

Abstract
Metabolism of glutamate, the main excitatory neurotransmitter and precursor of GABA, is exceedingly complex and highly compartmentalized in brain. Maintenance of these neurotransmitter pools is strictly dependent on the de novo synthesis of glutamine in astrocytes which requires both the anaplerotic enzyme pyruvate carboxylase and glutamine synthetase. Glutamate is formed directly from glutamine by deamidation via phosphate activated glutaminase a reaction that also yields ammonia. Glutamate plays key roles linking carbohydrate and amino acid metabolism via the tricarboxylic acid (TCA) cycle, as well as in nitrogen trafficking and ammonia homeostasis in brain. The anatomical specialization of astrocytic endfeet enables these cells to rapidly and efficiently remove neurotransmitters from the synaptic cleft to maintain homeostasis, and to provide glutamine to replenish neurotransmitter pools in both glutamatergic and GABAergic neurons. Since the glutamate-glutamine cycle is an open cycle that actively interfaces with other pathways, the de novo synthesis of glutamine in astrocytes helps to maintain the operation of this cycle. The fine-tuned biochemical specialization of astrocytes allows these cells to respond to subtle changes in neurotransmission by dynamically adjusting their anaplerotic and glycolytic activities, and adjusting the amount of glutamate oxidized for energy relative to direct formation of glutamine, to meet the demands for maintaining neurotransmission. This chapter summarizes the evidence that astrocytes are essential and dynamic partners in both glutamatergic and GABAergic neurotransmission in brain.
Keywords: Glutamate, Glutamine, GABA, Metabolic compartmentation, Astrocytes, Glutamate-glutamine cycle, Pyruvate carboxylase, Glutamine synthetase, Phosphate activated glutaminase, Glutamate dehydrogenase
Introduction
Glutamate plays a key role in intermediary metabolism in all organs and tissues linking carbohydrate and amino acid metabolism via the tricarboxylic acid (TCA) cycle. The reason is that it serves as a co-substrate in all reactions catalyzed by aminotransferases being converted to α-ketoglutarate, a key intermediate in the TCA cycle (McKenna et al. 2012). In the brain, glutamate metabolism extends beyond this general view as it serves as the immediate precursor for γ-aminobutyric acid (GABA) which is formed by decarboxylation of glutamate catalyzed by glutamate decarboxylase (GAD) as first shown by Roberts and Frankel (1950). The fact that GABA is metabolized by GABA-transaminase (GABA-T) and further to the TCA cycle intermediate succinate, catalyzed by succinic semialdehyde dehydrogenase, provides a deviation of the TCA cycle reactions called the GABA-shunt which circumvents succinyl CoA (see Schousboe et al. 2013). As both glutamate and GABA serve dual roles in the brain as metabolites and important neurotransmitters mediating excitatory and inhibitory signals, respectively (for references see Schousboe et al. 2013, Schousboe et al. 2012), their metabolic pathways are of significant interest. The immediate precursor for neuronal synthesis of glutamate is glutamine. This reaction is catalyzed by phosphate activated glutaminase (PAG) which hydrolytically deamidates glutamine to form glutamate and ammonia (for further details, see below). Interestingly, this enzymatic reaction was extensively investigated by Krebs (1935) in several tissues including the brain. Later, detailed studies of glutamate and glutamine metabolic pathways in the brain performed in the laboratories of H. Waelsch and J.H. Quastel (e.g. Quastel 1975, Berl & Clarke 1969) provided evidence that glutamate metabolism in the brain is extremely complex. It was noted that using radioactively labeled glucose as a precursor higher specific radioactivity was seen in glutamate; whereas, with the radioactive precursors leucine, acetate and bicarbonate, glutamine exhibited a higher specific radioactivity than its precursor glutamate (for references, see Berl & Clarke 1969, Quastel 1975). A higher specific radioactivity in a compound (e.g. glutamine) than that seen in its precursor (e.g. glutamate) indicates that the precursor exists in separate metabolic pools having different turn-over rates or in other words this is referred to as “metabolic compartmentation” (for further details, see McKenna et al. 2012). This finding led to the concept of metabolic compartmentation of glutamate in the brain, with at least two compartments (van den Berg & Garfinkel 1971, Garfinkel 1966) which were subsequently defined as representing neurons and astrocytes. This concept is based on the finding that glutamine synthetase (GS), the enzyme that converts glutamate to glutamine, is exclusively localized in astrocytes (Norenberg & Martinez-Hernandez 1979), together with the finding that higher specific radioactivity in glutamine is observed with the precursors acetate, bicarbonate and leucine (see above). It is of interest that the de novo synthesis of glutamate, the precursor of glutamine, is also restricted to astrocytes as the anaplerotic enzyme pyruvate carboxylase (PC) is exclusively localized in astrocytes (Shank et al. 1985, Yu et al. 1983). This will be discussed in further detail below.
Enzymatic reactions involving glutamate as substrate or product
Aspartate aminotransferase
This enzyme catalyzing the reversible interconversion of aspartate, α-ketoglutarate, oxaloacetate and glutamate (Fig.1) is present in all tissues, and has the highest specific activity among aminotransferases in the brain (for references, see Cooper 1988). The equilibrium constant for the enzyme is close to unity (Krebs 1953), hence, the reaction proceeds easily in both directions and is generally considered an exchange reaction. This is important considering the fact that the two keto acid substrates (oxaloacetate and α-ketoglutarate) are constituents of the TCA cycle. Thus, the corresponding amino acids aspartate and glutamate are in equilibrium with these TCA cycle intermediates and reflect the metabolic status of the keto acids at any given time due to the high activity of this enzyme (for references, see McKenna et al. 2012). This forms the basis for using the appearance of labeled carbon in glutamate and aspartate, from labeled precursors that provide substrates for the TCA cycle as a surrogate for determining the activity of the TCA cycle (see, McKenna et al. 2012). It should be noted, that aspartate aminotransferase (AAT) can reversibly bind to the inner mitochondrial membrane, which influences the functional activity of this enzyme (for futher details, see McKenna et al. 2000, 2006).
Figure 1.
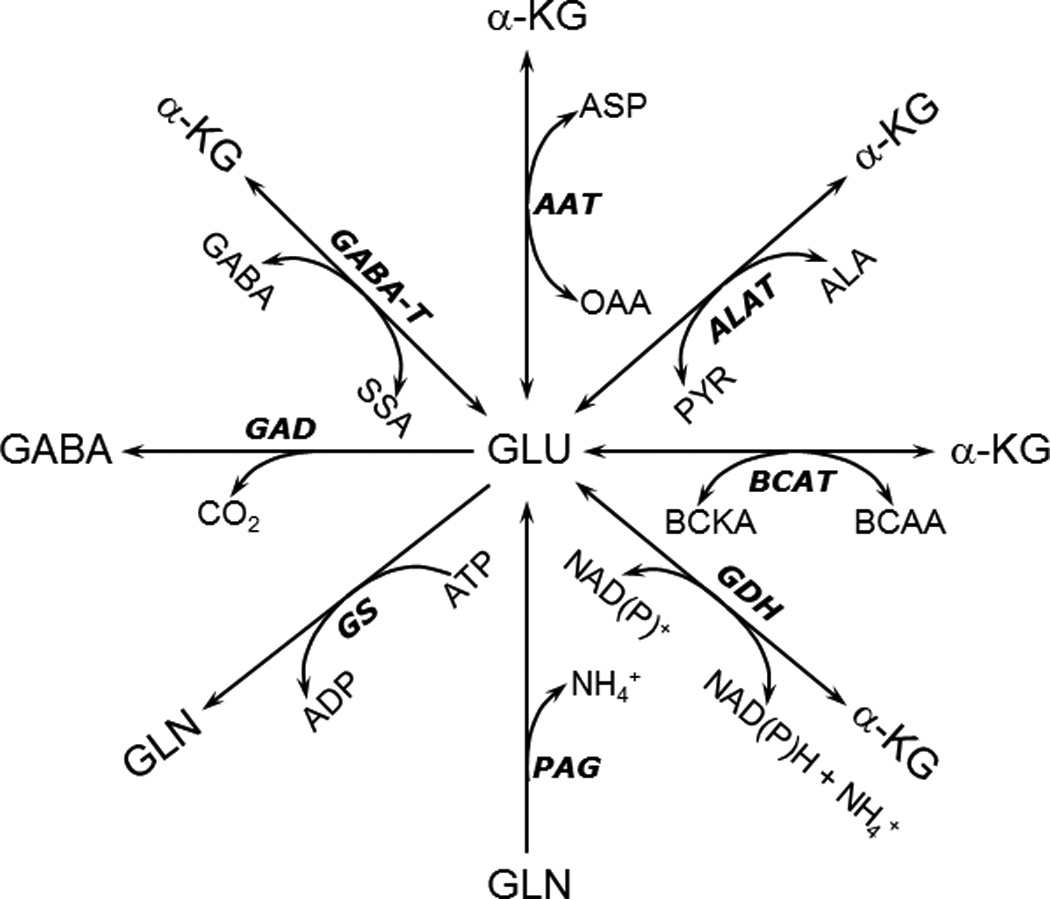
Reactions involving glutamate (Glu) in astrocytes. Ala, Alanine; α-KG, α-ketoglutarate; Asp, aspartate; BCAA, branched chain amino acid; Gln, glutamine. See text for other abbreviations.
Alanine aminotransferase
This aminotransferase catalyzes the reversible interconversion of alanine, α-ketoglutarate, pyruvate and glutamate (Fig.1) and its activity in the brain as well as in cultured brain cells is two orders of magnitude lower than that of aspartate aminotransferase (Benuck et al. 1972, Larsson et al. 1985, Westergaard et al. 1993). The equilibrium constant for alanine aminotransferase (ALAT) is close to unity (Krebs 1953). Since pyruvate, one of the substrates for this enzyme is a product of glycolysis, ALAT couples the glycolytic pathway to amino acid metabolism and carbon label from glucose can be monitored by its appearance in alanine, which has a slower turnover rate than pyruvate. This provides a method to monitor glycolytic activity although it should be kept in mind that this relationship is not a direct correlation since the pyruvate pool has been shown to be compartmentalized (for further details, see Bakken et al. 1998, Obel et al. 2012, Waagepetersen et al. 2000).
Alanine aminotransferase is present in both astrocytes and neurons and may have a role in transfer of ammonia nitrogen between these cells as suggested by Waagepetersen et al. (2000). The ALAT reaction in concert with glutamate dehydrogenase (GDH) may be a particularly important mechanism for fixation of ammonia during hyperammonemic conditions when the normal activity of glutamine synthetase (see below) is inhibited (Dadsetan et al. 2011, 2013). This aspect will be discussed in further detail below.
Branched chain amino acid aminotransferase
The three branched chain amino acids (valine, leucine and isoleucine) are metabolized by transamination with α-ketoglutarate, catalyzed by a common aminotransferase forming glutamate (Fig.1) and the three keto acids α-ketoisovalerate, α-ketoisocaproate and α-keto-β-methylvalerate, respectively (Cooper 1988). The branched chain amino acid aminotransferase isozymes (BCATs) are compartmentalized in brain cells; with the mitochondrial form being selectively localized in astrocytes and the cytosolic form localized in neurons (see, Lieth et al. 2001). This selective localization plays an important functional role as the branched chain amino acids also mediate shuttling of ammonia nitrogen between astrocytes and neurons (Bak et al. 2013, Lieth et al. 2001). This reaction may also contribute the amino group for de novo glutamate synthesis in astrocytes, which requires CO2 fixation (Lieth et al. 2001). The branched chain amino acids have also been proposed to play a role in removal of ammonia during hyperammonemic conditions as seen in hepatic encephalopathy (Ott et al. 2005). Additionally, since these amino acids ultimately are metabolized to propionyl-CoA, acetyl-CoA, succinyl-CoA and acetoacetate, the latter three compounds can fuel the TCA cycle and facilitate glutamine production during hyperammonemia thereby ameliorating some of the excess ammonia (Ott et al. 2005). This functional role has been questioned since studies using isolated neurons and astrocytes suggest that the branched chain amino acids are only modestly metabolized in brain (see, Bak et al. 2013). However, it should be noted that supplementation with branched chain amino acids was neuroprotective after traumatic brain injury (Cole et al. 2010).
Glutamate dehydrogenase
This mitochondrially localized (Salganicoff & Derobertis 1965) enzyme catalyzes the redox-based interconversion of glutamate and α-ketoglutarate using NAD(P)+ as coenzyme (Fig.1). The thermodynamic equilibrium constant (6 × 10−15 M) favors the reductive amination of glutamate (Engel & Dalziel 1967). However, in the brain the normal direction of the reaction favors oxidative deamination of glutamate since the NAD+/NADH ratio is high and the Km, for ammonia (14–26 mM) is orders of magnitude higher than the prevailing ammonia concentration (Zaganas et al. 2001, 2009). The enzyme is highly regulated by ADP and leucine both of which function as allosteric activators and by GTP, which acts as an allosteric inhibitor (Spanaki et al. 2012). Emerging evidence suggests that GDH can be allosterically inhibited by mitochondrial SIRT4 (silent information regulator 4) that is highly expressed during brain development (Komlos et al. 2013, Lavu et al. 2008). Humans express two isoforms of the enzyme (GDH1 and GDH2) that differ dramatically with regard to the allosteric regulation; other mammals express only the housekeeping isoform, GDH1 (Spanaki et al. 2012). The enzyme is present in both neurons and astrocytes albeit the expression may be higher in astrocytes (Lovatt et al. 2007), particularly those from brain regions with high glutamatergic activity (Aoki et al. 1987).
As pointed out above, glutamate dehydrogenase in combination with aminotransferases is important for transferring ammonia to form amino acids from keto acids. Since reductive amination can only occur when the ammonia concentration approaches the Km value for ammonia, it is likely that enzymatic complexes between GDH and the respective aminotransferases must exist to facilitate this reaction in vivo and there is some experimental evidence to suggest that this is indeed the case (Fahien et al. 1977, Islam et al. 2010, McKenna 2011).
Phosphate activated glutaminase
This enzyme which hydrolyzes glutamine to glutamate (Fig.1) was first described by Krebs (1935), and its phosphate dependence was reported shortly thereafter by Errera and Greenstein (1949). The name, phosphate activated glutaminase or PAG, originates from this latter publication. The brain enzyme has been purified and extensively characterized by Kvamme and co-workers and is highly enriched in neurons (for review, see Kvamme et al. 2001). Using a preparation of cultured astrocytes, which may reflect the properties of these cells in situ (Lange et al. 2012), it was shown by Schousboe et al. (1979) that this enzyme is also found in astrocytes. This finding has been controversial but a transcriptomic analysis of acutely isolated astrocytes has recently confirmed that PAG is indeed expressed by astrocytes (Lovatt et al. 2007). PAG is essential for synthesis of transmitter glutamate in glutamatergic neurons and it also plays a significant role in the synthesis of neurotransmitter GABA, since NMR studies revealed that glutamine synthesized in astrocytes and deamidated by neuronal PAG is preferentially used for GABA synthesis (see, Schousboe et al. 2013, Sonnewald et al. 1993c).
Glutamate decarboxylase
The presence of GABA in the brain and its synthesis by α-decarboxylation of glutamate (see Fig.1) was first described by Roberts and Frankel (1950). An enzyme, glutamic acid decarboxylase, catalyzing the conversion of glutamate to GABA was first identified in plants and bacteria. Therefore, it was concluded that formation of GABA from glutamate by α-decarboxylation in the brain was likely to be catalyzed by this enzyme. Glutamic acid decarboxylase (GAD) was finally purified to homogeneity from mouse brain more than twenty years later by Wu et al. (1973). Cloning studies subsequently showed that GAD exists in two isoforms with molecular weights of 65 kD and 67 kD; these isozymes are referred to as GAD65 and GAD67, respectively (for references, see Soghomonian & Martin 1998). In the brain these GAD isozymes are restricted to neurons and primarily GABAergic cells (Saito et al. 1974). The enzymes are also expressed in other tissues such as the pancreas (Okada et al. 1976). Both isoforms of the enzyme are located in the cytosol, but their subcellular localization differs, with the GAD67 isoform having a widespread expression compatible with a function related to general metabolism of the GABA pool; whereas the GAD65 isoform is mainly present in nerve endings and thought to be involved in the synthesis of neurotransmitter GABA (Walls et al. 2011, Martin & Rimvall 1993).
Glutamine synthetase
As already pointed out, glutamate metabolism in the brain is highly complex and compartmentalized with different enzymes located in different cell types. The enzyme glutamine synthetase (GS) is responsible for conversion of glutamate to glutamine (Fig.1) and is expressed exclusively in astrocytes (Norenberg & Martinez-Hernandez 1979). The functional importance of GS can most conveniently be assessed by use of the irreversible inhibitor methionine sulfoximine (MSO; Ronzio et al. 1969). The GS reaction adds a second nitrogen atom in the form of ammonia to glutamate to form glutamine and requires ATP for energy (Fig 1). The Km values for the three substrates glutamate, ammonia and ATP are 2.5 mM, 0.2 mM and 2.3 mM, respectively (Pamiljans et al. 1962), which means that the enzyme in situ is unlikely to be saturated with any of the substrates (see, Schousboe et al. 2012).
One of the most essential roles of astrocytes is removal of neurotransmitter glutamate from the synaptic cleft after depolarization (to maintain the low resting glutamate concentration of 1–10 µM) and conversion to glutamine (Bergles et al. 1999, Matsui et al. 2005). It should be noted, that the de novo synthesis of glutamine takes place only in astrocytes. De novo synthesis of glutamine from glucose requires the concerted action of glycolytic enzymes, pyruvate carboxylase, the TCA cycle and conversion of α-ketoglutarate to glutamate, which via the action of GS can be converted to glutamine (Fig. 2). Pyruvate carboxylase like GS is dependent on ATP and is only expressed in astrocytes (Yu et al. 1983). Pyruvate carboxylation adds a carbon to pyruvate to form oxaloacetate and serves to replenish intermediates that would otherwise be drained from the TCA cycle by the conversion of α-ketoglutarate to glutamate and subsequently glutamine. 13C-NMR studies show compartmentation in this process as the carbon added via pyruvate carboxylase is preferentially found in newly synthesized glutamine (Sonnewald et al. 1993a). Due to the selective cellular localization of glutamine synthetase and pyruvate carboxylase only astrocytes are able to perform a net synthesis of glutamine, which is the major precursor for the two most important neurotransmitters, glutamate and GABA (see, McKenna 2007, McKenna et al. 2012). A further discussion of this important aspect of brain function based on cellular specialization is provided below.
Figure 2.
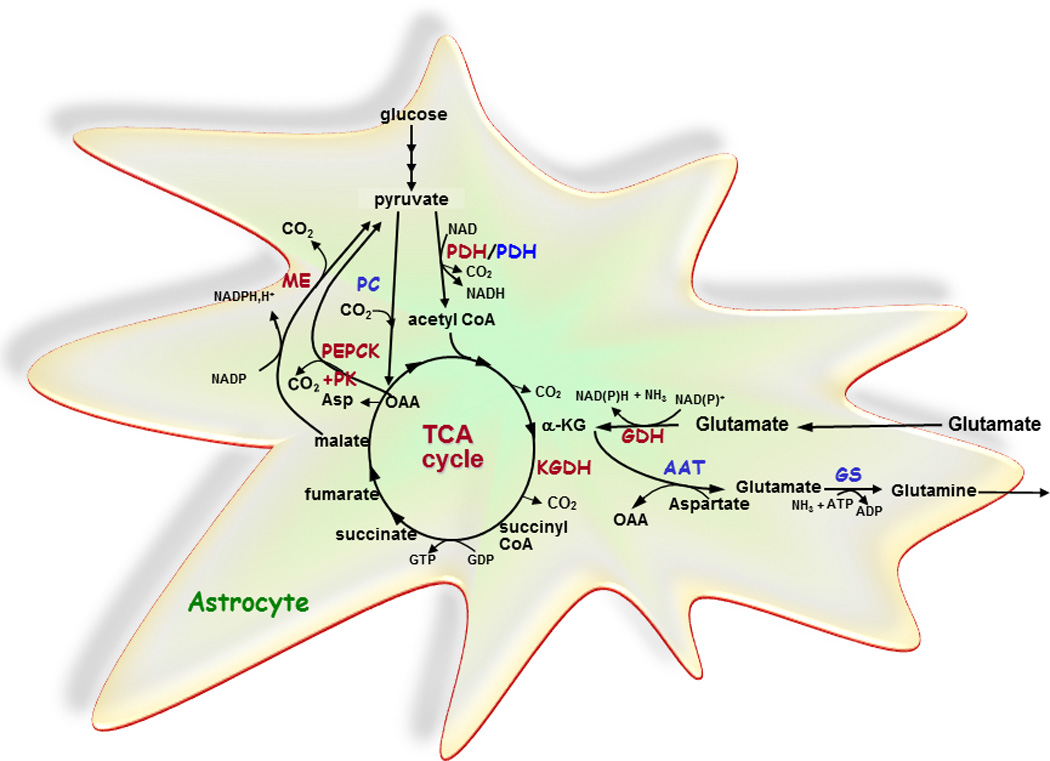
Schematic representation of complete oxidation of glutamate via the ”pyruvate recycling pathway” (essential enzymes in red) and de novo synthesis of glutamate and glutamine from glucose via pyruvate carboxylase (PC) (essential enzymes in blue) in astrocytes.
For de novo synthesis of glutamate and glutamine, glucose is metabolized to acetyl CoA and oxaloacetate via pyruvate dehydrogenase and pyruvate carboxylase, respectively. Acetyl CoA and oxaloacetate condenses to citrate and subsequently, α-ketoglutarate (α-KG) is formed. α-Ketoglutarate is converted to glutamate catalyzed by aspartate aminotransferase (AAT) and via an amidation, catalyzed by the energy requiring enzyme glutamine synthetase (GS), glutamine may be formed. The complete oxidation of glutamate is initiated by oxidative deamination catalyzed by glutamate dehydrogenase (GDH) and oxidative decarboxylation of α-ketoglutarate to succinyl CoA catalyzed by α-ketoglutarate dehydrogenase (KGDH). The carbon skeleton is subsequently converted into malate via multiple steps in the tricarboxylic acid (TCA) cycle and pyruvate via malic enzyme (ME). Alternatively, the concerted action of phosphoenolpyruvate carboxykinase (PEPCK) and pyruvate kinase (PK) converts oxaloacetate (OAA) into pyruvate. The operation of ME or PEPCK plus PK in the direction of pyruvate is necessary for complete oxidation of glutamate. Pyruvate re-enters the TCA cycle via pyruvate dehydrogenase (PDH) and the carbon skeleton originating from glutamate may through this pathway be completely oxidized to CO2 in the TCA cycle. Asp, aspartate.
Overview of the glutamate-glutamine cycle and its role in glutamatergic neurotransmission
Based on a series of elegant studies of radioactive labeling of glutamate and glutamine using different 14C-labeled precursors such as glucose, acetate and bicarbonate it was concluded that the brain contains at least two separate cellular compartments of these amino acids each having a distinct turn-over of the amino acid pools (for references, see McKenna et al. 2012, Schousboe 2012). The seminal demonstration of the astrocyte specific localization of glutamine synthetase (Norenberg & Martinez-Hernandez 1979) together with the multiple compartments of glutamate revealed by the labeling studies, led to the firm conclusion that the proposed glutamate-glutamine cycle (see Figure 3) must be the mechanism allowing inter-cellular exchange of these amino acids (see, Öz et al. 2012, and Schousboe et al. 2012 for references). The selective cellular localization of specific glutamate and glutamine transporters on neuronal and glial plasma membranes is consistent with the functional importance of this cycle (see, Schousboe et al. 2012).
Figure 3.
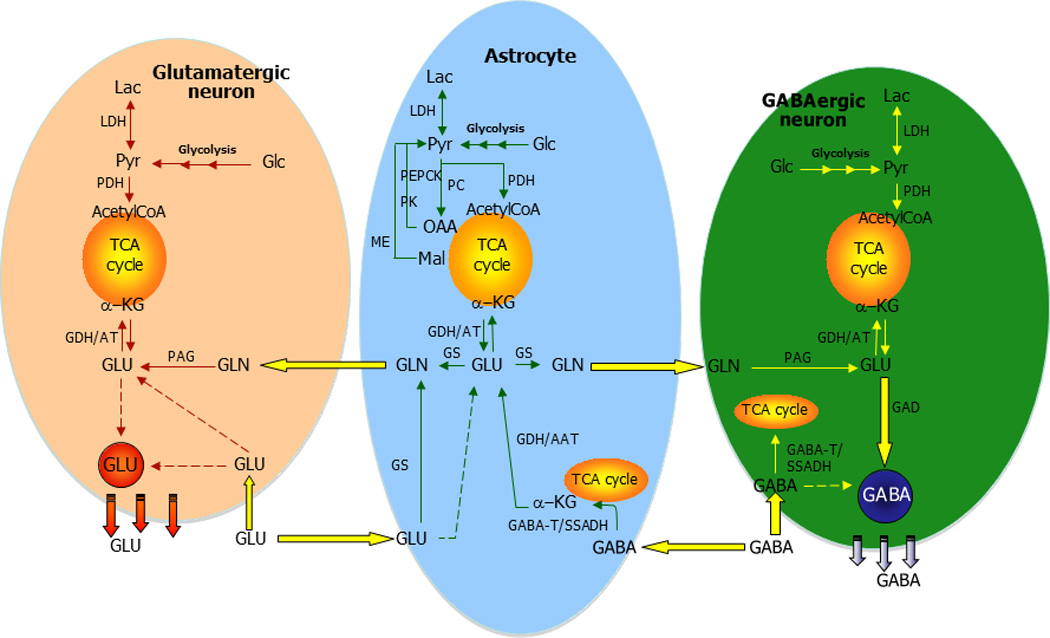
Neurotransmitter glutamate (GLU) is mainly taken up by surrounding astrocytes subsequent to interaction with receptors in the synapse. In the astrocyte, glutamate is either converted to glutamine (GLN) catalyzed by glutamine synthetase (GS) as part of the glutamate-glutamine cycle or metabolized in the tricarboxylic acid (TCA) cycle. Glutamine is transferred to the glutamatergic neuron to be used for synthesis of glutamate catalyzed by phosphate activated glutaminase (PAG). Glutamate enters the TCA cycle by the activity of glutamate dehydrogenase (GDH) or an aminotransferase (AT), and the carbon skeleton may either be completely oxidatively metabolized via pyruvate recycling including malic enzyme (ME) activity or phosphoenolpyruvate carboxykinase and pyruvate kinase. Alternative, the carbon skeleton supports the pool of TCA cycle intermediates and in that way potentially increases the oxidation of acetyl CoA in the TCA cycle. De novo synthesis of glutamate and glutamine from glucose occurs via the concerted action of pyruvate dehydrogenase (PDH) and pyruvate carboxylase (PC) making a net synthesis of TCA cycle intermediates. CIT, citrate; OAA, oxaloacetate; PYR, pyruvate.
When the concept of the glutamate-glutamine cycle was first introduced it was thought that essentially all glutamate being released as neurotransmitter would be transferred to the astrocytes where it would quantitatively be converted to glutamine and subsequently transferred back to the glutamatergic neurons (Cotman et al. 1981). The fact that exogenous glutamate taken up by astrocytes can be oxidatively metabolized (see below for further details) demonstrates that the glutamate-glutamine cycle does not operate in a stoichiometric manner as first proposed (see, McKenna 2007, McKenna et al. 2012). If the glutamate-glutamine cycle exclusively operated in a stoichiometric fashion, then none of the glutamate taken up would be used to offset the high energy cost of glutamate transport into astrocytes, i.e. 3 ATP are required for transport of each glutamate molecule and activity of Na+-K+-ATPase to restore the sodium gradient (Attwell & Laughlin 2001, McKenna 2013).
Since the glutamate-glutamine cycle is an open cycle that loses intermediates to other pathways, the de novo synthesis of glutamine in astrocytes helps to maintain the operation of this cycle. Numerous studies have been aimed at obtaining a quantitative measure of the anaplerotic de novo synthesis of glutamate and glutamine; this information has been summarized by Öz et al. (2012). The rate of anaplerosis mediated by pyruvate carboxylase ranges from 6 to 35 % of the rate of the TCA cycle and fulfills the need for de novo synthesis of glutamine (Öz et al. 2004, 2012). Since glutamine also serves as the main precursor for GABA (Sonnewald et al. 1993c) and some of the GABA released by neurons is taken up and metabolized by astrocytes, maintenance of GABAergic neurotransmission also relies on anaplerosis (Schousboe et al. 2013).
Influence of the concentration of glutamate on the metabolic pathways for glutamate metabolism
Early studies of glutamate metabolism in astrocytes using tracer concentrations of 14C-labeled precursors reported a high proportion of the glutamate was converted to glutamine (Farinelli & Nicklas 1992, Zielke et al. 1990). However, a study by Yu et al. (1982) showed that a substantial fraction of exogenous glutamate was metabolized via GDH and subsequently oxidized to CO2 in astrocytes. Sonnewald et al. (1993b) using 13C-NMR spectroscopy reported that a significant proportion of the label from glutamate metabolism in astrocytes was found in lactate. This labeling can only occur when glutamate is converted to α-ketoglutarate and further metabolized via the TCA cycle to malate, which can be converted to pyruvate and subsequently to lactate by a partial pyruvate recycling pathway (Fig. 4). These studies confirmed that a significant amount of glutamate was oxidatively metabolized for energy in astrocytes (Sonnewald et al. 1993b, McKenna 2013). McKenna et al. (1996) demonstrated that when the exogenous glutamate concentration was increased from 0.1 mM to 0.5 mM the proportion of glutamate oxidized by the TCA cycle in astrocytes greatly increased (from ~ 15% to 43%) and the percent converted to glutamine decreased correspondingly (from ~ 85% to 57%). This demonstrated that the metabolic fate of glutamate was shifted away from conversion to glutamine and towards oxidative metabolism as the concentration of glutamate in the extracellular milieu increased. Since the amount of glutamate in the synaptic cleft increases several orders of magnitude during depolarization (from ~ 10 µM to 1 mM) this provides astrocytes with the metabolic flexibility to readily form ATP from glutamate oxidation in the TCA cycle which offsets the high cost of glutamate transport into astrocytes (for further details, see McKenna 2013).
Figure 4.
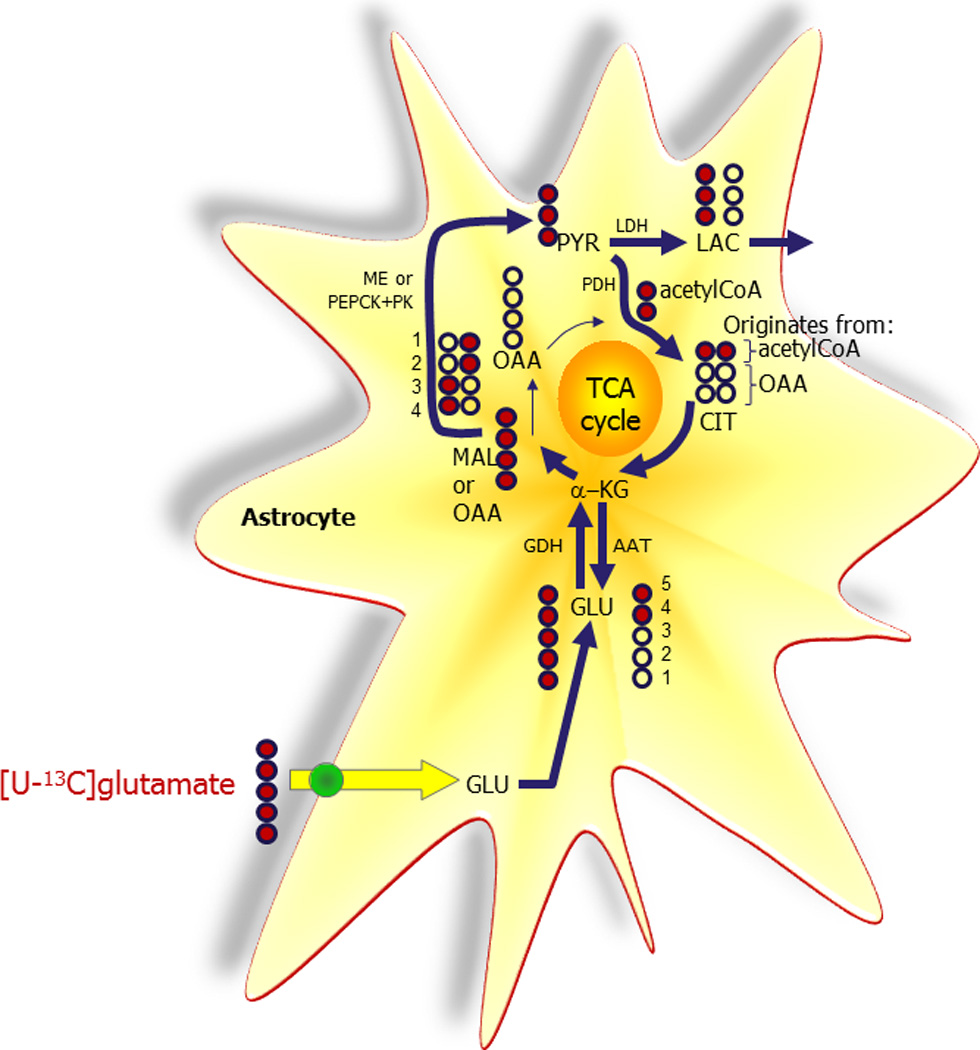
[U-13C]Glutamate (GLU) may be completely metabolized to CO2 via the “pyruvate recycling pathway”. Initially [U-13C]glutamate is converted to α-ketoglutarate (α-KG) catalyzed mainly by glutamate dehydrogenase (GDH). Via the tricarboxylic acid (TCA) cycle [U-13C]malate (MAL)/oxaloacetate (OAA) is formed. [U-13C]Malate may be oxidatively decarboxylated to [U-13C]pyruvate (PYR) catalyzed by malic enzyme (ME). Alternatively, [U-13C]oxaloacetate may be metabolized to pyruvate via [U-13C]phosphoenolpyruvate catalyzed by the concerted action of phosphoenolpyruvate carboxykinase (PEPCK) and pyruvate kinase (PK). The carbons originating from glutamate can subsequently re-enter the TCA cycle by oxidative decarboxylation of [U-13C]pyruvate to [1,2-13C]acetyl CoA catalyzed by pyruvate dehydrogenase (PDH). Unlabeled oxaloacetate may condense with [1,2-13C]acetyl CoA forming [1,2-13C]citrate (CIT). From metabolism in the TCA cycle [4,5-13C] α-ketoglutarate is formed and consequently [4,5-13C]glutamate due to the activity of aspartate aminotransferase (AAT). The symmetric succinate molecule causes scrambling of labeling and therefore both [1,2-13C] and [3,4-13C] oxaloacetate are formed. Oxaloacetate is in equilibrium with [1,2-13C] and [3,4-13C]aspartate due to the activity of aspartate aminotransferase. Alternative to oxidative decarboxylation of pyruvate, [U-13C]pyruvate may be converted to [U-13C]lactate (LAC) catalyzed by lactate dehydrogenase (LDH).
Recent studies show that the astrocyte glutamate transporter GLT-1 (in humans EAAT2) forms a complex with other proteins including hexokinase, several specific mitochondrial proteins including GDH and mitochondria that together serves to effectively channel glutamate towards energy producing oxidative metabolism rather than the energy utilizing reaction of conversion to glutamine in the cytosol (Genda et al. 2011).
Glutamate and ammonia homeostasis under normal and hyperammonemic conditions
Physiological conditions
In glutamatergic and to some extent in GABAergic neurons ammonia is generated by the action of PAG, which removes the amido nitrogen from glutamine to form glutamate and subsequently GABA in GABAergic neurons (Fig. 5). Since GS, the primary enzyme capable of ammonia fixation in the brain, is located in astrocytes there needs to be a way by which ammonia nitrogen can be transferred between the neurons and the surrounding astrocytes. The specific mechanism(s) for returning ammonia removed by PAG in neurons to astrocytes has not been defined. For years it was assumed that the ammonium ion (being the prevailing form of ammonia) can diffuse freely through plasma membranes or perhaps through channels (Cooper & Plum 1987). Recently, two shuttle mechanisms based on exchange of an amino acid have been proposed (Lieth et al. 2001, Waagepetersen et al. 2000, Yudkoff et al. 1996, Zwingmann et al. 2000). Both models operate on the basis of ammonia being fixed into the amino group of glutamate by the action of glutamate dehydrogenase in neurons, and subsequently being transferred to either alanine or a branched chain amino acid, which serves to return the amino group to astrocytes (Fig. 5). In astrocytes the amino groups are transferred to glutamate by the respective aminotransferases (see above and Fig. 1) forming branched chain keto acids and pyruvate from the branch chain amino acids and alanine, respectively. The amino group transferred to glutamate is subsequently removed by oxidative deamination via GDH making it available to glutamine synthetase for glutamine formation (see Figs. 1 and 5). A careful analysis of these mechanisms by modeling the rates of the reactions involved has, however, failed to provide evidence that the activity of these amino acid shuttles would be sufficient to match the actual need for transfer of ammonia back to astrocytes (Rothman et al. 2012). Hence, at the present time the quantitative importance of these proposed mechanisms for ammonia transfer in vivo has not been resolved.
Figure 5.
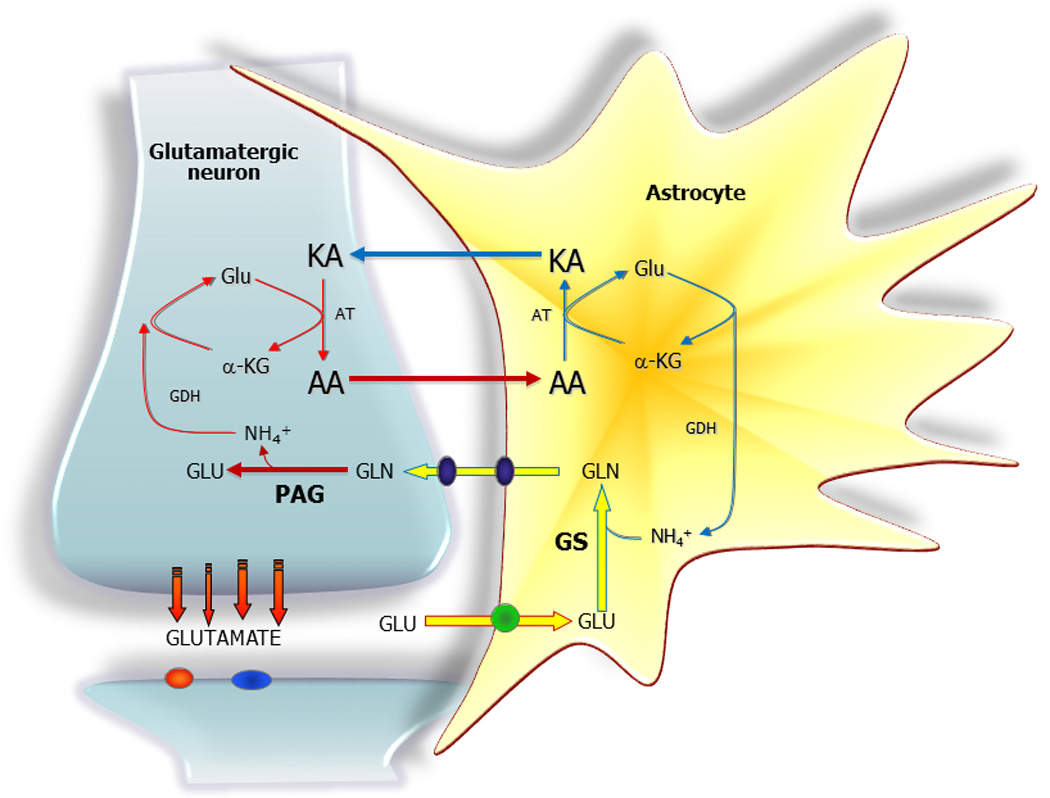
Neurotransmitter glutamate is subsequent to receptor interaction primarily taken up mainly by astrocytes. Glutamate (GLU) is amidated to glutamine (GLN) via glutamine synthetase (GS) and glutamine is subsequently released from the astrocyte via specific transporters followed by uptake into the neuron. In the neuron glutamine is deamidated by phosphate activated glutaminase (PAG) to glutamate, which completes the glutamate - glutamine cycle. A net transfer of nitrogen from the astrocyte to the neuron occurs as part of the glutamate - glutamine cycle. This nitrogen transfer may be counteracted by transport of a neuro-inactive amino acid (AA) likely either alanine or one of the branched chain amino acids. The glutamate - glutamine cycle is coupled to an amino acid – keto acid (KA) cycle via the action of glutamate dehydrogenase (GDH) and the relevant aminotransferase (AT).
Hyperammonemia
The most efficient pathway for ammonia fixation in the brain is the GS catalyzed conversion of glutamate to glutamine and a classical experiment by Cooper et al. (1979) using infusion of trace amounts of [13N]ammonia into the brain showed that at least 98% of the labeled ammonia could be found in the amide group of glutamine within seconds. When GS was inhibited by MSO, ammonia radioactivity was recovered in glutamate, albeit the amounts were small. This does, however, show that under appropriate conditions reductive amination mediated by GDH can occur (see above). In this context, it may be noted that during hyperammonemic conditions in neuronal-astrocytic co-cultures, the formation of both alanine and glutamine is increased (Leke et al. 2011). As pointed out above, de novo synthesis of glutamine requires CO2 fixation by pyruvate carboxylase to form oxaloacetate; interestingly, hyperammonemic conditions have been shown to stimulate this pathway (Leke et al. 2011). On the other hand, when GS was inhibited and ammonia was fixed in alanine, the glycolytic pathway was accelerated while the anaplerotic pathway did not increase (Dadsetan et al. 2011, 2013). This shows that astrocytes are able to switch back and forth between anaplerosis and glycolysis depending on whether pyruvate or a TCA cycle intermediate is needed. This concept has been confirmed in cultures and in the brain in vivo since inhibition of GS by MSO during hyperammonemic conditions leads to a decrease in glutamine and an increase in alanine production (Dadsetan et al. 2011, 2013, Fries et al. 2013). This may be due in part to the fact that alanine does not function as an intracellular osmolyte in astrocytes, whereas glutamine does perform this function. It has been proposed that inhibition of GS may be a potentially useful therapeutic strategy to prevent the glutamine-associated glial swelling and the brain edema observed in hepatic encephalopathy (Brusilow et al. 2010, Dadsetan et al. 2011, 2013).
Concluding remarks
It should be noted that glutamate metabolism is exceedingly complex and highly compartmentalized. Maintenance of the glutamate and GABA neurotransmitter pools is strictly dependent on the de novo synthesis of glutamine in astrocytes. The exquisite anatomical specialization of astrocytic endfeet enables these cells to rapidly and efficiently remove neurotransmitters from the synaptic cleft and provide glutamine to replenish neurotransmitter pools in glutamatergic and GABAergic neurons. This is possible due to the fine-tuned biochemical machinery including the distribution of pertinent transporters, as well as the presence of mitochondria and specific cytosolic enzymes in the astrocytic processes which ensheathe the synapses. This allows astrocytes to respond to subtle changes in neurotransmission by dynamically adjusting their anaplerotic and glycolytic activities to meet the demands for maintaining neurotransmission. Additionally, astrocytes can readily shift the metabolic fate of glutamate removed from the synaptic cleft towards increased oxidative energy metabolism relative to direct formation of glutamine.
In conclusion, this chapter has summarized the evidence that astrocytes are dynamic partners in both glutamatergic and GABAergic neurotransmission in brain. Protoplasmic astrocytes cover and actively interact with hundreds of thousands to millions synapses in human brain (Oberheim et al. 2006, 2009). Hence, astrocytes are essential partners in neurotransmission since they continuously monitor the extracellular milieu and modulate their metabolic activity in response to fluctuations in neuronal activity.
References
- Aoki C, Milner TA, Berger SB, Sheu KF, Blass JP, Pickel VM. Glial glutamate dehydrogenase: ultrastructural localization and regional distribution in relation to the mitochondrial enzyme, cytochrome oxidase. J. Neurosci. Res. 1987;18:305–318. doi: 10.1002/jnr.490180207. [DOI] [PubMed] [Google Scholar]
- Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 2001;21:1133–1145. doi: 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- Bak LK, Waagepetersen HS, Sorensen M, Ott P, Vilstrup H, Keiding S, Schousboe A. Role of branched chain amino acids in cerebral ammonia homeostasis related to hepatic encephalopathy. Metab. Brain Dis. 2013;28:209–215. doi: 10.1007/s11011-013-9381-7. [DOI] [PubMed] [Google Scholar]
- Bakken IJ, White LR, Aasly J, Unsgard G, Sonnewald U. [U-13C]Aspartate metabolism in cultured cortical astrocytes and cerebellar granule neurons studied by NMR spectroscopy. Glia. 1998;23:271–277. doi: 10.1002/(sici)1098-1136(199807)23:3<271::aid-glia9>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Benuck M, Stern F, Lajtha A. Regional and subcellular distribution of aminotransferases in rat brain. J. Neurochem. 1972;19:949–957. doi: 10.1111/j.1471-4159.1972.tb01416.x. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Diamond JS, Jahr CE. Clearance of glutamate inside the synapse and beyond. Curr. Opin Neurobiol. 1999;9:293–298. doi: 10.1016/s0959-4388(99)80043-9. [DOI] [PubMed] [Google Scholar]
- Berl S, Clarke DD. Compartmentation of amino acid metabolism. In: In: Lajtha A, editor. Handbook of neurochemistry. Vol. 2. New York, USA: Plenum Publishing Corporation; 1969. pp. 447–472. [Google Scholar]
- Brusilow SW, Koehler RC, Traystman RJ, Cooper AJ. Astrocyte glutamine synthetase: importance in hyperammonemic syndromes and potential target for therapy. Neurotherapeutics. 2010;7:452–470. doi: 10.1016/j.nurt.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JT, Mitala CM, Kundu S, Verma A, Elkind JA, Nissim I, Cohen AS. Dietary branched chain amino acids ameliorate injury-induced cognitive impairment. Proc. Nat. Acad. Sci. U.S.A. 2010;107:366–371. doi: 10.1073/pnas.0910280107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AJ. L-Glutamate(2-oxoglutarate) aminostransferases. In: Kvamme E, editor. Glutamine and glutamate in mammals. I. Boca Raton, FL, USA: CRC Press; 1988. pp. 123–152. [Google Scholar]
- Cooper AJ, McDonald JM, Gelbard AS, Gledhill RF, Duffy TE. The metabolic fate of 13N-labeled ammonia in rat brain. J. Biol. Chem. 1979;254:4982–4992. [PubMed] [Google Scholar]
- Cooper AJ, Plum F. Biochemistry and physiology of brain ammonia. Physiol. Rev. 1987;67:440–519. doi: 10.1152/physrev.1987.67.2.440. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Foster A, Lanthorn T. An overview of glutamate as a neurotransmitter. Advan. Biochem. Psychopharm. 1981;27:1–27. [PubMed] [Google Scholar]
- Dadsetan S, Bak LK, Sorensen M, Keiding S, Vilstrup H, Ott P, Leke R, Schousboe A, Waagepetersen HS. Inhibition of glutamine synthesis induces glutamate dehydrogenase-dependent ammonia fixation into alanine in co-cultures of astrocytes and neurons. Neurochem. Int. 2011;58:482–488. doi: 10.1016/j.neuint.2011.03.008. [DOI] [PubMed] [Google Scholar]
- Dadsetan S, Kukolj E, Bak LK, Sorensen M, Ott P, Vilstrup H, Schousboe A, Keiding S, Waagepetersen HS. Brain alanine formation as an ammonia-scavenging pathway during hyperammonemia: effects of glutamine synthetase inhibition in rats and astrocyte-neuron co-cultures. J. Cereb. Blood Flow Metab. 2013;33:1235–1241. doi: 10.1038/jcbfm.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel PC, Dalziel K. The equilibrium constants of the glutamate dehydrogenase systems. Biochem. J. 1967;105:691–695. doi: 10.1042/bj1050691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errera M, Greenstein JP. Phosphate-activated glutaminase in kidney and other tissues. J. Biol. Chem. 1949;178:495–502. [PubMed] [Google Scholar]
- Fahien LA, Hsu SL, Kmiotek E. Effect of aspartate on complexes between glutamate dehydrogenase and various aminotransferases. J. Biol. Chem. 1977;252:1250–1256. [PubMed] [Google Scholar]
- Farinelli SE, Nicklas WJ. Glutamate metabolism in rat cortical astrocyte cultures. J. Neurochem. 1992;58:1905–1915. doi: 10.1111/j.1471-4159.1992.tb10068.x. [DOI] [PubMed] [Google Scholar]
- Fries A, Dadsetan S, Keiding S, et al. Effect of glutamine synthetase inhibition on brain and interorgan ammonia metabolism in bile duct ligated rats. J. Cereb. Blood Flow Metab. 2013 doi: 10.1038/jcbfm.2013.218. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garfinkel D. A simulation study of the metabolism and compartmentation in brain of glutamate, aspartate, the Krebs cycle, and related metabolites. J. Biol. Chem. 1966;241:3918–3929. [PubMed] [Google Scholar]
- Genda EN, Jackson JG, Sheldon AL, et al. Co-compartmentalization of the astroglial glutamate transporter, GLT-1, with glycolytic enzymes and mitochondria. J. Neurosci. 2011;31:18275–18288. doi: 10.1523/JNEUROSCI.3305-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MM, Nautiyal M, Wynn RM, Mobley JA, Chuang DT, Hutson SM. Branched-chain amino acid metabolon: interaction of glutamate dehydrogenase with the mitochondrial branched-chain aminotransferase (BCATm) J. Biol. Chem. 2010;285:265–276. doi: 10.1074/jbc.M109.048777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komlos D, Mann KD, Zhuo Y, Ricupero CL, Hart RP, Liu AY, Firestein BL. Glutamate dehydrogenase 1 and SIRT4 regulate glial development. Glia. 2013;61:394–408. doi: 10.1002/glia.22442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs HA. Metabolism of amino-acids: The synthesis of glutamine from glutamic acid and ammonia, and the enzymic hydrolysis of glutamine in animal tissues. Biochem. J. 1935;29:1951–1969. doi: 10.1042/bj0291951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs HA. Equilibria in transamination systems. Biochem. J. 1953;54:82–86. doi: 10.1042/bj0540082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvamme E, Torgner IA, Roberg B. Kinetics and localization of brain phosphate activated glutaminase. J. Neurosci. Res. 2001;66:951–958. doi: 10.1002/jnr.10041. [DOI] [PubMed] [Google Scholar]
- Lange SC, Bak LK, Waagepetersen HS, Schousboe A, Norenberg MD. Primary Cultures of Astrocytes: Their Value in Understanding Astrocytes in Health and Disease. Neurochem. Res. 2012;37:2569–2588. doi: 10.1007/s11064-012-0868-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson OM, Drejer J, Kvamme E, Svenneby G, Hertz L, Schousboe A. Ontogenetic development of glutamate and GABA metabolizing enzymes in cultured cerebral cortex interneurons and in cerebral cortex in vivo. Int. J. Dev. Neurosci. 1985;3:177–185. doi: 10.1016/0736-5748(85)90008-5. [DOI] [PubMed] [Google Scholar]
- Lavu S, Boss O, Elliott PJ, Lambert PD. Sirtuins--novel therapeutic targets to treat age-associated diseases. Nat. Rev. Drug Discov. 2008;7:841–853. doi: 10.1038/nrd2665. [DOI] [PubMed] [Google Scholar]
- Leke R, Bak LK, Anker M, et al. Detoxification of Ammonia in Mouse Cortical GABAergic Cell Cultures Increases Neuronal Oxidative Metabolism and Reveals an Emerging Role for Release of Glucose-Derived Alanine. Neurotox. Res. 2011;19:496–510. doi: 10.1007/s12640-010-9198-7. [DOI] [PubMed] [Google Scholar]
- Lieth E, LaNoue KF, Berkich DA, Xu B, Ratz M, Taylor C, Hutson SM. Nitrogen shuttling between neurons and glial cells during glutamate synthesis. J. Neurochem. 2001;76:1712–1723. doi: 10.1046/j.1471-4159.2001.00156.x. [DOI] [PubMed] [Google Scholar]
- Lovatt D, Sonnewald U, Waagepetersen HS, et al. The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J. Neurosci. 2007;27:12255–12266. doi: 10.1523/JNEUROSCI.3404-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DL, Rimvall K. Regulation of gamma-aminobutyric acid synthesis in the brain. J. Neurochem. 1993;60:395–407. doi: 10.1111/j.1471-4159.1993.tb03165.x. [DOI] [PubMed] [Google Scholar]
- Matsui K, Jahr CE, Rubio ME. High-concentration rapid transients of glutamate mediate neural-glial communication via ectopic release. J. Neurosci. 2005;25:7538–7547. doi: 10.1523/JNEUROSCI.1927-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna M. Glutamate pays its own way in astrocytes. Front. Endocrinol. 2013 doi: 10.3389/fendo.2013.00191. In revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna M, Dienel GA, Sonnewald U, Waagepetersen HS, Schousboe A. Energy metabolism of the brain. In: In: Brady ST, Siegel GJ, Albers RW, Price DI, editors. Basic Neurochmistry. Waltham, MS, USA: Academic Press, Elsevier; 2012. pp. 200–231. [Google Scholar]
- McKenna MC. The glutamate-glutamine cycle is not stoichiometric: fates of glutamate in brain. J. Neurosci. Res. 2007;85:3347–3358. doi: 10.1002/jnr.21444. [DOI] [PubMed] [Google Scholar]
- McKenna MC. Glutamate dehydrogenase in brain mitochondria: do lipid modifications and transient metabolon formation influence enzyme activity? Neurochem. Int. 2011;59:525–533. doi: 10.1016/j.neuint.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna MC, Hopkins IB, Lindauer SL, Bamford P. Aspartate aminotransferase in synaptic and nonsynaptic mitochondria: differential effect of compounds that influence transient hetero-enzyme complex (metabolon) formation. Neurochem. Int. 2006;48:629–636. doi: 10.1016/j.neuint.2005.11.018. [DOI] [PubMed] [Google Scholar]
- McKenna MC, Sonnewald U, Huang X, Stevenson J, Zielke HR. Exogenous glutamate concentration regulates the metabolic fate of glutamate in astrocytes. J. Neurochem. 1996;66:386–393. doi: 10.1046/j.1471-4159.1996.66010386.x. [DOI] [PubMed] [Google Scholar]
- McKenna MC, Stevenson JH, Huang X, Hopkins IB. Differential distribution of the enzymes glutamate dehydrogenase and aspartate aminotransferase in cortical synaptic mitochondria contributes to metabolic compartmentation in cortical synaptic terminals. Neurochem. Int. 2000;37:229–241. doi: 10.1016/s0197-0186(00)00042-5. [DOI] [PubMed] [Google Scholar]
- Norenberg MD, Martinez-Hernandez A. Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res. 1979;161:303–310. doi: 10.1016/0006-8993(79)90071-4. [DOI] [PubMed] [Google Scholar]
- Obel LF, Andersen KMH, Bak LK, Schousboe A, Waagepetersen HS. Effects of adrenergic agents on intracellular ca2+ homeostasis and metabolism of glucose in astrocytes with an emphasis on pyruvate carboxylation, oxidative decarboxylation and recycling: implications for glutamate neurotransmission and excitotoxicity. Neurotox. Res. 2012;21:405–417. doi: 10.1007/s12640-011-9296-1. [DOI] [PubMed] [Google Scholar]
- Oberheim NA, Takano T, Han X, et al. Uniquely hominid features of adult human astrocytes. J. Neurosci. 2009;29:3276–3287. doi: 10.1523/JNEUROSCI.4707-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberheim NA, Wang X, Goldman S, Nedergaard M. Astrocytic complexity distinguishes the human brain. Trends Neurosci. 2006;29:547–553. doi: 10.1016/j.tins.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Okada Y, Taniguchi H, Schimada C. High concentration of GABA and high glutamate decarboxylase activity in rat pancreatic islets and human insulinoma. Science. 1976;194:620–622. doi: 10.1126/science.185693. [DOI] [PubMed] [Google Scholar]
- Ott P, Clemmesen O, Larsen FS. Cerebral metabolic disturbances in the brain during acute liver failure: from hyperammonemia to energy failure and proteolysis. Neurochem. Int. 2005;47:13–18. doi: 10.1016/j.neuint.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Öz G, Berkich DA, Henry PG, Xu Y, LaNoue K, Hutson SM, Gruetter R. Neuroglial metabolism in the awake rat brain: CO2 fixation increases with brain activity. J. Neurosci. 2004;24:11273–11279. doi: 10.1523/JNEUROSCI.3564-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Öz G, Okar DA, Henry PG. Glutamate-glutamine cycle and anaplerosis. In: Choi I, Gruetter R, editors. Neural metabolism in vivo, Adv. Neurobiol. Vol. 4. New York, USA: Springer; 2012. pp. 921–946. [Google Scholar]
- Pamiljans V, Krishnaswamy PR, Dumville G, Meister A. Studies on the mechanism of glutamine synthesis; isolation and properties of the enzyme from sheep brain. Biochemistry. 1962;1:153–158. doi: 10.1021/bi00907a023. [DOI] [PubMed] [Google Scholar]
- Quastel JH. Metabolic compartmentation in the brain and effects of metabolic inhibitors. In: Berl S, Clarke DD, Scheider D, editors. Metabolic compartmentation and neurotransmission. Vol. 6. New York, USA: Plenum Publishing Corporation; 1975. pp. 337–361. [Google Scholar]
- Roberts E, Frankel S. gamma-Aminobutyric acid in brain: its formation from glutamic acid. J. Biol. Chem. 1950;187:55–63. [PubMed] [Google Scholar]
- Ronzio RA, Rowe WB, Meister A. Studies on the mechanism of inhibition of glutamine synthetase by methionine sulfoximine. Biochemistry. 1969;8:1066–1075. doi: 10.1021/bi00831a038. [DOI] [PubMed] [Google Scholar]
- Rothman DL, De Feyter HM, Maciejewski PK, Behar KL. Is there in vivo evidence for amino acid shuttles carrying ammonia from neurons to astrocytes? Neurochem. Res. 2012;37:2597–2612. doi: 10.1007/s11064-012-0898-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Barber R, Wu J, Matsuda T, Roberts E, Vaughn JE. Immunohistochemical localization of glutamate decarboxylase in rat cerebellum. Proc. Nat. Acad. Sci. U.S.A. 1974;71:269–273. doi: 10.1073/pnas.71.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salganicoff L, Derobertis E. Subcellular Distribution of the Enzymes of the Glutamic Acid, Glutamine and Gamma-Aminobutyric Acid Cycles in Rat Brain. J. Neurochem. 1965;12:287–309. doi: 10.1111/j.1471-4159.1965.tb06766.x. [DOI] [PubMed] [Google Scholar]
- Schousboe A. Studies of brain metabolism: A historical perspective. In: Choi I, Gruetter R, editors. Neural metabolism in vivo, Adv. Neurobiol. Vol. 4. New York, USA: Springer; 2012. pp. 909–920. [Google Scholar]
- Schousboe A, Bak LK, Madsen KK, Waagepetersen HS. Amino acid neurotransmitter synthesis and removal. In: Kettenmann H, Ransom B, editors. Neuroglia. Oxford, UK: Oxford University Press; 2012. pp. 443–456. [Google Scholar]
- Schousboe A, Bak LK, Waagepetersen HS. Astrocytic Control of Biosynthesis and Turnover of the Neurotransmitters Glutamate and GABA. Front. Endocrinol. 2013;4:102. doi: 10.3389/fendo.2013.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schousboe A, Hertz L, Svenneby G, Kvamme E. Phosphate activated glutaminase activity and glutamine uptake in primary cultures of astrocytes. J. Neurochem. 1979;32:943–950. doi: 10.1111/j.1471-4159.1979.tb04579.x. [DOI] [PubMed] [Google Scholar]
- Shank RP, Bennett GS, Freytag SO, Campbell GL. Pyruvate carboxylase: an astrocyte-specific enzyme implicated in the replenishment of amino acid neurotransmitter pools. Brain Res. 1985;329:364–367. doi: 10.1016/0006-8993(85)90552-9. [DOI] [PubMed] [Google Scholar]
- Soghomonian JJ, Martin DL. Two isoforms of glutamate decarboxylase: why? Trends Pharmacol. Sci. 1998;19:500–505. doi: 10.1016/s0165-6147(98)01270-x. [DOI] [PubMed] [Google Scholar]
- Sonnewald U, Westergaard N, Hassel B, Muller TB, Unsgard G, Fonnum F, Hertz L, Schousboe A, Petersen SB. NMR spectroscopic studies of 13C acetate and 13C glucose metabolism in neocortical astrocytes: evidence for mitochondrial heterogeneity. Devel. Neurosci. 1993a;15:351–358. doi: 10.1159/000111355. [DOI] [PubMed] [Google Scholar]
- Sonnewald U, Westergaard N, Petersen SB, Unsgard G, Schousboe A. Metabolism of [U-13C]glutamate in astrocytes studied by 13C NMR spectroscopy: incorporation of more label into lactate than into glutamine demonstrates the importance of the tricarboxylic acid cycle. J. Neurochem. 1993b;61:1179–1182. doi: 10.1111/j.1471-4159.1993.tb03641.x. [DOI] [PubMed] [Google Scholar]
- Sonnewald U, Westergaard N, Schousboe A, Svendsen JS, Unsgard G, Petersen SB. Direct demonstration by [13C]NMR spectroscopy that glutamine from astrocytes is a precursor for GABA synthesis in neurons. Neurochem. Int. 1993c;22:19–29. doi: 10.1016/0197-0186(93)90064-c. [DOI] [PubMed] [Google Scholar]
- Spanaki C, Zaganas I, Kounoupa Z, Plaitakis A. The complex regulation of human glud1 and glud2 glutamate dehydrogenases and its implications in nerve tissue biology. Neurochem. Int. 2012;61:470–481. doi: 10.1016/j.neuint.2012.05.020. [DOI] [PubMed] [Google Scholar]
- van den Berg CJ, Garfinkel D. A stimulation study of brain compartments. Metabolism of glutamate and related substances in mouse brain. Biochem. J. 1971;123:211–218. doi: 10.1042/bj1230211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waagepetersen HS, Sonnewald U, Larsson OM, Schousboe A. A possible role of alanine for ammonia transfer between astrocytes and glutamatergic neurons. J. Neurochem. 2000;75:471–479. doi: 10.1046/j.1471-4159.2000.0750471.x. [DOI] [PubMed] [Google Scholar]
- Walls AB, Eyjolfsson EM, Smeland OB, Nilsen LH, Schousboe I, Schousboe A, Sonnewald U, Waagepetersen HS. Knockout of GAD65 has major impact on synaptic GABA synthesized from astrocyte-derived glutamine. J. Cereb. Blood Flow Metab. 2011;31:494–503. doi: 10.1038/jcbfm.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westergaard N, Varming T, Peng L, Sonnewald U, Hertz L, Schousboe A. Uptake, release, and metabolism of alanine in neurons and astrocytes in primary cultures. J. Neurosci. Res. 1993;35:540–545. doi: 10.1002/jnr.490350510. [DOI] [PubMed] [Google Scholar]
- Wu JY, Matsuda T, Roberts E. Purification and characterization of glutamate decarboxylase from mouse brain. J. Biol. Chem. 1973;248:3029–3034. [PubMed] [Google Scholar]
- Yu AC, Drejer J, Hertz L, Schousboe A. Pyruvate carboxylase activity in primary cultures of astrocytes and neurons. J. Neurochem. 1983;41:1484–1487. doi: 10.1111/j.1471-4159.1983.tb00849.x. [DOI] [PubMed] [Google Scholar]
- Yu AC, Schousboe A, Hertz L. Metabolic fate of 14C-labeled glutamate in astrocytes in primary cultures. J. Neurochem. 1982;39:954–960. doi: 10.1111/j.1471-4159.1982.tb11482.x. [DOI] [PubMed] [Google Scholar]
- Yudkoff M, Daikhin Y, Nelson D, Nissim I, Erecinska M. Neuronal metabolism of branched-chain amino acids: flux through the aminotransferase pathway in synaptosomes. J. Neurochem. 1996;66:2136–2145. doi: 10.1046/j.1471-4159.1996.66052136.x. [DOI] [PubMed] [Google Scholar]
- Zaganas I, Kanavouras K, Mastorodemos V, Latsoudis H, Spanaki C, Plaitakis A. The human GLUD2 glutamate dehydrogenase: localization and functional aspects. Neurochem. Int. 2009;55:52–63. doi: 10.1016/j.neuint.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Zaganas I, Waagepetersen HS, Georgopoulos P, Sonnewald U, Plaitakis A, Schousboe A. Differential expression of glutamate dehydrogenase in cultured neurons and astrocytes from mouse cerebellum and cerebral cortex. J. Neurosci. Res. 2001;66:909–913. doi: 10.1002/jnr.10058. [DOI] [PubMed] [Google Scholar]
- Zielke HR, Tildon JT, Landry ME, Max SR. Effect of 8-bromo-cAMP and dexamethasone on glutamate metabolism in rat astrocytes. Neurochem. Res. 1990;15:1115–1122. doi: 10.1007/BF01101713. [DOI] [PubMed] [Google Scholar]
- Zwingmann C, Richter-Landsberg C, Brand A, Leibfritz D. NMR spectroscopic study on the metabolic fate of [3-(13)C]alanine in astrocytes, neurons, and cocultures: implications for glia-neuron interactions in neurotransmitter metabolism. Glia. 2000;32:286–303. doi: 10.1002/1098-1136(200012)32:3<286::aid-glia80>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]