

Abstract
Background
Presynaptic terminals contribute to cognitive reserve, balancing the effects of age-related pathologies on cognitive function in the elderly. The presynaptic protein Munc18-1, alternatively spliced into long (M18L) or short (M18S) isoforms, is a critical modulator of neurotransmission. While subtle alterations in Munc18-1 have been shown to cause severe neuropsychiatric disorders with cognitive impairment, little information is known regarding the specific roles of Munc18-1 splice variants. We first investigated functional and anatomical features evidencing the divergent roles of M18L and M18S, and then evaluated their contribution to the full range of age-related cognitive impairment in the dorsolateral prefrontal cortex of a large sample of participants from a community-based aging study, including subjects with no-(NCI, n = 90), or mild-(MCI, n = 86) cognitive impairment, or with clinical dementia (n = 132). Finally, we used APP23 mutant mice to study the association between M18L/S and the time-dependent accumulation of common Alzheimer’s disease pathology.
Results
Using isoform-specific antibodies, M18L was localized to the synaptosomal fraction, with a distribution matching lipid raft microdomains. M18S was found widely across cytosolic and synaptosomal compartments. Immunocytochemical studies identified M18L in perisomatic, GABAergic terminals, while M18S was broadly distributed in GABAergic and glutamatergic terminals. Using regression models taking into account multiple age-related pathologies, age, education and sex, global cognitive function was associated with the level of M18L (p = 0.006) but not M18S (p = 0.88). Mean M18L in dementia cases was 51 % lower than in NCI cases (p < 0.001), and each unit of M18L was associated with a lower likelihood of dementia (odds ratio = 0.68, 95 % confidence interval = 0.50–0.90, p = 0.008). In contrast, M18S balanced across clinical and pathologically diagnosed groups. M18L loss may not be caused by age-related amyloid pathology, since APP23 mice (12- and 22-months of age) had unchanged cortical levels of M18L/S compared with wild-type animals.
Conclusions
M18L was localized to presynaptic inhibitory terminals, and was associated with cognitive function and protection from dementia in an elderly, community-based cohort. Lower M18L in inhibitory presynaptic terminals may be an early, independent contributor to cognitive decline.
Electronic supplementary material
The online version of this article (doi:10.1186/s13024-015-0061-4) contains supplementary material, which is available to authorized users.
Keywords: Syntaxin-binding protein, SNARE, Protein-protein interactions, VGAT, VGLUT1, Human postmortem brain, Aging study, Mild cognitive impairment, Synaptic pathology, Alzheimer’s disease
Background
Common age-related neuropathologies, including Alzheimer’s disease pathology (i.e. neuritic plaques and neurofibrillary tangles), cerebral infarcts, and Lewy bodies, accumulate in the human brain across the lifetime. The susceptibility of cognitive function to decline as growing amounts of neuropathology develop in the forebrain varies greatly between individuals, raising the concept of cognitive reserve [1–3]. Large community-based studies have been designed, at least in part, to investigate the biological substrates of cognitive reserve [4–7]. The initial model that people with mentally enriched lives may exhibit greater resistance against brain-damaging diseases [2] is not yet supported with clear neurochemical correlates. Synaptic pathology is common in aging, being first reported as a deficit of presynaptic markers [8–11]. In some studies, synaptic pathology showed stronger association with cognitive decline than other common age-related pathologies [12], suggesting that intact, functional synapses may contribute to cognitive reserve.
In this context, we previously reported that better cognitive performance, a reduced odds of dementia, and slower rate of cognitive decline over multiple years prior to death were associated with greater densities of specific presynaptic proteins, and enhanced presynaptic protein-protein interactions (PPIs) [13, 14]. Interestingly, these PPIs involved the associations between syntaxin-1, synaptosomal-associated protein-25 (SNAP-25), and vesicle-associated membrane protein (VAMP, or synaptobrevin), which together assemble the machinery facilitating vesicle trafficking and neurotransmitter release, known as the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) [15]. However, other frequent targets of synaptic pathology, such as synaptophysin, were not associated with cognitive function or Alzheimer’s disease pathology in the same community-based cohort study, which included subjects with the full range of cognition from normality, to mild cognitive impairment, to dementia. Of note, many of the studies related to synaptic pathology were based on subpopulations of hospital- or clinic-based patients displaying moderate-to-severe degrees of dementia, not necessarily representative of the spectrum of cognitive decline in aging [16]. Previous analyses in mixed postmortem brain series found biphasic alterations of brain presynaptic markers (including synaptophysin) across age-related cognitive decline and/or Alzheimer’s disease progression, with upregulated densities at intermediate stages and reduced levels in more severe cases [17–19]. Together, these observations suggest that an initial failure of presynaptic terminals to maintain synaptic function (reflected by SNARE disruption) might be compensated by upregulating the expression of certain presynaptic genes; further accumulation of neuropathologies may result in the observed synaptic degeneration at relatively more severe stages. The mechanisms underlying initial SNARE and presynaptic dysfunction could be directly linked to age-related cognitive impairment and progression to dementia. Consistent with these observations, murine models with impaired SNARE PPIs undergo related behavioral and physiological neurodegenerative syndromes [20, 21].
Although the biochemical and physiological nature of neurosecretion is well established [22–24], the association between SNARE dysregulation and the pathophysiology of aging-related diseases is unclear. Among the regulators potentially involved in SNARE dysfunction, mammal unc-18-1 (Munc18-1 or n-Sec1; herein M18) is a strong candidate. First identified by random mutagenesis screening for uncoordinated phenotypes in the worm Caenorhabditis elegans, M18 caught the attention of many research teams when described as a ‘syntaxin-binding protein’ (coining the M18 coding gene as STXBP1) twenty years later [25–27]. While still controversial, M18 is believed to have multiple activities during SNARE-regulated exocytosis, which may include trafficking and chaperoning syntaxin-1, transition from cis-to-trans SNARE conformations, and/or docking the presynaptic vesicle [23, 28, 29]. The observation that STXBP1 null mice lack neocortical synaptic activity [30] highlights the irreplaceable role of M18 for neurosecretion. Interestingly, M18 knockout mice also exhibit massive neuronal apoptosis and widespread neurodegeneration, and consequently do not survive after birth [30].
In mammals, the STXBP1 gene is alternatively spliced to yield either a long (M18L) or a short (M18S) variant [31, 32], also called isoforms a and b, respectively. Processing of the final exon in the STXBP1 primary transcript may include (M18L) or skip (M18S) a sequence of 110 bp containing a stop codon [32], resulting in two different C-terminal amino acid sequences for M18L/S (see Fig. 1a). To our knowledge, specific regulatory mechanisms of STXBP1 gene splicing have not been described. Although intensive research focuses on M18 activities in regulated exocytosis, the potentially divergent roles of the M18 variants are relatively neglected, assuming no substantial differences between them [32]. However, recent observations indicate that M18 variants may not have overlapping functions. Transgenic mice expressing a fluorophore-tagged, functional M18S were generated using a replacement strategy that erased the M18L splice variant [33, 34]. These animals show severe physical and behavioral deficits, and die prematurely a few weeks after birth. Conversely, mice overexpressing M18L in both glutamatergic and GABAergic neurons display aspects of a schizophrenia-like phenotype [35], compatible with alterations observed in human postmortem studies [36–38]. At a cellular level, transfection of either M18L or M18S to excitatory neuronal cell cultures from the hippocampus of M18 knockouts succeeded to a similar extent in rescuing basal synaptic activity [39]. However, M18S appeared to support high frequency stimulation more efficiently than the long variant, suggesting different roles in short-term plasticity. Altogether, these findings indicate that the functions of M18 splice variants may not be interchangeable, and the multiplicity of activities attributed to M18 may represent a composite of M18L + S.
Fig. 1.
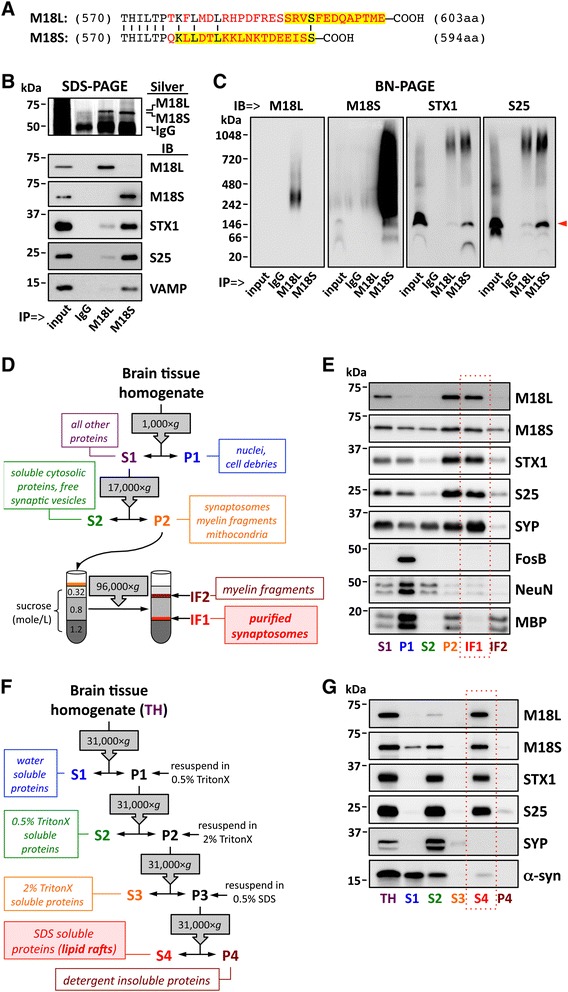
Biochemical characterization of M18 splice variants. a Alignment of M18L/S C-terminal amino acid (aa) sequences. Mismatching residues are in red. Amino acids highlighted (in yellow) represent the immunogenic sequences used for production of variant-specific antibodies. Omitted M18L/S N-terminal sequences are 100 % identical. b–c Immunoprecipitation (IP) of M18L/S with variant specific antibodies (and anti-mouse IgG as a negative control) using human brain homogenates. NCI control subjects (n = 3) were tested with similar results. IP products, along with input samples and negative controls (IgG), were resolved by (b) standard SDS- or (c) BN-PAGE, followed by either silver staining (Silver) or immunoblotting (IB) with specific antibodies against M18 variants, syntaxin-1 (STX1), SNAP-25 (S25), or VAMP. c M18S antibody recognized two bands at ~150 and ~70 kDa, putatively corresponding to a SNARE-M18 heterotetramer (pointed with a red arrowhead) and the monomeric form, respectively. d, f Schematic illustration depicting the sequential extraction of (d) synaptosomes and (f) lipid rafts from human cortical homogenates. g, relative centrifugal forces; IF, interface; P, pellet; S, supernatant. e Equivolumetric amounts of fractions obtained in (d) were resolved by SDS-PAGE and immunoblotted using antibodies targeting M18L/S, STX1, S25, VAMP, and markers for synaptic vesicles (synaptophysin [SYP]), nuclei (FosB), nuclei + cytosol (NeuN), and myelin fragments (myelin basic protein [MBP]). Note that MBP strongly labels P1, as heavy myelin fragments precipitate along with cell debris. g Equivolumetric amounts of fractions obtained in (f) were resolved by SDS-PAGE and immunoblotted using the antibodies above. α-Synuclein (α-syn) antibody was additionally used, showing similar distribution across fractions as previously reported [66]. e, g IF1 and S4 fractions were framed in a red, dashed box to highlight synaptosome and lipid raft-enriched proteins, respectively. b–g Masses (in kDa) of prestained markers are indicated on the left side of immunoblots
It has been reported that de novo mutations affecting STXBP1 gene can cause Ohtahara syndrome in humans [40–45], a devastating neurological disease characterized by early onset of epileptic seizures and a profound intellectual disability. Most STXBP1 mutations associated with Ohtahara syndrome impair M18–syntaxin-1 interaction (and thereby proper SNARE assembly), compromising cellular availability of functional M18 (haploinsufficiency) [41, 43]. Conversely, other mutations are predicted to confer aberrant alternative splicing of M18 RNA [44]. In addition, some STXBP1 de novo mutations were associated with similar mental retardation without epileptic seizures [42].
In the context of aging, two previous studies addressed alterations in M18 protein levels in Alzheimer’s disease postmortem brain. In the first, M18 immunodensity was lower in two cortical areas in n = 32 Alzheimer’s disease cases, although after synaptophysin normalization, the authors concluded that productive synapses in Alzheimer’s disease brains might be enriched in M18 [46]. A later proteomic study [47] found increased M18 levels in cell membrane extractions from cortical samples of n = 5 Alzheimer’s disease cases. These two studies did not examine the correlation of M18 with cognitive outcomes, or with the likelihood of dementia; nor were potential differences in M18 splice variants assessed. In addition, the small sample sizes limited the possibility of investigating possible effects of stage of illness, or potential confounders.
Altogether, clinical and preclinical data demonstrated that M18 is essential for neurosecretion, subtle alterations have a dramatic impact on normal synaptic function and cognition in humans and rodents, and could represent an early sign of synaptic pathology and cognitive impairment. For the present study we hypothesized that cortical downregulation of M18 long and/or short (M18L/S) splice variants could contribute to poorer cognitive performance and increased likelihood of dementia in old age. Due to the scarce data on specific functions of M18 isoforms, we first performed functional and anatomical characterization experiments that evidenced the complementary, rather than overlapping roles of M18L and M18S. Using tissue samples from the community-based Memory and Aging Project (MAP) [7], cortical expression of M18 variants was quantified in a large sample of MAP participants with and without clinical dementia, representing the range of cognitive impairment in an elderly, aging population. The potential effects of Alzheimer’s disease pathology on M18 levels were further modeled using APP23 transgenic mice, which overexpress a pathogenic variant of the human amyloid precursor protein (APP) [48].
Results
Biochemical and anatomical evidence indicates different roles of M18 splice variants
We first used immunoprecipitation (IP) followed by either sodium dodecyl sulfate (SDS) or blue native (BN) polyacrylamide gel electrophoresis (PAGE) to investigate functional binding affinities of M18 isoforms for the SNARE complex. As expected from their target immunogenic sequences (Fig. 1a), and also from previous data [36], variant-specific antibodies selectively immunoprecipitated M18L or M18S without cross-reacting, confirmed by immunoblot analyses (Fig. 1b). Surprisingly, anti-M18S IP products were 7–12 fold enriched in SNARE proteins (i.e. syntaxin-1, SNAP-25 and VAMP), compared to those of anti-M18L (Fig. 1b). This was not due to a lower affinity of the antibody for M18L, since comparable amounts of both isoforms were observed in silver stained gels (Fig. 1b). Rather, M18L displayed (under current assay conditions) a poorer ability to bind syntaxin-1. Accordingly, M18S formed a stable ~150-kDa complex with SNARE proteins, which was barely detected in M18L co-IP products, as revealed by BN-PAGE (Fig. 1c).
Substantial differences in the subcellular localization of M18 variants were also observed (Fig. 1d–g). While M18L was apparently restricted to the synaptosomal fraction (IF1), M18S showed the expected wide distribution across cytosolic (S2) and synaptosomal compartments (Fig. 1e). Furthermore, M18L was almost exclusively found within the Triton X-100 (TritonX)-insoluble, SDS-soluble protein fraction (S4), matching lipid raft microdomains, while M18S was present in water- and detergent-soluble subcellular compartments (Fig. 1g).
Immunohistochemical analyses of rat brain revealed a differential distribution of M18 variants, particularly in the hippocampus (Fig. 2a). M18L was highly abundant in the pyramidal cell layer of the Ammon's horn (CA) regions, as well as in the dentate gyrus granule cell layer. In marked contrast, M18S showed greatest immunostaining in pyramidal cell-flanking strata (with particularly strong labeling of the mossy fibers), but was barely present within the pyramidal layer. In cortical regions, M18L accumulated in neuronal perisomatic areas, whereas M18S showed the punctate staining of neuropil typically observed for presynaptic proteins (Fig. 2a). Despite their apparently distinct patterns of distribution (best characterized in CA3), confocal imaging showed some degree of overlap between splice variants in areas such as the granule cell layer (Fig. 2b). Of note, colocalization of M18 isoforms also overlapped with syntaxin-1. Similar results were observed in human hippocampus (Additional file 1: Figure S1A). In NeuN co-stained sections, preferential perisomatic accumulation of M18L was highlighted, and additional evidence of the cytosolic localization of M18S (but not M18L or syntaxin-1) obtained (Fig. 2c–d).
Fig. 2.
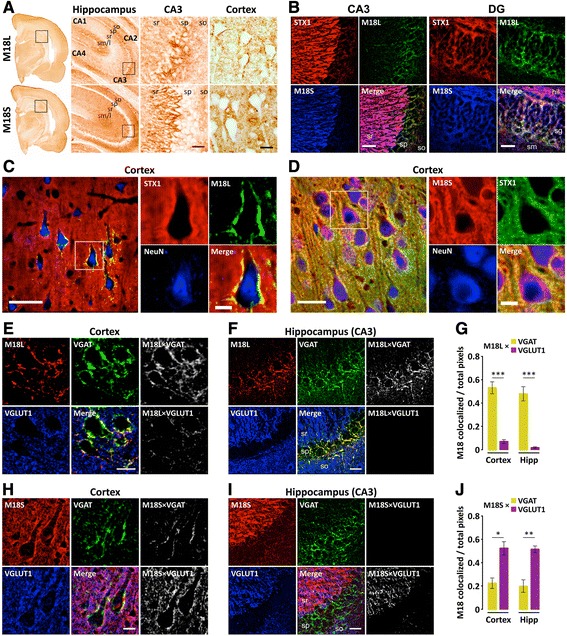
Immunohistochemical characterization of M18 splice variants in rat brain. (a) Photomicrographs showing M18L (upper panels) and M18S (bottom panels) immunostaining with variant specific antibodies at various magnifications. Hippocampus and CA3 are magnified captions from the framed areas at immediate left-side images. (b–j) Confocal images and analyses from triple co-immunolabeled sections with the antibodies indicated at the top-left corners. Colors were arbitrarily assigned to maximize overlap visualization. (e–j) Rat brain sections were co-immunolabeled with antibodies against vesicular GABA (VGAT) and glutamate (VGLUT1) transporters, along with (e–f) anti-M18L or (h–i) anti-M18S, and representative images from (e and h) cortical and (f and i) hippocampal CA3 are shown. In every 6-panel composite: left and middle panels correspond to single-labeled or merged-channel images; right panels are ImageJ-generated bitmaps resulting from colocalization analyses, in which pixels mirror the intensity of colocalization (in white) between VGAT (top) or VGLUT1 (bottom) and the corresponding M18 splice variant. (g and j) Quantitative colocalization analyses of (G) M18L or (J) M18S with each of the vesicular transporters (VGAT and VGLUT1) Bars represent mean ± standard error of n = 4 rats. *p < 0.05, **p < 0.01, and ***p < 0.001 (paired t-test). Abbreviations: DG, dentate gyrus; hil, hilus; Hipp, hippocampus sg; stratum granulosum (i.e. granule cell layer); sm/l, stratum moleculare/lacunosum; so, stratum oriens; sp, stratum pyramidale (i.e. pyralmidal cell layer); sr, stratum radiatum (in CA3 also contains stratum lucidum. Scale bars: a, 200 (CA3) or 20 (cortex) μm; b, 30 μm; c, 50 (left) or 10 (right) μm; d, 30 (left) or 10 (right) μm; e and h, 10 μm; f and i, 25 μm
Triple co-immunolabeling with antibodies against M18L, vesicle GABA (VGAT) and glutamate (VGLUT1) transporters showed a high degree of M18L localization within the inhibitory (VGAT-positive) synapses, with little (or possibly no) presence at excitatory (VGLUT1-positive) terminals, in both cortex and hippocampus (Fig. 2e–g). In contrast, overlap of M18S was greater with VGLUT1 than with VGAT (Fig. 3h–j), although this could be explained by the relative abundance of glutamatergic over GABAergic terminals. This difference was most remarkable in CA3, where a complete segregation of M18L and M18S with respectively, VGAT or VGLUT1 positive terminals was observed (Fig. 2f, i). In human hippocampus, similar distributions were seen (Additional file 1: Figure S1B–C).
Fig. 3.

Burden of Alzheimer’s disease pathology in MAP participants and relation to clinical diagnoses. a Composite Alzheimer’s disease (AD) pathology values were estimated for each participant and plotted by clinical diagnosis criteria into no- (NCI, n = 90), or mild-cognitive impairment (MCI, n = 86), or dementia (DEM, n = 132). Whiskers represent 10th and 90th percentiles and boxes enclose interquartile ranges crossed by the median of Alzheimer’s disease pathology scores within groups. As expected, Kruskal-Wallis test detected differences on the accumulated Alzheimer’s disease pathology across clinical diagnoses (KW-statistic = 57.0, p < 0.001; Mean rank differences: NCI vs MCI = 30.7; NCI vs DEM = 88.7; MCI vs DEM = 58.0). ns, not significant, ***p < 0.001, Kruskal-Wallis followed by Dunn’s multiple comparison test. b Bubble plot illustrating the distribution of MAP participants across clinically diagnosed (NCI/MCI/DEM) and neuropathologically graded [by NIA/Reagan, CERAD (Consortium to Establish a Registry for Alzheimer’s Disease), and Braak scales] groups
Characteristics of MAP participants and potential confounds
Demographic, cognitive and pathological variables of MAP participants are summarized in Table 1. No correlation was detected between M18 splice variants and potential confounders, including age at death, postmortem interval, years of education and brain weight. Similarly, M18L/S cortical immunodensity did not vary across sex, race, or ApoE genotype. Furthermore, tobacco and/or alcohol users did not present different M18L/S levels from those without these histories.
Table 1.
Demographic, cognitive and pathological characteristicsa of MAP participants included in the present study
| Variable | All participants (n = 308) | NCI (n = 90) | MCI (n = 86) | Dementia (n = 132) |
|---|---|---|---|---|
| Demographic | ||||
| Female, no. (%) | 194 (63 %) | 64 (71 %) | 48 (56 %) | 82 (62 %) |
| Age at death, years | 88.8 ± 6.0 | 86.8 ± 6.3 | 88.1 ± 6.4 | 90.7 ± 5.0 |
| Education, years | 14.4 ± 2.8 | 14.0 ± 2.4 | 14.7 ± 2.5 | 14.5 ± 3.2 |
| Race, no. W:AA:NA | 300:7:1 | 86:4:0 | 84:1:1 | 130:2:0 |
| APOE ε4 allele, no. (%) | 84 (27 %) | 15 (17 %) | 22 (26 %) | 47 (36 %) |
| PMI, hours | 7.2 ± 4.8 | 6.9 ± 4.4 | 8.1 ± 5.5 | 6.7 ± 4.6 |
| Cognitive function proximate to death | ||||
| Global cognition score | −0.85 ± 1.09 | 0.16 ± 0.41 | −0.43 ± 0.45 | −1.83 ± 0.88 |
| Episodic memory | −0.84 ± 1.19 | 0.34 ± 0.48 | −0.55 ± 0.70 | −1.84 ± 0.90 |
| Semantic memory | −0.70 ± 1.23 | 0.12 ± 0.56 | −0.19 ± 0.53 | −1.61 ± 1.31 |
| Working memory | −0.66 ± 1.14 | 0.16 ± 0.77 | −0.29 ± 0.79 | −1.50 ± 1.01 |
| Perceptual speed | −1.13 ± 1.13 | −0.28 ± 0.85 | −0.70 ± 0.88 | −2.02 ± 0.77 |
| Visuospatial ability | −0.63 ± 1.23 | 0.12 ± 0.62 | −0.16 ± 0.78 | −1.49 ± 1.29 |
| MMSE | 21.3 ± 8.8 | 28.1 ± 1.8 | 25.7 ± 3.8 | 13.6 ± 8.2 |
| Pathological | ||||
| NIA/Reagan scale, no. in 1:2:3:4b | 49:135:120:4 | 4:25:59:2 | 4:44:36:2 | 41:66:25:0 |
| CERAD scale, no. in 1:2:3:4c | 95:99:40:74 | 13:21:19:37 | 19:31:13:23 | 63:47:8:14 |
| Braak stage, no. in 0:I:II:III: | 4:20:33:91: | 2:14:16:33: | 2:2:14:28: | 0:4:3:30: |
| IV:V:VI | 88: 67:5 | 21:4:0 | 30:10:0 | 37:53:5 |
| Global AD pathology | 0.69 ± 0.62 | 0.38 ± 0.39 | 0.60 ± 0.57 | 0.97 ± 0.66 |
| Macroinfarcts, no. (%) | 108 (35 %) | 20 (22 %) | 29 (34 %) | 59 (45 %) |
| Microinfarcts, no. (%) | 71 (23 %) | 20 (22 %) | 14 (16 %) | 37 (28 %) |
| Lewy body disease, no. (%) | 27 (9 %) | 2 (2 %) | 4 (5 %) | 21 (16 %) |
| Hippocampal sclerosis, no. (%) | 26 (8 %) | 1 (1 %) | 5 (6 %) | 20 (15 %) |
Abbreviations: AA Afro-American, AD, Alzheimer’s disease, BL baseline, CERAD Consortium to establish a registry for AD, MCI mild cognitive impairment, MMSE mini mental state examination, NA Native-American, NCI no cognitive impairment, NIA National Institute on Aging, no number of subjects, PMI postmortem interval, SD standard deviation, W White
aValues are mean ± SD unless noted otherwise
bNIA/Reagan scale: high (1), intermediate (2), low (3), or no (4) likelihood of AD, according to neuritic plaques and tangles
cCERAD scale: definite (1), possible (2), probable (3), or no (4) AD, according to neuritic plaques
Similar numbers of clinically diagnosed MAP participants with no (NCI) or mild (MCI) cognitive impairment, and dementia were observed (Table 1). As expected, common Alzheimer’s disease pathology was largely abundant in dementia cases, compared to NCI and MCI participants (Fig. 3a), although some degree of variability was present in all clinically diagnosed groups, as documented previously in larger epidemiologic studies [5, 7]. Similarly, clinical diagnoses showed substantial, but not complete, overlap with CERAD and Braak rating scales (Fig. 3b).
Association of cortical M18L with cognitive function and dementia
We investigated possible alterations in the amounts of M18 splice variants, and their potential association with cognitive function, in samples from the dorsolateral prefrontal cortex (DLPFC) of MAP participants (n = 308). Initial inspection of quantitative immunoblotting datasets (normalized by β-actin content) revealed large variability in M18L immunodensities among MAP participants (interquartile range = 7–185 %), compared to that of M18S (86–122 %) or β-actin (85–117 %). The immunodensities of the M18 variants showed a non-linear association (Additional file 1: Figure S2A, Spearman rho = 0.53), best fitted by a semi-log curve (r = 0.56, p < 0.001). Due to the large number of values proximate to zero (see Additional file 1: Figure S2), and consequent skewness (skew = 1.712), the distribution of M18L values did not pass the Kolmogorov-Smirnov normality test. To construct linear models, we log-transformed M18L values, and then standardized (by subtracting the mean and dividing by the standard deviation) both variant datasets. Data transformations symmetrized the distribution of M18L values and rendered linear associations between the variants (r = 0.51, p < 0.001).
We initially conducted regression models (controlling for demographics) to address basic associations between M18 variants, Alzheimer’s disease pathology and global cognitive function. Lower cortical immunodensity of M18L, but not M18S, was associated with the severity of Alzheimer’s disease pathology, and lower cognitive scores (Fig. 4a). Importantly, for those participants above the 90th percentile of M18L the effect of higher levels of Alzheimer’s disease pathology on cognition was not statistically significant, although the number of cases with high pathology was limited, preventing definitive conclusions (Fig. 4b). In contrast, in subjects with M18L score below the 10th percentile, cognitive function was highly sensitive to Alzheimer’s disease pathology. While these observations suggested an interaction between Alzheimer’s disease pathology and M18L cortical levels, the inclusion of a statistical interaction term in the full sample analysis did not render significance. Decay curves for pathology-cognition associations were similar across M18S-ranked MAP participants (Fig. 4b).
Fig. 4.

Associations of M18 splice variants with Alzheimer’s disease pathology, global cognition and clinical dementia. a Contour plots illustrating MAP participants’ (n = 308) global cognition (z-score) as a function of M18L or M18S DLPFC immunodensities, and Alzheimer’s disease (AD) pathology. Only AD pathology × M18L (r =−0.251, p < 0.001), AD pathology × global cognition (r =−0.545, p < 0.001), and M18L × global cognition (r = 0.373, p < 0.001) were significantly correlated. b Regression analyses predicting global cognition as a function of AD pathology in groups of MAP participants having M18L or M18S immunodensities above 90th (green), between 10th and 90th (blue), or below 10th percentile (red). Lines represent best-fit curves and 95 % confident intervals. For M18L, significant correlations between AD pathology and global cognitive function were observed for participants among the lowest (r =−0.735, p < 0.001, n = 31) and intermediate (r =−0.512, p < 0.001, n = 243), but not the highest (r =−0.399, p = 0.074, n = 31), M18L groups. For M18S, statistically significant correlations were seen in all three M18S ranked groups: low (r =−0.460, p = 0.036, n = 31), intermediate (r =−0.559, p < 0.001, n = 245) and high (r =−0.509, p = 0.021, n = 29). c Whiskers represent 10th and 90th percentiles of M18L or M18S values, with boxed interquartile ranges crossed by the median, in participants with no- (NCI, n = 90), or mild-cognitive impairment (MCI, n = 86), or with dementia (DEM, n = 132). One-way ANOVA detected a significant effect only for M18L (F(2305) = 8.77, p < 0.001). *p < 0.05 and ***p < 0.001, ANOVA followed by Bonferroni’s test. d Representative immunoblots of M18L/S and β-actin, with various participants and standard (std) samples. Masses are indicated in kDa
Based on prior work showing that select presynaptic proteins were related to cognition in a relatively independent manner [13], we next generated linear regression models to evaluate the impact of M18L on cognitive function, accounting for age-related neuropathology. In these analyses, each of the common age-related neuropathologies was associated to a variable degree with global and/or domain-specific cognitive functions (Table 2, Model 1). Alzheimer’s disease pathology, cerebral macroinfarcts, hippocampal sclerosis, arteriolosclerosis, and atherosclerosis were all associated with poorer cognitive abilities. Lewy bodies and microinfarcts were not, in this subset of MAP participants. Overall, greater M18L cortical immunodensity was associated with better cognitive abilities (Model 2). Cortical amounts of M18L explained an additional 1.7 % of the variance in global cognition, relatively independent of any neuropathology. Of note, the frontal cortical M18L contribution was significant for episodic, semantic and working memory, but not for perceptual speed or visuospatial skills. Variations in M18S immunodensities were not related to global cognition or any of its domains (Model 3); when including both M18L and M18S (Model 4) only the M18L effect remained significant. In a final model (not shown), we analyzed the possible interaction between Alzheimer’s disease pathology and M18L cortical density by adding a statistical interaction term to the model. No significant effect was detected for this interaction term.
Table 2.
Linear regression modelsa predicting global or single-domain cognitive function
| Model no. and terms | Global cognition | Episodic memory | Semantic memory | Working memory | Perceptual speed | Visuospatial ability | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (n = 302) | (n = 302) | (n = 296) | (n = 301) | (n = 293) | (n = 289) | |||||||
| r 2 or βb | p-value | r 2 or β | p-value | r 2 or β | p-value | r 2 or β | p-value | r 2 or β | p-value | r 2 or β | p-value | |
| Model 1 | 0.328 | 0.394 | 0.208 | 0.174 | 0.189 | 0.121 | ||||||
| AD pathology | −0.872 | <0.001 | −1.091 | <0.001 | −0.713 | <0.001 | −0.634 | <0.001 | −0.477 | <0.001 | −0.362 | 0.014 |
| Macroinfarcts | −0.283 | 0.016 | −0.253 | 0.039 | −0.366 | 0.011 | −0.470 | 0.001 | −0.052 | 0.681 | −0.065 | 0.657 |
| Lewy bodies | −0.184 | 0.193 | −0.111 | 0.452 | −0.027 | 0.875 | −0.102 | 0.533 | −0.027 | 0.857 | −0.112 | 0.518 |
| Hipp sclerosis | −0.660 | 0.001 | −1.034 | <0.001 | −0.662 | 0.005 | −0.143 | 0.529 | −0.687 | 0.001 | −0.315 | 0.198 |
| Model 2 | 0.345 | 0.406 | 0.226 | 0.193 | 0.193 | 0.123 | ||||||
| AD pathology | −0.793 | <0.001 | −1.018 | <0.001 | −0.628 | <0.001 | −0.549 | <0.001 | −0.434 | 0.001 | −0.323 | 0.032 |
| Macroinfarcts | −0.269 | 0.021 | −0.240 | 0.048 | −0.347 | 0.014 | −0.455 | 0.001 | −0.043 | 0.731 | −0.058 | 0.693 |
| Lewy bodies | −0.141 | 0.314 | −0.071 | 0.630 | 0.021 | 0.900 | −0.055 | 0.735 | −0.004 | 0.981 | −0.088 | 0.611 |
| Hipp sclerosis | −0.658 | 0.001 | −1.032 | <0.001 | −0.665 | 0.005 | −0.141 | 0.530 | −0.686 | 0.001 | −0.317 | 0.196 |
| M18Lc | 0.0010 | 0.003 | 0.0009 | 0.009 | 0.0012 | 0.006 | 0.0011 | 0.006 | 0.0005 | 0.141 | 0.0005 | 0.226 |
| Model 3 | 0.328 | 0.392 | 0.209 | 0.177 | 0.188 | 0.120 | ||||||
| AD pathology | −0.865 | <0.001 | −1.090 | <0.001 | −0.707 | <0.001 | −0.624 | <0.001 | −0.470 | <0.001 | −0.357 | 0.016 |
| Macroinfarcts | −0.290 | 0.014 | −0.255 | 0.038 | −0.372 | 0.010 | −0.483 | <0.001 | −0.057 | 0.650 | −0.071 | 0.629 |
| Lewy bodies | −0.175 | 0.217 | −0.108 | 0.464 | −0.011 | 0.947 | −0.086 | 0.598 | −0.018 | 0.907 | −0.100 | 0.566 |
| Hipp sclerosis | −0.670 | 0.001 | −1.037 | <0.001 | −0.676 | 0.005 | −0.160 | 0.480 | −0.696 | 0.001 | −0.325 | 0.184 |
| M18Sc | 0.0016 | 0.331 | 0.0004 | 0.794 | 0.0024 | 0.247 | 0.0027 | 0.150 | 0.0015 | 0.389 | 0.0018 | 0.383 |
| Model 4 | 0.343 | 0.405 | 0.224 | 0.191 | 0.190 | 0.120 | ||||||
| AD pathology | −0.793 | 0.001 | −1.014 | <0.001 | −0.629 | <0.001 | −0.551 | <0.001 | −0.436 | 0.001 | −0.626 | 0.021 |
| Macroinfarcts | −0.267 | 0.022 | −0.232 | 0.057 | −0.348 | 0.014 | −0.460 | 0.001 | −0.047 | 0.714 | −0.062 | 0.673 |
| Lewy bodies | −0.141 | 0.313 | −0.074 | 0.614 | 0.022 | 0.896 | −0.053 | 0.745 | −0.002 | 0.990 | −0.085 | 0.625 |
| Hipp sclerosis | −0.657 | 0.001 | −1.023 | <0.001 | −0.667 | 0.005 | −0.146 | 0.515 | −0.690 | 0.001 | −0.321 | 0.189 |
| M18Lc | 0.0010 | 0.006 | 0.0011 | 0.006 | 0.0011 | 0.013 | 0.0010 | 0.017 | 0.0005 | 0.217 | 0.0004 | 0.343 |
| M18Sc | −0.0003 | 0.877 | −0.0015 | 0.419 | 0.0003 | 0.890 | 0.0009 | 0.669 | 0.0006 | 0.741 | 0.0010 | 0.658 |
Abbreviations: AD Alzheimer’s disease Hipp, hippocampal; M18L/S, Munc18-1 long/short splice variant
aAll models included terms (not shown) for age, sex, education, microinfarcts, arteriolosclerosis, and atherosclerosis
bValues are whole model adjusted r-squared, or individual β-coefficients for each term
cStandardized M18L/S values (z-scores) were used
Grouping MAP participants by clinical diagnosis revealed that dementia cases had lower M18L cortical immunodensity (−51 %, p < 0.001) than those with no dementia (Fig. 4c–d). Differences were also noted when subjects were graded using either NIA/Reagan or Braak, with lower M18L levels (−77 % and−64 %, respectively) in participants with high probability of Alzheimer’s disease (i.e. subjects falling into NIA/Reagan group 1 and/or Braak stage V–VI) (Additional file 1: Figure S2B). Minimal differences for M18S were observed across clinical diagnoses or postmortem rankings. In further multiple logistic regression analyses we aimed to evaluate the significance of M18L cortical downregulation to the likelihood of clinical dementia (Table 3). As expected, each unit of Alzheimer’s disease pathology, cerebrovascular disease (excluding microinfarcts), Lewy bodies and hippocampal sclerosis increased the odds of dementia (Table 3, Model 1). Each unit of M18L, however, was associated with lower odds of dementia, without altering the effects of neuropathologies (Model 2). The probability of dementia was not related to M18S cortical levels (Model 3), which in turn did not modify M18L effects (Model 4).
Table 3.
Logistic regression modelsa predicting likelihood of clinical dementia per unit of term
| Model terms | Model 1 | Model 2 | Model 3 | Model 4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Odds ratio | 95 % CI | p-value | Odds ratio | 95 % CI | p-value | Odds ratio | 95 % CI | p-value | Odds ratio | 95 % CI | p-value | |
| AD pathology | 3.15 | 1.80–5.73 | <0.001 | 2.84 | 1.60–5.22 | <0.001 | 3.16 | 1.81–5.75 | <0.001 | 2.83 | 1.59–5.21 | <0.001 |
| Macroinfarcts | 2.16 | 1.20–3.94 | 0.010 | 2.06 | 1.14–3.79 | 0.017 | 2.20 | 1.22–4.02 | 0.009 | 2.05 | 1.13–3.78 | 0.018 |
| Microinfarcts | 0.73 | 0.39–1.36 | 0.326 | 0.77 | 0.41–1.43 | 0.412 | 0.75 | 0.40–1.40 | 0.370 | 0.77 | 0.41–1.43 | 0.409 |
| Arteriolosclerosis | 1.30 | 0.95–1.78 | 0.098 | 1.34 | 0.98–1.84 | 0.068 | 1.32 | 0.97–1.82 | 0.078 | 1.33 | 0.98–1.84 | 0.071 |
| Atherosclerosis | 1.36 | 1.02–1.82 | 0.035 | 1.36 | 1.02–1.84 | 0.038 | 1.34 | 1.00–1.80 | 0.046 | 1.36 | 1.02–1.84 | 0.038 |
| Lewy bodies | 4.92 | 1.79–15.40 | 0.002 | 4.63 | 1.67–14.70 | 0.003 | 4.93 | 1.79–15.51 | 0.002 | 4.62 | 1.66–14.66 | 0.003 |
| Hipp sclerosis | 4.39 | 1.72–12.16 | 0.002 | 4.41 | 1.70–12.39 | 0.002 | 4.59 | 1.78–12.80 | 0.001 | 4.38 | 1.68–12.34 | 0.002 |
| M18Lb | 0.68 | 0.50–0.90 | 0.008 | 0.67 | 0.47–0.93 | 0.018 | ||||||
| M18Sb | 0.84 | 0.64–1.10 | 0.208 | 1.03 | 0.75–1.43 | 0.871 | ||||||
Abbreviations: AD Alzheimer’s disease, CI confidence interval Hipp, hippocampal, M18L/S Munc18-1 long/short splice variant
aAll models were controlled for age, sex and education
bStandardized M18L/S values (z-scores) were used
APP23 mice do not show alterations in M18L/S cortical amounts
We used APP23 transgenic mice to examine the association between M18L and a common Alzheimer’s disease pathology (i.e. amyloid plaques). These animals develop an Alzheimer’s disease-like syndrome via expression of a mutant APP that causes abnormal, age-dependent extra-synaptic amyloid-β accumulation [48]. M18L/S cortical immunodensities were compared between adult (12-month old) and aged (22-month old) APP23 mice, and age-matched wild type (WT) littermates. Although overt synaptic damage is reported in APP23 mice [48], cortical levels of M18L/S were not significantly different from those in WT controls, at either age (Fig. 5). However, M18L was slightly reduced in aged rats regardless of genotype (−13 to −16 %, p > 0.05), while APP23 mice tended to display lower M18S cortical immunodensities regardless of age (−21 to−22 %, p > 0.05). These marginal effects were considerably smaller than the M18L loss (−64 %) observed in the DLPFC of MAP participants graded according to Braak’s V–VI stages.
Fig. 5.

M18 splice variants are not altered in APP23 mice. a Immunodensities of M18L/S splice variants were quantified by Western blotting in brain homogenates (frontal cortex) from adult (12-month-old; T12) and aged (22-month-old; T22) wild type (WT) and APP23 transgenic mice. Columns are immunodensity mean values ± standard error (normalized by β-actin) of n = 6 mice per group, and represented in percentage to control (T12–WT) animals. Two-way ANOVA only detected a significant effect for genotype (but not age) on M18S (F(1,20) = 4.51, p = 0.0462), and a borderline age (but not genotype) effect on M18L (F(1,20) = 3.44, p = 0.0784), without specific between-group differences in the following Dunnett’s post hoc tests. b Representative immunoblots of M18L, M18S and β-actin, with one sample per group. Masses (in kDa) of proximal prestained markers are indicated on the left side of immunoblots
Discussion
In the present study, variations in frontal cortical M18 levels were associated with cognitive function and the likelihood of dementia in the elderly, beyond the effects of common age-related neuropathologies. Specific loss of M18L (but not M18S), which was found preferentially at GABAergic presynaptic terminals, may initiate synaptic dysfunction and consequent synaptic pathology.
We first characterized biochemical, cellular and anatomical aspects of M18 splice variants. Compared to M18S, M18L showed minimal binding affinity for syntaxin-1 (and hence for the SNARE complex) in two functional assays, co-IP and BN-PAGE. A previous study showed similar, but less pronounced, variant-specific binding affinities to syntaxin-1 [32]. Furthermore, in a yeast two-hybrid setting, depletion of any segment of the M18S (M18L was not reported) amino acid sequence (including the 25 C-terminal residues) completely abolished M18-syntaxin-1 association [49]. It is possible that the present assay conditions (e.g. absence of Ca2+) may induce a SNARE conformation that favors M18S relative to M18L binding in vitro. Since binding to syntaxin-1 is the major mechanism by which M18 exerts its functions [23, 28, 29], future studies should address this apparent difference in M18 variants in more detail. Given its globular, hydrophilic nature, M18 attachment to cell membranes is thought to occur through syntaxin-1 interaction [50]. Despite the lower affinity for syntaxin-1, M18L seemed confined to the highly hydrophobic, lipid raft-enriched fraction. While the role of these cholesterol-enriched microdomains in regulated exocytosis is unclear, some studies proposed that specific pools of target SNARE proteins may concentrate at lipid rafts, which in turn could discriminate between different kinds of neurotransmitter-containing vesicles [51]. Of note, the M18L C-terminal sequence does not display specific motifs for palmitoylation (cysteine enriched sequences), myristoylation (typically occurring at a N-terminal glycine), or prenylation (typically in a C-terminal cysteine), major post-translational modifications determining membrane localization for globular proteins. Possible explanations for M18L localization at lipid rafts could include a preferential M18L–syntaxin-1 interaction in the biochemical environment of these membrane microdomains, or alternatively, M18L may display greater affinity for another raft-attached protein. Munc18-interacting proteins (Mint1/2) may be key partners in this process, as they bear two membrane-attaching PDZ domains in addition to a M18-binding sequence [52]. Remarkably, Mint1/2 can regulate M18-syntaxin-1 interaction and were found also altered in Alzheimer's diseased postmortem brains [46]. Additionally, the presence of a putative phosphoserine site for Ca2+-calmodulin kinase II (CaMKII) activity in the C-terminal tail of M18L, but not M18S, may indicate a differential regulatory mechanism for the M18 variants [39]. This CaMKII activity was presumed to be responsible for the enhanced depression of synchronous vesicle release in M18 null pyramidal neurons rescued with M18L, compared to those carrying M18S only.
Immunohistochemical assays were consistent with tissue fractionation experiments. In contrast to the wide distribution of M18S across subcellular compartments, M18L was restricted to nerve endings and enriched in perisomatic areas. The M18S subcellular and histological distribution was similar to that previously reported for M18 [32, 53], and no cell-type specificity was presumed. The distribution of M18L was consistent with the localization expected for GABAergic presynaptic proteins [54]. Indeed, M18L closely matched VGAT distribution in all brain areas analyzed, especially in the hippocampus, where it was abundantly expressed in the perisomatic area of pyramidal cells. Interestingly, a previous in situ hybridization showed greatest expression of M18L in cells within the granule and pyramidal layers of mouse hippocampus [35]. Combined, these experiments indicate that M18L protein is mainly present in the presynaptic terminals of GABAergic interneurons within these hippocampal layers, although we cannot exclude some residual M18L expression in glutamatergic or other neuromodulatory terminals. This finding suggests a qualitative difference between the neurosecretory mechanisms of excitatory and inhibitory neurons. Several epileptic syndromes have been associated with firing abnormalities of dentate gyrus granule cells, likely due to an inappropriate inhibitory tone [55, 56]. This model is consistent with the mutations in M18 related to Ohtahara syndrome [41, 45], a disease accompanied by seizures and severe intellectual disability.
We found marked reductions of M18L, but not M18S, in DLPFC of MAP participants displaying clinical dementia and/or a large burden of Alzheimer’s disease pathology. Pairwise correlations between M18L levels, the burden of Alzheimer’s disease pathology, and cognitive function scores were all significant. Accordingly, participants having high M18L cortical density displayed greater cognitive resistance against the accumulation of Alzheimer’s disease pathology, which may directly implicate M18L in the molecular mechanisms of cognitive reserve. Statistical modeling indicated that loss of M18L contributed moderately to a higher likelihood of dementia and to cognitive impairment. Additionally, including synaptophysin as a covariate did not alter the results, suggesting that loss of M18L is an early event in synaptic pathology. The importance of maintaining appropriate splicing balance between M18L/S isoforms is probably best represented by the dramatic endophenotypes observed in murine strains either lacking or overexpressing the M18L variant [34, 35]. While specific regulatory splicing mechanisms of the STXBP1 transcript are unclear, other splicing deficits have been associated with aging related neuropathologies and dementia [57].
The predominant expression of M18L in GABAergic terminals suggests that synaptic deficits may be initiated in inhibitory, rather than excitatory synapses. This could be related to the surprising relative upregulation of glutamatergic terminals observed in MCI [58]. Indeed, there is compelling evidence involving GABAergic system in early stages of dementia [59, 60]. By contrast, previous observations in transgenic mouse models suggested that cholinergic and glutamatergic terminals may collapse first as a result of APP-mediated synaptic damage [61]. In the present study, APP23 mice did not display reduced levels of M18L/S, which agrees with previous findings [46]. Thus, cortical loss of M18L may be an independent event from amyloid-β accumulation, and perhaps other common age-related neuropathologies, and could represent a distinct contribution to cognitive impairment, possibly through erosion of cognitive reserve.
Conclusions
The present study identified the M18L isoform as a relatively specific component of the GABAergic presynaptic machinery. In contrast to the alternatively spliced M18S, which is highly abundant and ubiquitously expressed in inhibitory and excitatory neurons, M18L appears restricted to lipid raft microdomains within the presynaptic compartment, and may have a function independent from its interaction with syntaxin-1. Importantly, M18L, but not M18S, is associated with cognitive function in the elderly, and may contribute to the mechanisms of cognitive reserve that protect against dementia related to age-associated pathologies.
Methods
Participants, cognitive evaluations and neuropathological assessments
MAP recruits volunteers without known dementia, living in Chicago (IL, USA) [7]. Since 1997, this study enrolled over 1750 community-dwelling participants. At enrollment, all participants signed an informed consent and an Anatomic Gift Act for organ donation upon death. All protocols were approved by the Institutional Review Board of Rush University Medical Center. The overall follow-up rate is 95 % and the autopsy rate exceeds 80 %. Samples obtained in a consecutive series of autopsies of n = 308 participants were included in the present study. A summary of demographic, cognitive and pathological characteristics is listed in Table 1.
Methodological approaches to systematic cognitive, clinical and neuropathological evaluations were extensively reported [7, 62]. Annual cognitive evaluations comprised a battery of 21 standard tests, 19 of which are used to summarize one of the following domains: episodic memory, semantic memory, working memory, perceptual speed or visuospatial ability. For the present work, last valid cognitive tests were used in all analyses. The 19 tests could also be combined into a single variable, based on an average z-score to summarize global cognitive function nearest to death [7, 63]. The final clinical diagnoses of dementia followed the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association criteria [64], and were made by a board-certified neurologist blind to all pathological data.
Neuropathological examinations documented Alzheimer’s disease-related pathology (neuritic and diffuse plaques, and neurofibrillary tangles), vascular diseases (macroscopic and microscopic infarcts, arteriolosclerosis and atherosclerosis), Lewy bodies, and hippocampal sclerosis [7, 62]. A board-certified neuropathologist made all diagnoses blind to all clinical data. A composite measure of global Alzheimer’s disease pathology was created using a standardized average of neuritic and diffuse plaques and neurofibrillary tangles [62]. The burden of Alzheimer’s disease pathology was also categorized using Braak, CERAD and NIA/Reagan scales.
Animals
Approval from the UBC’s Animal Care Committee was obtained prior to experiments involving laboratory animals. Adult Sprague–Dawley rats were supplied by Charles-River (Montreal, QC, Canada). Novartis Pharma (Basel, Switzerland) provided APP23 transgenic mice, overexpressing a variant of human APP carrying the ‘Swedish double mutation’ KM670/671NL [48]. Pentobarbital-anaesthetized adult rats, as well as 12- or 22-month old APP23 mice and wild-type (WT) littermates, were killed by decapitation. Hemispheres were separated, and used for electrophoretic or immunohistochemical assays.
Purification of synaptosomes
Cortical synaptosomes were obtained following standard procedures (see cartoon in Fig. 1d), similar to those originally developed by Grey and Whittaker in the 1960’s [65]. All sucrose solutions described below were HEPES-buffered (4 mM, pH 7.4), supplemented with 1 % of a protease inhibitor cocktail (Sigma, St. Louis, MO, USA), and pre-chilled at 4 °C. Approximately 1 g of human inferior temporal cortex was homogenized in 10 ml of 0.32 M sucrose buffer, using a motorized 20-ml Potter-Elvehjem tissue grinder, with a clearance of 0.13–0.18 mm between the Teflon pestle and the glass chamber. The following centrifugal separations were all performed at 4 °C in an Avanti J-30I high performance centrifuge (Beckman Coulter, Fullerton, CA, USA), equipped with a JA-30.50 fixed angle rotor. Tissue debris, along with cell nuclei, were removed at 1000 × g for 1 min (P1). Supernatants (S1) were centrifuged at 17,000 × g for 15 min, and the resulting pellets (P2) washed in 0.32 M sucrose and re-centrifuged. P2 fractions were resuspended in 0.32 M sucrose and layered onto a sucrose discontinuous gradient, with 1.2 (bottom), 0.8 M (middle), and 0.32 M (topping) layers. After a 12-h centrifugation at 96,000 × g, the interfaces between 0.8 and 1.2 M (IF1, containing the crude synaptosomal fraction), and 0.32–0.8 M (IF2, mainly myelin-coated fragments) were collected. IF1 was again overlaid on 0.32 M sucrose buffer and centrifuged at 30,000 × g for 20 min. The resulting pellet, containing purified synaptosomes, was resuspended in 1 ml of homogenization buffer supplemented with 0.5 % TritonX. In all steps, aliquots of each fraction were separated for further SDS-PAGE and immunoblotting analyses.
Extraction of lipid rafts
Purification of lipid raft-enriched fraction was based on the TritonX-insoluble, SDS-soluble property of these membrane microdomains. The present method, initially designed for separating cytosolic from insoluble, raft-associated α-synuclein [66], offers a simple and quick assay to yield a fraction (S4) of TritonX-insoluble proteins highly enriched in lipid rafts through sequential centrifugation steps [67]. Figure 1f in the main text illustrates the procedure. Briefly, ~150 mg of cortical tissue were homogenized in 1.5 mL of ice-cold 10 mM Tris-buffer, pH 6.8, containing 1 % of a protease inhibitor cocktail (Sigma). Total homogenate (TH) was centrifuged at 31,000 × g for 1 h at 4 °C. Supernatant was collected (S1), and the pellet (P1) was resuspended in 1 ml of the same ice-cold Tris-buffered solution supplemented with 0.5 % TritonX. To achieve a homogenous solution, P1 was gently sonicated (F60 Sonic Dismembrator, Fisher Scientific, Waltham, MA, USA). The same centrifugation step was performed and the supernatant (S2) was kept. The pellet (P2) was again resuspended and sonicated in Tris-buffer containing 2 % TritonX, and the centrifugation step was repeated. The subsequent supernatant (S3) was separated and the pellet (P3) homogenized as above in ice-cold Tris-buffer containing 0.5 % SDS. P3 was incubated at 15 °C with rotation for 10 min before a final centrifugation at 31,000 × g and at 12 °C was performed for 1 h. The supernatant (S4), enriched in lipid-raft proteins, was collected, and the final pellet (P4) resuspended as above. All fractions were stored at−80 °C until resolved by SDS-PAGE and immunoblotting.
Antibodies
A list of primary antibodies used appears in Additional file 2: Table S1. Commercial antibodies selectively targeting M18L and M18S variants were raised against their respective C-terminal amino acidic sequences (Fig. 1a), which share 100 % homology across human and rodent orthologues. The isoform-selectivity of these antibodies was previously reported [36], and further characterized by co-immunoprecipitation. Production and characterization of mouse monoclonal antibodies against syntaxin-1, SNAP-25, VAMP and synaptophysin was described elsewhere [68, 69]. Peroxidase- and Alexa-Fluor 488/555/647-conjugated secondary antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA, USA) or Molecular Probes (Eugene, OR, USA), respectively.
Immunoprecipitation
Target proteins were immunoprecipitated using protein G-coated magnetic Dynabeads (Life Technologies, Carlsbad, CA, USA), as reported [38]. In each reaction, 50 μg of beads were incubated in phosphate-buffered saline (PBS), supplemented with 0.1 % TritonX, and containing 0.33 μg of anti-mouse IgG (negative control), or antibodies against either M18L or M18S splice variants. Nonspecific binding sites were blocked in the same buffer supplemented with 3 % bovine serum albumin (BSA). In parallel, human cortical samples were ground homogenized in PBS containing 1 % TritonX, and 1 % of protease inhibitors (Sigma), and solubilized for 1 h at 4 °C. Prior to IP reactions, solubilized brain proteins were pre-cleared with antibody-free beads. Antibody-conjugated magnetic beads were combined with excess (2 mg) of pre-cleared brain proteins, and incubated overnight at 4 °C. After washing, IP products were eluted in 25 μl of 20 mM tricine, pH 2.7 to preserve native protein structures and interactions.
Quantitative immunoblotting
Grey matter samples from the middle-frontal gyrus (Brodmann’s area 46/9) of the DLPFC were obtained at autopsies of MAP participants, following a standard atlas [70]. The DLPFC was selected for its central role in complex cognitive tasks and contribution to age-related cognitive decline [71]. Samples (40–80 mg) were ground-homogenized using a Teflon pestle in ice-cold PBS, pH 7.4, and stored at−80 °C until use [13]. Prior to immunoassays, protein concentrations were determined by DC assay (Bio-Rad, Hercules, CA, USA), and samples adjusted to equal concentrations with homogenization buffer.
Quantification and characterization of M18L/S variants and β-actin in MAP cortical samples was achieved by SDS-PAGE, using 10 % or 12 % minigels (Bio-Rad), followed by immunoblotting, as previously reported [38]. Quantification of other presynaptic proteins was previously reported [13]. Total brain homogenates (and also subcellular fractions or IP products) were combined with equal volumes of 2× loading buffer (100 mM Tris, pH 6.8, 4 % SDS, 0.2 % bromophenol blue, 20 % glycerol, 200 mM β-mercaptoethanol). All samples were boiled for 5 min prior to electrophoretical separations. Preliminary analyses determined that 10-μg protein aliquots of total brain homogenates from DLPFC were optimal for densitometric quantifications [36]. The standard sample (pool of n = 132 MAP participants) was spaced-loaded in triplicate in all gels to control for correct loading and protein transfer to the membranes (see Fig. 4d), and also to account for between-gel variability. For quality control, immunoblots were rejected when the coefficient of variation (CV) of in-gel standard samples exceeded 10 %, although typical CV < 5 % values were obtained. For the analysis of brain fractions, equivolumetric amounts were loaded onto the SDS-gels, while 5 μl of IP products were found optimal. All brain samples were resolved in 10–12 % polyacrylamide minigels (Bio-Rad, Hercules, CA, USA). After electrophoresis, proteins were transferred to polyvinylidene difluoride (PVDF) membranes, and subsequently blocked (1 h), and incubated with primary (overnight, 4 °C; see Additional file 2: Table S1) and secondary (1 h; 1:5000) antibodies, in TBS containing 5 % milk and 0.1 % Tween-20. Chemiluminescence was enhanced with commercial reagents (Perkin Elmer, Waltham, MA, USA), and images were digitized using a LAS-3000 Image Reader (Fujifilm, Tokyo, Japan).
In quantitative studies, membrane stripping and reprobing with anti-β-actin antibody was done for housekeeping and data normalization purposes. Densitometric analyses of the immunoblots were done with ImageGauge software, version 4.22 (Fujifilm). Every gel was run with 14 brain samples (including 11 MAP participants randomly selected, and the triplicate standard sample), and a molecular weight ladder (Bio-Rad). Gels containing the same subset of samples were assessed at least twice in different days. For quality control, a minimal between-gel Pearson’s correlation coefficient of r = 0.80 was required for M18L/S and β-actin immunoreactivity values. Gels not meeting this criterion were discarded and repeated. For each sample, immunoreactivity of M18L/S (in arbitrary optical density units) was first divided by that of β-actin within the same gel, and then calculated as a percentage of in-gel standards. Mean value between the two different, valid gels was used as a final estimate. This procedures were reported to reduce the experimental variability in studies that used immunoblotting as a quantitative technique for large cohorts of samples [36, 38].
BN-PAGE
Originally developed to study mitochondrial membrane complexes [72], separation of solubilized brain proteins by BN-PAGE was recently shown suitable to identify and quantify presynaptic complexes in human postmortem tissue [38]. Brain samples were combined with equal volumes of ice-cold 100 mM Bis-tris, pH 7.0, solubilization buffer, containing 50 mM NaCl, 2 mM EDTA, 4 mM 6-aminohexanoic acid, 1 % TritonX, and 1 % protease inhibitor cocktail, and incubated for 1 h at 4 °C with gentle rotation. After centrifugation (16,000 × g, 30 min, 4 °C), supernatants or IP products were combined with equal volumes of a loading buffer (0.5 % TritonX, 0.25 % Coomassie brilliant blue G-250, 10 % glycerol). Samples were loaded in 4–16 % gradient NativePAGE precast gels (Novex, Carlsbad, CA, USA). Electrophoresis was run under constant voltage (150 V), at 4 °C, with pre-chilled anode (50 mM Bis-tris, pH 7.0) and cathode (15 mM Bis-tris, 50 mM tricine, pH 7.0, 0.02 % Coomassie dye) buffers. Before transferring proteins and complexes to PVDF membranes, BN-gels were incubated an ice-cold 12 mM Tris, 96 mM glycine buffer, pH 8.3, containing 0.1 % SDS. To remove excess of Coomassie dye, membranes were rinsed in 100 % methanol, followed by immediate rehydration in TBS for 15 min. Following immunoblotting procedures were as described for SDS-PAGE. Molecular markers (NativeMARK, Novex, range 20–1240 kDa) were loaded in all gels to estimate protein complex sizes.
Immunohistochemistry and immunofluorescence
Forty-μm floating coronal sections (−3.30 to−4.20 relative to bregma) were obtained from paraformaldehyde-fixed rat brain hemispheres [73]. Other floating coronal sections were from the mid-hippocampus of selected cases with or without dementia [74]. Retrieval of the M18L epitope required 20-min incubation at 95 °C in 20 mM Tris buffer (pH 9.0), containing 1 mM EDTA and 0.05 % Tween-20, possibly because of the major localization in TritonX-insoluble subcellular compartments (see Results). Other antigen retrievals were done in 20 mM citrate buffer (pH 6.0) at 80 °C for 15 min. For procedures involving 3,3′–diaminobenzidine staining, we used commercially available kits (Vector Labs, Burlingame, CA, USA), with incubations as reported [75]. Images were acquired with an Olympus BX61 microscope (Olympus, Tokyo, Japan). Immunofluorescence assays were performed as described [74, 76]. A series of orthogonal images were obtained using a LSM 5 Pascal confocal microscope (Zeiss; Jena, Germany). Co-staining of M18L/S isoforms with VGAT and VGLUT1 was assessed to determine presence in inhibitory or excitatory presynaptic terminals, respectively. Appropriate negative controls were included in all experiments.
Statistical analyses
In quantitative immunoblotting assays, M18L/S immunodensities were first normalized to corresponding β-actin values, and calculated as a percentage of in-gel standards [36, 38]. Linear models were constructed with log-transformed (M18L) and/or standardized (M18L and M18S) values (z-scores). Data transformations did not alter the observed differences between groups.
Multivariate analyses were performed to survey putative associations between M18 isoforms and cognitive, pathological, neurochemical and/or putative confounding variables. An additional exploratory analysis was performed by ranking participants according to their M18L/S cortical immunodensities and comparing pathology-cognition decay curves across subjects with high (above the 90th percentile) and low (below 10th percentile) M18L/S values. Given the potential effect of cortical M18L levels on cognitive performance, we generated multiple linear regression models with cognitive measures as outcomes and pathological and neurochemical variables as predictors. Additionally, using the same predictors, we built logistic regression models with clinical diagnosis of dementia as the outcome. Linear and logistic models were controlled for sex, age and education. Differences in M18 immunodensities between clinically diagnosed or pathologically graded groups were assessed by ANOVA followed by Bonferroni’s post hoc test for standardized data, or Kruskal-Wallis followed by Dunn’s post hoc tests for non-transformed values. Experiments involving APP23 mice were analyzed with two-way ANOVA, with genotype and age as independent factors, followed by Dunnet’s post hoc test.
For colocalization analyses of confocal imaging, we used an ImageJ 2.0 (NIH, Bethesda, MA, USA) built-in method [76, 77]. Comparisons of M18L/S colocalizations across inhibitory versus excitatory compartments were performed with paired t-tests.
All tests were two-tailed, and p-values < 0.05 were considered significant. Datasets were analyzed and plotted with JMP 10.0.2 (SAS Institute, Cary, NC, USA), and/or GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA, USA).
Acknowledgements
We wish to express our gratitude to all participants in MAP, and to the staff in Rush Alzheimer's Disease Center. We also thank Hong-Ying Li and Jenny Yang for their skillful technical assistance. The study was supported by the Canadian Institutes of Health Research (MT-14037, MOP-81112), the Michael Smith Foundation for Health Research (Grant #5401), and the Jack Bell Chair in Schizophrenia. The Memory and Aging Project is a collaborative, multidisciplinary and translational research project subsidized by the National Institute on Aging (Grants R01AG17917, R01AG42210).
Abbreviations
- ANOVA
Analysis of variance
- ANCOVA
Analysis of covariance
- APP
Amyloid precursor protein
- BN-PAGE
Blue native-PAGE
- BSA
Bovine serum albumin
- CA
Ammon’s horn
- CERAD
Consortium to Establish a Registry for Alzheimer’s Disease
- CV
Coefficient of variation
- DC
Detergent compatible
- DG
Dentate gyrus
- DLPFC
Dorsolateral prefrontal cortex
- ECL
Enhanced chemiluminescence
- EDTA
Ethylenediaminetetraacetic acid
- ELISA
Enzyme-linked immunoadsorbant assay
- GABA
Gamma-aminobutyric acid
- GCL
Granule cell layer
- IP
Immunoprecipitation
- M18
Munc18-1
- M18L/S
M18 long/short splice variant
- MAP
Memory and aging project
- MCI
Mild cognitive impairment
- NCI
No cognitive impairment
- PAGE
Polyacrylamide gel electrophoresis
- PBS
Phosphate-buffered saline
- PMI
Postmortem interval
- PPIs
Protein-protein interactions
- PVDF
Polyvinylidene difluoride
- SDS
Sodium dodecyl sulfate
- SNAP-25
Synaptosome-associated protein of 25 kDa
- SNARE
soluble N-ethylmaleimide-sensitive factor attachment protein receptor
- STXBP1
Syntaxin-binding protein-1
- TBS
TRIS-buffered saline
- TritonX
Triton X-100
- VAMP
Vesicle-associated membrane protein
- VGAT
Vesicle GABA transporter
- VGLUT1
Vesicle glutamate transporter-1
- WT
Wild-type
Additional files
Immunohistochemical characterization of Munc18-1 splice variants in human dentate gyrus reveals similar cellular and subcellular distributions of M18L and M18S than those in rat brain. Confocal images show a preferential localization of M18L to inhibitory presynaptic terminals, as its immunofluorescence fully overlaps with that of VGAT, but not VGLUT1. M18S shows ubiquitous distribution. Figure S2. M18L, but not M18S, immunodensity is reduced in the DLPFC of MAP participants with clinical dementia, compared to those with no or mild cognitive impairment, as well as in those presenting high burden of Alzheimer’s disease pathology, using either NIA/Reagan or Braak scales. (PDF 986 kb)
List of both commercially available and locally produced primary antibodies used in the present study. References cited in Table S1 are listed below the table. (PDF 107 kb)
Footnotes
Competing interests
WGH has received consulting fees or sat on paid advisory boards for: In Silico, Lundbeck/Otsuka, Eli Lilly, and Roche. AMB is on the advisory board or received consulting fees from Roche Canada, and received educational grant support from BMS Canada. The Organizations cited above had no role in (and therefore did not influence) the design of the present study, the interpretation of results, and/or preparation of the manuscript. All other authors have no financial interest on the reported data and declare that no competing interests exist.
Authors’ contributions
WGH, DAB, AR-M and AMB, designed the study. AR-M performed all characterization and quantification experiments of M18L/S splice variants in human and rat brain tissues. CH and CLB participated in the immunohistochemical and immunofluorescence assays in rat and human brain sections. TAB and PF contributed to the APP23 mice study. DAB and JAS conceived the Memory and Aging Project, performed all clinical and pathological exams, and procured human tissue samples. SEL compiled all participants’ demographic, clinical and pathological data and, with the collaboration of AR-M and WGH, run all statistical analyses. AR-M, WGH and DAB wrote the first draft of the manuscript. All authors critically contributed to the discussion of the results and approved the final version of the manuscript.
Authors’ information
AR-M is a Post-Doctoral Fellow of the BC Schizophrenia Society Foundation and the Michael Smith Foundation for Health Research.
Contributor Information
Alfredo Ramos-Miguel, Email: alfredo.ramos@ubc.ca.
Christa Hercher, Email: christahercher@yahoo.ca.
Clare L. Beasley, Email: cbeasley@mail.ubc.ca
Alasdair M. Barr, Email: al.barr@ubc.ca
Thomas A. Bayer, Email: thomas.bayer@medizin.uni-goettingen.de
Peter Falkai, Email: peter.falkai@med.uni-muenchen.de.
Sue E. Leurgans, Email: Sue_E_Leurgans@rush.edu
Julie A. Schneider, Email: julie_a_schneider@rush.edu
David A. Bennett, Email: david_a_bennett@rush.edu
William G. Honer, Email: william.honer@ubc.ca
References
- 1.Satz P. Brain reserve capacity on symptom onset after brain injury: A formulation and review of evidence for threshold theory. Neuropsychology. 1993;7:273–295. doi: 10.1037/0894-4105.7.3.273. [DOI] [Google Scholar]
- 2.Stern Y. What is cognitive reserve? Theory and research application of the reserve concept. J Int Neuropsychol Soc. 2002;8:448–460. doi: 10.1017/S1355617702813248. [DOI] [PubMed] [Google Scholar]
- 3.Katzman R, Terry R, DeTeresa R, Brown T, Davies P, Fuld P. Clinical, pathological, and neurochemical changes in dementia: A subgroup with preserved mental status and numerous neocortical plaques. Ann Neurol. 1988;23:138–144. doi: 10.1002/ana.410230206. [DOI] [PubMed] [Google Scholar]
- 4.White L, Small BJ, Petrovitch H, Ross GW, Masaki K, Abbott RD, et al. Recent clinical-pathologic research on the causes of dementia in late life: update from the Honolulu-Asia Aging Study. J Geriatr Psychiatry Neurol. 2005;18:224–227. doi: 10.1177/0891988705281872. [DOI] [PubMed] [Google Scholar]
- 5.Bryne C, Matthews FE, Xuereb JH, Broome JC, McKenzie J, Rossi M, et al. Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Lancet. 2001;357:169–175. doi: 10.1016/S0140-6736(00)03589-3. [DOI] [PubMed] [Google Scholar]
- 6.Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA. 1997;277:813–817. doi: 10.1001/jama.1997.03540340047031. [DOI] [PubMed] [Google Scholar]
- 7.Bennett DA, Schneider JA, Buchman AS, Barnes LL, Boyle PA, Wilson RS. Overview and findings from the Rush Memory and Aging Project. Curr Alzheimer Res. 2012;9:646–663. doi: 10.2174/156720512801322663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lassmann H, Weiler R, Fischer P, Bancher C, Jellinger K, Floor E, et al. Synaptic pathology in Alzheimer’s disease: immunological data for markers of synaptic and large dense-core vesicles. Neuroscience. 1992;46:1–8. doi: 10.1016/0306-4522(92)90003-K. [DOI] [PubMed] [Google Scholar]
- 9.Honer WG, Dickson DW, Gleeson J, Davies P. Regional synaptic pathology in Alzheimer’s disease. Neurobiol Aging. 1992;13:375–382. doi: 10.1016/0197-4580(92)90111-A. [DOI] [PubMed] [Google Scholar]
- 10.Masliah E, Hansen L, Albright T, Mallory M, Terry RD. Immunoelectron microscopic study of synaptic pathology in Alzheimer’s disease. Acta Neuropathol. 1991;81:428–433. doi: 10.1007/BF00293464. [DOI] [PubMed] [Google Scholar]
- 11.Lassmann H, Fischer P, Jellinger K. Synaptic pathology of Alzheimer’s disease. Ann N Y Acad Sci. 1993;695:59–64. doi: 10.1111/j.1749-6632.1993.tb23028.x. [DOI] [PubMed] [Google Scholar]
- 12.Honer WG. Pathology of presynaptic proteins in Alzheimer’s disease: more than simple loss of terminals. Neurobiol Aging. 2003;24:1047–1062. doi: 10.1016/j.neurobiolaging.2003.04.005. [DOI] [PubMed] [Google Scholar]
- 13.Honer WG, Barr AM, Sawada K, Thornton AE, Morris MC, Leurgans SE, et al. Cognitive reserve, presynaptic proteins and dementia in the elderly. Transl Psychiatry. 2012;2:e114. doi: 10.1038/tp.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boyle PA, Wilson RS, Yu L, Barr AM, Honer WG, Schneider JA, et al. Much of late life cognitive decline is not due to common neurodegenerative pathologies. Ann Neurol. 2013;74:478–489. doi: 10.1002/ana.23964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Söllner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- 16.Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol. 2009;66:200–208. doi: 10.1002/ana.21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Minger SL, Honer WG, Esiri MM, McDonald B, Keene J, Nicoll JA, et al. Synaptic pathology in prefrontal cortex is present only with severe dementia in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:929–936. doi: 10.1093/jnen/60.10.929. [DOI] [PubMed] [Google Scholar]
- 18.Harrington CR, Wischik CM, Honer W, Jakes RG, McGee M, Paykel ES, et al. Staging of Cytoskeletal and β-Amyloid Changes in Human Isocortex Reveals Biphasic Synaptic Protein Response during Progression of Alzheimer’s Disease. Am J Pathol. 2000;157:623–636. doi: 10.1016/S0002-9440(10)64573-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berchtold NC, Sabbagh MN, Beach TG, Kim RC, Cribbs DH, Cotman CW. Brain gene expression patterns differentiate mild cognitive impairment from normal aged and Alzheimer’s disease. Neurobiol Aging. 2014;35:1961–1972. doi: 10.1016/j.neurobiolaging.2014.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakata Y, Yasuda T, Fukaya M, Yamamori S, Itakura M, Nihira T, et al. Accumulation of α-synuclein triggered by presynaptic dysfunction. J Neurosci. 2012;32:17186–17196. doi: 10.1523/JNEUROSCI.2220-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma M, Burre J, Sudhof TC. Proteasome Inhibition Alleviates SNARE-Dependent Neurodegeneration. Sci Transl Med. 2012;4:147ra113. doi: 10.1126/scitranslmed.3004028. [DOI] [PubMed] [Google Scholar]
- 22.Südhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toonen RFG, Verhage M. Munc18-1 in secretion: lonely Munc joins SNARE team and takes control. Trends Neurosci. 2007;30:564–572. doi: 10.1016/j.tins.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 24.Südhof TC. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron. 2013;80:675–690. doi: 10.1016/j.neuron.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hata Y, Slaughter CA, Südhof TC. Synaptic vesicle fusion complex contains unc-18 homologue bound to syntaxin. Nature. 1993;366:347–351. doi: 10.1038/366347a0. [DOI] [PubMed] [Google Scholar]
- 26.Hosono R, Hekimi S, Kamiya Y, Sassa T, Murakami S, Nishiwaki K, et al. The unc-18 gene encodes a novel protein affecting the kinetics of acetylcholine metabolism in the nematode Caenorhabditis elegans. J Neurochem. 1992;58:1517–1525. doi: 10.1111/j.1471-4159.1992.tb11373.x. [DOI] [PubMed] [Google Scholar]
- 27.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rizo J, Südhof TC. The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices--guilty as charged? Annu Rev Cell Dev Biol. 2012;28:279–308. doi: 10.1146/annurev-cellbio-101011-155818. [DOI] [PubMed] [Google Scholar]
- 29.Han GA, Malintan NT, Collins BM, Meunier FA, Sugita S. Munc18-1 as a key regulator of neurosecretion. J Neurochem. 2010;115:1–10. doi: 10.1111/j.1471-4159.2010.06900.x. [DOI] [PubMed] [Google Scholar]
- 30.Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, et al. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- 31.Swanson DA, Steel JM, Valle D. Identification and characterization of the human ortholog of rat STXBP1, a protein implicated in vesicle trafficking and neurotransmitter release. Genomics. 1998;48:373–376. doi: 10.1006/geno.1997.5202. [DOI] [PubMed] [Google Scholar]
- 32.Garcia EP, McPherson PS, Chilcote TJ, Takei K, De Camilli P. rbSec1A and B colocalize with syntaxin 1 and SNAP-25 throughout the axon, but are not in a stable complex with syntaxin. J Cell Biol. 1995;129:105–110. doi: 10.1083/jcb.129.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cijsouw T, Weber JP, Broeke JH, Broek JAC, Schut D, Kroon T, et al. Munc18-1 redistributes in nerve terminals in an activity- and PKC-dependent manner. J Cell Biol. 2014;204:759–775. doi: 10.1083/jcb.201308026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cijsouw T. Protein and vesicle dynamics in regulated secretion. Vrije Universiteit Amsterdam; 2014. http://hdl.handle.net/1871/52074
- 35.Leza JC, Pazos A, Garzón J, Meana JJ, Micó JA, Desco M, et al. Behavioral, neurochemical and morphological changes induced by the overexpression of munc18-1a in brain of mice: relevance to schizophrenia. Transl Psychiatry. 2013;3:e221. doi: 10.1038/tp.2012.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gil-Pisa I, Munarriz-Cuezva E, Ramos-Miguel A, Urigüen L, Meana JJ, García-Sevilla JA. Regulation of munc18-1 and syntaxin-1A interactive partners in schizophrenia prefrontal cortex: down-regulation of munc18-1a isoform and 75 kDa SNARE complex after antipsychotic treatment. Int J Neuropsychopharmacol. 2012;15:573–588. doi: 10.1017/S1461145711000861. [DOI] [PubMed] [Google Scholar]
- 37.Behan AT, Byrne C, Dunn MJ, Cagney G, Cotter DR. Proteomic analysis of membrane microdomain-associated proteins in the dorsolateral prefrontal cortex in schizophrenia and bipolar disorder reveals alterations in LAMP, STXBP1 and BASP1 protein expression. Mol Psychiatry. 2009;14:601–613. doi: 10.1038/mp.2008.7. [DOI] [PubMed] [Google Scholar]
- 38.Ramos-Miguel A, Beasley CL, Dwork AJ, Mann JJ, Rosoklija G, Barr AM, et al. Increased SNARE Protein-Protein Interactions in Orbitofrontal and Anterior Cingulate Cortices in Schizophrenia. Biol Psychiatry. 2014;78:361–373. doi: 10.1016/j.biopsych.2014.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meijer M, Cijsouw T, Toonen RF, Verhage M. Synaptic Effects of Munc18-1 Alternative Splicing in Excitatory Hippocampal Neurons. PLoS One. 2015;10:e0138950. doi: 10.1371/journal.pone.0138950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saitsu H, Kato M, Mizuguchi T, Hamada K, Osaka H, Tohyama J, et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet. 2008;40:782–788. doi: 10.1038/ng.150. [DOI] [PubMed] [Google Scholar]
- 41.Saitsu H, Kato M, Matsumoto N. Haploinsufficiency of STXBP1 and Ohtahara syndrome. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. 4. Bethesda (MD), USA: National Center for Biotechnology Information; 2012. pp. 1–12. [PubMed] [Google Scholar]
- 42.Hamdan FF, Gauthier J, Dobrzeniecka S, Lortie A, Mottron L, Vanasse M, et al. Intellectual disability without epilepsy associated with STXBP1 disruption. Eur J Hum Genet. 2011;19:607–609. doi: 10.1038/ejhg.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamdan FF, Piton A, Gauthier J, Lortie A, Dubeau F, Dobrzeniecka S, et al. De novo STXBP1 mutations in mental retardation and nonsyndromic epilepsy. Ann Neurol. 2009;65:748–753. doi: 10.1002/ana.21625. [DOI] [PubMed] [Google Scholar]
- 44.Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- 45.Deprez L, Weckhuysen S, Holmgren P, Suls A, Van Dyck T, Goossens D, et al. Clinical spectrum of early-onset epileptic encephalopathies associated with STXBP1 mutations. Neurology. 2010;75:1159–1165. doi: 10.1212/WNL.0b013e3181f4d7bf. [DOI] [PubMed] [Google Scholar]
- 46.Jacobs EH, Williams RJ, Francis PT. Cyclin-dependent kinase 5, Munc18a and Munc18-interacting protein 1/X11alpha protein up-regulation in Alzheimer’s disease. Neuroscience. 2006;138:511–522. doi: 10.1016/j.neuroscience.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 47.Donovan LE, Higginbotham L, Dammer EB, Gearing M, Rees HD, Xia Q, et al. Analysis of a membrane-enriched proteome from postmortem human brain tissue in Alzheimer’s disease. PROTEOMICS – Clin Appl. 2012;6:201–211. doi: 10.1002/prca.201100068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A. 1997;94(November):13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hata Y, Sudhof TC. A novel ubiquitous form of Munc-18 interacts with multiple syntaxins. Use of the yeast two-hybrid system to study interactions between proteins involved in membrane traffic. J Biol Chem. 1995;270:13022–13028. doi: 10.1074/jbc.270.22.13022. [DOI] [PubMed] [Google Scholar]
- 50.de Wit H, Cornelisse LN, Toonen RFG, Verhage M. Docking of secretory vesicles is syntaxin dependent. PLoS One. 2006;1:e126. doi: 10.1371/journal.pone.0000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Salaun C, James DJ, Chamberlain LH. Lipid rafts and the regulation of exocytosis. Traffic. 2004;5:255–264. doi: 10.1111/j.1600-0854.2004.0162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Okamoto M, Sudhof TC. Mints, Munc18-interacting proteins in synaptic vesicle exocytosis. J Biol Chem. 1997;272:31459–31464. doi: 10.1074/jbc.272.50.31459. [DOI] [PubMed] [Google Scholar]
- 53.Zhu Y, Ning D, Wang F, Liu C, Xu Y, Jia X, et al. Effect of thyroxine on munc-18 and syntaxin-1 expression in dorsal hippocampus of adult-onset hypothyroid rats. Eur J Histochem. 2012;56:135–141. doi: 10.4081/ejh.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chaudhry FA, Reimer RJ, Bellocchio EE, Danbolt NC, Osen KK, Edwards RH, et al. The vesicular GABA transporter, VGAT, localizes to synaptic vesicles in sets of glycinergic as well as GABAergic neurons. J Neurosci. 1998;18:9733–9750. doi: 10.1523/JNEUROSCI.18-23-09733.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sloviter RS. Decreased hippocampal inhibition and a selective loss of interneurons in experimental epilepsy. Science. 1987;235:73–76. doi: 10.1126/science.2879352. [DOI] [PubMed] [Google Scholar]
- 56.Wittner L, Magloczky Z, Borhegyi Z, Halasz P, Toth S, Eross L, et al. Preservation of perisomatic inhibitory input of granule cells in the epileptic human dentate gyrus. Neuroscience. 2001;108:587–600. doi: 10.1016/S0306-4522(01)00446-8. [DOI] [PubMed] [Google Scholar]
- 57.Bai B, Hales CM, Chen P-C, Gozal Y, Dammer EB, Fritz JJ, et al. U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc Natl Acad Sci. 2013;110:16562–16567. doi: 10.1073/pnas.1310249110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bell KFS, Bennett DA, Cuello AC. Paradoxical upregulation of glutamatergic presynaptic boutons during mild cognitive impairment. J Neurosci. 2007;27:10810–10817. doi: 10.1523/JNEUROSCI.3269-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koliatsos VE, Kecojevic A, Troncoso JC, Gastard MC, Bennett DA, Schneider JA. Early involvement of small inhibitory cortical interneurons in Alzheimer’s disease. Acta Neuropathol. 2006;112:147–162. doi: 10.1007/s00401-006-0068-6. [DOI] [PubMed] [Google Scholar]
- 60.Limon A, Reyes-Ruiz JM, Miledi R. Loss of functional GABA(A) receptors in the Alzheimer diseased brain. Proc Natl Acad Sci U S A. 2012;109:10071–10076. doi: 10.1073/pnas.1204606109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Canas PM, Simoes AP, Rodrigues RJ, Cunha RA. Predominant loss of glutamatergic terminal markers in a beta-amyloid peptide model of Alzheimer’s disease. Neuropharmacology. 2014;76(Pt A):51–56. doi: 10.1016/j.neuropharm.2013.08.026. [DOI] [PubMed] [Google Scholar]
- 62.Schneider JA, Wilson RS, Bienias JL, Evans DA, Bennett DA. Cerebral infarctions and the likelihood of dementia from Alzheimer disease pathology. Neurology. 2004;62:1148–1155. doi: 10.1212/01.WNL.0000118211.78503.F5. [DOI] [PubMed] [Google Scholar]
- 63.Bennett DA, Schneider JA, Tang Y, Arnold SE, Wilson RS. The effect of social networks on the relation between Alzheimer’s disease pathology and level of cognitive function in old people: a longitudinal cohort study. Lancet Neurol. 2006;5:406–412. doi: 10.1016/S1474-4422(06)70417-3. [DOI] [PubMed] [Google Scholar]
- 64.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939–944. doi: 10.1212/WNL.34.7.939. [DOI] [PubMed] [Google Scholar]
- 65.Gray EG, Whittaker VP. The isolation of nerve endings from brain: an electron-microscopic study of cell fragments derived by homogenization and centrifugation. J Anat. 1962;96:79–88. [PMC free article] [PubMed] [Google Scholar]
- 66.Deramecourt V, Bombois S, Maurage C-A, Ghestem A, Drobecq H, Vanmechelen E, et al. Biochemical staging of synucleinopathy and amyloid deposition in dementia with Lewy bodies. J Neuropathol Exp Neurol. 2006;65:278–288. doi: 10.1097/01.jnen.0000205145.54457.ea. [DOI] [PubMed] [Google Scholar]
- 67.Ohrfelt A, Zetterberg H, Andersson K, Persson R, Secic D, Brinkmalm G, et al. Identification of novel alpha-synuclein isoforms in human brain tissue by using an online nanoLC-ESI-FTICR-MS method. Neurochem Res. 2011;36:2029–2042. doi: 10.1007/s11064-011-0527-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Barakauskas VE, Beasley CL, Barr AM, Ypsilanti AR, Li H-Y, Thornton AE, et al. A novel mechanism and treatment target for presynaptic abnormalities in specific striatal regions in schizophrenia. Neuropsychopharmacology. 2010;35:1226–1238. doi: 10.1038/npp.2009.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Honer WG, Hu L, Davies P. Human synaptic proteins with a heterogeneous distribution in cerebellum and visual cortex. Brain Res. 1993;609:9–20. doi: 10.1016/0006-8993(93)90848-H. [DOI] [PubMed] [Google Scholar]
- 70.Mai JK, Assheuer J, Paxinos G. Atlas of the Human Brain. 3. San Diego, CA: Academic; 1997. [Google Scholar]
- 71.Morrison JH, Baxter MG. The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci. 2012;13:240–250. doi: 10.1038/nrn3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wittig I, Braun H-P, Schägger H. Blue native PAGE. Nat Protoc. 2006;1:418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- 73.Barr AM, Wu CH, Wong C, Hercher C, Töpfer E, Boyda HN, et al. Effects of chronic exercise and treatment with the antipsychotic drug olanzapine on hippocampal volume in adult female rats. Neuroscience. 2013;255:147–157. doi: 10.1016/j.neuroscience.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 74.Sawada K, Barr AM, Nakamura M, Arima K, Young CE, Dwork AJ, et al. Hippocampal complexin proteins and cognitive dysfunction in schizophrenia. Arch Gen Psychiatry. 2005;62:263–272. doi: 10.1001/archpsyc.62.3.263. [DOI] [PubMed] [Google Scholar]
- 75.Hercher C, Chopra V, Beasley CL. Evidence for morphological alterations in prefrontal white matter glia in schizophrenia and bipolar disorder. J Psychiatry Neurosci. 2014;39:376–385. doi: 10.1503/jpn.130277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ramos-Miguel A, Honer WG, Boyda HN, Sawada K, Beasley CL, Procyshyn RM, et al. Exercise prevents downregulation of hippocampal presynaptic proteins following olanzapine-elicited metabolic dysregulation in rats: Distinct roles of inhibitory and excitatory terminals. Neuroscience. 2015;301:298–311. doi: 10.1016/j.neuroscience.2015.06.022. [DOI] [PubMed] [Google Scholar]
- 77.Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J. 2004;86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]