

Abstract
Currently, there is a trend of an increasing number of Plasmodium vivaxmalaria cases in China that are imported across its Southeast Asia border, especially in the China-Myanmar border area (CMB). To date, little is known about the genetic diversity of P. vivax in this region. In this paper, we report the first genome sequencing of a P. vivaxisolate (CMB-1) from a vivax malaria patient in CMB. The sequencing data were aligned onto 96.43% of the P. vivax Salvador I reference strain (Sal I) genome with 7.84-fold coverage as well as onto 98.32% of 14 Sal I chromosomes. Using the de novo assembly approach, we generated 8,541 scaffolds and assembled a total of 27.1 Mb of sequence into CMB-1 scaffolds. Furthermore, we identified all 295 known virgenes, which is the largest subtelomeric multigene family in malaria parasites. These results provide an important foundation for further research onP. vivax population genetics.
Keywords: Plasmodium vivax, genome, vir
As the most common human malaria species with the widest geographic distribution,Plasmodium vivax is mostly found outside of Africa and is especially prevalent in Southeast Asia and America (Price et al. 2007). The P. vivax parasite is now considered the cause of severe malaria syndromes that have been blamed on P. falciparum (Price et al. 2009). It was estimated that half of the world’s population is at risk of P. vivax malaria (Guerra et al. 2010).
In China, P. vivax was the major species for a relatively long time. The Yunnan province remains the highest transmission area in China, particularly in the southern border areas adjacent to Myanmar, a highly endemic area for P. vivax malaria in the Greater Mekong Subregion countries (Zhou et al. 2014). Due to the increasing numbers of Chinese working abroad, the number of imported P. vivax cases has exhibited an increasing trend in recent years (Feng et al. 2015). The imported P. vivax malaria may lead to high malaria risk in malaria-free localities where the Anopheles sinensismosquito is prevalent, particularly in central China, such as in Anhui and Henan provinces (Gao et al. 2004). In 2012, 1,143P. vivax malaria cases were reported in China, accounting for 41.9% of the total malaria cases.
Hundreds of P. falciparum isolates have been sequenced or genotyped (Winzeler 2008), but less information onP. vivax isolates has been reported. The first complete genome ofP. vivax was published in 2008 (Carlton et al. 2008), revealing that P. vivax resembles other Plasmodium spp in gene content and metabolic traits, but it possesses novel gene families and potential alternative invasion pathways not previously been recognised. Although they were started more recently, there are several P. vivax isolate genome sequencing projects underway and more sequence data have been made available (Neafsey et al. 2012). Previously, 23 isolates genome data were accessible in the National Center for Biotechnology Information (NCBI) database. However, no isolates from China or China’s borders with Southeast Asian countries have previously been included.
In this paper, we report the first P. vivax genome sequence of a clinical isolate obtained in the China-Myanmar border area (CMB-1) as well as in the China-Southeast Asia border areas. The genomic DNA of the P. vivaxCMB-1 for sequencing was extracted from a whole blood sample from a patient with microscopically positive for P. vivax and polymerase chain reaction confirmed sole infection with P. vivax. The Ethical Committee of the National Institute of Parasitic Diseases, China Center for Disease Control and Prevention approved the study (NIPD 2013-010). The study protocol, potential risks and potential benefits were explained to the patient and informed consent was verbally obtained. The genomic DNA was used to construct the Illumina sequencing library with insert sizes of 360 bp. The library was sequenced on a HiSeq 2000 sequencer. After filtering the sequences as for the Homo sapiens genome, the reads werede novo assembled using an A5 assembly pipeline (Coil et al. 2015). The Illumina sequencing reads have been submitted to the NCBI Short Read Archive (accession SRS941624).
The whole genome sequencing generated 31,471,932 paired-end reads with an average read length of 125 bp. Low-quality bases and adapters were trimmed using Trimmomatic v.0.30 (Bolger et al. 2014). The sequence reads were aligned to the P. vivax Salvador I reference strain (Sal I) genome using BWA-0.7.1 (Li & Durbin 2009). In total, 5.86% of 26 million quality-evaluated reads were aligned onto 96.43% of the Sal I genome with 7.84-fold coverage as well as onto 98.32% of the 14 chromosomes of the Sal I strain covering 95.96-99.05% for each chromosome.
The de novo assembly yielded a database with 8,541 scaffolds (10,639 contigs) and an average guanine-cytosine content of 39.1%. A total sequence coverage of 10.26-fold produced this assembly with N50 scaffold lengths of 5.9 kb. A total of 27.1 Mb of sequence was assembled in the CMB-1 scaffolds (Table).
TABLE . De novo assembly statistics of the Plasmodium vivax China-Myanmar border area-1 genome.
| Attribute | |
|---|---|
| Raw reads | 15,105,614 paired |
| Unmapped reads to Homo sapiens | 1,268,041 paired |
| After quality control | 1,267,552 paired |
| Contigs (n) | 10,639 |
| Scaffolds (n) | 8,541 |
| Longest scaffold (bp) | 125,157 |
| N50 (bp) | 5,936 |
| Genome size (bp) | 27,164,492 |
| Coverage | 10.26 |
Genomic map of Plasmodium vivax China-Myanmar border area (CMB)-1 isolate. The outermost circle shows the whole genome ofP. vivax Salvador I reference strain. The next ring represents the location of 295 vir genes, each subfamily marked in different colour. The third ring shows the coding sequences zone of CMB-1 isolate scaffolds and vir gene orthologs are indicated by black. The inner circle shows the genomic map of P. vivax CMB-1 isolate and the histogram represents the degree of similarity (blue: identity > 99%). The figure was drawn using Circos (Krzywinski et al. 2009) and alignment was performed using MUMmer 3.0 (Kurtz et al. 2004).
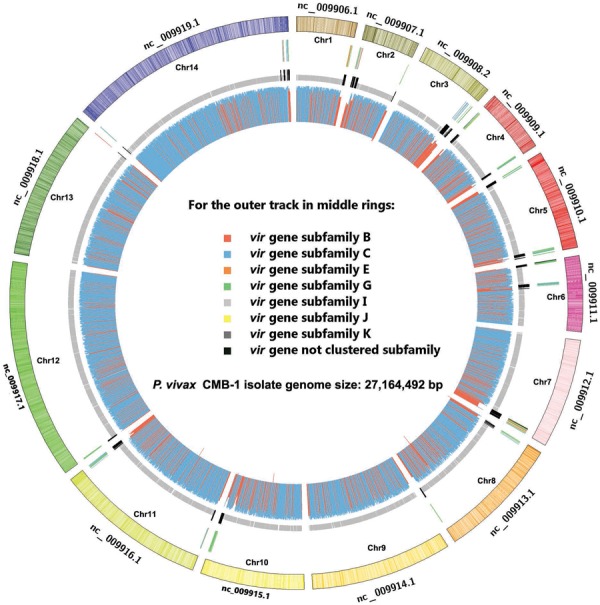
An overall comparative genomic analysis was conducted using the complete genome of theP. vivax reference strain Sal I, as shown in Figure. It is widely known that the vir super-family is variably expressed and encodes proteins that are exported to the host cell surface to evade the host adaptive immune response (Fernandez-Becerra et al. 2009). As the largest subtelomeric multigene family of malaria parasites, the virsuper-family consists of seven different subfamilies. In the CMB-1 de novo assembled sequences, we identified all published 295vir genes (Lopez et al. 2013) based on their sequence similarity in BLASTX.
The findings in this paper provide whole genomic information on the current epidemiological scenario of vivax malaria in the CMB, where the number of P. vivax cases imported from Southeast Asia is increasing and accompanied by growing concern. The results of this work contribute to a better understanding ofP. vivax evolution and provide an informative basis for further study of the population genomics of this parasite.
ACKNOWLEDGEMENTS
To the staff of the Yunnan Institute of Parasitic Diseases, for collection of the blood sample from P. vivax-infected individual, and to Prof Zheng Feng, for critical reading of the paper.
Footnotes
Financial support: International S&T Cooperation Program (2014DFA31130), Foundation of National Science and Technology Major Program (2012ZX10004-220), Special Fund for Health Research in the Public Interest (201202019)
REFERENCES
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton JM, Adams JH, Silva JC, Bidwell SL, Lorenzi H, Caler E, Crabtree J, Angiuoli SV, Merino EF, Amedeo P. Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature. 2008;455:757–763. doi: 10.1038/nature07327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coil D, Jospin G, Darling AE. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics. 2015;31:587–589. doi: 10.1093/bioinformatics/btu661. [DOI] [PubMed] [Google Scholar]
- Feng J, Xiao H, Zhang L, Yan H, Feng X, Fang W, Xia Z. The Plasmodium vivax in China: decreased in local cases but increased imported cases from Southeast Asia and Africa. Sci Rep. 2015;5: doi: 10.1038/srep08847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Becerra C, Yamamoto MM, Vencio RZ, Lacerda M, Rosanas-Urgell A, del Portillo HA. Plasmodium vivax and the importance of the subtelomeric multigene vir superfamily. Trends Parasitol. 2009;25:44–51. doi: 10.1016/j.pt.2008.09.012. [DOI] [PubMed] [Google Scholar]
- Gao Q, Beebe N, Cooper R. Molecular identification of the malaria vectors Anopheles anthropophagus and Anopheles sinensis (Diptera: Culicidae) in central China using polymerase chain reaction and appraisal of their position within the Hyrcanus group. J Med Entomol. 2004;41:5–11. doi: 10.1603/0022-2585-41.1.5. [DOI] [PubMed] [Google Scholar]
- Guerra CA, Howes RE, Patil AP, Gething PW, Van Boeckel TP, Temperley WH, Kabaria CW, Tatem AJ, Manh BH, Elyazar IR. The international limits and population at risk of Plasmodium vivax transmission in 2009. PLoS Negl Trop Dis. 2010;4: doi: 10.1371/journal.pntd.0000774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5: doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez FJ, Bernabeu M, Fernandez-Becerra C, del Portillo HA. A new computational approach redefines the subtelomeric vir superfamily of Plasmodium vivax. BMC Genomics. 2013;14: doi: 10.1186/1471-2164-14-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neafsey DE, Galinsky K, Jiang RH, Young L, Sykes SM, Saif S, Gujja S, Goldberg JM, Young S, Zeng Q. The malaria parasite Plasmodium vivax exhibits greater genetic diversity than Plasmodium falciparum. Nat Genet. 2012;44:1046–1050. doi: 10.1038/ng.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price RN, Douglas NM, Anstey NM. New developments in Plasmodium vivax malaria: severe disease and the rise of chloroquine resistance. Curr Opin Infect Dis. 2009;22:430–435. doi: 10.1097/QCO.0b013e32832f14c1. [DOI] [PubMed] [Google Scholar]
- Price RN, Tjitra E, Guerra CA, Yeung S, White NJ, Anstey NM. Vivax malaria: neglected and not benign. Am J Trop Med Hyg. 2007;77:79–87. [PMC free article] [PubMed] [Google Scholar]
- Winzeler EA. Malaria research in the post-genomic era. Nature. 2008;455:751–756. doi: 10.1038/nature07361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Huang JL, Njuabe MT, Li SG, Chen JH, Zhou XN. A molecular survey of febrile cases in malaria-endemic areas along China-Myanmar border in Yunnan province, People’s Republic of China. Parasite. 2014;21: doi: 10.1051/parasite/2014030. [DOI] [PMC free article] [PubMed] [Google Scholar]