

Abstract
Hereditary Long QT Syndrome is caused by deleterious mutation in one of several genetic loci, including locus LQT2 that contains the KCNH2 gene (or hERG), causing faulty cardiac repolarization. Here, we describe and characterize a novel mutation, p.Asp219Val in the hERG channel, identified in an 11 year old male with syncope and prolonged QT interval. Genetic sequencing showed a non-synonymous variation in KCNH2 (c.656A>T: amino acid p.Asp219Val). p.Asp219Val resides in a region of the channel predicted to be unstructured and flexible, located between the PAS (Per-Arnt-Sim) domain and its interaction sites in the transmembrane domain. The p.Asp219Val hERG channel produced K+ current that activated with modest changes in voltage dependence. Mutant channels were also slower to inactivate, recovered from inactivation more readily and demonstrated a significantly accelerated deactivation rate compared to the slow deactivation of WT channels. The intermediate nature of the biophysical perturbation is consistent with the degree of severity in the clinical phenotype. The findings of this study demonstrate a previously unknown role of the proximal N-terminus in deactivation and support the hypothesis that the proximal N-terminal domain is essential in maintaining slow hERG deactivation.
Keywords: KCNH2, hERG, ventricular arrhythmia, deactivation, LQT2
Introduction
Long QT Syndrome is a cardiac arrhythmia disorder caused by delayed cardiac repolarization that manifests as a prolonged QT interval on an electrocardiogram (ECG). This slowed cardiac repolarization increases the risk for polymorphic ventricular tachycardia (also known as Torsades de Pointes, TdP) that can degenerate into ventricular fibrillation and sudden death. Long QT Syndrome (LQTS) can be acquired, where delayed cardiac repolarization is due to drugs or other physiological triggers negatively impacting the repolarization forces in the heart, or it can be inherited through mutation, causing proteins that facilitate cardiac repolarization to malfunction.
At least 1 in 2,500 people in the United States have congenital Long QT Syndrome (Schwartz et al., 2009) and 13 genetic loci have been identified as causative for the syndrome (Shimizu et al., 2009). However, a majority of hereditary LQTS patients have mutations in either KCNQ1 (Locus LQT1) or KCNH2 (locus LQT2; MIM# 152427). These two genes encode proteins that form potassium channels that provide the bulk of repolarization for the ventricular cardiomyocyte membrane at the end of each action potential. KCNQ1 encodes the alpha subunit of the potassium channel that, when complexed with its beta subunit KCNE1, forms the slowly-activating delayed rectifier potassium current (IKs) (Sanguinetti et al., 1996). KCNH2 (also known as hERG—human Ether-a-go-go Related Gene) encodes the alpha subunit of the channel that forms the rapidly-activating delayed rectifier potassium current (IKr) (Curran et al., 1995),(Sanguinetti et al., 1995). Throughout this discussion, KCNH2 will refer to the gene and hERG will be used to refer to the channel protein product of the KCNH2 gene. Mutations in the KCNQ1 protein causing LQTS usually lead to a gating or permeation problem in the channel that perturbs proper ion flux. In contrast, most mutations in KCNH2 result in a trafficking defect, preventing the channel from reaching the cell surface (Anderson et al., 2006, 2014). In either case, the mutations usually compromise potassium current in a dominant fashion due to the tetrameric assembly of both the KCNQ1 and hERG potassium channels. Here we describe a novel missense KCNH2 mutation (rs587777907, c.656A>T, p.Asp219Val) that resides in an area of the hERG channel that has received scant attention due to lack of structural data or prior LQTS mutation. In contrast to most KCNH2 mutations, p.Asp219Val causes malfunction of the biophysical behavior of the potassium channel. Functional characterization of this mutant reveals an isolated and selective perturbation in the kinetics of channel gating that is commensurate with the moderate degree of clinical phenotype.
Materials and Methods
Genetic Analysis
Genetic testing for LQTS mutations was carried out by Transgenomic, Inc. (New Haven, CT, USA). Genomic DNA was amplified by polymerase chain reaction (PCR) to generate templates for direct sequencing of the targeted exons, splice junctions, and flanking regions of the genes KCNQ1, KCNH2, SCN5A, KCNE1, KCNE2, KCNJ2, CACNA1C, CAV3, SCN4B, AKAP9, and SNTA1. Seven known Andersen-Tawil syndrome associated mutations in KCNJ2 were not analyzed for in the genetic testing due to an intellectual property issue at the time of the patient's testing. However, the patient did not demonstrate any phenotypic characteristics of Andersen-Tawil syndrome (see Case Report), and we do not believe the lack of testing impacts the patient diagnosis. Sequencing results were compared to a >1300 member cohort of multi-ethnic, normal healthy volunteers to determine common polymorphisms (Kapa et al., 2009). A novel SNP: c.656A>T, p.Asp219Val was noted in the KCNH2 locus (MIM# 152427) and is the subject of this study. Nucleotide numbering uses +1 as the A of the ATG translation initiation codon in the reference sequence NM_000238.3, with the initial codon as codon 1. This novel SNP was submitted to the dbSNP database (NM_000238.3:c.656A>T, SNP_ID: rs587777907, SubSNP_ID: ss1457253113).
Plasmids, Cell Culture and Transfection
The c.656A>T (p.Asp219Val) mutation was introduced into KCNH2 and inserted in the pCMV-tag-3a vector (Agilent Technologies, Santa Clara, CA, USA) using site-directed mutagenesis with the following primers: forward 5′- gacagccatggtcaaccacgtg-3′ and reverse 5′-cacgtggttgaccatggctgtc-3′ (IDT, Coralville, IA, USA). PCR was performed using cloned Pfu enzyme (Agilent Technologies, Inc, Santa Clara, CA) and the conditions were as follows: 98°C for 2 minutes for one cycle, followed by 96°C for 1 minute, 65°C for 1 minute and 72°C for 16 minutes, for a total of 18 cycles. Mutated cDNA vectors were verified by automated bidirectional DNA sequencing.
For electrophysiology, Human Embryonic Kidney (HEK) 293 cells (American Type Culture Collection, Manassas, VA, USA) were maintained in RPMI 1640 media (HyClone, Logan, UT, USA) supplemented with 10% Fetal Bovine Serum (HyClone, Logan, UT, USA) and 10,000 IU Penicillin/ Streptomycin under 5% CO2 conditions at 37°C. Transiently transfected cells were maintained at 37°C for 48 hours, split onto glass slides in 35 mm dishes (ThermoFisher Scientific, Pittsburgh, PA, USA) and then kept at 31°C for ∼12 hours prior to electrophysiology. Stably transfected cells were kept at 37°C, split onto glass cover slides in 35 mm dishes and moved to 31°C for ∼12 hours prior to electrophysiology. We have used 31°C prior to electrophysiology to prevent cells from over-growing and making cell-cell contact that prohibits adequate voltage clamp. Initial patch clamp studies at 37°C and 31°C verified a lack of difference in current amplitude or kinetics. Transient transfections were done using Fugene 6 (Roche, Basel, Switzerland) at a DNA KCNH2:GFP ratio of 3:1. All recordings were performed 48-72 hours post transfection at room temperature (approximately 22°C). Stable line transfections were established through transient transfection followed by a 2-week positive selection for Neomycin resistance with G418 (2 mg/mL), and resistant cells were then clonally expanded.
For the immunoblots, HEK 293 cells were transfected in a 1:4 DNA to Fugene 6 ratio and maintained as described in the cell culture section of this materials and methods section. Experiments were performed at 48 hours post-transfection.
Immunoblots
Transiently transfected HEK 293 cells were plated on 60 mm dishes (Fisher Scientific, Pittsburgh, PA, USA). After a 48-hour transfection, HEK 293 cells were lysed with NDET buffer (1% NP-40, 0.4% Deoxycholic acid, 5 mM EDTA, 25 mM Tris, 150 mM NaCl, pH7.5) with complete protease inhibitor (Roche) for 15 minutes at 4°C with gentle agitation. The lysates were then centrifuged at 13,000 RPM for 5 minutes at 4°C in a microcentrifuge and the supernatants were mixed with SDS-PAGE loading buffer and incubated at 37°C for 30 minutes. Proteins were then separated on a 6-7.5% SDS-PAGE and transferred to a nitrocellulose membrane (BioRad Laboratories, Hercules, CA, USA) by a semi-dry blotting unit (FisherBiotech, Thermo Fisher Scientific, Waltham, MA, USA). The membranes were blocked with Tris-Buffered Saline with 0.5% Tween-20 (TBS-T) and 5% Nonfat dry milk for 30 minutes at room temperature. The membranes were then washed 3 times in TBS-T for 5 minutes per wash. Then, the membranes were subject to overnight staining in primary antibody at 4°C with gentle agitation. Rabbit polyclonal H175 was used to detect hERG protein (Santa Cruz Biotechnology, Inc, Santa Cruz CA, USA) and mouse polyclonal A6F (Developmental Studies Hybridoma Bank, Iowa City, Iowa, USA) was used to detect Na+/K+-ATPase as a loading control. The membrane was then washed 3 times, 5 minutes per wash, in TBS-T, and stained with the corresponding infrared-fluorescence IRDye® 800 conjugated goat anti-rabbit, or IRDye®700 conjugated donkey anti mouse secondary antibodies at a dilution of 1:10,000 in 5% non-fat milk. Membranes were covered and stained in the dark for 30 minutes at room temperature with gentile agitation, and then imaged using the Odyssey Detection System (Li-COR biosciences, Lincoln, NE, USA) and densitometry of the bands was done using Li-COR software. Statistical analysis and images were done using OriginLabs software (OriginLabs, Northhampton, MA, USA). ANOVA statistical analysis was used to determine differences between groups, with a p-value <0.05 counted as significant. Post-hoc analysis was done with the Turkey's test at a confidence interval of 95%, and the details are included in Supp. Table S1.
Electrophysiology
For electrophysiology experiments, transiently or stably transfected HEK 293 cells were used as indicated. Cells were transfected with either WT KCNH2, 50/50 mix of WT and p.Asp219Val or p.Asp219Val mutated KCNH2. Channel-expressing cells were plated and grown on sterile glass cover slips and then placed into an acrylic/ polystyrene perfusion chamber (Warner Instruments, Hamden, CT, USA). Chambers were mounted on an inverted microscope equipped with fluorescence optics and patch pipette micromanipulators. All recordings were done at room temperature (∼22°C) and at 48-72 hours post transfection. Cells were bathed in extracellular solution (150 mM NaCl, 1.8 mM CaCl2, 4 mM KCl, 1 mM MgCl2, 5 mM glucose, and 10 mM HEPES buffer pH 7.4) at room temperature. Intracellular solution filled the micropipette (126 mM KCl, 4 mM Mg-ATP, 2 mM MgSO4, 5 mM EGTA, 0.5 mM CaCl2, and 25 mM HEPES buffer pH 7.2). Patch clamp pipettes with a tip resistance of approximately 2-3 mega-ohms were used to obtain the whole-cell configuration (Hamill et al., 1981). A MultiClamp 700B amplifier (Molecular Devices, LLC, Sunnyvale, CA, USA) was utilized and patch-clamp protocols were executed through the pCLAMP10 acquisition and analysis software (Molecular Devices). Pipette offset potential was zeroed prior to gigaseal formation on the cell. Whole-cell capacitance was electronically compensated for through the amplifier. Whole cell resistance was compensated to 75-85%. A standard holding potential of -80 mV was used for patching and whole-cell configurations, and figure insets illustrate the voltage command protocol. The data filtering (8-pole Bessel) at 1.4 kHz and sampling at 5 kHz was done for both the activation and deactivation protocols. For inactivation protocols, data were filtered at 2 kHz with sampled at 10 kHz. All data were analyzed with CLAMPFIT software (Molecular Devices) and figures were created and statistics were performed using Origin7.5 software (OriginLab, Northampton, MA). ANOVA statistical analysis was used to determine differences between groups, with a p-value <0.05 counted as significant. Post-hoc analysis was done with the Turkey's test at a confidence interval of 95%, and the details are included in Supp. Table S2 (activation), Supp. Table S3 (inactivation) and Supp. Table S4 (deactivation).
Prediction of Protein Damage due to Mutation
Three different software programs were used to predict the impact of the KCNH2 c.656A>T (p.Asp219Val) mutation on the hERG channel protein. SIFT (Kumar et al., 2009, http://sift.jcvi.org/) PolyPHEN2 (Adzhubei et al., 2010, http://genetics.bwh.harvard.edu/pph2/) and KvSNP (Stead et al., 2011, http://www.bioinformatics.leeds.ac.uk/KvDB/KvSNP.html) were all used. To generate predictions from SIFT, first the SIFT Human Protein program was selected. Then the Ensembl transcript identifier (ENST00000262186) was entered, along with the mutation of interest (D219V), and a prediction and SIFT score were obtained. For PolyPHEN2, the Ensembl gene identifier for KCNH2 (ENSG00000055118) along with the amino acid substitution (D219V) was entered, and the software program generated a prediction of how damaging the mutation would be to the protein. For KvSNP, the P0386; KCNH2 protein was selected. Then, the wild-type residue D (asparagine) was selected, residue number 219 was entered and the changed amino acid V (valine) was selected. KvSNP then generated a prediction for the disease-impact of the mutation (both predicted to be disease-causing (y/n) and probability of causing disease (from 0 to 1)). This was also repeated for the p.Asp144Val mutation discussed in the results section.
Protein Structure Prediction
Amino acids 140-280 were entered into the Robetta structure prediction software for 3-D modeling, using default parameters (Kim et al., 2004, http://robetta.bakerlab.org) for both Wild-Type and p.Asp219Val hERG. The Robetta-generated PDB files were analyzed in CHIMERA (Resource for Biocomputing, Visualization, and Information at the University of California, San Francisco)(Pettersen et al., 2004, http://www.cgl.ucsf.edu/chimera/, Version 1.9). The amino acid contacts were calculated using the CHIMERA program for overlap at > 0.4 angstroms.
Case Presentation
A previously healthy 11-year-old male from Southeast Asia was referred for cardiac evaluation following a syncopal episode. The episode occurred while the patient was experiencing fever and nasopharyngeal congestion. He rose from a seated position, felt nauseated and dizzy, and then collapsed. He reportedly lost consciousness for 3 to 4 minutes but recovered spontaneously. Emergency medical technicians initially recorded normal vital signs. His medications at the time of the syncopal episode were Zyrtec and Singulair. These medications are not known to cause arrythmia or prolongation of the QT interval. An electrocardiogram demonstrated normal sinus rhythm with a maximal recorded resting QTc of approximately 580msec with bifid T-waves (Figure 1A). Holter monitoring showed sinus rhythm ranging from 51 to 140 beats per minute (bpm) with no arrhythmia. Additional QTc intervals were reported as long as 553 msec during the hospitalization. An exercise test was performed with a Bruce protocol (a specified protocol of escalating treadmill grade and speed of the exercise) and the conclusion from this test was an abnormal response due to the increased QT interval seen on ECG while exercise was performed, characteristic of Long QT syndrome. Nadolol 20 mg was started prior to discharge. His physical examination was normal for an 11-year-old boy and he exhibited no physical manifestations of Andersen-Tawil syndrome or history of skeletal muscle weakness. The patient was adopted at age 3 months, and his biological family and birth history are unknown.
Figure 1. Patient ECG and Location of p.Asp219Val hERG.
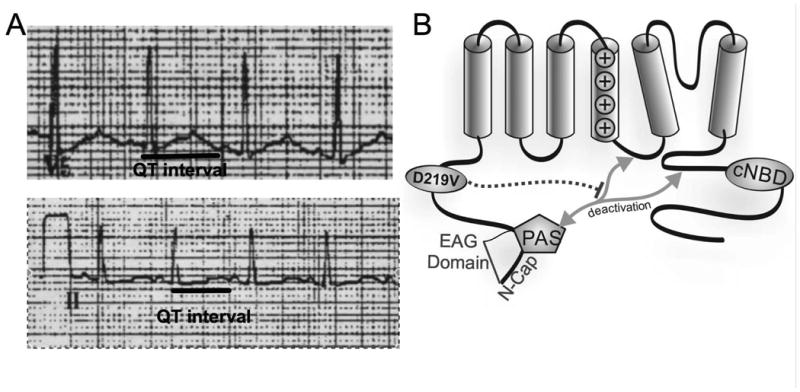
A) Leads V5 and II of a 12 lead ECG obtained at rest from the proband following a syncopal episode and showing a prolonged QT interval. B) Representation of the hERG protein, with mutation location denoted by the circle within the proximal N terminus. Grey arrows indicate interacting regions that affect channel gating. Dashed line shows proposed influence of the p.Asp219Val mutation.
Six months following his initial presentation, the patient experienced another brief syncopal episode that occurred after rising from bed. The EEG done by the neurologist demonstrated left posterotemporal epileptiform discharges, indicating a potential epileptogenic focus. However, further neurological evaluation failed to demonstrate seizure disorder and seizures have not occurred in the subsequent 5 years. Nadolol was increased to 40 mg. Subsequent outpatient QTc intervals have ranged from 400 to 440msec. Genetic screening for LQTS was performed. A point mutation, c.656A>T, was identified in the KCNH2 exon 4, in the cytoplasmic N-terminal region of hERG, resulting in the p.Asp219Val mutation. This mutation was reported as a class II variant, a variant of uncertain significance.
Nadolol 40 mg had been continued, and in the 5 years since the second presentation there have been no further symptoms of syncope or palpitations. A recent exercise stress test was performed, and while there were no arrhythmias, the recovery phase demonstrated an increased QT interval. At baseline, the QT response was 390msec, at 2min 50sec recovery, the QT response was 470msec and at 10 min recovery, the QT response was 400msec. The patient has continued to have normal QT intervals on resting ECGs.
Results
Channel Protein Expression
On an immunoblot, the hERG channel protein is visualized as a double band due to post-translational modifications of the channel that occur in the trans-golgi as the channel is trafficked to the cell membrane. The 135kD band represents the immature, core glycosylated form of hERG localized to the ER and cis-golgi. The 155kD band represents the complexly glycosylated, mature form of hERG located at the cell membrane. The ratio of 155/135kD bands, therefore, has been accepted as a surrogate marker for surface expression of the channel (Zhou et al., 1998). The comparison of the 155kD band to the 135 kD band is done when investigating hERG mutations, as most mutations causative of LQT2 induce a trafficking defect, significantly reducing the amount of 155kD protein visualized (Anderson, 2006, 2014). However, there were no significant differences between wild type hERG channels, a 50/50 mixed transfection of p.Asp219Val and WT hERG, and p.Asp219Val hERG total protein expression (Figure 2B) and no significant differences in the ratio of mature to immature hERG between these samples (Figure 2C). This result indicates that the p.Asp219Val mutation in the hERG channel does not impact protein synthesis or channel trafficking, an unusual characteristic for hERG channel mutations causing Long QT Syndrome, type 2. This conclusion was also confirmed through the lack of significant difference in current density for each of the channel types (WT, 50/50 mix and p.Asp219Val).
Figure 2. Heterologous protein expression of p.Asp219Val hERG.
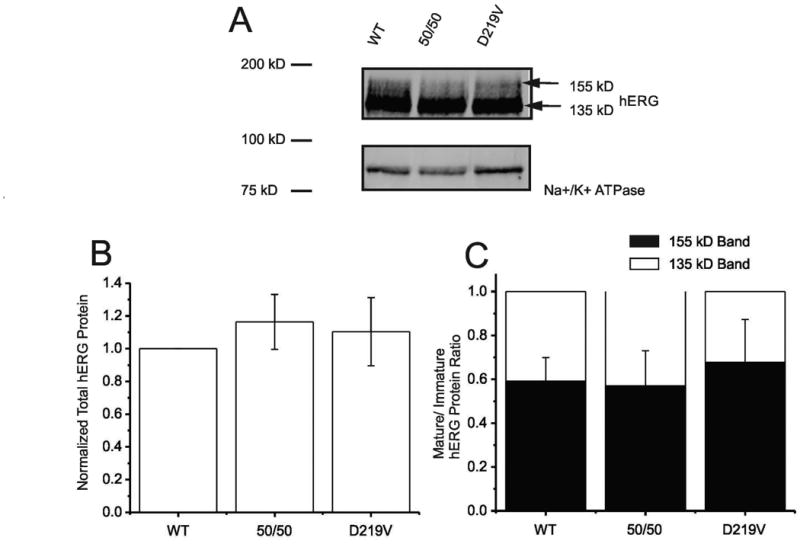
A) Representative Immunoblot shows whole cell lysate of HEK 293 cells transiently transfected with WT hERG, 50/50 mix or p.Asp219Val hERG plasmid DNA. Antibodies targeted against hERG and Na+/K+-ATPase (loading control) were used. hERG channel protein appears as a double band (a discrete 135 kD species and 155 kD broad smear of heavily glycosylated protein) representing immature and mature hERG, respectively. These data are a summary of 5 immunoblots performed and analyzed. B) Histogram showing summary of total hERG protein from 5 immunoblots, normalized to Na+/K+-ATPase. Graph shows densitometry quantification of WT hERG and p.Asp219Val hERG and demonstrates that there was no significant difference between total protein expression. C) Histogram illustrates relative distribution of immature hERG (135 kD) and mature hERG (155 kD), normalized to 1 for WT, 50/50 mix of WT and mutant, and p.Asp219Val hERG. This reflects the ratio of channel protein expressed on the cell membrane to the channel present in the ER/Golgi. The ratio indicates trafficking success to the surface of the cell. There was no significant difference between these ratios for each of the samples.
Electrophysiology
To investigate the functional characteristics of the p.Asp219Val mutant hERG channel, the whole cell patch clamping technique was used. HEK 293 cells were transiently transfected with either wild-type KCNH2, a 50/50 mix of mutant and wild-type KCNH2, or c.656A>T KCNH2 plasmid DNA. Approximately 48-hours post transfection, the cells were studied. For channel activation, the membrane was stepped from -60 to 60 mV for 1.5s, then repolarized to -40 mV for 0.5s and finally hyperpolarized to -120 mV for 0.5s. Current density was determined by normalizing for cell capacitance. The p.Asp219Val mutation did not significantly impact current density (Figure 3B). Voltage-dependence of activation, as measured by plotting the peak tail current against the previous voltage step, was fit by a Boltzmann function: I = 1/(1+exp[(V½-V)/k]), where I is the relative tail current amplitude, V is the applied membrane voltage, V½ is the voltage at half-maximal activation, and k is the slope factor (Figure 3D). The V1/2 for WT was –7.83±0.2mV; for 50/50 mix was -10.66±0.68mV; and for the p.Asp219Val mutant was -0.57±0.47mV. ANOVA analysis indicated that the p.Asp219Val mutation had a modest but significant (p=<0.001) depolarizing effect on the voltage dependence of the channel.
Figure 3. Effect of p.Asp219Val mutation on hERG channel activation.
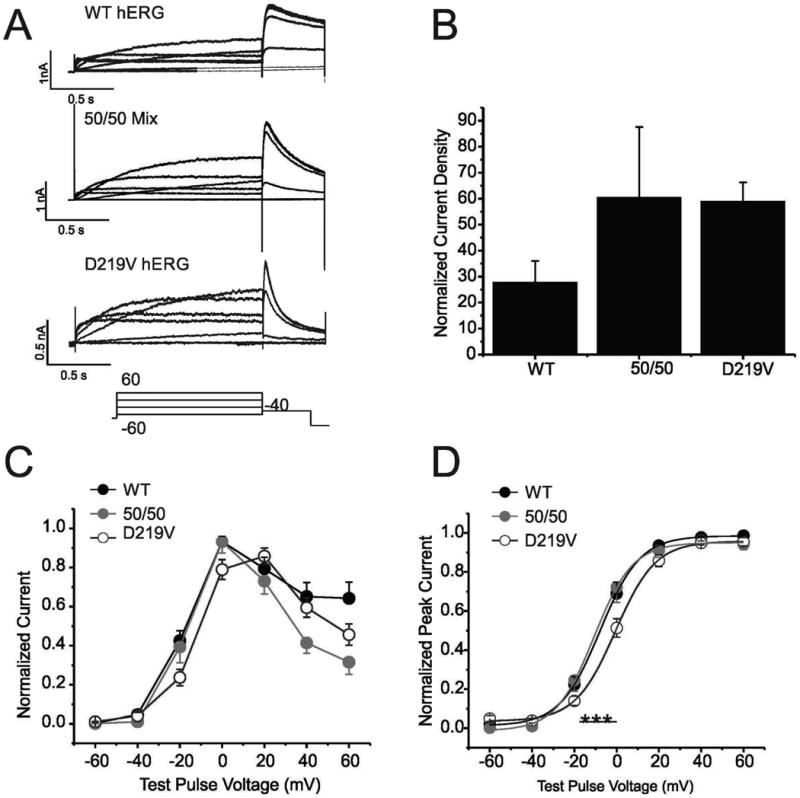
A) Traces are families of whole cell K+ currents from cells expressing the indicated cDNAs in response to a series of depolarizing steps (Voltage clamp protocol is illustrated below). HEK 293 cells were transiently transfected with WT hERG, a 50/50 mix of WT and p.Asp219Val hERG, and p.Asp219Val hERG alone. 9, 8 and 32 cells respectively were sampled. B) Histogram demonstrates summary of current density for each of the samples. Current density was measured at peak amplitude at the -40 tail current/cell capacitance. There is no significant difference between these current densities. C). Graph shows the current-voltage relationship of WT, 50/50 mix and p.Asp219Val. Measurements were taken at the end of the depolarizing phase. D) Normalized voltage-dependent activation curves are shown for all 3 samples. Curves were fitted using a Boltzman function. V½ for WT hERG was -7.83±0.2mV, 50/50 mix was -10.6±0.68mV and p.Asp219Val hERG was -0.57±0.47mV. These values were significantly different, with a P value of > 0.001.
To investigate steady-state inactivation of channels with p.Asp219Val mutation the membrane was held at +20mV and was stepped to test voltages ranging from -120 to +40mV for 20msec and then back to +20mV(Smith et al., 1996). The peak current at the onset of the final depolarization step was measured and plotted against the previous step potential. Rapid deactivation of channels at the negative voltages was corrected by the method of Smith et al (Smith et al., 1996). A Boltzmann function was then fitted to the data. The V1/2 for WT hERG was -43.46±2.24mV , for 50/50 mix was -18.42 ± 1.88 and for the p.Asp219Val hERG was -19.46±2.11mV. The difference was statistically different (ANOVA, p=<0.001), indicating that the voltage dependence for steady-state inactivation was also shifted in a depolarizing direction by the p.Asp219Val mutation alone, and when expressed in combination with the WT hERG as seen in the 50/50 mix.
To assess the onset of inactivation, the membrane was held at +20mV and a hyperpolarizing step to -120mV for 6msec was applied to release inactivation, followed by voltage steps from 70mV to -20mV for 125 msec (Bian et al., 2004). To correct for more rapid deactivation at negative potentials a separate set of depolarizing/repolarizing steps were performed to determine the fraction of channel deactivated and correct for a reduction of current negative to -60mV (Smith et al., 1996). The decline in subsequent current to steady-state was fitted by a single exponential that reflected the onset of inactivation (Figure 4B). The p.Asp219Val hERG channel exhibited a trend of slower inactivation onset at all points that was statistically different from WT between the voltages +30mv to +70mV (unpaired T-test, p=<0.05). In the 50/50 mix, the mutation seemed to have an intermediate impact. The differences between all 3 current traces were only significant at 40 mV and 60 mV (ANOVA, p<0.05).
Figure 4. Effect of p.Asp219Val mutation on hERG channel inactivation.
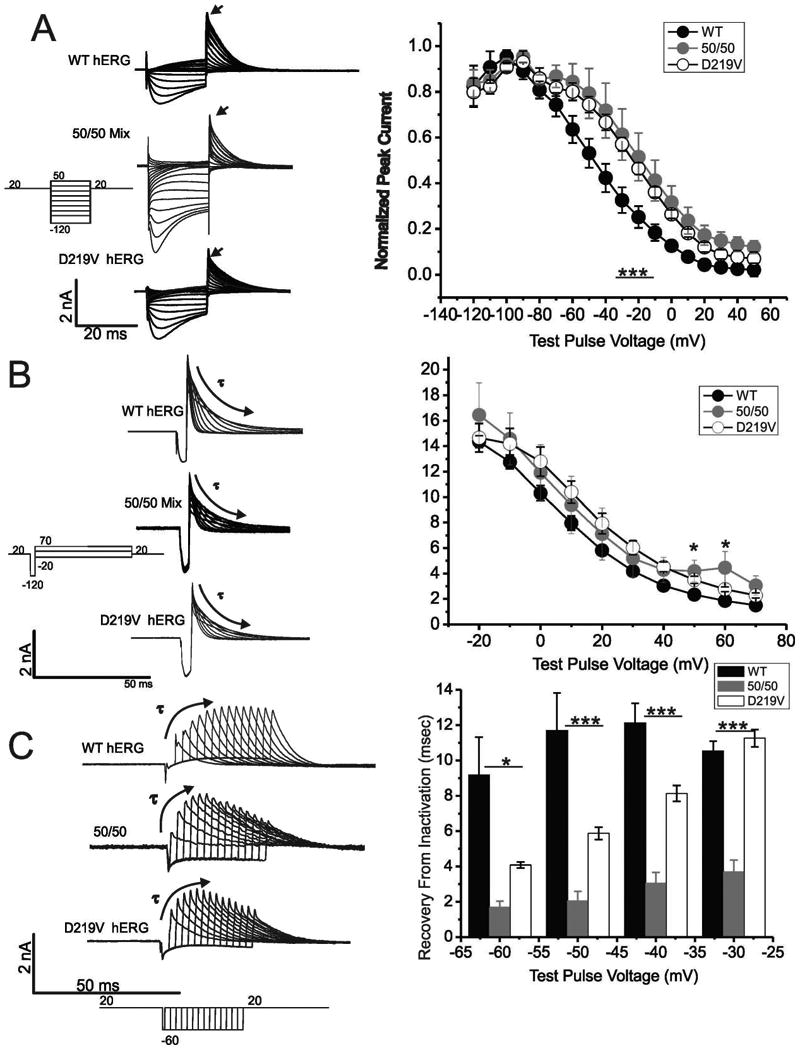
A) Data traces show a family of whole cell current recordings of WT hERG, 50/50 mix or p.Asp219Val hERG undergoing a protocol for steady-state inactivation (protocol is below traces). Stably transfected WT-hERG and p.Asp219Val hERG HEK cells were used, and HEK cells transiently transfected with WT hERG and p.Asp219Val hERG plasmids were used for the 50/50 mix electrophysiology. Peak current was measured at the depolarization to +20 mV after the test pulses (indicated by the arrowhead), and was graphed against voltage. Summary data is displayed graphically in the panel to the right. The 50/50 mix and p.Asp219Val hERG steady-state of inactivation was right-shifted, and was significantly different than WT hERG (p= <0.0001). B) Families of whole cell current recordings demonstrating onset of inactivation are shown for WT, 50/50 mix or p.Asp219Val hERG. The lines were fitted by a single exponential curve to determine time of inactivation onset, and time constant was graphed against each respective voltage. The graph demonstrates a trend that p.Asp219Val hERG and the 50/50 mix have a longer onset of inactivation, with significantly different points at 40 and 60 mV mV (p=<0.05). C) Whole cell traces of WT, 50/50 mix or p.Asp219Val hERG demonstrating recovery from inactivation. Peak current at each time was obtained, and a single exponential curve was fitted to these data (to the peak, indicated by the curved arrow). The resulting time constant was plotted against the voltage. This protocol was repeated at -60 mV, -50 mV, -40 mV and -30 mV. There was a significant difference between the time constants for recovery from inactivation at -60- -50 (p<0.05) and between -40 - -30 mV (p=<0.001) for WT, 50/50 mix and and p.Asp219Val hERG. N=5 for WT hERG, 4 for 50/50 mix and 6 for p.Asp219Val.
The recovery from inactivation was examined from a holding potential of +20mV with steps to -60mV for varying duration (between 1 and 24.5msec) followed by return to +20mV (Figure 4C). This was repeated with step potentials to -50mV, -40mV and -30mV. The peak outward current after each step was plotted against time until the data reached a peak, and a single exponent was fitted to the these data points. The 50/50 mix and p.Asp219Val hERG channel had a significantly faster recovery from inactivation. (ANOVA, p<0.05 for -60 and -50 mV and p<0.001 for -40 and -30 mV). The p.Asp219Val mutation had a drastic impact on the recovery from inactivation when co-expressed with the WT hERG as seen in the 50/50 mix traces and graphs, demonstrating a more significant impact on current when co-expressed than when only mutant channel is expressed. This may have clinical ramifications, as this mutation was heterogeneous.
To investigate deactivation, membrane potential was held at -70mV and a pulse depolarizing the membrane to +20mV for 1.7s was applied, followed by voltage steps from -40 to -100mV for 4.9s. Deactivation of the current during the final voltage steps was fitted with a double-exponential function (Itail = Ao + AMPF*expt/τF + AMPS*expt/τS , where Ao is the initial amplitude, AMPF and AMPS are the relative amplitudes of the fast and slow components and τF and τS are the time constants for the Fast Tau and Slow Tau, respectively) (Sanguinetti, 1995). Compared to the WT channel, p.Asp219Val hERG deactivates very quickly. In the individual traces seen in Figure 5A, the dramatic difference in deactivation rate between WT hERG and p.Asp219Val can be seen. The time constants tau fast and tau slow are shown in Figure 5B, and while there are not significant differences between the WT, 50/50 mix and mutant slow time constants, there is significant acceleration of the fast time constants for mutant and 50/50 mix channels (ANOVA, p=<0.05). Furthermore, relative contribution of the fast time constant to the total deactivation time was significantly greater in mutant channels and the 50/50 mix when compared to WT channels (Figure 5C). The differences in contribution of tau fast when compared to the total time were significant at voltages -70- -40 mV (ANOVA, p<0.05).
Figure 5. p.Asp219Val mutation effects on channel deactivation.
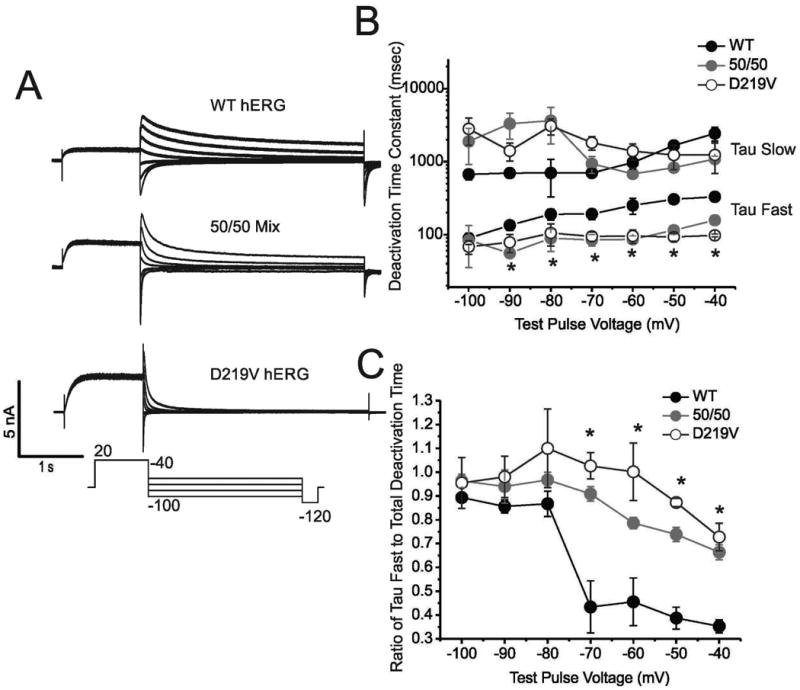
A) Whole cell current traces of hERG channels in repose to a deactivation voltage clamp protocol (protocol is shown below the traces). A combination of stably transfected WT hERG and p.Asp219Val hERG HEK cells and transiently transfected WT, 50/50 mix and p.Asp219Val HEK cells were used for these traces. B) Log10 graph demonstrating the time constants (Tau Fast and Tau Slow) for the WT hERG, 50/50 mix and p.Asp219Val hERG deactivation. There is no significant difference between the Tau Slow constants of the samples. The differences in Tau Fast between WT hERG, 50/50 Mix and p.Asp219Val is significant at all points except -100 (N=5, 5, and 11 for WT, 50/50 and p.Asp219Val respectively), each with a P-value of <0.05. C) Graphical illustration of relative contribution of Tau Fast to total deactivation time constant in WT, 50/50 mix and p.Asp219Val. This shows a decrease in Tau Fast contribution at -70 mV through -40 mV in WT hERG that is not seen in the p.Asp219Val and 50/50 mix samples.
Protein Structure Modeling
To investigate the potential structures of the proximal N-terminus of the WT and p.Asp219Val channels, the amino acid sequences140-280 of both WT and mutant hERG channel were predicted using the ROBETTA server and numerous potential structural conformations were examined (Raman et al., 2009). Each of the 5 predicted conformations demonstrated that the sequence immediately adjacent to the Asp219 site has a high likelihood of being unstructured. The UCSF CHIMERA structural analysis package (Pettersen et al., 2004) was used to analyze potential contacts and clashes of the p.Asp219Val substitution with other amino acids in the predicted structure. While numerous contacts were predicted in each structure, only a few potential interactions were predicted between amino acids not immediately adjacent to p.Asp219. One of these potential contacts, p.Arg269 when mutated to p.Arg269Trp (Itoh et al., 2010) has been implicated in LQTS. Another potential contact, p.Asp144, when mutated to p.Asp144Val, is a rare variant of unknown significance, but is predicted to be disease-causing with a probability of 0.877 (KvSNP, Stead et al., 2011). These contacts may be important in maintaining flexibility of the region surrounding p.Asp219 thereby allowing for correct movement of the N-terminal PAS domain (Amino acids 25-135, see Figure 1b) such that it may interact with the transmembrane and C-terminal regions, and the p.Asp219Val mutation may disrupt this interaction. This is supported by the Robetta generated structures of the mutant sequence that indicate the introduction of alpha helices in each of the 5 predicted conformations of the mutant protein (Figure 6A).
Figure 6. Protein secondary structure modeling.
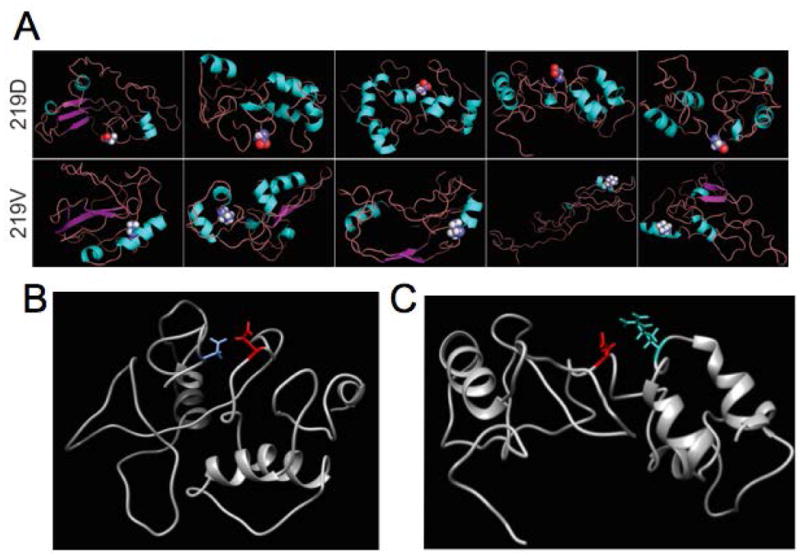
Ab inititio modeling of the segment surrounding p.Asp219 of hERG was performed using ROBETTA software and analyzed in CHIMERA. A) Demonstration of the top 5 predicted structures for both the wild-type p.Asp219 (top row) and the mutant p.Asp219Val hERG (bottom row). The 219 residue side chain is shown in spheres. All WT hERG structures demonstrated an unstructured area for the segment of the protein surrounding p.Asp219, while the mutant structures predict that p.Asp219Val lies in a helical formation in the protein. B) Model of wild type hERG segment adjacent to residue 219 with amino acid p.Asp144 (blue) predicted as a potential contact for p. Asp219. C) p.Arg269 (cyan) was also predicted as a potential contact for p.Asp219 in the wild type structure.
Discussion
Here we present the functional characterization of the patient-derived p.Asp219Val mutation in the hERG channel. The p.Asp219Val variant had not been described in dbSNP or the Exome variant server, but was documented in the ExAC database in 2 separate individuals of South Asian decent. This allele is a very rare variant, and there is no clinical data with this documentation of this mutation. Since Southeast Asians are poorly represented in these databases, it was of particular interest to pursue functional characterization of this variant to assess its pathogenic potential. Mutation-phenotype prediction databases yielded variable results [SIFT (Kumar et al., 2009) predicted the mutation to be tolerated (SIFT score 0.11); Polyphen2 (Adzhubei et al., 2010) predicted a 0.65 probability of damaging effect and KvSNP (Stead et al., 2011) predicted 0.97 probability of damaging effects]. Since the proband's clinical phenotype was intermediate in severity, full characterization of the genetic variant was indicated prior to embarking on life-long therapy.
The hERG channel has unique gating characteristics. Depolarization-dependent activation occurs at a moderately fast rate (hundreds of milliseconds) (Bian et al., 2001). Repolarization-dependent deactivation occurs relatively slowly (Curran et al., 1995). Inactivation takes place during depolarization and releases during repolarization at a very rapid rate (Smith et al., 1996). Through precise timing of these voltage-dependent processes, hERG quickly accumulates a population of channels in an open but inactivated state during the plateau of the depolarized cardiac action potential. As the cell membrane repolarizes and inactivation is rapidly released, there is a resurgent outward current (due to the slower process of deactivation) that hastens the membrane repolarization once it has initiated (Sanguinetti et al., 1995). Through this method, hERG controls the steepness of the ventricular myocyte repolarizing phase and if this hERG is perturbed, it may lead to an LQTS phenotype.
The p.Asp219Val mutation did not appreciably alter expression or trafficking of the hERG channel. This finding is supported by the robust current density carried by the mutant channels. The functional consequences include a depolarizing “right-shift” in the voltage dependence of activation. This activation change leads to less current generated for a given voltage step in the mutant channels. The p.Asp219Val mutation also produced a right shift in voltage dependence of inactivation, as well as a slower onset of and more rapid recovery from inactivation. These results suggest that the mutated channel has a less stable inactivation state than the wild-type channel. The inactivation changes would predict more current during depolarization for the mutant channel but less resurgent current properties during the repolarizing period. The most prominent gating effect of the mutation however, was acceleration of deactivation. Taken together, these data demonstrate that accelerated deactivation overrides the less stable inactivation effects for mutant and mutant/WT heteromultimeric channels with an end result of less effective repolarizing force during the latter part of the action potential where IKr activity is most important, leading to a LQT2 phenotype.
hERG deactivation has been shown to be regulated by the PAS domain (amino acids 25-135), which makes contacts with the C-terminal domain and in the transmembrane domains (Wang et al., 1998; Chen et al.,1999; Viloria et al.,2000; Brown et al.,2008; Fernandez-Trillo et al.,2011; Ng et al., 2011, 2012, 2014; Gianulis and Trudeau, 2011, 2013; de la Pena et al., 2013, 2014). The whole EAG domain (N-cap + PAS domain, Figure 1B) also impacts inactivation (Barros et al., 2012; de la Pena et al., 2013; Gustina and Trudeau, 2013). The mechanism for the complex voltage-dependence of hERG has not been fully elucidated, but there is evidence that the N-terminal cap (amino acids 1-25) plays a role in transducing channel activation to the voltage sensing domain (Barros et al., 2012; Tan et al., 2012). It has also been suggested that the proximal N-terminus may regulate voltage-dependence of steady-state activation and inactivation (Saenen et al., 2006; Gustina and Trudeau, 2013). Our results with the p.Asp219Val hERG mutation are similar to mutations and truncations involving PAS and EAG domains (Chen et al., 1999; Gustina and Trudeau, 2009, 2011, 2013; Gianulis et al., 2011, 2013). However, since it was shown that adding in a soluble PAS or EAG domain restored WT-like deactivation and inactivation properties to the truncated protein or LQT2 mutants, it is believed that the region between the EAG domain and the transmembrane segments was not functionally important in these processes (Gustina and Trudeau, 2009, 2013; Gianulis and Trudeau, 2011).
How can a mutation in the intervening, unstructured N-terminal segment affect hERG gating kinetics nearly identical to mutations disrupting PAS and/or EAG domain interactions? There are several possibilities that can reconcile our observations with prior results. hERG gating kinetics are dependent on numerous allosteric contacts, and when one of these contacts is disrupted, the channel may display a mutant phenotype. A change in charge or size of these amino acids may change these potential interactions, and prevent the proximal N-terminus from correct movement, contacts or necessary flexibility, inhibiting potential distal N-terminal movement and interaction with the rest of the channel protein. The hypothesis that we favor is that the region surrounding p.Asp219 is unstructured and flexible, and that it facilitates rapid movement of the EAG domain into the necessary contacts to maintain correct gating. If the substituted amino acid residues in the region surrounding p.Asp219 impose structure with less flexibility or cause changes in intra-molecular side-chain interactions, the EAG region contacts may be hampered. The ultimate result would be similar to mutations directly within the regulatory EAG domain that perturb interactions with the C-terminus and/or the transmembrane domains. This hypothesis is consistent both with the electrophysiology data and the protein structure modeling presented here.
Although the p.Asp219Val mutation demonstrates a clear importance of the region between the EAG domain and the transmembrane segments in terms of hERG activation, inactivation and deactivation, the overall biophysical impact of this mutation is not severe. Mutant channels and hybrids with co-expressed wild type channels carry ample potassium current, albeit with temporal differences in gating. This mutation does not demonstrate dominant effects in its impact on steady-state activation or inactivation. It does have a dominant effect in deactivation, aligning with the hypothesis that deactivation is a concerted “all or none” event (Thomson et al., 2014). The clinical presentation of this p.Asp219Val mutation has been rather mild, to date. Studies from the Robertson and Trudeau groups increasingly point to contributions of the hERG1b splice isoform to the IKr current, which has an abbreviated N-terminus and faster deactivation (Jones, et.al, 2014). The hERG1b isoforms would not exhibit the p.Asp219Val mutation and thus would not be functionally affected by this mutation. It is difficult to predict with accuracy the in vivo impact of the p.Asp219Val mutant in a complex comprised of hERG1a and hERG1b due to the many possible allelic expression differences, subunit stoichiometries and relative differences in isoform expression. Nevertheless, if the hERG1b splice isoform were preferentially of the p.Asp219Val allele, the phenotype might be milder. However if the hERG1a isoform is more represented by the mutant allele, we would predict that there would be a more pronounced phenotype. The patient has only experienced 2 self-limited syncopal episodes in 6 years. Thus, the biophysical phenotype is in good concordance with the clinical phenotype. This demonstrates the importance of functionally characterizing the genetic variants of unknown significance found in LQT patients undergoing genetic screening tests. Such data can inform the medical approach to the disease in a “personalized” manner (Zhao et al., 2009).
Conclusion
The functional implications of the p.Asp219Val mutation in the hERG channel highlight the importance of a previously under-studied segment of hERG. Prior to this study, it was suspected that this region of the protein did not play an impactful role in the gating of the hERG channel. Our results, however, suggest that this region is of biophysical and clinical importance since mutation caused significant changes in voltage-dependence, inactivation and deactivation. We propose that the flexibility of this predicted unstructured region is important in facilitating key functional interactions between the EAG domain and other portions of the channel. Ultimately, this model will require future structural determination to resolve the varied mechanisms by which LQTS mutations cause biophysical and clinical phenotypes.
Supplementary Material
Acknowledgments
The authors are funded in part by the Albert Einstein College of Medicine MSTP training grant, T32-GM007288 (MLO) and R21 HL120782 (TVM). Molecular graphics and analyses were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco. The Na+/K+-atpase polyclonal antibody used as a control in the immunoblots was obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242.
Grant Sponsors: MLO- T32-GM007288
NIGMS- MSTP Grant Funding
TVM- R21 HL120782
NHLBI- R21 PI funding
Footnotes
The authors have no conflicts of interest.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CL, Delisle BP, Anson BD, Kilby JA, Will ML, Tester DJ, Gong Q, Zhou Z, Ackerman MJ, January CT. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006;113:365–373. doi: 10.1161/CIRCULATIONAHA.105.570200. [DOI] [PubMed] [Google Scholar]
- Anderson CL, Kuzmicki CE, Childs RR, Hintz CJ, Delisle BP, January CT. Large-scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome. Nature communications. 2014;5:5535. doi: 10.1038/ncomms6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros F, Dominguez P, de la Pena P. Cytoplasmic domains and voltage-dependent potassium channel gating. Frontiers in pharmacology. 2012;3:49. doi: 10.3389/fphar.2012.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian J, Cui J, McDonald TV. HERG K(+) channel activity is regulated by changes in phosphatidyl inositol 4,5-bisphosphate. Circulation research. 2001;89:1168–1176. doi: 10.1161/hh2401.101375. [DOI] [PubMed] [Google Scholar]
- Bian JS, Cui J, Melman Y, McDonald TV. S641 contributes HERG K+ channel inactivation. Cell biochemistry and biophysics. 2004;41:25–40. doi: 10.1385/CBB:41:1:025. [DOI] [PubMed] [Google Scholar]
- Brown S, Sonntag DP, Sanguinetti MC. A highly conserved alanine in the S6 domain of the hERG1 K+ channel is required for normal gating. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2008;22:601–610. doi: 10.1159/000185544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zou A, Splawski I, Keating MT, Sanguinetti MC. Long QT syndrome-associated mutations in the Per-Arnt-Sim (PAS) domain of HERG potassium channels accelerate channel deactivation. The Journal of biological chemistry. 1999;274:10113–10118. doi: 10.1074/jbc.274.15.10113. [DOI] [PubMed] [Google Scholar]
- Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- de la Pena P, Machin A, Fernandez-Trillo J, Dominguez P, Barros F. Mapping of interactions between the N- and C-termini and the channel core in HERG K+ channels. The Biochemical journal. 2013;451:463–474. doi: 10.1042/BJ20121717. [DOI] [PubMed] [Google Scholar]
- de la Pena P, Machin A, Fernandez-Trillo J, Dominguez P, Barros F. Interactions between the N-terminal tail and the gating machinery of hERG K channels both in closed and open/inactive states. Pflugers Archiv : European journal of physiology. 2014 doi: 10.1007/s00424-014-1612-1. [DOI] [PubMed] [Google Scholar]
- Fernandez-Trillo J, Barros F, Machin A, Carretero L, Dominguez P, de la Pena P. Molecular determinants of interactions between the N-terminal domain and the transmembrane core that modulate hERG K+ channel gating. PloS one. 2011;6:e24674. doi: 10.1371/journal.pone.0024674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianulis EC, Liu Q, Trudeau MC. Direct interaction of eag domains and cyclic nucleotide-binding homology domains regulate deactivation gating in hERG channels. The Journal of general physiology. 2013;142:351–366. doi: 10.1085/jgp.201310995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianulis EC, Trudeau MC. Rescue of aberrant gating by a genetically encoded PAS (Per-Arnt-Sim) domain in several long QT syndrome mutant human ether-a-go-go-related gene potassium channels. The Journal of biological chemistry. 2011;286:22160–22169. doi: 10.1074/jbc.M110.205948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustina AS, Trudeau MC. A recombinant N-terminal domain fully restores deactivation gating in N-truncated and long QT syndrome mutant hERG potassium channels. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13082–13087. doi: 10.1073/pnas.0900180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustina AS, Trudeau MC. hERG potassium channel gating is mediated by N- and C-terminal region interactions. The Journal of general physiology. 2011;137:315–325. doi: 10.1085/jgp.201010582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustina AS, Trudeau MC. The eag domain regulates hERG channel inactivation gating via a direct interaction. The Journal of general physiology. 2013;141:229–241. doi: 10.1085/jgp.201210870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Archiv : European journal of physiology. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Itoh H, Shimizu W, Hayashi K, Yamagata K, Sakaguchi T, Ohno S, Makiyama T, Akao M, Ai T, Noda T, Miyazaki A, Miyamoto Y, et al. Long QT syndrome with compound mutations is associated with a more severe phenotype: a Japanese multicenter study. Heart rhythm : the official journal of the Heart Rhythm Society. 2010;7:1411–1418. doi: 10.1016/j.hrthm.2010.06.013. [DOI] [PubMed] [Google Scholar]
- Jones DK, Liu F, Vaidyanathan R, Eckhardt LL, Trudeau MC, Robertson GA. hERG 1b is critical for human cardiac repolarization. Proceedings of the National Academy of Sciences of the United States of America. 2014 doi: 10.1073/pnas.1414945111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, Wilde AA, Ackerman MJ. Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation. 2009;120:1752–1760. doi: 10.1161/CIRCULATIONAHA.109.863076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DE, Chivian D, Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic acids research. 2004;32:W526–531. doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Ng CA, Hunter MJ, Perry MD, Mobli M, Ke Y, Kuchel PW, King GF, Stock D, Vandenberg JI. The N-terminal tail of hERG contains an amphipathic alpha-helix that regulates channel deactivation. PloS one. 2011;6:e16191. doi: 10.1371/journal.pone.0016191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CA, Perry MD, Tan PS, Hill AP, Kuchel PW, Vandenberg JI. The S4-S5 linker acts as a signal integrator for HERG K+ channel activation and deactivation gating. PloS one. 2012;7:e31640. doi: 10.1371/journal.pone.0031640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CA, Phan K, Hill AP, Vandenberg JI, Perry MD. Multiple Interactions between Cytoplasmic Domains Regulate Slow Deactivation of Kv11.1 Channels. The Journal of biological chemistry. 2014;289:25822–25832. doi: 10.1074/jbc.M114.558379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. Journal of computational chemistry. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Raman S, Vernon R, Thompson J, Tyka M, Sadreyev R, Pei J, Kim D, Kellogg E, DiMaio F, Lange O, Kinch L, Sheffler W, et al. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins. 2009;77(Suppl 9):89–99. doi: 10.1002/prot.22540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saenen JB, Labro AJ, Raes A, Snyders DJ. Modulation of HERG gating by a charge cluster in the N-terminal proximal domain. Biophysical journal. 2006;91:4381–4391. doi: 10.1529/biophysj.106.087247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, Gabbarini F, Goulene K, Insolia R, Mannarino S, Mosca F, Nespoli L, et al. Prevalence of the congenital long-QT syndrome. Circulation. 2009;120:1761–1767. doi: 10.1161/CIRCULATIONAHA.109.863209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu W, Moss AJ, Wilde AA, Towbin JA, Ackerman MJ, January CT, Tester DJ, Zareba W, Robinson JL, Qi M, Vincent GM, Kaufman ES, et al. Genotype-phenotype aspects of type 2 long QT syndrome. Journal of the American College of Cardiology. 2009;54:2052–2062. doi: 10.1016/j.jacc.2009.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PL, Baukrowitz T, Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- Stead LF, Wood IC, Westhead DR. KvSNP: accurately predicting the effect of genetic variants in voltage-gated potassium channels. Bioinformatics (Oxford, England) 2011;27:2181–2186. doi: 10.1093/bioinformatics/btr365. [DOI] [PubMed] [Google Scholar]
- Tan PS, Perry MD, Ng CA, Vandenberg JI, Hill AP. Voltage-sensing domain mode shift is coupled to the activation gate by the N-terminal tail of hERG channels. The Journal of general physiology. 2012;140:293–306. doi: 10.1085/jgp.201110761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson SJ, Hansen A, Sanguinetti MC. Concerted All-or-none Subunit Interactions Mediate Slow Deactivation of Human ether-a-go-go-related Gene K+ Channels. The Journal of biological chemistry. 2014;289:23428–23436. doi: 10.1074/jbc.M114.582437. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Viloria CG, Barros F, Giraldez T, Gomez-Varela D, de la Pena P. Differential effects of amino-terminal distal and proximal domains in the regulation of human erg K(+) channel gating. Biophysical journal. 2000;79:231–246. doi: 10.1016/S0006-3495(00)76286-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Trudeau MC, Zappia AM, Robertson GA. Regulation of deactivation by an amino terminal domain in human ether-a-go-go-related gene potassium channels. The Journal of general physiology. 1998;112:637–647. doi: 10.1085/jgp.112.5.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao JT, Hill AP, Varghese A, Cooper AA, Swan H, Laitinen-Forsblom PJ, Rees MI, Skinner JR, Campbell TJ, Vandenberg JI. Not all hERG pore domain mutations have a severe phenotype: G584S has an inactivation gating defect with mild phenotype compared to G572S, which has a dominant negative trafficking defect and a severe phenotype. Journal of cardiovascular electrophysiology. 2009;20:923–930. doi: 10.1111/j.1540-8167.2009.01468.x. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Gong Q, Epstein ML, January CT. HERG channel dysfunction in human long QT syndrome. Intracellular transport and functional defects. The Journal of biological chemistry. 1998;273:21061–21066. doi: 10.1074/jbc.273.33.21061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.