii
Background
Green algae hold great promise for the manufacture of renewable biofuels [1]. The relatively low yield of algae-based biofuels, however, does not currently make them an economically viable replacement for fossil fuels [1]. Several strategies have been used to improve biofuel production, from strain design to improvements in growth, harvesting and refining techniques [2], but a large gap still needs to be bridged. Green algae accumulate lipids when subjected to nutrient depletion [3–10] and other stresses [11–13]. In parallel, nutrient starvation also causes growth arrest, thus limiting biomass accumulation, which is a major disadvantage for large-scale biofuel production. Consequently, an obvious strategy for the improvement of microalgae biofuel yield would be the decoupling of lipid accumulation from growth arrest at the molecular level. Such a rational strain design strategy requires a comprehensive mechanistic understanding of the transcriptional regulatory network (TRN) controlling lipid accumulation in order to identify the key regulatory elements (e.g., transcription factors (TFs) or metabolic bottlenecks), which coordinate the transition from nutrient starvation to growth arrest and lipid accumulation.
Our understanding of lipid accumulation in green algae has recently benefited from the use of high-throughput technologies to track genome-wide transcriptional changes during state transitions. In this regard, C. reinhardtii has emerged as the de facto model organism for algal biofuels research [12]. The early analyses of whole transcriptome changes in C. reinhardtii [6] uncovered the main hallmarks of nitrogen (N) starvation and lipid accumulation, i.e. the downregulation of protein synthesis and photosynthetic apparatus and redirection of primary carbon (C) metabolism. Subsequent high-resolution time series transcriptome experiments revealed that the TF nitrogen-responsive regulator-1 (NRR1) is in part responsible for the transcriptional changes that result in lipid accumulation during N starvation [14]. More integrative views of the phenotype transition during N starvation [15, 16] highlighted the importance of early transcriptional responses and gradual upregulation of alternative pathways of N assimilation and C metabolism. In addition to transcriptomic approaches, quantitative proteomic methods have provided unique insights into the global metabolic adjustments that drive N starvation-induced lipid accumulation, uncovering complex activity changes in the enzymes responsible for C and N metabolism [17–19]. For instance, proteomics analysis of lipid bodies revealed lipid accumulation to be a complex process involving lipid synthesis and recycling, as well as lipid trafficking and signaling to maintain homeostasis in microalgae oil bodies [20]. Further comprehensive analysis integrating transcriptome and proteome measurements revealed the multilevel responses of N-sparing mechanisms [21]. Metabolomics studies have also been conducted to characterize C. reinhardtii response to N starvation, which revealed differential flux signatures under N deprivation [22–24]. Finally, other macronutrient starvation studies, such as for sulfur (S) and phosphorus (P), have allowed for comparative transcriptome analyses. While several characteristic physiological changes are shared across N, S and P starvations (e.g., photosynthesis apparatus downregulation, reduced C fixation and lipid accumulation), other acclimation responses are nutrient-specific, such as the upstream controllers of the starvation response signaling circuit (e.g., SNRK2.1 or PSR1 [25, 26]), triacylglycerol (TAG) accumulation temporal profiles [27] and thylakoid membrane conservation [28].
The availability of high-throughput data sets provides an opportunity to use computational modeling approaches to obtain added insight into the process of lipid accumulation. In particular, the computational inference of TRNs from genome-wide gene expression datasets should be achievable, as a suite of methods are available to construct such network models [29–31]. For instance, the cMonkey algorithm [32], a semi-supervised biclustering algorithm that uses gene expression data guided by biologically informative priors and de novocis-acting gene regulatory element (GRE) detection, has been successfully applied to numerous organisms across all domains of life to build accurate and predictive models of TRNs [33–38]. High-throughput data sets also provide an opportunity to study the behavior of metabolism at a systems level. In this regard, constraint-based modeling approaches such as flux balance analysis [39] have proved to be particularly valuable for genome-scale investigation of metabolic flux distributions and for the development of metabolic engineering strategies in many species [40–45] while they can also serve as valuable platforms for data integration [46].
To obtain mechanistic insight into the transcriptional response of C. reinhardtii to N starvation, we employed a systems-level approach to unravel the program responsible for coordinating transcriptional changes that occur from the onset of N starvation to lipid accumulation. Through the integration of multiple publicly available data sets and resources, we used cMonkey to build the first mechanistic TRN model for the transition from N starvation into lipid accumulation in C. reinhardtii. To assess the impact of transcriptional regulation affecting metabolism, we integrated the TRN model with a genome-scale metabolic model for C. reinhardtii [47]. This enabled prediction of metabolic flux distributions during N starvation, as well as the identification of putative targets for increasing lipid yield. Furthermore, we conducted a comparative analysis of the expression pattern of key metabolic genes between N, S and P starvation experiments to identify common and condition-specific responses to those macronutrient starvations. Thus, through the integration of co-regulated modules identified from the time course analysis into a comprehensive metabolic model of C. reinhardtii, we obtained a systems-level understanding of how genome-wide transcriptional changes induced by N starvation drive the metabolic shift into a lipid accumulation state.
Results and discussion
To infer a N starvation-specific TRN model for C. reinhardtii, we integrated complementary data from: (1) the C. reinhardtii genome annotation [48] (version 5.5, available from Phytozome [49]), (2) the highest resolution time series transcriptome data set available for C. reinhardtii N starvation [14], and (3) a network of functional protein–protein interactions [50]. Analysis of the publicly available transcriptome data resulted in the identification of a set of 2,147 transcripts that showed significant difference in abundance between the onset of N starvation and lipid accumulation (see “Methods”; Fig. 1a). This set of transcripts constitutes the core network of transcriptionally regulated genes responsible for sensing N starvation and orchestrating the subsequent physiological shift to the lipid accumulation phenotype. We analyzed this set of 2147 transcripts with cMonkey [32] and identified a total of 215 putatively co-regulated modules, which were organized into a high confidence TRN model that can be explored through the interactive Chlamy Network Portal [51] (Fig. 1b).
Fig. 1.
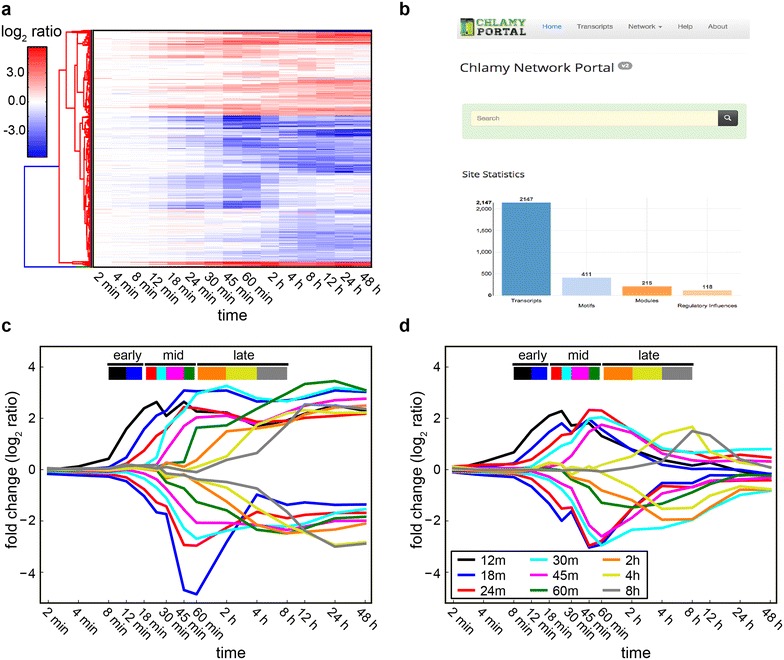
Transcriptional response of C. reinhardtii to N starvation and lipid accumulation. a Heatmap representation for the hierarchical clustering of the log2 expression changes in 2,147 post-filtered set of transcripts. Red (blue) indicates a relative increase (decrease) of expression. Color intensities are proportional to fold change magnitude. b Default view of the front page of the Chlamy Network Portal. In the portal, 2,147 transcripts are organized into 215 modules, 118 regulatory influences and 411 motifs. The site includes a powerful Apache Solr-based faceted search and navigation tools. The portal content is also linked to other information resources like Phytozome, STRING, GO categories and relevant literature. c, d Differentially expressed transcripts organized as a sequential set of transcriptional waves: 17 monotonic transcriptional waves composed of 125 transcriptional modules and 1,482 transcripts (c), and 15 transient transcriptional waves of 55 transcriptional modules and 758 transcripts (d). Each line represents average fold change for a given transcriptional wave. The timestamp on each wave reflects the timepoint at which transcript level change crosses a twofold threshold
The 215 co-regulated transcriptional modules were combined based on their temporal profiles to generate an ordered sequence of 31 transcriptional waves of two types: monotonic, in which transcript abundances changed and achieved new steady state levels; and transient, in which transcript abundances changed transiently before returning to the pre-starvation levels (Fig. 1c, d, Additional file 1: Table S1). The sizes of these transcriptional waves ranged from 17 to 340 transcripts, while their temporal schedules ranged from 12 min (min), for the earliest responding transcriptional modules, through 8 h (h) for late-stage responses. We labeled each wave by the timestamp at which the mean gene expression level of the corresponding transcriptional module(s) crossed the twofold threshold relative to the pre-starvation condition. The overall schedule of changes can be divided into three major stages of transcriptome transitions: early-stage response from 0 to 18 min, which was characterized by changes in transcripts for N transport, cellular signaling, ionic composition and protein translation; mid-stage response from 18 to 60 min, marked by the reorganization of metabolism; and late-stage transition between 1 and 8 h just before cells undergo a phenotypic state transition marked by lipid accumulation. We describe below a temporally organized summary of changes, with selected highlights of major events that occur at each of the three stages of the N starvation response (Table 1 serves as a reference for summary and highlights, and Additional file 1: Table S1 contains a comprehensive list of all transcripts associated with each stage, wave and module).
Table 1.
Summary of the organization of the transcriptional changes
| Stage | Time | Trend | Dynamics | Waves, modules, transcripts | Functional highlights (corrected p value) |
|---|---|---|---|---|---|
| Early | 0–18 min | Up | Monotonic | 2, 7, 105 | Ammonium transport (1.7e−9) Potassium ion transmembrane transport (1.4e−5) Ion channel activity (2.8e−3) Ion transport (3.4e−3) Protein kinase activity (1.3e−2) |
| Transient | 2, 7, 118 | ||||
| Down | Monotonic | 1, 1, 17 | Ribosome biogenesis (6.3e−8) Transcription (3.7e−6) rRNA processing (2.9e−5) Pseudouridine synthesis (7.0e−5) Methyltransferase activity (9.0e−5) |
||
| Transient | 1, 17, 202 | ||||
| Mid | 18–60 min | Up | Monotonic | 4, 24, 304 | Mitochondrial pyruvate transport (1.0e−6) Glutamine biosynthetic process (3.0e−5) Lipid metabolic process (5.2e−5) Amine metabolic process (7.5e−5) Proteolysis (9.6e−3) |
| Transient | 3, 7, 110 | ||||
| Down | Monotonic | 4, 44, 552 | Purine nucleotide biosynthetic process (5.1e−5) Fatty acid beta-oxidation (4.9e−4) Pseudouridine synthesis (8.7e−4) Malate dehydrogenase activity (1.6e−3) Fatty acid biosynthetic process (1.9e−3) |
||
| Transient | 4, 15, 237 | ||||
| Late | 1–8 h | Up | Monotonic | 3, 12, 177 | DNA replication (8.6e−7) Microtubule cytoskeleton organization (1.1e−4) NADP binding (3.5e−3) Lipid metabolic process (1.8e−2) Nucleotide binding (2.4e−2) |
| Transient | 2, 8, 139 | ||||
| Down | Monotonic | 3, 47, 489 | Photosynthesis (7.7e−31) Protein folding (1.0e−13) Fructose-bisphosphate aldolase activity (8.4e−6) Cell redox homeostasis (4.9e−5) Fatty acid biosynthetic process (7.5e−3) |
||
| Transient | 2, 5, 95 |
Transcriptional transition is organized within three time stages according to their temporal schedules. Transcripts are organized into co-regulated modules, which in turn are compiled into transcriptional waves. Selected significantly enriched functional categories (GO terms) are shown
The early-stage response: sensing a new environment
The early-stage response occurred from 0 to 18 min, with the earliest consistent transcriptional change (>twofold) initiating at 12 min. This stage included 442 transcripts in 32 co-regulated modules that were organized into 6 waves –122 transcripts were in 3 waves that changed monotonically (105 upregulated (2 waves), 17 downregulated (1 wave)), while 320 transcripts were in 3 waves that changed transiently [118 upregulated (2 waves), 202 downregulated (1 wave)] (Table 1, Additional file 1: Table S1). As expected, the earliest transcriptional changes were directly related to the N starvation response and included up to an 8-fold upregulation of 4 ammonium transporters and permeases (AMT3-5 and AMT8). In addition, transcripts encoding the mitochondrial carbonic anhydrase CAH5 and the ABC transporter HLA3, key metabolic proteins involved in the regulation of the C/N ratio homeostasis [52, 53], were among the earliest changing metabolic transcripts. The early-stage response also appeared to be devoted to modulating intracellular ionic composition through the upregulation of 12 membrane ion transporters, including Ca2+, Na+, K+, and Zn2+ transporters. Nineteen transcripts associated with signaling and modulation of growth and stress response were also upregulated, including transcripts of the mitogen-activated protein kinase (MAPK) pathway (e.g., PP2C, STK24, MAPK7), brassinosteroids biosynthesis pathway (e.g., DET2 [54]), and the auxin pathway (e.g., Ran-binding protein 9 (Cre01.g007150) [55]). Transcripts encoding proteins involved in protein degradation were also upregulated at this stage including at least 10 transcripts associated with the ERAD/ubiquitination pathway (e.g., CDC48 [56] and Cre01.g038950, a Sep15 domain protein [57]). Overall, 92 % of the transcriptional repression in the early-stage occurred transiently and mainly impacted ribosome biogenesis and RNA processing proteins, including RNA methylation (Supplementary Table S1). These early-stage transcriptional changes likely set the stage for the major metabolic restructuring that occurs during the mid-stage response.
Mid-stage response: metabolic state transition
The mid-stage response was comprised of 1203 differentially regulated transcripts within 90 co-regulated modules. These modules were organized into 15 waves that changed between 24 and 60 min after N starvation (Fig. 1c, d). Of these transcripts, 856 changed monotonically (304 were upregulated (4 waves), and 552 were downregulated (4 waves)) and 347 changed transiently (110 upregulated (3 waves), 237 downregulated (4 waves)). A significant fraction of the mid-stage transcriptional response was devoted to metabolic N scavenging, as evidenced by the upregulation of 14 transcripts encoding key metabolic enzymes involved in N scavenging from amino acids, nucleotides and polyamines (Table 2, Additional file 2: Table S2). Concomitantly, 12 transcripts responsible for the key steps of purine, pyrimidine, amino acids and polyamines biosynthesis were all downregulated (Additional file 2: Table S2). Targeted ubiquitin-mediated protein degradation during the early-stage response might feed amino acids into these N-scavenging and recycling processes. Consistent with reduced biosynthesis of amino acids, 41 transcripts encoding components of the protein synthesis and folding machinery were also downregulated (Table 2, Additional file 2: Table S2). Collectively these changes serve to mitigate the consequences of N starvation by conserving internal N resources for cellular sustenance upon growth arrest. Furthermore, the downregulation of the chaperones may indicate a higher rate of protein misfolding [58], and as a consequence higher endoplasmic reticulum stress which has been directly associated with the induction of lipid bodies [59].
Table 2.
Transcriptional changes on the cellular hallmarks of N starvation
| Trend | Pathway | Representative examples | Timestamp |
|---|---|---|---|
| Up | Pentose phosphate pathway | Glucose-6-phosphate dehydrogenase | 18 min |
| Purine catabolism | Xanthine dehydrogenase Adenosine deaminase |
18–30 min | |
| Urea metabolism | Carbamoyl phosphate synthetase Ornithine transcarbamylase Agmatine deiminase Allophanate hydrolase Urea carboxylase |
24–30 min | |
| Glutamine metabolism | Glutamine synthetase | 30 min | |
| Polyamines oxidation | Copper amine oxidase | 18 min–2 h | |
| TAG biosynthesis | Diacylglycerol acyltransferase | 24–45 min | |
| Down | FA β-oxidation | Acyl-CoA oxidases 2,4 dienoyl-CoA reductase |
24 min |
| Purine and pyrimidine biosynthesis | Adenylosuccinate synthase Uridine 5′-monophosphate synthase |
24 min | |
| FA biosynthesis | 3-ketoacyl-CoA-synthase Biotin synthase Acetyl-CoA carboxylase, 3-ketoacyl-ACP reductase |
24–45 min | |
| Translation | Translation initiation factors Translation elongation factors Release factors |
24 min–4 h | |
| Amino acid biosynthesis | Threonine deaminase Shikimate dehydrogenase Dihydrodipicolinate synthase 3-Phosphoglycerate dehydrogenase |
24–30 min | |
| Protein folding | Chaperonins | 45 min | |
| Calvin cycle | Triose phosphate isomerase Transketolase Ribose-5-phosphate isomerase |
45 min | |
| Carbon concentrating mechanism | Malate dehydrogenase | 45 min | |
| Polyamines biosynthesis | Ornithine decarboxylase Adenosylmethionine decarboxylase Spermidine synthase |
45 min–4 h | |
| Glycerolipid metabolism | Phosphatidate cytidylyltransferase Glycerol-3-phosphate acyltransferase |
45 min–4 h | |
| Chlorophyll biosynthesis | Chlorophyll synthase Geranylgeranyl diphosphate synthase Geranylgeranyl reductase Uroporphyrinogen decarboxylase |
60 min–4 h | |
| Two-component peroxide-detoxifying system | NADPH-dependent thioredoxin reductase 2-Cys peroxiredoxin Peroxidase |
2 h | |
| Photosynthesis | Ferredoxin Photosystem I and II proteins ATP synthase Cytochrome b6f complex subunits PsbP |
2–8 h | |
| Glutathione-ascorbate cycle | Ascorbate peroxidase Prohibitin, thioredoxin |
4 h | |
| Glycolysis | Glyceraldehyde-3-phosphate Dehydrogenase | 4 h |
Summary of key cellular pathways differentially regulated with representative transcript examples. Timestamp and direction of regulation (trend) are indicated
Transcripts encoding proteins involved in many carbon sequestration processes were downregulated during the mid-stage response including those implicated in carbon concentrating mechanisms and those encoding several key enzymes of the Calvin cycle (5 transcripts in 10 co-regulated modules in 1 wave, see Additional file 2: Table S2). Other C metabolism enzymes that were downregulated include glyceraldehyde-3-phosphate dehydrogenase (Cre01.g010900), which has been recently implicated in lipid accumulation in Arabidopsis [60] and the periplasmic carbonic anhydrase 1 (CAH1), which exhibited a dramatic drop in abundance from a maximum level of ~1500 to 0 FPKM (fragments per kilobase of transcript per million mapped reads). The overall repression of C fixation is an indication of transition into a lowered metabolic state as a consequence of the cessation of growth. In addition, alternative sources of carbon such as recycled C-backbones from degraded protein and acetate influx might also compensate for the downregulation of C fixation.
While lipids start to accumulate at 8 h after N starvation [14], within 60 min post N starvation we observed the upregulation of transcripts encoding enzymes responsible for maintaining the cellular redox balance (high NADPH/NADP+) required for lipid biosynthesis [61] and synthesis of lipid precursor metabolites (e.g., GLD2 [62]). Similarly and as previously described by Boyle et al. [14], transcripts for TAG biosynthesis DGAT1 and DGTT1 were upregulated. Importantly, DGAT1, which catalyzes the rate-limiting step in the de novo TAG biosynthesis, increased up to fourfold, 45 min after N starvation. Membrane remodeling [63] was enhanced through the upregulation of membrane lipases such as saposin (Cre05.g235700), indicative of catabolism of glycosphingolipids, which could facilitate membrane remodeling and in extreme cases membrane lysis [64]. Notably, transcripts encoding key enzymes for the de novo fatty acid (FA) biosynthesis, such as the three β-ketoacyl-(acyl-carrier-protein) synthases (KAS1, KAS2 and KAS3; Cre11.g467723, Cre07.g335300 and Cre04.g216950, respectively), which are responsible for the first enzymatic step catalyzed by the multi-enzyme complex fatty acid synthase (FAS) were all downregulated. Importantly, the downregulation of lipid biosynthesis transcripts continued through the late-stage of the N starvation response.
Late-stage response: transition into the lipid accumulation state
The observed commencement of significant lipid accumulation 8 h after N starvation was set up by transcriptional changes occurring in the late-stage response, which began after 1 h from N starvation. Comprised of 665 monotonic (177 up-, 489 downregulated) and 234 transient (139 up-, 95 downregulated) transcripts organized into 72 co-regulated modules in 10 waves, the late-stage response was characterized by lipid metabolism remodeling, downregulation of photosynthesis, oxidative stress and protein folding stress responses, and an upregulation of sugar catabolism. While an increased metabolic flux into FA biosynthesis would be expected to support TAG production, the late-stage response involved the downregulation of many transcripts associated with FA biosynthesis such as the chloroplastic pyruvate dehydrogenase complex (PDC), acetyl-CoA carboxylase (ACC), and malonyl-CoA:ACP transacylase, all involved in the biosynthesis of FA precursors (Fig. 2a, b). Interestingly, upregulation of some of these enzymes such as PDC is known to be strongly correlated with lipid accumulation [65]. Additionally, the downregulation of FA biosynthesis-related transcripts during the mid-stage response (i.e., downregulation of KAS1-3), continued into the late-stage response with the transcriptional downregulation of the 3 enzymes of the FAS complex involved in the subsequent steps of FA biosynthesis: 3-ketoacyl-ACP reductase (Cre03.g172000), β-hydroxyacyl-ACP dehydratase (Cre03.g208050) and enoyl-ACP-reductase (Cre06.g294950). The FAs 16:0 and 18:1 are the most abundant of the TAG fractions in N starved cells [28], reflecting the importance of de novo FA synthesis for TAG accumulation, which accounts for 79 % of the TAG content in N starved cells [63], providing further evidence that the lipid accumulation response is a time-dependent additive process. Indeed, transcriptional downregulation of FA biosynthesis enzymes during lipid accumulation is consistent with previous observations in C. reinhardtii [14, 21, 23] and other microalgae [66, 67], but not in diatoms [68, 69]. Downregulation at the transcriptional level is consistent with a strong reduction in protein levels for enzymes of the early steps of the FA biosynthesis, particularly ACC [21, 70]. It is of note that although FA biosynthesis enzymes were transcriptionally downregulated, their absolute abundance remained relatively high at this stage, ranging from a minimum value of 3 FPKM for KAS3 to 572 FPKM for ACP2, presumably to maintain flux towards FA biosynthesis (Fig. 2c). Furthermore, while transcript abundance provides insights into changes in metabolic activity, actual enzyme levels can deviate from transcript levels based on other factors such as protein stability and turnover. Therefore, long-term lipid accumulation might be accomplished by increasing the overall carbon flux towards the FA pathway [71] or, as previously noted, by limiting the catabolism of lipids through the FA β-oxidation pathway during lipid accumulation [72]. Besides, several FA desaturases (FAB2, FAD3, FAD7), which incorporate double bonds into FAs at the cost of reducing equivalents, were downregulated, potentially to conserve energy while producing lower amounts of unsaturated FAs.
Fig. 2.
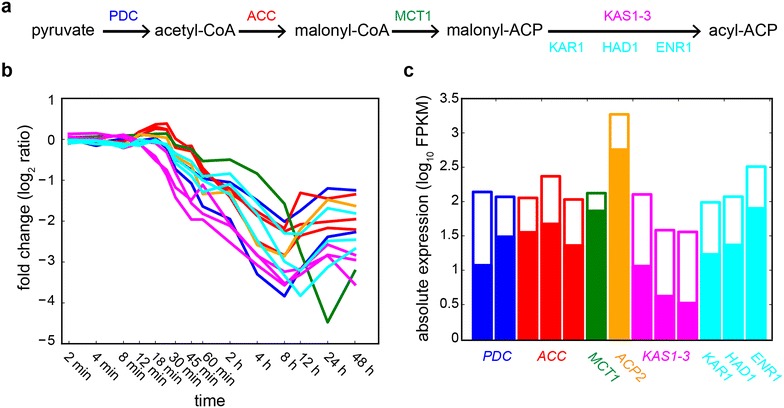
FA biosynthesis pathway is transcriptionally downregulated during N starvation. a Schematic view of the reaction steps for FA biosynthesis and the enzymes that catalyze them. b Expression profile for transcripts encoding enzymes highlighted in (a). c Absolute expression values for FA biosynthesis transcripts in log10 FPKM (open bars represent absolute expression level at time t = 0 while solid bars represent levels at t = 8 h). PDC pyruvate dehydrogenase complex, ACC acetyl-CoA carboxylase, MCT1 malonyl-CoA:ACP transacylase, ACP2 acyl carrier protein, KAS β-ketoacyl-(ACP) synthase, KAR1 3-ketoacyl-ACP reductase, HAD1 β-hydroxyacyl-ACP dehydratase, ENR1 enoyl-ACP-reductase
The transcript encoding the mitochondrial glycerol-3-phosphate dehydrogenase (GPD2, Cre01.g053000), which serves as a major link between carbohydrate metabolism and lipid metabolism, is upregulated as part of the late-stage response, in agreement with previous studies of the C. reinhardtii transcriptome during N starvation [18, 21]. Interestingly, Phaeodactylum tricornutum cells overexpressing GPD had a 60 % increase in neutral lipid content, reaching 39.7 % of dry cell weight in stationary phase [73], plus overexpression of GPD has also been reported to increase lipid content in plants [74]. By contrast, the cytoplasmic isoform GPD1 (Cre12.g511150) was downregulated as part of a late-stage response, highlighting the importance of cellular compartmentalization to achieve modularity in metabolism. Additionally, consistent with the previously described role for lipases in lipid accumulation [6], the TAG lipase expression profiles changed, with TGL9 and Cre09.g391838 upregulated, while TGL15 was downregulated up to 18-fold. Undoubtedly, TAG lipases are key modulators of TAG accumulation in C. reinhardtii as previous works have demonstrated [75, 76]. The extensive transcriptional changes in genes encoding lipid composition enzymes underscore the importance of regulatory mechanisms for the overall lipid increase and changed lipid composition during N starvation in C. reinhardtii.
Other pathways associated with biosynthesis of membrane components that were downregulated in the late-stage response (see Table 2, Additional file 2: Table S2) included biosynthesis of glycerolipids and isoprenoids (at least 7 key enzymes), porphyrin and chlorophyll (at least 4 key enzymes). Downregulation of the photosynthetic function and apparatus is a major hallmark of N starvation [77]. Our model accounts for the entire chloroplast light reaction chain, including photosystem I and II, cytochrome b6f, plastocyanin, ferredoxin, ferredoxin-NADP+ reductase and ATP synthase, comprising 54 transcripts distributed in 35 co-regulated modules and 4 waves (see Table 2, Additional file 2: Table S2). This downregulation during the late-stage response likely results in lower production of reactive oxygen species, and is consistent with the observation that many oxidative stress response systems were also downregulated (at least 9 transcripts of key oxidative protection enzymes).
The TRN for the N starvation response
In order to construct the network of transcriptional regulators (i.e., TFs and chromatin regulators) that coordinate the N starvation response, we computed the distances between temporally ordered, smoothed expression profiles of the transcriptional regulators (TRs) and the transcriptionally co-regulated modules. We selected the first fifth percentile of all distances within the physiologically relevant window of 15–90 min time lag between a change in the TR and its putative target transcriptional module. This time window was selected based on the time it takes for a differentially regulated TR to exert an observable effect on transcription ([75]; see “Methods” for details). Despite the possible contribution of post-transcriptional regulatory events in modulating transcriptional activity, this analysis uncovered a TRN in which 17 of the 34 differentially regulated TRs exerted 118 putative TR-module regulatory influences, coordinating 815 transcripts within 60 co-regulated modules (Table 3, Additional file 3: Table S3). This TRN model encompasses 17 waves across the three stages of the N starvation response (Fig. 3).
Table 3.
List of the TRs predicted to orchestrate the transcriptional response during N starvation
| Stage | Name | Description | Timestamp (min) | Trend | Predicted regulated modules |
|---|---|---|---|---|---|
| Early | Cre13.g573000 | SET domain methyltransferase | 12 | Down | 40, 113, 162, 197, 127 |
| BLZ8 | Basic region leucine zipper | 13 | Down | 105, 121 | |
| RWP11 | RWP-RK TF | 15 | Up | 150 | |
| Cre17.g746547 | bZIP TF | 15 | Down | 105, 121 | |
| Mid | CGL86 | Nuclear inhibitor of protein phosphatase-1 | 26 | Down | 129, 130, 99, 172, 13, 45, 82, 148 |
| NAT11 | Acetyltransferase (GNAT) | 30 | Down | 129 | |
| RWP1 | RWP-RK domain protein | 36 | Up | 130, 99, 13, 45, 82, 148 | |
| NAT1 | Acetyltransferase (GNAT) | 37 | Down | 200, 178, 93, 174 | |
| NRR1 | SBP domain | 44 | Up | 82, 67 | |
| RWP4 | RWP-RK domain-containing protein | 48 | Up | 193, 98, 35, 37, 72, 170, 45, 142, 175, 59, 99, 53, 87, 27, 95 | |
| GSM1 | Gamete-specific minus 1 | 50 | Up | 193, 98, 35, 37, 166, 172, 45, 175, 99, 59, 67, 53, 54, 87, 27, 149 | |
| Cre12.g523000 | Zinc finger CCCH domain containing protein | 62 | Up | 149, 193, 98, 35, 37, 102, 170, 172, 45, 142, 175, 99, 59, 67, 53, 54, 87, 185, 27, 211, 95 | |
| Late | CGL107 | Histone-like transcription factor (CBF/NF-Y) | 109 | Up | 193, 8, 9, 141, 50, 24, 154, 59, 92, 158 |
| Cre09.g386753 | DNA binding protein S1FA | 109 | Down | 98, 35, 166, 172, 175, 67, 149, 27 | |
| NAB1 | Nucleic acid binding protein | 146 | Down | 33, 34, 133, 104, 76, 144, 136, 210, 21, 151, 57, 186 | |
| NAT31 | Acetyltransferase (GNAT) | 371 | Up | 104, 136, 133 | |
| Cre03.g152150 | Zinc finger C2H2 type domain | 418 | Down | 129, 13 |
Seventeen differentially expressed TRs during N starvation have a predicted significant influence on at least one transcriptional module. TR name and description, first time point of differential expression (timestamp) and predicted influenced modules are indicated
Fig. 3.
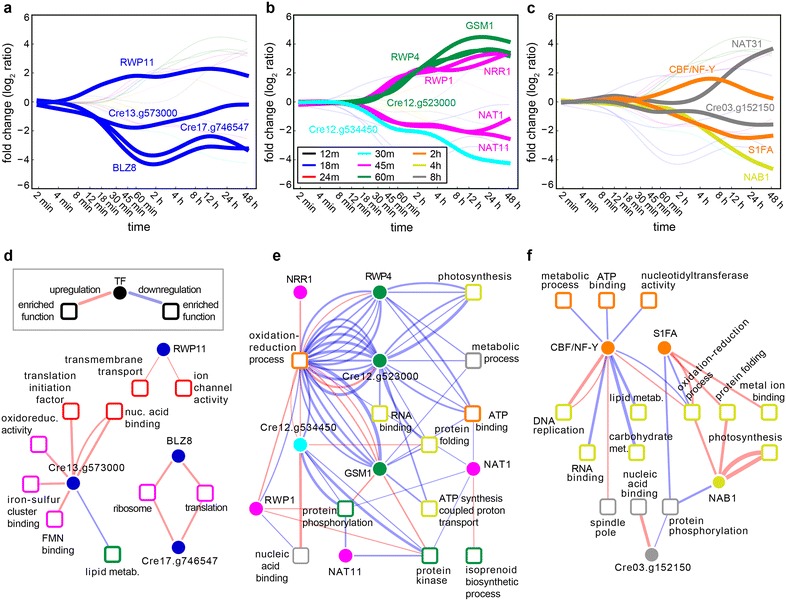
Expression dynamics and network of predicted transcriptional influences. a–c Top panels show TR expression dynamics: each profile is labeled with the relevant TR it represents. d–f Network of predicted transcriptional influences from TRs (circle nodes) on enriched GO terms (square nodes) through transcriptional modules (edges). Edge color indicates transcriptional activation (red) or repression (blue) influenced of a TR on the transcriptional module. Edge thickness is inversely proportional to the distance between the TR and the transcriptional module. Nodes are colored using the same pattern as in Fig. 1c, d, e.g., blue represents a differential expression acquired at time 18 min after N starvation
Based on this TRN model, the transcriptional cascade started within 12 min after N starvation with the twofold upregulation of a previously described N response regulator in C. reinhardtii and plants, RWP11 [78]. By 18 min, three other TRs were downregulated: BLZ8 and Cre17.g746547, both bZIP TFs, and Cre13.g573000, a SET domain methyltransferase (Fig. 3a, d). The subsequent remodeling of metabolism during the mid- and late-stage response was coordinated by at least 8 TRs that acted from 24 min to 2 h after N starvation. One of these TRs is NRR1, the squamosa promoter binding protein described by Boyle et al. [14] which reaches twofold upregulation 44 min after N starvation (Fig. 3c, e). Interestingly, RWP1, a RWP-RK domain TF [79], showed a very similar temporal expression pattern as NRR1. In the 48–68 min time frame, three other TFs crossed the twofold upregulation threshold: Cre12.g523000 (a C3H Zinc finger TF), RWP4 and GSM1. While GSM1 is known to function as a regulator of the minus mating type gametogenesis response, the function of these other two TFs is less well understood. Additionally, other TRs downregulated at this point include a FHA domain nuclear inhibitor of protein phosphatase-1 (Cre12.g534450) and several acetyltransferases (NAT1 and NAT11). The late transcriptional wave was putatively orchestrated by the transient upregulation of the subunit D of NF-Y TF (Cre07.g341800) [80], the transcriptional downregulation of the DNA binding protein S1FA [81], and the C2H2-family TF Cre03.g152150 (Fig. 3c, f). Other changes in TRs during the late-stage transcriptional waves include the upregulation of the NAT31 acetyltransferase and the downregulation of NAB1 (Cre06.g268600), which plays an important role in controlling the expression of the light-harvesting antenna of photosystem II in C. reinhardtii [82]. Finally, we found 17 additional transcriptional regulators with differential expression during the phenotype transition, but their expression profile did not match significantly any of the transcriptional modules, suggesting a more cooperative mode of action involving multiple TRs regulating the same transcriptional module. Additional file 4: Fig. S1 shows the dynamics of such TRs, with notable examples including the early downregulation of the AP2-family TF Cre06.g275500, up to 33-fold increase of the bHLH TF Cre01.g011150 during the mid-stage response and the upregulation of RWP8 during the late stage. While incomplete, the TRN model organized the complex set of transcriptional changes during the N starvation response into a modular framework of co-regulated transcripts that were temporally coordinated by as few as 34 TRs, allowing for model-based systematic analysis of metabolic consequences.
Metabolic network analysis
Using the available genome-scale metabolic network of C. reinhardtii [47], we performed a variation of flux balance analysis to assess the impact of knocking down the metabolic flux through reactions catalyzed by enzymes that were transcriptionally downregulated within specific co-regulated modules. In order to estimate the TAG content per biomass, we computed a dimensionless ratio (ρ) of relative TAG production per biomass accumulation, which allowed us to predict the impact of knocking down a transcript on the amount of lipids per unit biomass (see “Methods” for details). From the 1,058 monotonically downregulated transcripts, this approach led to the prediction that the knockdown (KD) of 57 transcripts would result in a relative increase of TAG production per biomass with respect to wild type conditions (i.e., ρ > 1) (Fig. 4a, Additional file 5: Table S4). Investigation of the expression patterns for these 57 transcripts revealed that they were not uniformly distributed along the whole time domain of transcriptional changes, but were mainly confined to the 30 min to 4 h range, coinciding with the metabolic state transition (Fig. 4b, Additional file 5: Table S4). The 57 transcripts were distributed across 51 modules and this TRN mapping further implicated guilt-by-association of 671 additional genes in driving lipid accumulation. The 12 TRs of these modules are predicted by the TRN to be putatively responsible for coordinating down regulation of these modules with other transcriptional changes during the N-starvation response. Notable functions represented among these modules (protein synthesis, signaling and photosynthesis) suggest a potential mechanistic linkage between transcriptional programs for growth arrest, the photosynthetic machinery and lipid accumulation during N-starvation (Fig. 4c).
Fig. 4.
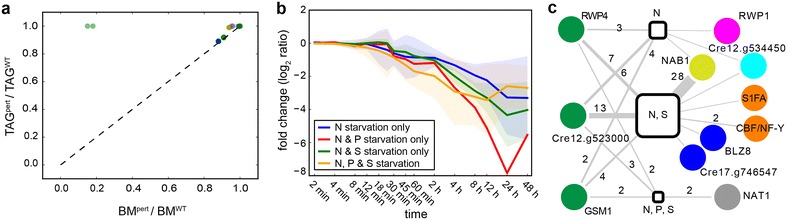
Metabolic targets for higher relative TAG to biomass ratio. a Model-predicted consequences of transcriptional downregulation on relative TAG per unit biomass. Dots represent prediction for 57 metabolic enzymes whose downregulation resolves in increased TAG per unit biomass (above the diagonal, ρ > 1). Among these 57 metabolic targets, only 10 genes (blue dots) are uniquely downregulated in N starvation, and 40 genes are also downregulated during S starvation (green dots). Five genes are downregulated in N, S and P (orange dots) and just one, Cre02.g082750, is downregulated in N and P only (red dot). b Average expression profiles during N starvation for the metabolic targets grouped as in a. c Network of transcriptional regulatory influences on metabolic targets. Each circle node represents a TR (colored as in Fig. 3). Edges represent the predicted regulatory influence of a given TR on specific genes across the different starvation responses (squared nodes). Edge labels indicate the number of genes regulated by each TR, one if no label is present
Metabolic targets changing earliest fall into the ATP homeostasis category, particularly control of ATP/NADPH ratios, like the nucleoside-diphosphate kinase (Cre07.g325734), the cytochrome b6f complex (responsible for creating the proton gradient that drives the synthesis of ATP in chloroplasts) and plastocyanin (Cre03.g182551), which is responsible for the flow of electrons between cytochrome b6f and photosystem I. Furthermore, we identified several targets from central metabolism like triose phosphate isomerase (TPI). TPI catalyzes the reversible conversion of dihydroxyacetone phosphate (DHAP) to glyceraldehyde-3-phosphate (GA3P) and coordinates many pathways including glycolysis, Calvin cycle, and glycerol metabolism. Notably, recent studies in Arabidopsis have uncovered the crucial role of TPI in general lipid metabolism [83]. Indeed, a TPI mutant in Arabidopsis with a fivefold reduction in transcript levels accumulated DHAP and glycerol, both byproducts of TAG mobilization and precursors for glycerolipid biosynthesis. Another key target identified from this analysis was Cre02.g080200, which encodes for the transketolase TRK1 which is an essential enzyme of both the Calvin cycle and the non-oxidative pentose phosphate pathway (PPP). TRK1 has also been found to play a key role in the regulation of C allocation through calcium-dependent phosphorylation regulation [84]. Our metabolic modeling approach also predicted that the KD of the next enzyme of the non-oxidative PPP, ribulose phosphate-3-epimerase (Cre12.g511900) results in a ρ > 1. The metabolic targets predicted to affect TAG production during the late-stage response between 2 and 4 h after N starvation were strongly dominated by genes involved in photosynthesis. Subunits of photosystem I and II, including light-harvesting proteins, chlorophyll binding proteins, thylakoid lumen proteins and ferredoxins, were all predicted to result in a ρ > 1 upon KD, in agreement with cell growth arrest and recycling of the photosynthetic apparatus. Interestingly, other metabolic targets downregulated in the 2–4 h period whose KD resulted in a ρ > 1, were involved in lipid biosynthesis functions, including enzymes like acyl-carrier-protein dehydratase (Cre03.g208050), farnesyl pyrophosphate synthase (Cre03.g207700) or geranylgeranyl diphosphate reductase (Cre01.g050950), which may be responsible of the observed changes in lipid composition in C. reinhardtii during N starvation. These observations may reflect both network redundancy on TAG production and how different categories of lipids affect biomass, as defined in the metabolic model biomass function.
Finally, we used the model to identify the metabolic genes predicted to be involved in the biosynthesis of acetyl-CoA, malonyl-CoA and glycerol 3-phosphate, key precursors for lipid biosynthesis. Out of the 100 genes predicted to be involved in this process, 26 are downregulated and only four are transcriptionally upregulated (Additional file 6: Table S5). The upregulated genes include glycerol-3-phosphate dehydrogenase and glyceraldehyde 3-phosphate dehydrogenase, key enzymes for the FA biosynthesis [73, 85], involved in the production of glycerol and reducing equivalents, respectively. The 26 downregulated enzymes participate at different levels of FA biosynthesis, ranging from carbon fixation and the acetyl-CoA and malonyl-CoA synthesis to CoA biosynthesis (Additional file 6: Table S5). Furthermore, we compared the pattern of expression of these metabolic targets under other nutrient starvations, e.g., S and P [25]. Virtually all the metabolic targets were also downregulated in S starvation conditions. We observed, however, a remarkably different expression pattern in P starvation, where the vast majority of these metabolic targets were not downregulated (Additional file 6: Table S5, Fig. 4c). Indeed, FA composition of accumulated TAG differs from N to P starvation, specially in 16:0 and 18:2 fractions [28], reflecting different routes of TAG production. These observations evidence how the metabolic and regulatory changes that occur during lipid accumulation in response to N and S starvation are similar, while P starvation induces a different lipid accumulation program.
Conclusion
Analysis of expression dynamics at the whole transcriptome scale for C. reinhardtii has uncovered a complex but structured transcriptional response during N starvation and subsequent transition into a state of lipid accumulation. The application of the biclustering algorithm cMonkey revealed that the transcriptional response was modular and organized as 31 temporally ordered waves that were triggered as early as 12 min and continued changing until 8 h after N starvation. Based on the timing and functional composition of the waves the N starvation response could be divided into three categories: the early response characterized by distinct cellular signaling, the mid response recapitulating how the cell metabolism transitions into a different state and the late response with hallmarks of the acquisition of the final phenotype, dominated by transcripts for photosynthesis, lipid metabolism, oxidative stress and protein folding. Furthermore, we predicted a network of transcriptional regulators, at least 17 TFs and chromatin remodeling proteins that putatively orchestrate the transcriptional response. Certainly, the phenotypic transition is not the consequence of regulation by a single TF, but the coordinated response of several factors. In fact, integration of the TRN model with a genome-scale metabolic flux model demonstrated that there are 57 metabolic bottlenecks that are distributed across different modules of the transcriptional program. The TRN mediates the coordinated downregulation of these 57 metabolic steps to drive increased lipid per unit biomass. Similar expression patterns of the 57 enzymes under S starvation are suggesting that the same TRN might oversee both the S- and N-starvation responses. In contrast, different metabolic and transcriptional networks appear to be responsible for the P-starvation response, wherein only 6 of the 57 enzymes were differentially expressed. Ultimately, to ensure the integration of the TRN model with other existing resources and broader dissemination among the scientific community, we developed an accessible web-based resource, the Chlamy Network Portal [51], which incorporates the TRN model together with the processed expression data and offers filtering and visualization interfaces for further analysis. Well-known resources such as Phytozome [49], STRING [50], and Gene Ontology (GO) terms [86] are seamlessly integrated at the Chlamy Network Portal.
Recent studies in C. reinhardtii [21, 70, 79] and other microalgae [87] have integrated various omics data sets to reveal that cellular response to N starvation is controlled by the structure of a TRN, and cannot be explained by individual action of a handful of TRs. Schmollinger et al. [21] performed a very comprehensive study of the cellular response to N starvation at different regulatory levels, quantifying the transcriptome, proteome and metabolome for both wild type and mutant strains. They uncovered several N-sparing mechanisms and listed a set of differentially regulated TFs as candidate regulators of the cellular response. In order to identify the key regulatory genes involved in the control of lipid metabolism, Gargouri et al. [79] used a time-lagged correlation analysis to identify putative TRs of cellular metabolism, before and after the onset of lipid accumulation. Our approach is distinct in that it integrates different types of evidence for co-regulation to infer a mechanistic and predictive TRN. Furthermore, by integrating this model with a metabolic model we were able to predict consequences of specific transcriptional changes on biomass production as well as metabolic flux towards TAG accumulation. The modular architecture of this TRN predicts TR regulation of not single transcripts or metabolites, but rather co-regulated modules enabling a robust assessment of the functional consequences of TR changes. Importantly, Valledor et al. [70] combined transcriptomics, proteome and metabolome measurements during N starvation and repletion to demonstrate how such integrative approaches reveal new understandings of molecular regulation. A beautiful example is the discovery of a central role for the major lipid droplet protein within such regulatory and metabolic network model in linking signaling cascades (GTPases downregulated during N starvation) to vesicle formation (COP II) and lipid body formation.
There are several possible limitations to our study. First, our TRN is based on expression data and transcriptional changes do not fully account for changes at the protein level, much less for functional activity of post-transcriptionally regulated proteins [88, 89], which may result in phenotypic changes not recapitulated by the transcriptional response. Therefore, integration of proteome and metabolome measurements would represent a powerful complement to our approach. At the level of the integration of the metabolic and TRN models, potential missing links may occur from the incomplete nature of the currently available metabolic models for C. reinhardtii consisting on 1080 genes and 2190 reactions, which is far from the complete metabolome set. Indeed, in a parallel effort, our group recently extended and improved the metabolic model for C. reinhardtii, upgrading it to 1355 genes (1460 transcripts), 2394 reactions and 1133 metabolites, enabling very accurate predictions of gene deletion growth phenotypes, with an area under the receiver operating characteristic curve of 0.92 [90]. Also, the lack of an exhaustive annotation for the TF set is another source for missing interactions between potential regulators of the transcriptional modules. Additionally, other potential regulators of gene expression like miRNAs are not currently included in our TRN model. Finally, the experimental validation of de novo detected GREs and predicted TF to transcriptional module interactions would produce a more accurate picture of the functional interactions recapitulated by our TRN.
We foresee two main future directions of this study: first, the expansion of the TRN model with expression data from other phenotypic transitions, e.g., starvation on other nutrients, different growing conditions (light, trophic conditions, CO2 levels, etc.), and different mutant strains. A second direction would be to refine the accuracy and specificity of the transcriptional regulatory influences with the integration of quantitative data that includes additional influencers, like miRNAs [91–95] or post-translational modifications [96]. Moreover, a genome-wide chromatin state map would constitute a resourceful informational tool for detecting and associating GREs to the transcriptional response [97, 98]. Recent experimental evolution work with C. reinhardtii [99–101] has shown the adaptive plasticity of this organism to quickly generate new phenotypes with increased fitness to novel environments. Experimental evolution approaches and gene editing [102] would be complementary and a good synergistic combination to TRN modeling to better understand the process of lipid accumulation in microalgae, with the ultimate goal of engineering new strains with desired phenotypes.
Given the complexity underlying the phenotypic transitions in C. reinhardtii, we are confident that the constructed TRN model presented here represents a relevant predictive tool that would uniquely guide the rational selection of candidate gene targets for improved biofuel production. The dissemination of the TRN through the Chlamy Network Portal platform will especially ensure an efficient broadcast of the model to the growing biofuel research community.
Methods
Expression data
Raw RNA-Seq expression data obtained from a N starvation time course experiment on C. reinhardtii wild type CC-3269 [14] was aligned with STAR [103] to the current genome annotation (version 5.5 from Phytozome [49]). Next, using cuffdiff [104], we computed for each transcript i at each time point t, the log2 expression ratio, ,
| 1 |
where represents the expression in fragments per kilobase of transcript per million mapped reads (FPKM) of transcript i at time t. We filtered out unchanging transcripts and noisy expression measurements by selecting any transcript that complied with any of the following two rules: (1) during the lipid accumulation time points, i.e., at time points 8 h, 12 h, 24 h and 48 h after N starvation [14] or (2) consistent for a single period of at least 30 min.
TRN inference
We used cMonkey [32] version 4.9.11 to build the TRN model. We set the searching and scanning window as (−10,2000) base pairs upstream from the transcriptional start site of common cis-acting gene regulatory elements (GREs). We obtained the TRs annotation from Schmollinger et al. [21], which contained 491 unique transcriptional regulator genes for the current version of C. reinhardtii annotation. Protein–protein functional interaction network was retrieved from STRING [50], version v9.05.
Inference of transcriptional regulators influences
Transcript expression trajectories were first smoothed with the csaps routine from MATLAB. Smoothed data was then divided by its maximum absolute value to account for non-linear relationships between transcriptional regulators and transcriptional modules. A matrix of distances between TRs and transcriptional modules was calculated allowing for a defined best time lag between the expression of the TR and the corresponding transcriptional module. For eukaryotic genes, it can take from several minutes up to several hours since the expression of a TF changes, to transcriptional changes on its target genes [105]. We used a time window of 15–90 min as time lag, based on reported values in literature [106]. Specifically, we allowed first for 5–20 min for the nuclear phase: transcription, transport and export (Ref. [107] and Bionumbers BNID:105650). As for the transcription time, we allowed 3–8 min; such time window is supported by the ~3 min mean translation time previously reported in yeast [108] in conjunction with additional data about translation rates of 5 to 11 aa/sec (Bionumbers BNID:104598 and BNID:109527, respectively [109, 110]). Then, we included 10 additional min into the time lag for protein translocation to the nucleus and binding to promoter sequences (BNID:109955), which sets our time lag ideally into the expected range of 18–38 min. Still, we searched for TR to transcriptional module interactions as late as 90 min, allowing for the effect of other factors like long sequences, protein maturation and other post-transcriptional regulatory mechanisms. We predicted as true TR to transcriptional module influences those whose best distance was smaller than the first fifth percentile of all the distances and also had a time delay within the defined time window of 15–90 min.
Metabolic analysis of genome-scale metabolic model
We used the genome-scale metabolic reconstruction iRC1080, the hitherto most comprehensive metabolic model of C. reinhardtii [47]. We simulated mixotrophic growth by allowing for the uptake of acetate as well as light via warm and cool fluorescent light sources (reactions 4 and 5 of the model) to mimic the experimental setup of Boyle et al. [14]. N is available only via ammonium uptake, whereas starch uptake was prohibited. Consequently, the lower bounds of the respective exchange reactions were either left at their published values or set to zero (to prohibit uptake). To separate TAG production from the biomass production, we subtracted all TAG producing compounds from the mixotrophic biomass definition and created a separate exchange reaction EX_TAG with the same TAG composition as in the biomass definition and a new biomass reaction BM_TAG. Without further flux bound perturbations we termed this model iRC1080WT. We conducted multiple Flux Balance Analysis (FBA) [39] runs to investigate the influence of reaction or module perturbation on TAG production in a dimensionless form. We first computed the FBA with BM_TAG as objective function from iRC1080_WT to derive the unperturbed biomass flux BM_TAG_WT. To avoid numerical problems, we considered a KD as a reduction of the flux to at least 1/16th of the original flux. based on the particular reaction flux from the flux distribution that generates BM_TAG_WT. Cell viability was ascertained by requiring at least 10 % biomass production with respect to the original wild type predictions. We again conducted an FBA to derive the BM_TAG_pert from the perturbed network iRC1080_pert. The ratio BM_TAG_pert/BM_TAG_WT provides then the relative change of BM_TAG flux. Next, we computed two further FBAs, this time with EX_TAG as objective function for iRC1080_WT and iRC1080_pert to derive TAG_WT and TAG_pert, respectively. The ratio TAG_pert/TAG_WT consequently provides the relative change in TAG production upon gene and affected reaction perturbation (taking the value of one for no change). Finally, we computed a ratio of these ratios in the following form:
| 2 |
The interpretation of Eq. 2 is thus straightforward: if ρ > 1 the model perturbation affects the biomass production more severely and causes, thus, more TAG content per biomass. Consequently, a value smaller than one relates to a perturbation that causes less TAG per biomass content. Note that Eq. 2 is dimensionless, because units cancel out in the TAG and BM ratios.
Authors’ contributions
ALGL analyzed and interpreted the data, designed the study and drafted the manuscript. SS carried out the metabolic analysis, interpreted the data and drafted the manuscript. JV designed the study, interpreted the data and drafted the manuscript. SI participated in the analysis and interpretation of the data and revised the manuscript. WC participated in the analysis and interpretation of the data and revised the manuscript. DDB participated in the conception of the study and the interpretation of the data and helped to draft the manuscript. CBY participated in the conception of the study and the interpretation of the data and helped to draft the manuscript. ST designed the web portal, participated in the conception of the study and the interpretation of the data and revised the manuscript. DJR participated in the conception of the study and the analysis of the data and revised the manuscript. MVO participated in the conception of the study and the interpretation of the data and revised the manuscript. NDP participated in the design of the study and revised the manuscript. NSB conceived the study, participated in its design and coordination and drafted the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We would like to thank Julie Kerns for a careful reading of the manuscript and for useful comments. We would also like to thank Gustavo Glusman, Max Robinson and Chris Lausted for useful comments. This work was partially funded by DOE-ABY (DEEE0006315) (SI, JV, ALGL, WC, NSB, NDP), NIH Center for Systems Biology (grant P50 GM076547) (ALGL, NSB, NDP) and the Camille Dreyfus Teacher-Scholar program (NDP). We also acknowledge financial support from the German Ministry for Research and Education within the framework of the GerontoSys (grant FKZ 0315581D) and e:Med (grant FKZ 01ZX1402C) initiative (SS).
Competing interests
The authors declare that they have no competing interests.
Abbreviations
- GRE
gene regulatory element
- TRN
transcriptional regulatory network
- KD
knockdown
- TAG
triacylglycerol
- FA
fatty acid
- TR
transcriptional regulator
- TF
transcription factor
Additional files
10.1186/s40064-015-1541-2 Comprehensive list of the transcriptional changes. As in Table 1, each transcriptional stage is hierarchically itemized into transcriptional waves, co-regulated modules and finally transcripts. Each wave has a link to a different sheet where each of the transcripts are defined, including their co-regulated module membership. Timestamp of co-regulated module is highlighted with different colors, using the same color map as in Fig. 1c-d.
10.1186/s40064-015-1541-2 Transcriptional changes on the cellular hallmarks of N starvation. For each functional hallmark, each differentially regulated transcript is grouped into a distinctive pathway. Phytozome locus name, gene names and description, co-regulated module membership, timestamp and direction of regulation are indicated.
10.1186/s40064-015-1541-2 List of the TRs predicted to orchestrate the transcriptional response during N starvation. Seventeen differentially expressed TRs during N starvation with a predicted significant influence on at least one transcriptional module. TR annotation (locus name, gene name and description), first time point of differential expression, predicted influenced modules and the enriched functions of those modules are indicated. Numbers in parenthesis indicate the number of network-predicted metabolic targets included within each module.
{kind=link}
10.1186/s40064-015-1541-2 Expression dynamics for additional TRs. Each panel shows the expression dynamics for the early (a), mid (b) and late (c) responses of the TRs for which no significant match to a co-regulated module was found.
10.1186/s40064-015-1541-2 List of predicted genes with ρ > 1. List of transcripts downregulated during N starvation predicted to result in higher TAG to biomass ratio upon gene knockdown. For each gene, we present the computed ρ value, the co-regulated module membership, the time of module downregulation and the predicted TR(s) regulating such module(s).
10.1186/s40064-015-1541-2 List of metabolic TAG production related genes varying during N starvation and LAP. At least 30 metabolic genes that have a direct functional role in TAG production are differentially expressed during N starvation. Expression pattern of these metabolic targets in S starvation is much more similar than in P starved cells. N starvation co-regulated module membership and predicted TRs are also shown.
Contributor Information
Adrián López García de Lomana, Email: alomana@systemsbiology.org.
Sascha Schäuble, Email: sascha.schaeuble@uni-jena.de.
Jacob Valenzuela, Email: jvalenzu@systemsbiology.org.
Saheed Imam, Email: simam@systemsbiology.org.
Warren Carter, Email: wcarter@systemsbiology.org.
Damla D. Bilgin, Email: damla.bilgin@sapphireenergy.com
Christopher B. Yohn, Email: chris.yohn@sapphireenergy.com
Serdar Turkarslan, Email: sturkarslan@systemsbiology.org.
David J. Reiss, Email: dreiss@systemsbiology.org
Mónica V. Orellana, Email: morellana@systemsbiology.org
Nathan D. Price, Email: nprice@systemsbiology.org
Nitin S. Baliga, Phone: 206.732.1266, Email: nbaliga@sytemsbiology.org
References
- 1.Wijffels RH, Barbosa MJ. An outlook on microalgal biofuels. Science. 2010;329:796–799. doi: 10.1126/science.1189003. [DOI] [PubMed] [Google Scholar]
- 2.Gimpel JA, Specht EA, Georgianna DR, Mayfield SP. Advances in microalgae engineering and synthetic biology applications for biofuel production. Curr Opin Chem Biol. 2013;17:489–495. doi: 10.1016/j.cbpa.2013.03.038. [DOI] [PubMed] [Google Scholar]
- 3.Weers PMM, Gulati RD. Growth and reproduction of Daphnia galeata in response to changes in fatty acids, phosphorus, and nitrogen in Chlamydomonas reinhardtii. Limnol Oceanogr. 1997;42:1584–1589. doi: 10.4319/lo.1997.42.7.1584. [DOI] [Google Scholar]
- 4.Matthew T, Zhou W, Rupprecht J, Lim L, Thomas-Hall SR, Doebbe A, et al. The metabolome of Chlamydomonas reinhardtii following induction of anaerobic H2 production by sulfur depletion. J Biol Chem. 2009;284:23415–23425. doi: 10.1074/jbc.M109.003541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang ZT, Ullrich N, Joo S, Waffenschmidt S, Goodenough U. Algal lipid bodies: stress induction, purification, and biochemical characterization in wild-type and starchless Chlamydomonas reinhardtii. Eukaryot Cell. 2009;8:1856–1868. doi: 10.1128/EC.00272-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller R, Wu G, Deshpande RR, Vieler A, Gärtner K, Li X, et al. Changes in transcript abundance in Chlamydomonas reinhardtii following nitrogen deprivation predict diversion of metabolism. Plant Physiol. 2010;154:1737–1752. doi: 10.1104/pp.110.165159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moellering ER, Benning C. RNA interference silencing of a major lipid droplet protein affects lipid droplet size in Chlamydomonas reinhardtii. Eukaryot Cell. 2010;9:97–106. doi: 10.1128/EC.00203-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goodson C, Roth R, Wang ZT, Goodenough U. Structural correlates of cytoplasmic and chloroplast lipid body synthesis in Chlamydomonas reinhardtii and stimulation of lipid body production with acetate boost. Eukaryot Cell. 2011;10:1592–1606. doi: 10.1128/EC.05242-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kropat J, Hong-Hermesdorf A, Casero D, Ent P, Castruita M, Pellegrini M, et al. A revised mineral nutrient supplement increases biomass and growth rate in Chlamydomonas reinhardtii. Plant J. 2011;66:770–780. doi: 10.1111/j.1365-313X.2011.04537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fan J, Cui Y, Wan M, Wang W, Li Y. Lipid accumulation and biosynthesis genes response of the oleaginous Chlorella pyrenoidosa under three nutrition stressors. Biotechnol Biofuels. 2014;7:17. doi: 10.1186/1754-6834-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhekisheva M, Boussiba S, Khozin-Goldberg I, Zarka A, Cohen Z. Accumulation of oleic acid in Haematococcus pluvialis (Chlorophyceae) under nitrogen starvation or high light is correlation with that of astaxanthin esters. J Phycol. 2002;38:325–331. doi: 10.1046/j.1529-8817.2002.01107.x. [DOI] [Google Scholar]
- 12.Siaut M, Cuiné S, Cagnon C, Fessler B, Nguyen M, Carrier P, et al. Oil accumulation in the model green alga Chlamydomonas reinhardtii: characterization, variability between common laboratory strains and relationship with starch reserves. BMC Biotechnol. 2011;11:7. doi: 10.1186/1472-6750-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Min SK, Yoon GH, Joo JH, Sim SJ, Shin HS. Mechanosensitive physiology of Chlamydomonas reinhardtii under direct membrane distortion. Sci Rep. 2014;4:4675. doi: 10.1038/srep04675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boyle NR, Page MD, Liu B, Blaby IK, Casero D, Kropat J, et al. Three acyltransferases and nitrogen-responsive regulator are implicated in nitrogen starvation-induced triacylglycerol accumulation in Chlamydomonas. J Biol Chem. 2012;287:15811–15825. doi: 10.1074/jbc.M111.334052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blaby IK, Glaesener AG, Mettler T, Fitz-Gibbon ST, Gallaher SD, Liu B, et al. Systems-level analysis of nitrogen starvation-induced modifications of carbon metabolism in a Chlamydomonas reinhardtii starchless mutant. Plant Cell. 2013;25:4305–4323. doi: 10.1105/tpc.113.117580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goodenough U, Blaby I, Casero D, Gallaher SD, Goodson C, Johnson S, et al. The path to triacylglyceride obesity in the sta6 strain of Chlamydomonas reinhardtii. Eukaryot Cell. 2014;13:591–613. doi: 10.1128/EC.00013-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Longworth J, Noirel J, Pandhal J, Wright PC, Vaidyanathan S. HILIC- and SCX-based quantitative proteomics of Chlamydomonas reinhardtii during nitrogen starvation induced lipid and carbohydrate accumulation. J Proteome Res. 2012;11:5959–5971. doi: 10.1021/pr300692t. [DOI] [PubMed] [Google Scholar]
- 18.Lv H, Qu G, Qi X, Lu L, Tian C, Ma Y. Transcriptome analysis of Chlamydomonas reinhardtii during the process of lipid accumulation. Genomics. 2013;101:229–237. doi: 10.1016/j.ygeno.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 19.Wase N, Black PN, Stanley BA, DiRusso CC. Integrated quantitative analysis of nitrogen stress response in Chlamydomonas reinhardtii using metabolite and protein profiling. J Proteome Res. 2014;13:1373–1396. doi: 10.1021/pr400952z. [DOI] [PubMed] [Google Scholar]
- 20.Nguyen HM, Baudet M, Cuiné S, Adriano J-M, Barthe D, Billon E, et al. Proteomic profiling of oil bodies isolated from the unicellular green microalga Chlamydomonas reinhardtii: with focus on proteins involved in lipid metabolism. Proteomics. 2011;11:4266–4273. doi: 10.1002/pmic.201100114. [DOI] [PubMed] [Google Scholar]
- 21.Schmollinger S, Mühlhaus T, Boyle NR, Blaby IK, Casero D, Mettler T, et al. Nitrogen-sparing mechanisms in Chlamydomonas affect the transcriptome, the proteome, and photosynthetic metabolism. Plant Cell. 2014;26:1410–1435. doi: 10.1105/tpc.113.122523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bölling C, Fiehn O. Metabolite profiling of Chlamydomonas reinhardtii under nutrient deprivation. Plant Physiol. 2005;139:1995–2005. doi: 10.1104/pp.105.071589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Msanne J, Xu D, Konda AR, Casas-Mollano JA, Awada T, Cahoon EB, et al. Metabolic and gene expression changes triggered by nitrogen deprivation in the photoautotrophically grown microalgae Chlamydomonas reinhardtii and Coccomyxa sp. C-169. Phytochemistry. 2012;75:50–59. doi: 10.1016/j.phytochem.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 24.Lee DY, Park J-J, Barupal DK, Fiehn O. System response of metabolic networks in Chlamydomonas reinhardtii to total available ammonium. Mol Cell Proteomics. 2012;11:973–988. doi: 10.1074/mcp.M111.016733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.González-Ballester D, Casero D, Cokus S, Pellegrini M, Merchant SS, Grossman AR. RNA-seq analysis of sulfur-deprived Chlamydomonas cells reveals aspects of acclimation critical for cell survival. Plant Cell. 2010;22:2058–2084. doi: 10.1105/tpc.109.071167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wykoff DD, Grossman AR, Weeks DP, Usuda H, Shimogawara K. Psr1, a nuclear localized protein that regulates phosphorus metabolism in Chlamydomonas. Proc Natl Acad Sci USA. 1999;96: 5336–15341. Available: http://www.ncbi.nlm.nih.gov/pubmed/10611385. [DOI] [PMC free article] [PubMed]
- 27.Cakmak T, Angun P, Demiray YE, Ozkan AD, Elibol Z, Tekinay T. Differential effects of nitrogen and sulfur deprivation on growth and biodiesel feedstock production of Chlamydomonas reinhardtii. Biotechnol Bioeng. 2012;109:1947–1957. doi: 10.1002/bit.24474. [DOI] [PubMed] [Google Scholar]
- 28.Iwai M, Ikeda K, Shimojima M, Ohta H. Enhancement of extraplastidic oil synthesis in Chlamydomonas reinhardtii using a type-2 diacylglycerol acyltransferase with a phosphorus starvation-inducible promoter. Plant Biotechnol J. 2014;12:808–819. doi: 10.1111/pbi.12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Babu MM, Lang B, Aravind L. Methods to reconstruct and compare transcriptional regulatory networks. Methods Mol Biol. 2009;541:163–180. doi: 10.1007/978-1-59745-243-4_8. [DOI] [PubMed] [Google Scholar]
- 30.De Smet R, Marchal K. Advantages and limitations of current network inference methods. Nat Rev Microbiol. 2010;8:717–729. doi: 10.1038/nrmicro2419. [DOI] [PubMed] [Google Scholar]
- 31.Marbach D, Costello JC, Küffner R, Vega NM, Prill RJ, Camacho DM, et al. Wisdom of crowds for robust gene network inference. Nat Methods. 2012;9:796–804. doi: 10.1038/nmeth.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reiss DJ, Baliga NS, Bonneau R. Integrated biclustering of heterogeneous genome-wide datasets for the inference of global regulatory networks. BMC Bioinform. 2006;7:280. doi: 10.1186/1471-2105-7-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonneau R, Facciotti MT, Reiss DJ, Schmid AK, Pan M, Kaur A, et al. A predictive model for transcriptional control of physiology in a free living cell. Cell. 2007;131:1354–1365. doi: 10.1016/j.cell.2007.10.053. [DOI] [PubMed] [Google Scholar]
- 34.Plaisier CL, Pan M, Baliga NS. A miRNA-regulatory network explains how dysregulated miRNAs perturb oncogenic processes across diverse cancers. Genome Res. 2012;22:2302–2314. doi: 10.1101/gr.133991.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoon SH, Turkarslan S, Reiss DJ, Pan M, Burn JA, Costa KC, et al. A systems level predictive model for global gene regulation of methanogenesis in a hydrogenotrophic methanogen. Genome Res. 2013;23:1839–1851. doi: 10.1101/gr.153916.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peterson EJR, Reiss DJ, Turkarslan S, Minch KJ, Rustad T, Plaisier CL, et al. A high-resolution network model for global gene regulation in Mycobacterium tuberculosis. Nucleic Acids Res. 2014;42:11291–11303. doi: 10.1093/nar/gku777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Danziger SA, Ratushny AV, Smith JJ, Saleem RA, Wan Y, Arens CE, et al. Molecular mechanisms of system responses to novel stimuli are predictable from public data. Nucleic Acids Res. 2014;42:1442–1460. doi: 10.1093/nar/gkt938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brooks AN, Reiss DJ, Allard A, Wu W-J, Salvanha DM, Plaisier CL, et al. A system-level model for the microbial regulatory genome. Mol Syst Biol. 2014;10:740. doi: 10.15252/msb.20145160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Orth JD, Thiele I, Palsson BØ. What is flux balance analysis? Nat Biotechnol. 2010;28:245–248. doi: 10.1038/nbt.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trinh CT, Srienc F. Metabolic engineering of Escherichia coli for efficient conversion of glycerol to ethanol. Appl Environ Microbiol. 2009;75:6696–6705. doi: 10.1128/AEM.00670-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lewis NE, Hixson KK, Conrad TM, Lerman JA, Charusanti P, Polpitiya AD, et al. Omic data from evolved E. coli are consistent with computed optimal growth from genome-scale models. Mol Syst Biol. 2010;6:390. doi: 10.1038/msb.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Curran KA, Crook NC, Alper HS. Using flux balance analysis to guide microbial metabolic engineering. Methods Mol Biol. 2012;834:197–216. doi: 10.1007/978-1-61779-483-4_13. [DOI] [PubMed] [Google Scholar]
- 43.Kaleta C, Schäuble S, Rinas U, Schuster S. Metabolic costs of amino acid and protein production in Escherichia coli. Biotechnol J. 2013;8:1105–1114. doi: 10.1002/biot.201200267. [DOI] [PubMed] [Google Scholar]
- 44.Agren R, Otero JM, Nielsen J. Genome-scale modeling enables metabolic engineering of Saccharomyces cerevisiae for succinic acid production. J Ind Microbiol Biotechnol. 2013;40:735–747. doi: 10.1007/s10295-013-1269-3. [DOI] [PubMed] [Google Scholar]
- 45.Khodayari A, Chowdhury A, Maranas CD. Succinate overproduction: a case study of computational strain design using a comprehensive Escherichia coli kinetic model. Front Bioeng Biotechnol. 2014;2:76. doi: 10.3389/fbioe.2014.00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chandrasekaran S, Price ND. Probabilistic integrative modeling of genome-scale metabolic and regulatory networks in Escherichia coli and Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2010;107:17845–17850. doi: 10.1073/pnas.1005139107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang RL, Ghamsari L, Manichaikul A, Hom EFY, Balaji S, Fu W, et al. Metabolic network reconstruction of Chlamydomonas offers insight into light-driven algal metabolism. Mol Syst Biol. 2011;7:518. doi: 10.1038/msb.2011.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Merchant SS, Prochnik SE, Vallon O, Harris EH, Karpowicz SJ, Witman GB, et al. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science. 2007;318:245–250. doi: 10.1126/science.1143609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;40:D1178–D1186. doi: 10.1093/nar/gkr944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, et al. STRING 8–a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37:D412–D416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chlamy Network Portal [Internet]. 2015. Available: http://networks.systemsbiology.net/chlamy-portal/.
- 52.Giordano M, Norici A, Forssen M, Eriksson M, Raven JA. An anaplerotic role for mitochondrial carbonic anhydrase in Chlamydomonas reinhardtii. Plant Physiol. 2003;132:2126–2134. doi: 10.1104/pp.103.023424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Duanmu D, Miller AR, Horken KM, Weeks DP, Spalding MH. Knockdown of limiting-CO2-induced gene HLA3 decreases HCO3− transport and photosynthetic Ci affinity in Chlamydomonas reinhardtii. Proc Natl Acad Sci USA. 2009;106:5990–5995. doi: 10.1073/pnas.0812885106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nemhauser JL, Mockler TC, Chory J. Interdependency of brassinosteroid and auxin signaling in Arabidopsis. PLoS Biol. 2004;2:E258. doi: 10.1371/journal.pbio.0020258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim SH, Arnold D, Lloyd A, Roux SJ. Antisense expression of an Arabidopsis ran binding protein renders transgenic roots hypersensitive to auxin and alters auxin-induced root growth and development by arresting mitotic progress. Plant Cell. 2001;13: 2619–2630. Available: http://www.ncbi.nlm.nih.gov/pubmed/11752376. [DOI] [PMC free article] [PubMed]
- 56.Baek GH, Cheng H, Choe V, Bao X, Shao J, Luo S, et al. Cdc48: a swiss army knife of cell biology. J Amino Acids. 2013;2013:183421. doi: 10.1155/2013/183421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Labunskyy VM, Hatfield DL, Gladyshev VN. The Sep15 protein family: roles in disulfide bond formation and quality control in the endoplasmic reticulum. IUBMB Life. 2007;59:1–5. doi: 10.1080/15216540601126694. [DOI] [PubMed] [Google Scholar]
- 58.Fink AL. Chaperone-mediated protein folding. Physiol Rev. 1999;79: 425–449. Available: http://www.ncbi.nlm.nih.gov/pubmed/10221986. [DOI] [PubMed]
- 59.Kim S, Kim H, Ko D, Yamaoka Y, Otsuru M, Kawai-Yamada M, et al. Rapid induction of lipid droplets in Chlamydomonas reinhardtii and Chlorella vulgaris by Brefeldin A. PLoS One. 2013;8:e81978. doi: 10.1371/journal.pone.0081978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo L, Ma F, Wei F, Fanella B, Allen DK, Wang X. Cytosolic phosphorylating glyceraldehyde-3-phosphate dehydrogenases affect Arabidopsis cellular metabolism and promote seed oil accumulation. Plant Cell. 2014;26:3023–3035. doi: 10.1105/tpc.114.126946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dal Santo S, Stampfl H, Krasensky J, Kempa S, Gibon Y, Petutschnig E, et al. Stress-induced GSK3 regulates the redox stress response by phosphorylating glucose-6-phosphate dehydrogenase in Arabidopsis. Plant Cell. 2012;24: 3380–3392. doi:10.1105/tpc.112.101279. [DOI] [PMC free article] [PubMed]
- 62.Wakao S, Andre C, Benning C. Functional analyses of cytosolic glucose-6-phosphate dehydrogenases and their contribution to seed oil accumulation in Arabidopsis. Plant Physiol. 2008;146:277–288. doi: 10.1104/pp.107.108423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fan J, Andre C, Xu C. A chloroplast pathway for the de novo biosynthesis of triacylglycerol in Chlamydomonas reinhardtii. FEBS Lett. 2011;585:1985–1991. doi: 10.1016/j.febslet.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 64.Darmoise A, Maschmeyer P, Winau F. The immunological functions of saposins. Adv Immunol. 2010;105:25–62. doi: 10.1016/S0065-2776(10)05002-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ke J, Behal RH, Back SL, Nikolau BJ, Wurtele ES, Oliver DJ. The role of pyruvate dehydrogenase and acetyl-coenzyme A synthetase in fatty acid synthesis in developing Arabidopsis seeds. Plant Physiol. 2000;123: 497–508. Available: http://www.ncbi.nlm.nih.gov/pubmed/10859180. [DOI] [PMC free article] [PubMed]
- 66.Liang C, Cao S, Zhang X, Zhu B, Su Z, Xu D, et al. De novo sequencing and global transcriptome analysis of Nannochloropsis sp. (Eustigmatophyceae) following nitrogen starvation. Bioenergy Res. 2012;6:494–505. doi: 10.1007/s12155-012-9269-0. [DOI] [Google Scholar]
- 67.Li J, Han D, Wang D, Ning K, Jia J, Wei L, et al. Choreography of transcriptomes and lipidomes of Nannochloropsis reveals the mechanisms of oil synthesis in microalgae. Plant Cell. 2014;26:1645–1665. doi: 10.1105/tpc.113.121418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Valenzuela J, Mazurie A, Carlson RP, Gerlach R, Cooksey KE, Peyton BM, et al. Potential role of multiple carbon fixation pathways during lipid accumulation in Phaeodactylum tricornutum. Biotechnol Biofuels. 2012;5:40. doi: 10.1186/1754-6834-5-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ge F, Huang W, Chen Z, Zhang C, Xiong Q, Bowler C, et al. Methylcrotonyl-CoA carboxylase regulates triacylglycerol accumulation in the model diatom Phaeodactylum tricornutum. Plant Cell. 2014;26:1681–1697. doi: 10.1105/tpc.114.124982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Valledor L, Furuhashi T, Recuenco-Muñoz L, Wienkoop S, Weckwerth W. System-level network analysis of nitrogen starvation and recovery in Chlamydomonas reinhardtii reveals potential new targets for increased lipid accumulation. Biotechnol Biofuels. 2014;7:171. doi: 10.1186/s13068-014-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bourgis F, Kilaru A, Cao X, Ngando-Ebongue G-F, Drira N, Ohlrogge JB, et al. Comparative transcriptome and metabolite analysis of oil palm and date palm mesocarp that differ dramatically in carbon partitioning. Proc Natl Acad Sci USA. 2011;108:12527–12532. doi: 10.1073/pnas.1106502108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Trentacoste EM, Shrestha RP, Smith SR, Glé C, Hartmann AC, Hildebrand M, et al. Metabolic engineering of lipid catabolism increases microalgal lipid accumulation without compromising growth. Proc Natl Acad Sci USA. 2013;110:19748–19753. doi: 10.1073/pnas.1309299110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yao Y, Lu Y, Peng K-T, Huang T, Niu Y-F, Xie W-H, et al. Glycerol and neutral lipid production in the oleaginous marine diatom Phaeodactylum tricornutum promoted by overexpression of glycerol-3-phosphate dehydrogenase. Biotechnol Biofuels. 2014;7:110. doi: 10.1186/1754-6834-7-110. [DOI] [Google Scholar]
- 74.Vigeolas H, Waldeck P, Zank T, Geigenberger P. Increasing seed oil content in oil-seed rape (Brassica napus L.) by over-expression of a yeast glycerol-3-phosphate dehydrogenase under the control of a seed-specific promoter. Plant Biotechnol J. 2007;5:431–441. doi: 10.1111/j.1467-7652.2007.00252.x. [DOI] [PubMed] [Google Scholar]
- 75.Li X, Benning C, Kuo M-H. Rapid triacylglycerol turnover in Chlamydomonas reinhardtii requires a lipase with broad substrate specificity. Eukaryot Cell. 2012;11:1451–1462. doi: 10.1128/EC.00268-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li X, Moellering ER, Liu B, Johnny C, Fedewa M, Sears BB, et al. A galactoglycerolipid lipase is required for triacylglycerol accumulation and survival following nitrogen deprivation in Chlamydomonas reinhardtii. Plant Cell. 2012;24:4670–4686. doi: 10.1105/tpc.112.105106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Juergens MT, Deshpande RR, Lucker BF, Park J-J, Wang H, Gargouri M, et al. The regulation of photosynthetic structure and function during nitrogen deprivation in Chlamydomonas reinhardtii. Plant Physiol. 2015;167:558–573. doi: 10.1104/pp.114.250530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chardin C, Girin T, Roudier F, Meyer C, Krapp A. The plant RWP-RK transcription factors: key regulators of nitrogen responses and of gametophyte development. J Exp Bot. 2014;65:5577–5587. doi: 10.1093/jxb/eru261. [DOI] [PubMed] [Google Scholar]
- 79.Gargouri M, Park J-J, Holguin FO, Kim M-J, Wang H, Deshpande RR, et al. Identification of regulatory network hubs that control lipid metabolism in Chlamydomonas reinhardtii. J Exp Bot. 2015;66:4551–4566. doi: 10.1093/jxb/erv217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Siefers N, Dang KK, Kumimoto RW, Bynum WE, 4th, Tayrose G, Holt BF., 3rd Tissue-specific expression patterns of Arabidopsis NF-Y transcription factors suggest potential for extensive combinatorial complexity. Plant Physiol. 2009;149:625–641. doi: 10.1104/pp.108.130591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhou DX, Bisanz-Seyer C, Mache R. Molecular cloning of a small DNA binding protein with specificity for a tissue-specific negative element within the rps1 promoter. Nucleic Acids Res. 1995;23: 1165–1169. Available: http://www.ncbi.nlm.nih.gov/pubmed/7739894. [DOI] [PMC free article] [PubMed]
- 82.Mussgnug JH, Wobbe L, Elles I, Claus C, Hamilton M, Fink A, et al. NAB1 is an RNA binding protein involved in the light-regulated differential expression of the light-harvesting antenna of Chlamydomonas reinhardtii. Plant Cell. 2005;17:3409–3421. doi: 10.1105/tpc.105.035774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen M, Thelen JJ. The plastid isoform of triose phosphate isomerase is required for the postgerminative transition from heterotrophic to autotrophic growth in Arabidopsis. Plant Cell. 2010;22:77–90. doi: 10.1105/tpc.109.071837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rocha AG, Mehlmer N, Stael S, Mair A, Parvin N, Chigri F, et al. Phosphorylation of Arabidopsis transketolase at Ser 428 provides a potential paradigm for the metabolic control of chloroplast carbon metabolism. Biochem J. 2014;458:313–322. doi: 10.1042/BJ20130631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Song P, Li L, Liu J. Proteomic analysis in nitrogen-deprived Isochrysis galbana during lipid accumulation. PLoS One. 2013;8:e82188. doi: 10.1371/journal.pone.0082188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.The Gene Ontology Consortium Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–D1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hu J, Wang D, Li J, Jing G, Ning K, Xu J. Genome-wide identification of transcription factors and transcription-factor binding sites in oleaginous microalgae Nannochloropsis. Sci Rep. 2014;4:5454. doi: 10.1038/srep05454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Maier T, Güell M, Serrano L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009;583:3966–3973. doi: 10.1016/j.febslet.2009.10.036. [DOI] [PubMed] [Google Scholar]
- 89.Gunawardana Y, Niranjan M. Bridging the gap between transcriptome and proteome measurements identifies post-translationally regulated genes. Bioinformatics. 2013;29:3060–3066. doi: 10.1093/bioinformatics/btt537. [DOI] [PubMed] [Google Scholar]
- 90.Imam S, Schäuble S, Valenzuela J, Garcia Lopez, de Lomana A, Carter W, Price ND, et al. A refined genome-scale reconstruction of Chlamydomonas metabolism provides a platform for systems-level analyses. Plant J. 2015 doi: 10.1111/tpj.13059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhao T, Li G, Mi S, Li S, Hannon GJ, Wang X-J, et al. A complex system of small RNAs in the unicellular green alga Chlamydomonas reinhardtii. Genes Dev. 2007;21:1190–1203. doi: 10.1101/gad.1543507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Molnár A, Schwach F, Studholme DJ, Thuenemann EC, Baulcombe DC. miRNAs control gene expression in the single-cell alga Chlamydomonas reinhardtii. Nature. 2007;447:1126–1129. doi: 10.1038/nature05903. [DOI] [PubMed] [Google Scholar]
- 93.Molnar A, Bassett A, Thuenemann E, Schwach F, Karkare S, Ossowski S, et al. Highly specific gene silencing by artificial microRNAs in the unicellular alga Chlamydomonas reinhardtii. Plant J. 2009;58:165–174. doi: 10.1111/j.1365-313X.2008.03767.x. [DOI] [PubMed] [Google Scholar]
- 94.Shu L, Hu Z. Characterization and differential expression of microRNAs elicited by sulfur deprivation in Chlamydomonas reinhardtii. BMC Genom. 2012;13:108. doi: 10.1186/1471-2164-13-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Voshall A, Kim E-J, Ma X, Moriyama EN, Cerutti H. Identification of AGO3-associated miRNAs and computational prediction of their targets in the green alga Chlamydomonas reinhardtii. Genetics. 2015;200:105–121. doi: 10.1534/genetics.115.174797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Del Campo EM. Post-transcriptional control of chloroplast gene expression. Gene Regul Syst Bio. 2009;3: 31–47. Available: http://www.ncbi.nlm.nih.gov/pubmed/19838333. [DOI] [PMC free article] [PubMed]
- 97.Winck FV, Vischi Winck F, Arvidsson S, Riaño-Pachón DM, Hempel S, Koseska A, et al. Genome-wide identification of regulatory elements and reconstruction of gene regulatory networks of the green alga Chlamydomonas reinhardtii under carbon deprivation. PLoS One. 2013;8:e79909. doi: 10.1371/journal.pone.0079909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sullivan AM, Arsovski AA, Lempe J, Bubb KL, Weirauch MT, Sabo PJ, et al. Mapping and dynamics of regulatory DNA and transcription factor networks in A. thaliana. Cell Rep. 2014;8:2015–2030. doi: 10.1016/j.celrep.2014.08.019. [DOI] [PubMed] [Google Scholar]
- 99.Ratcliff WC, Herron MD, Howell K, Pentz JT, Rosenzweig F, Travisano M. Experimental evolution of an alternating uni- and multicellular life cycle in Chlamydomonas reinhardtii. Nat Commun. 2013;4:2742. doi: 10.1038/ncomms3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Perrineau M-M, Gross J, Zelzion E, Price DC, Levitan O, Boyd J, et al. Using natural selection to explore the adaptive potential of Chlamydomonas reinhardtii. PLoS One. 2014;9:e92533. doi: 10.1371/journal.pone.0092533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Velmurugan N, Sung M, Yim SS, Park MS, Yang JW, Jeong KJ. Systematically programmed adaptive evolution reveals potential role of carbon and nitrogen pathways during lipid accumulation in Chlamydomonas reinhardtii. Biotechnol Biofuels. 2014;7:117. doi: 10.1186/s13068-014-0117-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jiang W, Brueggeman AJ, Horken KM, Plucinak TM, Weeks DP. Successful transient expression of Cas9 and single guide RNA genes in Chlamydomonas reinhardtii. Eukaryot Cell. 2014;13:1465–1469. doi: 10.1128/EC.00213-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pérez-Ortín JE, Alepuz PM, Moreno J. Genomics and gene transcription kinetics in yeast. Trends Genet. 2007;23:250–257. doi: 10.1016/j.tig.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 106.Milo R, Jorgensen P, Moran U, Weber G, Springer M. BioNumbers–the database of key numbers in molecular and cell biology. Nucleic Acids Res. 2010;38:D750–D753. doi: 10.1093/nar/gkp889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mor A, Suliman S, Ben-Yishay R, Yunger S, Brody Y, Shav-Tal Y. Dynamics of single mRNP nucleocytoplasmic transport and export through the nuclear pore in living cells. Nat Cell Biol. 2010;12:543–552. doi: 10.1038/ncb2056. [DOI] [PubMed] [Google Scholar]
- 108.Siwiak M, Zielenkiewicz P. A comprehensive, quantitative, and genome-wide model of translation. PLoS Comput Biol. 2010;6:e1000865. doi: 10.1371/journal.pcbi.1000865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Olofsson SO, Boström K, Carlsson P, Borén J, Wettesten M, Bjursell G, et al. Structure and biosynthesis of apolipoprotein B. Am Heart J. 1987;113: 446–452. Available: http://www.ncbi.nlm.nih.gov/pubmed/3812204. [DOI] [PubMed]
- 110.Siwiak M, Zielenkiewicz P. Transimulation—protein biosynthesis web service. PLoS One. 2013;8:e73943. doi: 10.1371/journal.pone.0073943. [DOI] [PMC free article] [PubMed] [Google Scholar]