

Abstract
Low-grade gliomas (LGG) constitute grade I and grade II tumors of astrocytic and grade II tumors of oligodendroglial lineage. Although these tumors are typically slow growing, they may be associated with significant morbidity and mortality due to recurrence and malignant progression, even in the setting of optimal resection. LGG in pediatric and adult age groups are currently classified by morphologic criteria. Recent years have heralded a molecular revolution in understanding brain tumors, including LGG. Next generation sequencing has definitively demonstrated that pediatric and adult LGG fundamentally differ in their underlying molecular characteristics, despite being histologically similar. Pediatric LGG show alterations in FGFR1 and BRAF in pilocytic astrocytomas and FGFR1 alterations in diffuse astrocytomas, each converging on the MAP kinase-signaling pathway. Adult LGG are characterized by IDH1/2 mutations and ATRX mutations in astrocytic tumors and IDH1/2 mutations and 1p/19q codeletions in oligodendroglial tumors. TERT promoter mutations are also noted in LGG and are mainly associated with oligodendrogliomas. These findings have considerably refined approaches to classifying these tumors. Moreover, many of the molecular alterations identified in LGG directly impact on prognosis, tumor biology, and the development of novel therapies.
Keywords: Glioma, Low-grade glioma, glioblastoma, pilocytic astrocytoma, IDH1, BRAF, ATRX, 1p/19q codeletion
Introduction
Gliomas constitute the most common primary central nervous system tumor variants. The current World Health Organization (WHO) classification system uses two basic morphologic criteria to delineate individual diagnostic entities. The first defines tumor type on the basis of presumed histiogenesis into either astrocytic or oligodendroglial groupings. Further classification by grade, anticipating biological behavior in the absence of treatment, yields the final classification scheme: Low-grade gliomas - (1) grade I astrocytomas – pilocytic astrocytomas, (2) grade II diffuse astrocytomas, and (3) grade II oligodendrogliomas; High-grade gliomas - (1) Grade III anaplastic astrocytomas, (2) Grade III anaplastic oligodendrogliomas and (3) grade IV glioblastomas (astrocytic tumors). Additional morphologic subtypes, such as angiocentric glioma, have also been described, although their underlying pathogenesis remains relatively obscure.
The past decade has seen a molecular revolution driven by high-throughput sequencing technology, yielding penetrating insights into glioma pathogenesis. Several new driver mutations have been described in gliomas and the field has rapidly grown to reflect the emerging complexity of these tumors. Many of these insights carry prognostic and therapeutic implications and have impacted how we approach the diagnosis and classification of gliomas. As such, histopathologic criteria may no longer be solely sufficient to appropriately classify these tumors and it is increasingly recognized that molecular information should be integrated into standard diagnostic interpretations (1).
While glioblastomas have undergone extensive genomic characterization, low-grade gliomas (LGG) remain comparatively less understood. In contrast to high-grade gliomas, LGG frequently exhibit extend periods of relative indolence in their growth and clinical behavior. Nevertheless, LGG that cannot be surgically resected are often associated with significant morbidity and mortality, with diffuse variants invariably progressing through recurrence to high-grade status. Thus, better molecular characterization of these tumors is critical to developing novel therapeutic targets for effective treatment. Recent years have witnessed significant advances in understanding the molecular characteristics of LGG. Moreover, while histopathologic features characterizing gliomas in adults and children are similar, it is becoming increasingly apparent that tumors in these two age groups have distinct underlying biological foundations. In this article we review the various molecular alterations identified and characterized in pediatric and adult LGG and discuss the implications of recent discoveries from both diagnostic and biologic perspectives.
Molecular aspects of pediatric low-grade gliomas
In recent years, findings from a number of studies have greatly clarified the molecular events likely driving pediatric gliomas. These discoveries include histone mutations in pediatric high-grade astrocytomas and alterations in BRAF, FGFR1 and MYB in astrocytic tumors (2–9). LGG are generally characterized by a lower frequency of somatic mutations and structural variations per tumor, suggesting that their pathogenesis is driven by fewer genetic changes overall. We discuss these molecular events below. Since many of these findings represent new or relatively new discoveries, data regarding prognosis and potential mechanism of pathogenesis are just beginning to emerge
BRAF alterations in LGG
BRAF-KIAA1549 fusions
The most frequent alterations in pediatric LGG involve the v-raf murine sarcoma viral oncogene homolog B1 (BRAF). BRAF is member of the Raf kinase family of proteins involved in the Mitogen-activated protein (MAP) kinase pathway. Pilocytic astrocytomas (grade I) show tandem duplications at chromosome 7q34 resulting in the formation of a fusion gene between the kinase domain of BRAF and KIAA1549 (BRAF-KIAA1549 fusion gene) (4–9). Rare BRAF fusions such as BRAF-FAM131B, BRAF-RNF130, BRAF-CLCN6, BRAF-MKRN1 and BRAF-GNAI1 have also been reported but at a much lower frequency (9, 10) (11). Interestingly, pilocytic astrocytomas show variable rates of BRAF fusion depending on their location in the central nervous system (CNS). For example ~75% of cerebellar pilocytic astrocytomas exhibit BRAF-KIAA1549 fusions. By contrast, only 33% of supratentorial and ~50% of optic nerve pilocytic astrocytomas harbor BRAF fusions (12–14). Data regarding the prognostic effects of BRAF fusion in pediatric LGG are variable, with no clear consensus, and thus may warrant further investigation (12, 13, 15, 16).
BRAF V600E mutation
Nonsynonymous point mutations in BRAF resulting in a valine to glutamic acid substitution at position 600 (V600E) were first described in melanocytic lesions. Subsequently, BRAF V600E mutations were identified in specific subtypes of gliomas. BRAF V600E mutations are most frequent in pleomorphic xanthoastrocytomas (PXA, ~70%) and gangliogliomas (GG, ~20%) and occur at lower frequencies in pilocytic astrocytomas, diffuse astrocytomas and pilomyxoid astrocytomas (8, 17–21). BRAF V600E mutant LGG exhibited a trend towards increased risk for progression (16) and in gangliogliomas was associated with shorter recurrence-free survival (22).
Receptor tyrosine kinase (RTK) FGFR1 alterations in LGG
RTK are cell surface receptors that play a key role in signal transduction. These proteins bear an extracellular domain, a transmembrane domain and an intracellular tyrosine kinase domain (TKD) (23), and have been extensively implicated in the pathogenesis of both adult and pediatric high-grade glioma (24). Two groups simultaneously reported LGG alterations in fibroblast growth factor receptor 1 (FGFR1), an RTK and member of the FGF receptor family that binds to the fibroblast growth factor family of proteins (8, 9).
In diffuse cerebral LGG, intragenic duplications of a portion of the TKD of FGFR1 were noted in 24% of cases (8). These alterations are thought to constitutively activate the receptor independent of ligand activation. Jones et al reported mutations affecting the TKD of FGFR1 in 14/141 (two sample sets including 5/96 and 9/45 tumors negative for BRAF alterations) pilocytic astrocytomas (9). Mutations involved two hotspot codons, Asn546 and Lys656. Moreover, FGFR1 mutant pilocytic astrocytomas tended toward extracerebellar and midline localization.
Interestingly, rare alterations involving the kinase domain of NTRK2 (TrkB), an RTK that binds BDNF, were noted in 2/49 pilocytic astrocytomas resulting in the formation of the fusion genes (QKI-NTRK2 and NACC2-NTRK2) (9). Similar to FGFR1 alterations, the fusion proteins include receptor dimerization domains and are hypothesized to dimerize independent of ligand.
MYB and MYBL1 mutations in LGG
MYB (V-Myb Avian Myeloblastosis Viral Oncogene Homolog) copy number alterations were initially described in 2 diffuse astrocytomas (WHO grade II) and 1 angiocentric glioma (WHO grade II) using Affymetrix SNP arrays and interphase FISH analyses (25). Subsequent efforts using whole genome sequencing, transcriptome and targeted high-throughput sequencing showed rearrangement of MYBL1 (V-Myb Avian Myeloblastosis Viral Oncogene Homolog-Like 1) in one diffuse astrocytoma and rearrangement or copy number alterations of MYB in 5 diffuse astrocytomas, 2 angiocentric gliomas and 1 oligodendroglioma (8). Together, MYBL1 and MYB alterations were present in 25% of diffuse grade II LGG. Alterations in MYB manifested as episome formation, deletion of the negative regulatory region of MYB or deletion of microRNA-binding sites (8).
Another group identified gains on chromosome 8q13.1 involving MYBL1 in 28% (5/18) of pediatric diffuse astrocytomas (26), where they resulted in tandem duplication and truncation of the negative-regulatory C-terminal domain of the protein. MYBL1 truncation products, but not wild type MYBL1, when transduced into NIH-3T3 cells, were able to generate tumors in vitro in soft agar and as xenografts in vivo. Interestingly, this group did not identify MYB alterations in diffuse astrocytomas, although two angiocentric gliomas showed 6q23.3 deletions resulting in truncated MYB.
Genetic alterations in BRAF and FGFR1 give rise to aberrant Mitogen-activated protein kinase (MAP Kinase) pathway signaling
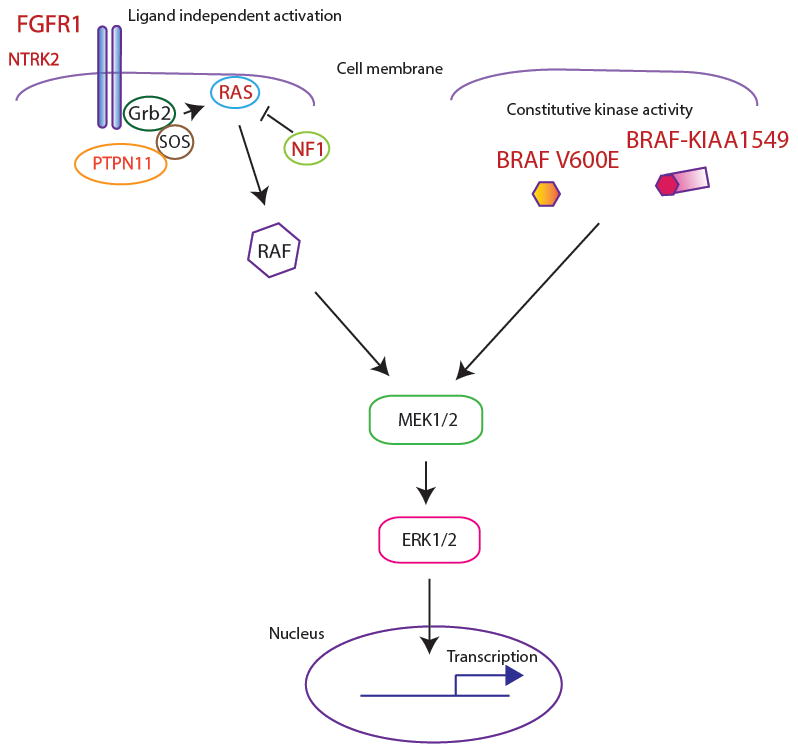
The MAP kinase pathway is critical to normal development and deregulated in a multitude of cancers. Situated downstream of receptor tyrosine kinase activation, pathway signaling is initiated by recruitment of the G-protein Ras (through Shc and Sos), which in turn activates the Raf/MEK/ERK cascade, ultimately leading to protein phosphorylation and activation of multiple protein targets (FIG. 1.).
FIG. 1. IDH 1/2, ATRX, and TERT promoter mutations and 1p/19q codeletion in adult LGG.
IDH1/2 mutations are thought to be an early event in adult LGG pathogenesis in a common precursor cell. Mutations in IDH1 (cytoplasmic) and IDH2 (mitochondrial) result in the generation of the oncometabolite 2-hydroxyglutarate (2-HG). 2-HG is thought to inhibit α-ketoglutarate dependent demethylases resulting in histone and DNA-CpG island methylation (G-CIMP phenotype). Mutations in ATRX are seen in astrocytic tumors. ATRX is a helicase belonging to the SWI/SNF family involved in H3.3 deposition (along with its partner DAXX). Its deficiency induces alternative lengthening of telomeres. 1p/19q codeletion is seen in oligodendrogliomas. CIC and FUBP1 alterations are associated with 1p/19q codeletion in a variable percentage of oligodendrogliomas. TERT promoter mutations are also noted in oligodendrogliomas and are thought to be important for telomere maintenance. Text in red indicates mutations.
In the context of LGG, BRAF fusion proteins and the BRAF V600E mutation each result in aberrant activation of the MAP kinase pathway. BRAF fusion proteins bear the kinase domain, but not the auto-inhibitory N-terminus and thus harbor constitutive kinase activity (27) (FIG. 1.). Similarly, intragenic duplications involving FGFR1 result in a constitutively active receptor protein, with downstream activation of the MAP kinase pathway. Indeed, expression of TKD-duplicated FGFR1 in vitro resulted in activation of the MAP kinase and phosphoinositide 3-kinase (PI3K) pathways and was reversed by FGFR1 inhibitors. Furthermore, Tp53-null astrocytes bearing TKD-duplicated FGFR1 when xenografted into mouse brains generated high-grade gliomas. The authors hypothesize that TKD-duplicated FGFR1 brings two TKD together overriding the requirement for receptor dimerization by FGF for signal transduction (8).
That deregulated MAP kinase pathway signaling serves as a major driver in these tumors is underscored by the identification in LGG, albeit at lower frequency, of mutations in other core pathway components. For instance, mutations in the tyrosine phosphatase PTPN11 were noted in 2/49 pilocytic astrocytomas with FGFR1 mutations (9). PTPN11 encodes a RAS-MAPK–related adaptor protein, suggesting cooperativity with activating FGFR1 alterations (FIG. 1.). Along these lines, mutations in RAS and NF1 (encoding the neurofibromin protein, a negative regulator of RAS) were noted at similar frequencies (8, 9). Together, these findings suggest that MAP kinase pathway activation is required for development of these tumors and, moreover, serves as a unifying pathogenic concept in the broad classification of well-encapsulated WHO grade I neuroepithelial neoplasms like PXA, GG, and pilocytic astrocytoma. Also consistent with these conjectures, conditionally deleted Mek1/2 significantly reduced glial progenitors in mice, leading to failed gliogenesis. Moreover, animals that survived to postnatal stages were nearly devoid of astrocytes and oligodendroglia (28). Similarly, conditional deletion of ERK1/2 in mice resulted in abnormal glial development and reduced progenitor proliferation (29). These factors underscore the importance of MAP kinase signaling in glial development and suggest that aberrant activation of this pathway mediates tumorigenesis in pediatric LGG.
Molecular characteristics of adult low-grade gliomas
IDH 1/2 mutations
The discovery of isocitrate dehydrogenase (IDH) 1/2 mutations in gliomas heralded the genomic era of glioma research. Large profiling studies have identified IDH1/2 mutations in >70% of grade II and grade III gliomas and more than 90% of secondary GBM (30–32). Wild type IDH proteins form core components of the TCA cycle, where they catalyze the conversion of isocitrate to α-ketoglutarate (α-KG) in the cytoplasm (IDH1) and mitochondria (IDH2). IDH mutations are invariably missense and heterozygous, with IDH1 mutations predominating (more than 90%), and involve active site arginine residues, either R132 in IDH1 or R172 in IDH2 (31–34). Mutant IDH1/2 catalyze the generation of the oncometabolite D-2HG from α-KG (35, 36) (FIG. 2.), whose physiologic effects are profound (see below). IDH-mutant gliomas tend to be occur more frequently in young adult patients (31, 37–40). Moreover, the association of IDH1/2 mutation with astrocytomas, oligodendrogliomas and oligoastrocytomas strongly suggests that the event arises early in the pathogenesis of LGG (37, 40, 41) (FIG. 2.). IDH-mutant tumors confer favorable prognosis relative to wild type counterparts regardless of WHO grade (42–46).
FIG. 2. FGFR1 and BRAF alterations in pediatric LGG converge on the MAP kinase pathway.

Alterations in FGFR1 result in constitutive activation of the receptor resulting in activation of the MAP kinase pathway. A subset of pediatric LGG also shows mutations in the receptor tyrosine kinase NTRK2. Rare mutations involve other members of this pathway including RAS, NF1 (negative regulator of RAS) and PTPN11, a tyrosine phosphatase adaptor protein. BRAF V600E mutations and the BRAF-KIAA1549 fusion (other rare fusions not illustrated) also result in constitutive kinase activity and aberrant MAP kinase activation. Text in red indicates mutations/alterations.
A subset of gliomas exhibits widespread DNA hypermethylation across the genome, referred to as the CpG island hypermethylator phenotype (G-CIMP) (47)). G-CIMP is strongly associated with mutations in IDH1/2 in LGG (47–51). Moreover, in both immortalized astrocytes and colon cancer cell lines, expression of IDH1 R132H, the most common glioma-associated IDH mutation, fully recapitulates G-CIMP (50, 51). The oncometabolite D-2HG generated in IDH-mutant tumors inhibits a variety of α-KG-dependent enzymes (52, 53) involved not only in DNA demethylation but also in carnitine synthesis, hypoxic sensing, collagen modifications and histone modification (reviewed in (54)). Indeed, astrocytic cell lines expressing mutant IDH1 R132H, and IDH-mutant oligodendrogliomas show increased trimethylation of histone marks such as H3K9, H3K27 and H3K36 (50, 55, 56) (FIG. 2.). Both DNA and histone hypermethylation, occurring as a consequence of elevated D-2HG, are thought to arrest cellular differentiation by repressing a broad spectrum of target genes (50, 55) (FIG. 2.).
CIC and FUBP1 mutations
Co-incident whole-arm loss of chromosomes 1p and 19q is observed in approximately 70% of oligodendrogliomas and is a significantly favorable prognostic factor (57–59). 1p/19q codeletion results from an unbalanced translocation involving the centromeric regions of chromosomes 1p and 19q (58) (FIG. 2.). Precisely how these genetic abnormalities contribute to oligodendroglial pathogenesis remains unestablished. However, it has been postulated for some time that these regions might harbor potential tumor suppressor genes.
Mutations in CIC (homolog of the Drosophila gene capicua) on chromosome 19q and FUBP1 (FUSE binding protein 1) on chromosome 1p have been recently described in oligodendrogliomas (60–63) (FIG. 2.). CIC mutations were observed in 17–79% of tumors diagnosed at WHO grade II oligodendrogliomas (5/14 (60), 7/9 (63), 11/62 (61), 8/21 (62)). This frequency rose to 25–75% when only 1p/19q codeleted tumors were considered (3/4 (60), 7/9 (63), 9/36 (61), 7/12 (64)). Consistent with a loss-of-function phenotype, CIC mutations are distributed throughout the coding region of the gene (with a predilection for exon 5) and include nonsense, insertions/deletions, missense and frame shift variants. Also consistent with loss-of-fuction, FUBP1 mutations are mainly frameshift and nonsense variants, and occur at lower frequencies (14–22%) than CIC mutations in low-grade oligodendrogliomas (2/14 (60), 2/9 (63), 3/21 (62), 3/17 (64)). CIC is known to functionally repress genes normally activated by RTK signaling by a mechanism called “default repression” (65), and FUBP1 is a DNA-binding protein that activates c-MYC transcription (66). Thus, both CIC and FUBP1 appear to serve as negative regulators of established oncogenic pathways. Nevertheless, establishing the precise mechanisms by which either CIC or FUBP1 mutations contribute to oligodendroglioma pathogenesis will require further study.
ATRX mutations
ATRX (α thalassemia/mental retardation syndrome X-linked) is a DNA helicase and chromatin remodeling protein, belonging to the SWI/SNF family (67). Germline loss-of-function mutations in ATRX are associated with alpha thalassemia mental retardation X-linked (ATR-X) syndrome (68). A primary function of ATRX is incorporation of histone H3.3 monomers into chromatin in collaboration with the histone chaperone protein DAXX (Death-associated protein 6) (69, 70) (FIG. 2.). In 2012, ATRX mutations were described in adult and pediatric astrocytic gliomas (3, 62, 71–73) where they exhibited a strong association with the alternate lengthening of telomeres (ALT) phenotype, a pathological telomere maintenance mechanism though to promote cellular immortality (3, 71, 74, 75) (FIG. 2.). In total, ATRX mutations were found in 33%–67% of grade II astrocytic tumors, and occurred in 75–80% of IDH-mutant LGG that did not also exhibit 1p/19q codeletion (62, 71, 73). In fact, ATRX mutation was mutually exclusive with 1p/19q codeletion in glioma and strongly associated with TP53 mutation (3, 62, 71–73) (FIG. 2.). These data suggest that ATRX mutation, together with TP53 mutation, may delineate a distinct pathogenic route operative in the majority of diffuse astrocytic LGG. That being said, the precise mechanism(s) by which ATRX mutations drive gliomagenesis remain unclear.
TERT promoter mutations
Point mutations in the promoter region of the telomerase reverse transcriptase (TERT) gene were first discovered in melanoma, and are thought to increase telomerase expression, thereby maintaining telomere length and enabling repeated cell division (76, 77). These mutations have been identified in many CNS tumors, including glioblastomas, medulloblastomas and LGG. They occur exclusively at positions −228 and −250 in the TERT promoter region, substituting a cytosine for a thymine in either case to unmask a binding site for ETS family transcription factors (78–82). In LGG, TERT promoter mutations are predominantly observed in oligodendrogliomas (63–78%, 12/19 (78); 25/34 (79) and 29/37 (83)) and less frequently (0–32%) in diffuse astrocytomas (0/8 (78), 10/52 (79) 8/25 (83)). Intriguingly, astrocytic tumors with TERT promoter mutations show an inverse relationship with IDH mutations (83). Additionally, TERT promoter mutations are tightly associated (98–100%) with 1p/19q co-deletion and are mutually exclusive with ATRX mutations in LGG (78–80, 83) (FIG. 2.). These findings emphasize the importance of pathological telomere maintenance in LGG, whether by way of TERT promoter mutations in 1p/19q codeleted tumors (predominantly oligodendroglioma), or ALT in ATRX-mutant tumors (predominantly astrocytoma).
Diagnostic and therapeutic implications of molecular genetics in pediatric and adult LGG
Diagnostic implications
As detailed above, high-throughput molecular profiling has dramatically altered conceptions of glioma biology and, in doing so, has led to refinements of well-established classification schemes (FIG. 1 and 2). Perhaps most notably, the identification of IDH1/2 mutations in both low and high-grade adult gliomas is now of considerable importance, due to the significant prognostic benefit conferred by the genomic alteration. The standard initial approach of many pathology practices is immunohistochemical, using an antibody specifically directed against IDH1 R132H (accounting for more than 95% of all glioma-associated IDH mutations), followed by sequencing-based genotyping in immunonegative cases. The availability of a robust immunohistochemical reagent recognizing most IDH-mutant tumors also facilitates the differentiation of true glial neoplasms from non-neoplastic glioma mimics such as reactive astrogliosis (33, 84–88).
The mutual exclusivity of ATRX mutation and 1p/19q codeletions in LGG has prompted the proposal that all diffuse gliomas be classified on the basis of IDH and ATRX mutational status—or a negative staining pattern by IHC (FIG. 3A)—combined with 1p/19q codeletion. This approach may lend better clarity to the often-subjective diagnosis of mixed lineage gliomas (oligoastrocytomas), as these tumors have been shown to nicely segregate into ATRX-mutant and 1p/19q codeleted subgroups (62, 71, 73). Moreover, prognostic stratification among gliomas delineated by these molecular criteria outperforms that seen following conventional histopathological classification (62) (89). CIC and FUBP1 mutations do not bear 100% concurrence with 1p/19q codeletions (60–63). Thus, from a diagnostic and prognostic view, assessment of 1p/19q deletion remains superior to CIC and FUBP1 genotyping in the establishment of oligodendroglial lineage. As an aside, the near universal occurrence of TERT mutations in 1p/19q-codeleted LGG suggests that combined IDH1/2 and TERT genotyping might also support the rendering of an oligodendroglioma diagnosis (78).
FIG. 3. Immunohistochemical assessment of mutations in ATRX and BRAF.

A, Loss-of-function mutations in ATRX lead to loss of nuclear expression. H & E stained and ATRX-immunostained micrographs of ATRX-mut and ATRX-wt tumors are shown. B, positive BRAF V600E immunostaining in a mutant-harboring PXA. H& E staining also shown.
In pediatric LGG, the identification of BRAF alterations—by molecular techniques or the BRAF V600E antibody (FIG. 3B)—is of significance due to the potential for targeted therapies (see below). Identifying BRAF alterations may also help in diagnostically challenging cases by designating encapsulated WHO grade I astrocytic variants (e.g. pilocytic astrocytoma) from other neoplastic entities (19, 90). That being said, more studies are required to fully assess the impact of BRAF, FGFR1 and MYB/MYBL1 alterations on current classification of pediatric LGG.
Therapeutic implications
The advances in the molecular characterization of LGG discussed above have provided numerous insights into potential pathogenic mechanisms. Indeed, these studies have yielded an array of therapeutic targets that can be leveraged to design novel therapies. Many of these efforts are still in experimental stages. However, pharmaceutically targeting BRAF V600E has already achieved considerable success in melanoma (91–93) and V600E inhibitors were effective in preclinical animal models of high-grade glioma (94, 95) and a single case of a 12-year-old patient with GBM (96). However, these successes should be treated with cautious optimism due to the established existence of multiple mechanisms of resistance to BRAF inhibitors in other tumor types (97). Recent studies have also shown that mutant IDH1 inhibition is partially effective in xenograft glioma models, although blood-brain barrier permeability remains an issue (98). Alternatively, vaccine-based approaches against IDH1 R132H have elicited anti-tumor immune responses in tumors bearing the mutation (99). The utility of these and other therapeutic strategies in targeting LGG remains unclear and will be the subject of extensive research effort in the immediate future.
Acknowledgments
S.V. is supported by NCI-K08 CA181475.
Footnotes
The authors have no conflicts of interest.
References
- 1.Louis DN, Perry A, Burger P, et al. International Society of Neuropathology-Haarlem Consensus Guidelines, for Nervous System Tumor Classification and Grading. Brain Pathol. 2014 doi: 10.1111/bpa.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu G, Broniscer A, McEachron TA, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nature genetics. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwartzentruber J, Korshunov A, Liu XY, et al. Driver mutations in histone H3. 3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 4.Bar EE, Lin A, Tihan T, et al. Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. Journal of neuropathology and experimental neurology. 2008;67:878–887. doi: 10.1097/NEN.0b013e3181845622. [DOI] [PubMed] [Google Scholar]
- 5.Jones DT, Kocialkowski S, Liu L, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer research. 2008;68:8673–8677. doi: 10.1158/0008-5472.CAN-08-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pfister S, Janzarik WG, Remke M, et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. The Journal of clinical investigation. 2008;118:1739–1749. doi: 10.1172/JCI33656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sievert AJ, Jackson EM, Gai X, et al. Duplication of 7q34 in pediatric low-grade astrocytomas detected by high-density single-nucleotide polymorphism-based genotype arrays results in a novel BRAF fusion gene. Brain pathology. 2009;19:449–458. doi: 10.1111/j.1750-3639.2008.00225.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J, Wu G, Miller CP, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nature genetics. 2013;45:602–612. doi: 10.1038/ng.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones DT, Hutter B, Jager N, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nature genetics. 2013;45:927–932. doi: 10.1038/ng.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cin H, Meyer C, Herr R, et al. Oncogenic FAM131B-BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta neuropathologica. 2011;121:763–774. doi: 10.1007/s00401-011-0817-z. [DOI] [PubMed] [Google Scholar]
- 11.Roth JJ, Santi M, Pollock AN, et al. Chromosome band 7q34 deletions resulting in KIAA1549-BRAF and FAM131B-BRAF fusions in pediatric low grade gliomas. Brain Pathol. 2014 doi: 10.1111/bpa.12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tihan T, Ersen A, Qaddoumi I, et al. Pathologic characteristics of pediatric intracranial pilocytic astrocytomas and their impact on outcome in 3 countries: a multi-institutional study. The American journal of surgical pathology. 2012;36:43–55. doi: 10.1097/PAS.0b013e3182329480. [DOI] [PubMed] [Google Scholar]
- 13.Horbinski C, Hamilton RL, Nikiforov Y, et al. Association of molecular alterations, including BRAF, with biology and outcome in pilocytic astrocytomas. Acta neuropathologica. 2010;119:641–649. doi: 10.1007/s00401-009-0634-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horbinski C. To BRAF or not to BRAF: is that even a question anymore? Journal of neuropathology and experimental neurology. 2013;72:2–7. doi: 10.1097/NEN.0b013e318279f3db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hawkins C, Walker E, Mohamed N, et al. BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:4790–4798. doi: 10.1158/1078-0432.CCR-11-0034. [DOI] [PubMed] [Google Scholar]
- 16.Horbinski C, Nikiforova MN, Hagenkord JM, et al. Interplay among BRAF, p16, p53, and MIB1 in pediatric low-grade gliomas. Neuro-oncology. 2012;14:777–789. doi: 10.1093/neuonc/nos077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schindler G, Capper D, Meyer J, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta neuropathologica. 2011;121:397–405. doi: 10.1007/s00401-011-0802-6. [DOI] [PubMed] [Google Scholar]
- 18.Koelsche C, Sahm F, Wohrer A, et al. BRAF-Mutated Pleomorphic Xanthoastrocytoma is Associated with Temporal Location, Reticulin Fiber Deposition and CD34 Expression. Brain pathology. 2013 doi: 10.1111/bpa.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ida CM, Vrana JA, Rodriguez FJ, et al. Immunohistochemistry is highly sensitive and specific for detection of BRAF V600E mutation in pleomorphic xanthoastrocytoma. Acta neuropathologica communications. 2013;1:20. doi: 10.1186/2051-5960-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dias-Santagata D, Lam Q, Vernovsky K, et al. BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: diagnostic and therapeutic implications. PloS one. 2011;6:e17948. doi: 10.1371/journal.pone.0017948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chappe C, Padovani L, Scavarda D, et al. Dysembryoplastic neuroepithelial tumors share with pleomorphic xanthoastrocytomas and gangliogliomas BRAF(V600E) mutation and expression. Brain Pathol. 2013;23:574–583. doi: 10.1111/bpa.12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dahiya S, Haydon DH, Alvarado D, et al. BRAF(V600E) mutation is a negative prognosticator in pediatric ganglioglioma. Acta neuropathologica. 2013;125:901–910. doi: 10.1007/s00401-013-1120-y. [DOI] [PubMed] [Google Scholar]
- 23.Goetz R, Mohammadi M. Exploring mechanisms of FGF signalling through the lens of structural biology. Nat Rev Mol Cell Bio. 2013;14:166–180. doi: 10.1038/nrm3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huse JT, Aldape KD. The molecular landscape of diffuse glioma and prospects for biomarker development. Expert Opin Med Diagn. 2013;7:573–587. doi: 10.1517/17530059.2013.846321. [DOI] [PubMed] [Google Scholar]
- 25.Tatevossian RG, Tang B, Dalton J, et al. MYB upregulation and genetic aberrations in a subset of pediatric low-grade gliomas. Acta Neuropathol. 2010;120:731–743. doi: 10.1007/s00401-010-0763-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramkissoon LA, Horowitz PM, Craig JM, et al. Genomic analysis of diffuse pediatric low-grade gliomas identifies recurrent oncogenic truncating rearrangements in the transcription factor MYBL1. Proc Natl Acad Sci U S A. 2013;110:8188–8193. doi: 10.1073/pnas.1300252110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodriguez FJ, Lim KS, Bowers D, et al. Pathological and molecular advances in pediatric low-grade astrocytoma. Annual review of pathology. 2013;8:361–379. doi: 10.1146/annurev-pathol-020712-164009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li XY, Newbern JM, Wu YH, et al. MEK Is a Key Regulator of Gliogenesis in the Developing Brain. Neuron. 2012;75:1035–1050. doi: 10.1016/j.neuron.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Imamura O, Pages G, Pouyssegur J, et al. ERK1 and ERK2 are required for radial glial maintenance and cortical lamination. Genes to cells : devoted to molecular & cellular mechanisms. 2010;15:1072–1088. doi: 10.1111/j.1365-2443.2010.01444.x. [DOI] [PubMed] [Google Scholar]
- 30.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balss J, Meyer J, Mueller W, et al. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008;116:597–602. doi: 10.1007/s00401-008-0455-2. [DOI] [PubMed] [Google Scholar]
- 33.Capper D, Zentgraf H, Balss J, et al. Monoclonal antibody specific for IDH1 R132H mutation. Acta Neuropathol. 2009;118:599–601. doi: 10.1007/s00401-009-0595-z. [DOI] [PubMed] [Google Scholar]
- 34.Hartmann C, Meyer J, Balss J, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 2009;118:469–474. doi: 10.1007/s00401-009-0561-9. [DOI] [PubMed] [Google Scholar]
- 35.Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2010;465:966. doi: 10.1038/nature09132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watanabe T, Nobusawa S, Kleihues P, et al. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol. 2009;174:1149–1153. doi: 10.2353/ajpath.2009.080958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mellai M, Piazzi A, Caldera V, et al. IDH1 and IDH2 mutations, immunohistochemistry and associations in a series of brain tumors. J Neurooncol. 2011;105:345–357. doi: 10.1007/s11060-011-0596-3. [DOI] [PubMed] [Google Scholar]
- 39.Metellus P, Coulibaly B, Colin C, et al. Absence of IDH mutation identifies a novel radiologic and molecular subtype of WHO grade II gliomas with dismal prognosis. Acta Neuropathol. 2010;120:719–729. doi: 10.1007/s00401-010-0777-8. [DOI] [PubMed] [Google Scholar]
- 40.Hartmann C, Meyer J, Balss J, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta neuropathologica. 2009;118:469–474. doi: 10.1007/s00401-009-0561-9. [DOI] [PubMed] [Google Scholar]
- 41.Lai A, Kharbanda S, Pope WB, et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J Clin Oncol. 2011;29:4482–4490. doi: 10.1200/JCO.2010.33.8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Houillier C, Wang X, Kaloshi G, et al. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology. 2010;75:1560–1566. doi: 10.1212/WNL.0b013e3181f96282. [DOI] [PubMed] [Google Scholar]
- 43.Gorovets D, Kannan K, Shen RL, et al. IDH Mutation and Neuroglial Developmental Features Define Clinically Distinct Subclasses of Lower Grade Diffuse Astrocytic Glioma. Clinical Cancer Research. 2012;18:2490–2501. doi: 10.1158/1078-0432.CCR-11-2977. [DOI] [PubMed] [Google Scholar]
- 44.Leu S, von Felten S, Frank S, et al. IDH/MGMT-driven molecular classification of low-grade glioma is a strong predictor for long-term survival. Neuro Oncol. 2013;15:469–479. doi: 10.1093/neuonc/nos317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun HR, Yin LH, Li SW, et al. Prognostic significance of IDH mutation in adult low-grade gliomas: a meta-analysis. J Neurooncol. 2013;113:277–284. doi: 10.1007/s11060-013-1107-5. [DOI] [PubMed] [Google Scholar]
- 46.Sabha N, Knobbe CB, Maganti M, et al. Analysis of IDH mutation, 1p/19q deletion, and PTEN loss delineates prognosis in clinical low-grade diffuse gliomas. Neuro Oncol. 2014;16:914–923. doi: 10.1093/neuonc/not299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laffaire J, Everhard S, Idbaih A, et al. Methylation profiling identifies 2 groups of gliomas according to their tumorigenesis. Neuro Oncol. 2011;13:84–98. doi: 10.1093/neuonc/noq110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Christensen BC, Smith AA, Zheng S, et al. DNA methylation, isocitrate dehydrogenase mutation, and survival in glioma. Journal of the National Cancer Institute. 2011;103:143–153. doi: 10.1093/jnci/djq497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Turcan S, Rohle D, Goenka A, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Duncan CG, Barwick BG, Jin G, et al. A heterozygous IDH1R132H/WT mutation induces genome-wide alterations in DNA methylation. Genome Res. 2012 doi: 10.1101/gr.132738.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chowdhury R, Yeoh KK, Tian YM, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12:463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Loenarz C, Schofield CJ. Expanding chemical biology of 2-oxoglutarate oxygenases. Nat Chem Biol. 2008;4:152–156. doi: 10.1038/nchembio0308-152. [DOI] [PubMed] [Google Scholar]
- 55.Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell Metab. 2012;16:9–17. doi: 10.1016/j.cmet.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Venneti S, Felicella MM, Coyne T, et al. Histone 3 lysine 9 trimethylation is differentially associated with isocitrate dehydrogenase mutations in oligodendrogliomas and high-grade astrocytomas. J Neuropathol Exp Neurol. 2013;72:298–306. doi: 10.1097/NEN.0b013e3182898113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith JS, Perry A, Borell TJ, et al. Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. Journal of Clinical Oncology. 2000;18:636–645. doi: 10.1200/JCO.2000.18.3.636. [DOI] [PubMed] [Google Scholar]
- 58.Jenkins RB, Blair H, Ballman KV, et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer research. 2006;66:9852–9861. doi: 10.1158/0008-5472.CAN-06-1796. [DOI] [PubMed] [Google Scholar]
- 59.Cairncross G, Jenkins R. Gliomas with 1p/19q codeletion: a.k.a. oligodendroglioma. Cancer journal. 2008;14:352–357. doi: 10.1097/PPO.0b013e31818d8178. [DOI] [PubMed] [Google Scholar]
- 60.Bettegowda C, Agrawal N, Jiao Y, et al. Mutations in CIC and FUBP1 contribute to human oligodendroglioma. Science (New York, NY. 2011;333:1453–1455. doi: 10.1126/science.1210557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yip S, Butterfield YS, Morozova O, et al. Concurrent CIC mutations, IDH mutations, and 1p/19q loss distinguish oligodendrogliomas from other cancers. J Pathol. 2012;226:7–16. doi: 10.1002/path.2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jiao Y, Killela PJ, Reitman ZJ, et al. Frequent ATRX, CIC, and FUBP1 mutations refine the classification of malignant gliomas. Oncotarget. 2012;3:709–722. doi: 10.18632/oncotarget.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sahm F, Koelsche C, Meyer J, et al. CIC and FUBP1 mutations in oligodendrogliomas, oligoastrocytomas and astrocytomas. Acta Neuropathol. 2012;123:853–860. doi: 10.1007/s00401-012-0993-5. [DOI] [PubMed] [Google Scholar]
- 64.Eisenreich S, Abou-El-Ardat K, Szafranski K, et al. Novel CIC point mutations and an exon-spanning, homozygous deletion identified in oligodendroglial tumors by a comprehensive genomic approach including transcriptome sequencing. PLoS One. 2013;8:e76623. doi: 10.1371/journal.pone.0076623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jimenez G, Shvartsman SY, Paroush Z. The Capicua repressor--a general sensor of RTK signaling in development and disease. Journal of cell science. 2012;125:1383–1391. doi: 10.1242/jcs.092965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang J, Chen QM. Far upstream element binding protein 1: a commander of transcription, translation and beyond. Oncogene. 2013;32:2907–2916. doi: 10.1038/onc.2012.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Picketts DJ, Higgs DR, Bachoo S, et al. ATRX encodes a novel member of the SNF2 family of proteins: mutations point to a common mechanism underlying the ATR-X syndrome. Hum Mol Genet. 1996;5:1899–1907. doi: 10.1093/hmg/5.12.1899. [DOI] [PubMed] [Google Scholar]
- 68.Gibbons RJ, Picketts DJ, Villard L, et al. Mutations in a putative global transcriptional regulator cause X-linked mental retardation with alpha-thalassemia (ATR-X syndrome) Cell. 1995;80:837–845. doi: 10.1016/0092-8674(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 69.Goldberg AD, Banaszynski LA, Noh KM, et al. Distinct Factors Control Histone Variant H3. 3 Localization at Specific Genomic Regions. Cell. 2010;140:678–691. doi: 10.1016/j.cell.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lewis PW, Elsaesser SJ, Noh KM, et al. Daxx is an H3. 3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci U S A. 2010;107:14075–14080. doi: 10.1073/pnas.1008850107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kannan K, Inagaki A, Silber J, et al. Whole-exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget. 2012;3:1194–1203. doi: 10.18632/oncotarget.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Khuong-Quang DA, Buczkowicz P, Rakopoulos P, et al. K27M mutation in histone H3. 3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012;124:439–447. doi: 10.1007/s00401-012-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu XY, Gerges N, Korshunov A, et al. Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol. 2012;124:615–625. doi: 10.1007/s00401-012-1031-3. [DOI] [PubMed] [Google Scholar]
- 74.Nguyen DN, Heaphy CM, de Wilde RF, et al. Molecular and Morphologic Correlates of the Alternative Lengthening of Telomeres Phenotype in High-Grade Astrocytomas. Brain Pathol. 2012 doi: 10.1111/j.1750-3639.2012.00630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Abedalthagafi M, Phillips JJ, Kim GE, et al. The alternative lengthening of telomere phenotype is significantly associated with loss of ATRX expression in high-grade pediatric and adult astrocytomas: a multi-institutional study of 214 astrocytomas. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2013;26:1425–1432. doi: 10.1038/modpathol.2013.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Horn S, Figl A, Rachakonda PS, et al. TERT promoter mutations in familial and sporadic melanoma. Science (New York, NY. 2013;339:959–961. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- 77.Huang FW, Hodis E, Xu MJ, et al. Highly recurrent TERT promoter mutations in human melanoma. Science (New York, NY. 2013;339:957–959. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Killela PJ, Reitman ZJ, Jiao Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110:6021–6026. doi: 10.1073/pnas.1303607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Arita H, Narita Y, Fukushima S, et al. Upregulating mutations in the TERT promoter commonly occur in adult malignant gliomas and are strongly associated with total 1p19q loss. Acta Neuropathol. 2013;126:267–276. doi: 10.1007/s00401-013-1141-6. [DOI] [PubMed] [Google Scholar]
- 80.Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu X, Wu G, Shan Y, et al. Highly prevalent TERT promoter mutations in bladder cancer and glioblastoma. Cell Cycle. 2013;12:1637–1638. doi: 10.4161/cc.24662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lindsey JC, Schwalbe EC, Potluri S, et al. TERT promoter mutation and aberrant hypermethylation are associated with elevated expression in medulloblastoma and characterise the majority of non-infant SHH subgroup tumours. Acta Neuropathol. 2014;127:307–309. doi: 10.1007/s00401-013-1225-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Koelsche C, Sahm F, Capper D, et al. Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol. 2013;126:907–915. doi: 10.1007/s00401-013-1195-5. [DOI] [PubMed] [Google Scholar]
- 84.Capper D, Weissert S, Balss J, et al. Characterization of R132H mutation-specific IDH1 antibody binding in brain tumors. Brain Pathol. 2010;20:245–254. doi: 10.1111/j.1750-3639.2009.00352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Capper D, Sahm F, Hartmann C, et al. Application of mutant IDH1 antibody to differentiate diffuse glioma from nonneoplastic central nervous system lesions and therapy-induced changes. Am J Surg Pathol. 2010;34:1199–1204. doi: 10.1097/PAS.0b013e3181e7740d. [DOI] [PubMed] [Google Scholar]
- 86.Horbinski C, Kofler J, Yeaney G, et al. Isocitrate dehydrogenase 1 analysis differentiates gangliogliomas from infiltrative gliomas. Brain Pathol. 2011;21:564–574. doi: 10.1111/j.1750-3639.2011.00480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Camelo-Piragua S, Jansen M, Ganguly A, et al. A sensitive and specific diagnostic panel to distinguish diffuse astrocytoma from astrocytosis: chromosome 7 gain with mutant isocitrate dehydrogenase 1 and p53. J Neuropathol Exp Neurol. 2011;70:110–115. doi: 10.1097/NEN.0b013e31820565f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Camelo-Piragua S, Jansen M, Ganguly A, et al. Mutant IDH1-specific immunohistochemistry distinguishes diffuse astrocytoma from astrocytosis. Acta Neuropathol. 2010;119:509–511. doi: 10.1007/s00401-009-0632-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wiestler B, Capper D, Holland-Letz T, et al. ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta neuropathologica. 2013;126:443–451. doi: 10.1007/s00401-013-1156-z. [DOI] [PubMed] [Google Scholar]
- 90.Ida CM, Lambert SR, Rodriguez FJ, et al. BRAF alterations are frequent in cerebellar low-grade astrocytomas with diffuse growth pattern. J Neuropathol Exp Neurol. 2012;71:631–639. doi: 10.1097/NEN.0b013e31825c448a. [DOI] [PubMed] [Google Scholar]
- 91.Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–714. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nicolaides TP, Li H, Solomon DA, et al. Targeted therapy for BRAFV600E malignant astrocytoma. Clin Cancer Res. 2011;17:7595–7604. doi: 10.1158/1078-0432.CCR-11-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Huillard E, Hashizume R, Phillips JJ, et al. Cooperative interactions of BRAFV600E kinase and CDKN2A locus deficiency in pediatric malignant astrocytoma as a basis for rational therapy. Proc Natl Acad Sci U S A. 2012;109:8710–8715. doi: 10.1073/pnas.1117255109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Robinson GW, Orr BA, Gajjar A. Complete clinical regression of a BRAF V600E-mutant pediatric glioblastoma multiforme after BRAF inhibitor therapy. BMC cancer. 2014;14:258. doi: 10.1186/1471-2407-14-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Menzies AM, Long GV. Systemic treatment for BRAF-mutant melanoma: where do we go next? The lancet oncology. 2014;15:e371–e381. doi: 10.1016/S1470-2045(14)70072-5. [DOI] [PubMed] [Google Scholar]
- 98.Rohle D, Popovici-Muller J, Palaskas N, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science (New York, NY. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schumacher T, Bunse L, Pusch S, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. 2014;512:324–327. doi: 10.1038/nature13387. [DOI] [PubMed] [Google Scholar]