

Abstract
The aim of this study was to assess antidepressant efficacy and safety of venlafaxine extended release in Japanese patients with major depressive disorder (MDD). We carried out a double-blinded, placebo-controlled, randomized study using fixed (75 mg/day) and flexible (75–225 mg/day, most patients attained to 225 mg/day) doses, followed by the long-term, open-labeled, extension study. Outpatients aged at least 20 years diagnosed with MDD were included. The primary efficacy measure was change from baseline in the Hamilton Rating Scale for Depression (HAM-D17) score at week 8; secondary efficacy measures included the Montgomery–Åsberg Depression Rating Scale, the Quick Inventory of Depressive Symptomatology self-report version, HAM-D6, and Clinical Global Impression scales in the double-blinded study. Overall, 538 patients were randomized; significant differences were observed in the primary efficacy variable in the fixed-dose group (−10.76; P=0.031), but not in the flexible-dose (−10.37; P=0.106) group compared with placebo (−9.25). However, the flexible-dose group showed significant efficacy in several secondary measures. Treatment-related adverse events in the treatment period were 51.7 and 67.8% in the fixed-dose and flexible-dose groups, respectively, versus 38.8% with placebo. Throughout the study period, no Japanese-specific adverse events were observed. Thus, venlafaxine extended release was efficacious and safe for MDD treatment in Japan.
Keywords: Hamilton Rating Scale For Depression, Japan, major depressive disorder, Montgomery–Åsberg Depression Rating Scale, placebo controlled, venlafaxine
Introduction
Major depressive disorder (MDD) is a serious disabling condition associated with significant morbidity and mortality, and affects more than 350 million individuals worldwide (World Health Organization, 2012). However, its occurrence varies considerably worldwide; the lifetime and 12-month prevalence of major depressive episodes defined by the Diagnostic and Statistical Manual of Mental Disorders, 4th ed. (DSM-IV), using the World Health Organization Composite International Diagnostic Interview for Japan is 6.6 and 2.2%, respectively (Bromet et al., 2011). Although both psychological and pharmacological treatments have been used, antidepressant drugs remain the treatment mainstay. According to the guidelines of the American Psychiatric Association, the Japanese Society of Mood Disorders, and the International Consensus Statement on MDD, an antidepressant medication is recommended as an initial treatment choice for patients with moderate-to-severe MDD (American Psychiatric Association, 2010; Nutt et al., 2010; Japanese Society of Mood Disorders, 2013). Selective serotonin reuptake inhibitors and serotonin–norepinephrine reuptake inhibitors (SNRIs) are the first-line treatment options among various types of antidepressants. Although two SNRIs (milnacipran and duloxetine) and four selective serotonin reuptake inhibitors (fluvoxamine, paroxetine, sertraline, and escitalopram) have been approved in Japan as of August 2015, the maximum dosages for most of these are considerably lower than those used in western countries on the basis of balance between the benefits and risks in the Japanese population. For example, the maximum dosages approved in the US and Japan, respectively, for the treatment of MDD are as follows: milnacipran (200 mg/100 mg), duloxetine (120 mg/60 mg), fluvoxamine (300 mg/150 mg), paroxetine (50 mg/40 mg), sertraline (200 mg/100 mg), and escitalopram (20 mg/20 mg). The approved maximum daily dosage of venlafaxine extended release (ER) is 225 mg in most countries. In addition, the remission rate associated with the initial treatment of MDD is quite low (Trivedi and Daly, 2008). Thus, additional options, with a wide range of dosages matching those in western countries, for the first-line treatment of MDD in Japan must be explored to fulfill the unmet medical needs.
Venlafaxine was the first SNRI approved by the FDA in 1993 for the treatment of MDD in adults (Papakostas, 2009a) as an immediate-release (IR) formulation. Venlafaxine ER, an oral once-a-day formulation of venlafaxine HCl (1-[2-(dimethylamino)-1-(4-methoxyphenyl) ethyl] cyclohexanol-HCl), has shown the same exposure as venlafaxine IR formulation with a dosing of two or three times a day. The robust acute efficacy of venlafaxine IR and ER has been established over placebo-controlled studies that involved both fixed-dose and flexible-dose regimens (fixed-dose regimens: Mendels et al., 1993; Khan et al., 1998; Rudolph et al., 1998, flexible-dose regimens: Cunningham, 1997; Thase, 1997; Rudolph and Feiger, 1999; Silverstone and Ravindran, 1999) at doses ranging from 75 to 375 mg/day. In addition, the long-term efficacy of venlafaxine ER has been reported in several studies, including the Prevention of Recurrent Episodes of Depression with Venlafaxine ER for Two Years (PREVENT) study (Montgomery et al., 2004; Simon et al., 2004; Keller et al., 2007; Kornstein et al., 2008).
The present study is a randomized, placebo-controlled investigation of the benefits and risks of venlafaxine treatment in Japanese patients with MDD. More specifically, the primary aim was to compare the antidepressant efficacy after 8 weeks of double-blinded treatment with a fixed dose of 75 mg/day of venlafaxine ER, flexible doses of 75–225 mg/day of venlafaxine ER, or placebo. The secondary objectives were to evaluate the safety and tolerability of venlafaxine ER in these patients. We also report long-term findings on the safety, tolerability, and efficacy of venlafaxine ER during an extended 44-week, open-labeled extension study subsequent to the double-blinded study period.
Methods
Study design
This was a phase 3, multicenter, randomized, double-blinded, placebo-controlled, parallel-group study (http://www.clinicaltrials.gov; NCT01441440) and a long-term, open-labeled, extension study (http://www.clinicaltrials.gov; NCT01485887) to evaluate the efficacy and safety of venlafaxine ER 75 mg/day (fixed dose) and venlafaxine ER 75–225 mg/day (flexible dose) compared with placebo.
The double-blinded study comprised a 2-week screening period, an 8-week treatment period, and a 2-week tapering period.
Patients
In the double-blinded study, outpatients aged at least 20 years with a primary diagnosis of MDD on the basis of the DSM-IV criteria, who experienced single or recurrent episodes without psychotic features, were eligible for the study. In addition, patients should have experienced depressive symptoms for at least 90 days in a single episode and for at least 28 days in a recurrent episode before the screening visit and have a Montgomery–Åsberg Depression Rating Scale (MADRS) total score of at least 26 at the screening and baseline visits with a change in MADRS total scores at baseline not beyond 25% from the screening visit, a 16-item Quick Inventory of Depressive Symptomatology self-report version (QIDS16-SR-J) total score of at least 16 at the screening and baseline visits (Rush et al., 2003), and a score of at least 4 on the Clinical Global Impressions Scale-Severity (CGI-S) at the screening and baseline visits. Moreover, they should have provided a personally signed and dated informed consent document indicating that they had been informed of all pertinent aspects of the study and were willing and able to comply with scheduled visits, treatment plan, laboratory tests, and other study procedures. All female and male patients who were biologically capable of having children had to agree and commit to the use of a reliable method of birth control during the study period and for 28 days after the last dose of study medication. Patients who had received treatment with venlafaxine or desvenlafaxine in the past; a history of personality disorder or mental retardation, substance abuse, psychotic disorders, dementia, obsessive compulsive disorder, post-traumatic stress disorder, bipolar disorder, anxiety disorder, or active suicidal tendency; other clinically important medical conditions as determined by the investigators; or any other unstable medical condition such as cardiovascular disease were excluded. Patients who had been nonresponsive to two antidepressant treatments in the past, had a history of chronic treatment with benzodiazepines for longer than 6 months before the screening visit, or had depression associated with the presence of an organic mental disorder because of a general medical condition or a neurologic disorder were also excluded.
Patients could withdraw from the study at will or at the discretion of the investigator on emergence of a serious adverse event (SAE) or adverse event (AE); if the patient could not take the study drug at week 1 and the following weeks; if dose adjustment was needed after week 4; if suicidal risk was observed; if the patient became pregnant; or if other difficulties were encountered in continuation of the study protocol. Patients who completed the double-blinded study, with no clinically significant safety findings during this part of the study, were included in the open-labeled extension study.
Settings
The double-blinded study was carried out at 62 investigational sites (27 in the long-term study) in Japan between November 2011 and March 2014. The study protocols and other relevant documentations were approved by the Institutional Review Board/Independent Ethics Committee. The studies were carried out in accordance with legal and regulatory requirements as well as the general principles set forth in the International Ethical Guidelines for Biomedical Research Involving Human Subjects (Council for International Organizations of Medical Sciences, 2002), Guidelines for Good Clinical Practice, and the Declaration of Helsinki. All patients provided written informed consent to participate in the study.
Treatment
After screening (visit 1), eligible patients were randomized equally (1 : 1 : 1) to double-blinded treatment with placebo, venlafaxine ER 75 mg/day (fixed dose), or venlafaxine ER 75–225 mg/day (flexible dose). Venlafaxine was initiated at 37.5 mg/day, which could be increased to 75 mg/day (week 1), followed by 75 and 150 mg/day in the fixed-dose and flexible-dose groups, respectively (week 2), if well tolerated. The dose was further escalated to 225 mg/day (week 3) in the flexible-dose group even if the patients responded well at a lower dose, assuming no tolerability concerns arose (forced dose titration). The patients were allowed to be treated at the same or a reduced dose if any tolerability concerns arose. Patients were withdrawn from the study if they could not be administered the doses at and beyond week 1. No dose adjustment was permitted from week 4.
During the tapering period, the doses were de-escalated to 37.5 mg/day for 1 week and placebo for another week in patients who received 75 mg/day at the end of the treatment period (week 8), followed by complete discontinuation. Patients who received 150 or 225 mg/day at the end of the treatment period (week 8) received a de-escalated dose of 75 mg/day for 1 week, followed by 37.5 mg/day for another week. Patients who were enrolled into the open-labeled extension study were dispensed the study drug for the long-term study at week 8, but not for the tapering period. Follow-up visits occurred after 2 weeks of the last study dose for all patients who received the study drug, irrespective of the treatment duration, except those who were enrolled into the open-labeled extension period. Patients who discontinued before week 8 were followed up for a further 2 weeks after the last study medication dose.
Among those patients who completed the double-blinded study, 50 were eligible for further treatment in the open-labeled extension study, which consisted of a 10-month treatment phase and a 1- to 3-week tapering phase. The two follow-up visits were performed at the end of 2 and 4 weeks after the last study medication dose. Eligible patients received a flexible once-daily dose (75, 150, or 225 mg/day). All study medications were self-administered orally: three capsules once daily after dinner at home. Once-daily morning administration was allowed in case of any concern in terms of tolerability or compliance for evening administration.
Assessments
In both the studies, treatment compliance was determined through capsule-counting procedures and patient–physician interviews at follow-up visits. In the double-blinded study, patients’ background information, physical examination, and vital signs were collected at screening (visit 1). The QIDS16-SR-J, Hamilton Rating Scale for Depression (HAM-D), MADRS, CGI-S, and Clinical Global Impression of Improvement (CGI-I) were the primary tools used to assess efficacy by measuring the intensity and the course of the depressive disorder. The primary efficacy measure of change from baseline in the HAM-D17 total score at week 8 was compared among both venlafaxine groups and the placebo group, and HAM-D17 was used as the primary endpoint to be consistent with western and previous Japanese venlafaxine clinical studies. The total score of HAM-D6 (Bech et al., 1975, 1981) was measured using the formula, Σ (items 1, 2, 7, 8, 10, and 13) and the sleep disturbance score was measured as Σ (items 4, 5, and 6) of the HAM-D scale (Cleary and Guy, 1977).
In the open-labeled extension study, QIDS16-SR-J, HAM-D, CGI-S, and CGI-I were used for the efficacy measurements. In both studies, for safety assessment, AEs, weight, vital signs, 12-lead ECG, clinical laboratory testing, and the Columbia-Suicide Severity Rating Scale were recorded.
Statistical analysis
The efficacy analysis was based on the full analysis set, which was defined as all patients who received at least one dose of the study drug during the double-blinded treatment period and had both baseline and at least one postbaseline measurement of the primary efficacy variable. The primary efficacy analysis consisted of two pairwise comparisons of the mean primary efficacy variable: fixed dose versus placebo and flexible dose versus placebo, which used the analysis of covariance model with treatment groups as a factor and baseline measurement of HAM-D17 total score as a covariate. The flexible dose versus placebo comparison was carried out only after statistical significance was observed in fixed dose versus placebo comparison for control of the familywise error rate. For patients who discontinued before study completion, the last observation was carried forward to week 8. The secondary efficacy analysis for change from baseline at all visits in HAM-D17 total score was carried out using a mixed-effect model for repeated measures including treatment group, visit, interaction of group with visit, and respective baseline measurement. For secondary efficacy endpoints, including mean change from baseline at week 8 for the MADRS total score, the QIDS16-SR-J total score, the HAM-D6 total score, and the CGI-S, all treatment groups were compared using an analysis of covariance model with treatment group as a factor and respective baseline measurement as a covariate. In hypothesis testing, a significance level of 0.05 for a two-sided test and 0.025 for a one-sided test was used. The safety analysis set included all patients who received at least one dose of the study drug; the safety measures were summarized descriptively. The sample size was determined to provide a 90% chance of statistical significance in both two pairwise comparisons of the primary efficacy analyses.
To examine some of the reasons for the inconsistent results found among the efficacy endpoints, the change from baseline at all visits in the sleep disturbance score derived from HAM-D17 was analyzed using a mixed-effect model for repeated measures set forth above. For the same purpose, the mean change from baseline to week 8 (last observation carried forward) was also calculated in each item score of MADRS.
Besides, to investigate the effect of an increase in the dosage, changes in the CGI-S and the CGI-I scores were summarized for before and after prescription changes from 75 mg/day or corresponding placebo to 150 mg/day or corresponding placebo and from 150 mg/day or corresponding placebo to 225 mg/day or corresponding placebo by treatment group. Reductions of the CGI-S and CGI-I scores were classified as ‘improved’, no change as ‘Unchanged’, and increase as ‘Worsened’. The scores were summarized only for patients with CGI-S at least 4 or CGI-I at least 4.
For the open-labeled extension study, the HAM-D17 total score at each visit was summarized using descriptive statistics for the observed cases. Remission rate was defined by the proportion of patients with HAM-D17 total scores up to 7 at each visit.
Results
Patient disposition
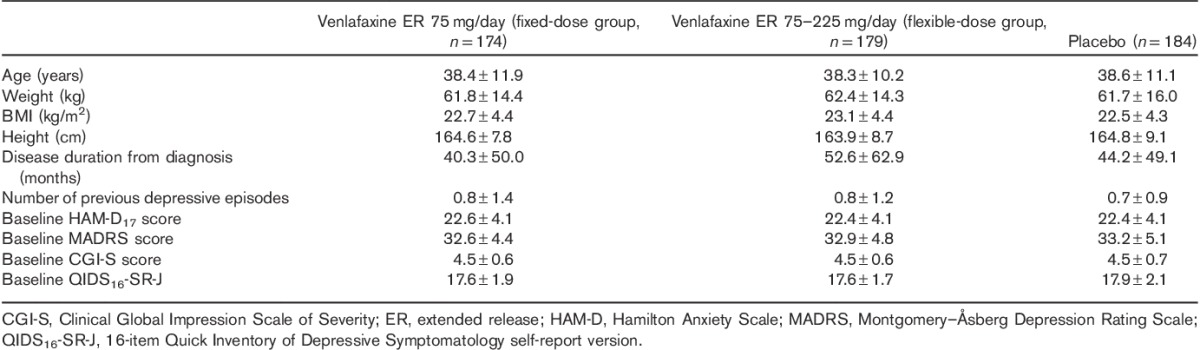
Of the 538 patients randomized, 537 patients received the study drug (174 and 179 patients in the fixed-dose and flexible-dose groups, respectively, and 184 patients in the placebo group; Fig. 1). In total, 475 patients completed the double-blinded study period; nine patients in each of the fixed-dose and flexible-dose groups and two in the placebo group discontinued the study drug because of AEs, although the total numbers of patients who discontinued the study were similar between the three treatment groups (13.2, 11.7 and 9.8% in the fixed-dose, flexible-dose, and placebo groups, respectively). All patients were Asian in origin, with a mean (SD) age of 38.4 (11.1) years. All study groups were comparable in terms of demographic characteristics, disease duration from diagnosis, and number of previous depressive episodes (Table 1). In the flexible-dose group, the distribution of the last dose during the 8-week treatment period was 4.5% for 37.5 mg/day, 4.5% for 75 mg/day, 10.6% for 150 mg/day, and 80.4% for 225 mg/day.
Fig. 1.
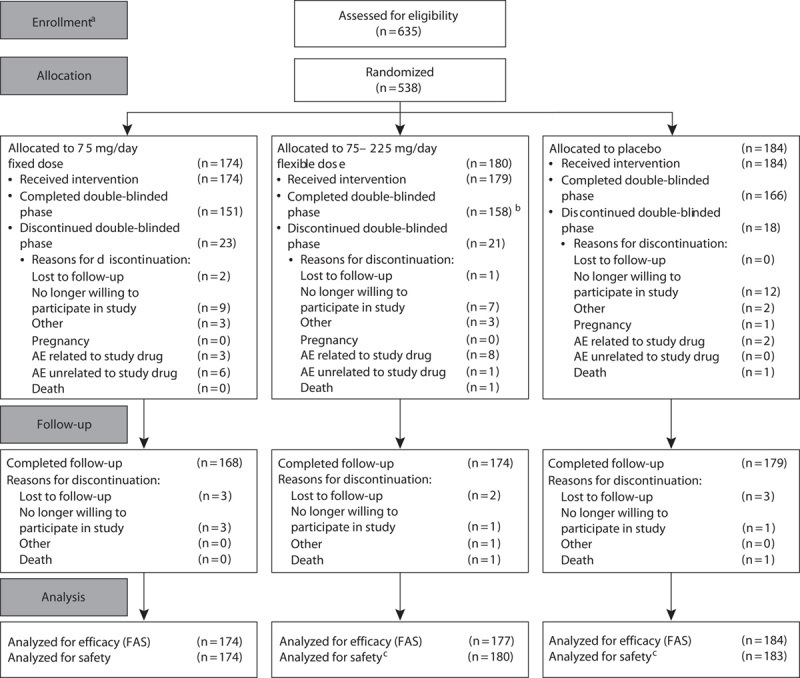
Patient disposition. aThe study was initiated on 17 November 2011; b80.0% patients titrated up to the maximum dose of 225 mg/day; cone patient randomized to placebo received incorrect treatment and reported under venlafaxine ER 75–225 mg flexible-dose group. AE, adverse event; FAS, full analysis set.
Table 1.
Demographic and disease characteristics presented as mean±SD
In the open-labeled extension study, 50 patients were enrolled, of whom 38 patients (76.0%) completed the treatment and tapering phases, and 12 patients (24.0%) discontinued the study [AEs (eight patients), no willingness to participate (two patients), or other reasons (two patients)]. A total of 46 (92.0%) patients completed the follow-up phase and four patients discontinued the follow-up phase [AEs (two patients) or other reasons (two patients)]. Of the patients enrolled in the open-labeled extension study, 35 and 15 patients had been in the venlafaxine and the placebo groups, respectively, in the double-blinded study.
Efficacy
The mean change from baseline in HAM-D17 total score at week 8 was −10.76, −10.37, and −9.25 in the fixed-dose, flexible-dose, and placebo groups, respectively. The adjusted mean difference compared with placebo (95% confidence intervals) in the primary efficacy variable was statistically significant in the fixed-dose group [1.50 (0.14–2.87); P=0.031], but not in the flexible-dose group [1.12 (−0.24 to 2.48); P=0.106]. The fixed-dose and flexible-dose group showed greater reduction in HAM-D17 total scores over time than the placebo group (Fig. 2a). However, all treatment groups showed a reduction in sleep disturbance score until week 4, following which the flexible-dose group deviated from the trend shown by the fixed-dose and placebo groups (Fig. 2b). However, further improvement in depressive symptomatology as a result of venlafaxine treatment was shown by the change from baseline at week 8 in MADRS, HAM-D6, and CGI-S total scores, which was significantly greater in both fixed-dose and flexible-dose groups than in the placebo group (P<0.05; Table 2). The mean change from baseline in MADRS total score at week 8 was −15.30, −15.05, and −12.41 in the fixed-dose, flexible-dose, and placebo groups, respectively. The adjusted mean difference compared with placebo (confidence interval) for the MADRS total score was significant in both fixed-dose [2.88 (0.77–5.00); P=0.008] and flexible [2.64 (0.54–4.74); P=0.014]-dose groups. In addition, Fig. 3 showed consistently greater reduction in all the items of MADRS in both the venlafaxine groups compared with the placebo group. The reduction at week 8 in QIDS16-SR-J total score was significantly greater in the fixed-dose group (P=0.004), but not the flexible-dose group (P=0.137), than in the placebo group. Overall improvement in disease condition measured by CGI-I scores at week 8 was marginally greater in the fixed-dose group (2.32; P=0.073) and significantly greater in the flexible-dose group (2.28; P=0.034) than in the placebo group (2.53). Figure 4 shows the relationship between improvements in the CGI (CGI-I and CGI-S) scales and specific changes in dosages among patients with CGI-I/CGI-S scores of at least 4 before dose escalation. Patients with dose escalation from 150 to 225 mg showed greater improvement than did those with dose escalation from 75 to 150 mg; in addition, a majority of patients showed neither improvement nor worsening of CGI scores when the dose was escalated from 75 to 150 mg.
Fig. 2.
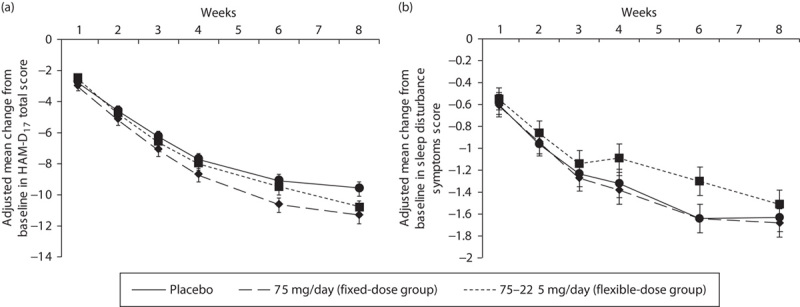
Adjusted mean change from baseline at each visit in (a) HAM-D17 total score and (b) sleep disturbance symptoms measured by HAM-D (full analysis set, MMRM). HAM-D, Hamilton Rating Scale for Depression; MMRM, mixed-effect model for repeated measures. *P<0.05 for fixed dose versus placebo group.
Table 2.
Summary of efficacy: mean change from baseline to week 8 in efficacy measure (full analysis set, last observation carried forward, ANCOVA model)
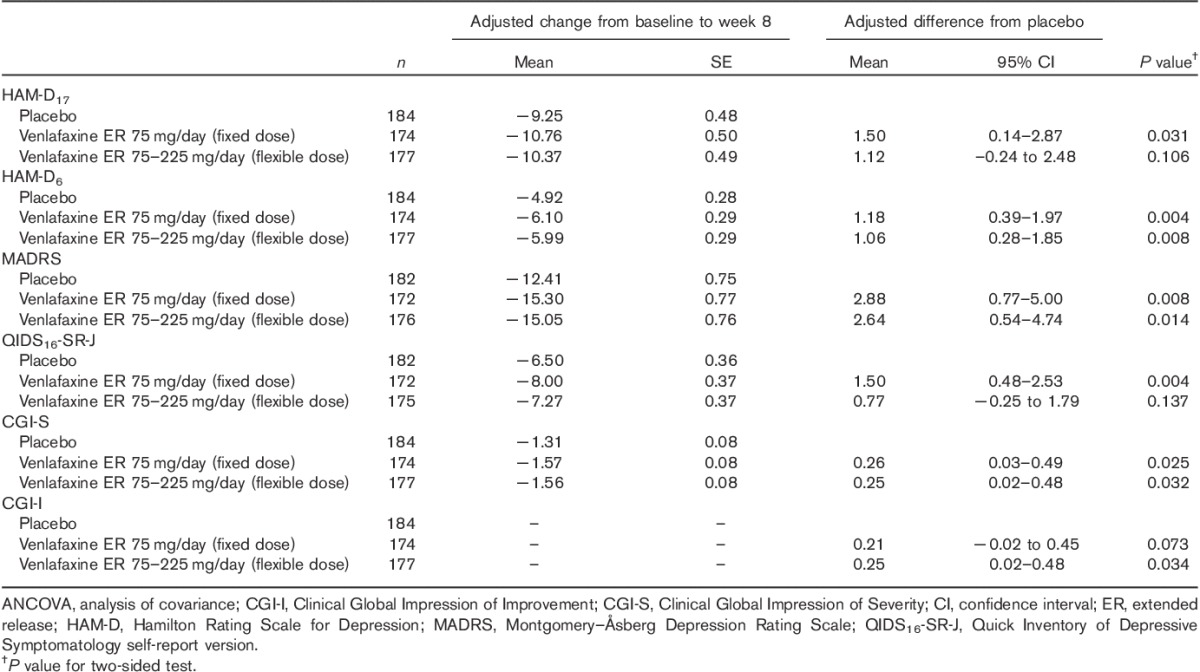
Fig. 3.
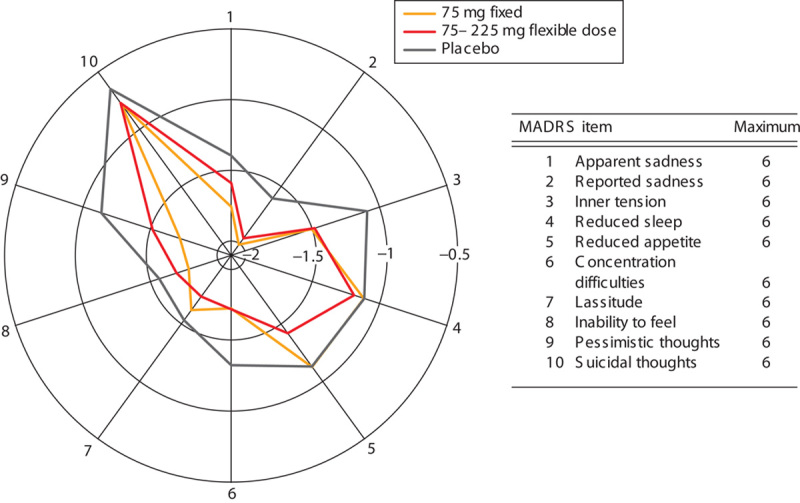
Mean change from baseline at week 8 (LOCF) in MADRS for each item. LOCF, last observation carried forward; MADRS, Montgomery–Åsberg Depression Rating Scale.
Fig. 4.
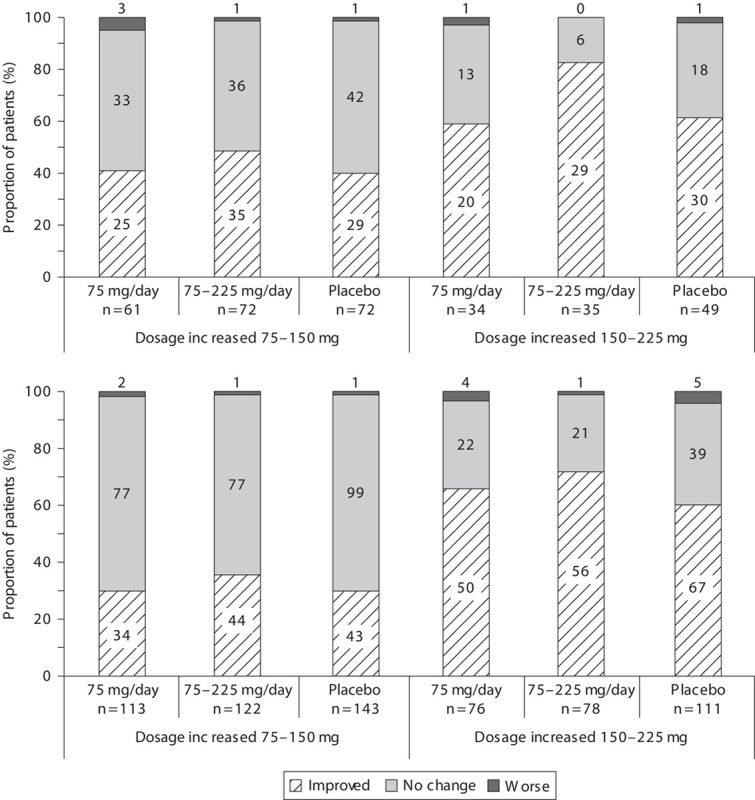
Distribution of patients rated to have improved, not changed, or worsened on (a) CGI-I and (b) CGI-S scales in all treatment groups categorized by specific change in venlafaxine dosages. Values specified on the bars signify n. CGI-I, Clinical Global Impression of Improvement; CGI-S, Clinical Global Impression of Severity.
For patients enrolled in the open-labeled extension study, the mean HAM-D17 total score for 50 patients on entry to the extension phase was 12.0 (SD=6.1). The mean HAM-D17 total score for 40 patients who completed the 44-week treatment was 5.7 (SD=6.0) and the remission rate was 75.0% (30/40). Findings from other secondary endpoints, including CGI-S, CGI-I, and QIDS16-SR-J, confirmed the sustained long-term efficacy of venlafaxine (data not shown).
Safety
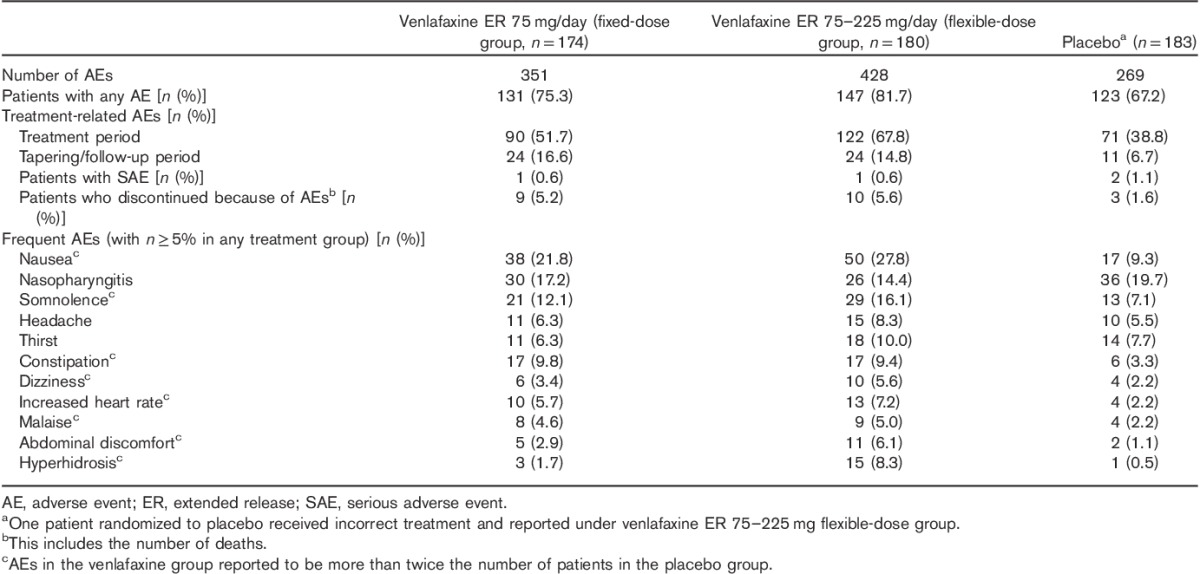
During the treatment period, a comparable proportion of patients experienced AEs in all study groups: 118 (67.8%), 138 (76.7%), and 108 (59.0%) patients in the fixed-dose, flexible-dose, and placebo groups, respectively. In the tapering/follow-up periods, AEs occurred in 49 (33.8%), 50 (30.9%), and 46 (28.0%) patients in the fixed-dose, flexible-dose, and placebo groups, respectively. Treatment-related AEs during the treatment period were reported for 90 (51.7%), 122 (67.8%), and 71 (38.8%) patients in the fixed-dose, flexible-dose, and placebo groups, respectively. Frequently reported AEs in the venlafaxine groups, experienced by more than twice the number of patients than in the placebo group, were nausea, somnolence, constipation, dizziness, increased heart rate, malaise, abdominal discomfort, and hyperhidrosis (Table 3); most AEs were mild or moderate in severity.
Table 3.
Summary of adverse events
Sustained hypertension (defined as systolic blood pressure≥140 mmHg with change from baseline≥20 mmHg, diastolic blood pressure≥90 mmHg with change from baseline≥10 mmHg, or pulse rate≥100 bpm with change from baseline≥15 bpm observed over three consecutive visits) was experienced by less than 3% patients and was comparable across all treatment groups. There were no clinically significant AEs in laboratory tests.
SAEs were rarely experienced by patients in any treatment group: however, one death was reported in each of the flexible-dose and placebo groups. The cause of both deaths was suicide; however, the relationship with the study drug could not be ruled out. Other SAEs included one case each of anemia and Ménière’s disease in the placebo and fixed-dose groups, respectively; these SAEs were considered unrelated to the study drug. Among patients who discontinued the study drug because of AEs, a causal relationship with the study drug could not be ruled out for three patients in each of the fixed-dose and placebo groups, and nine patients in the flexible-dose group.
After study treatment initiation, self-injurious behavior was reported in one patient in the fixed-dose group, who discontinued the study because of agitation, which occurred concurrently with the self-injurious behavior. Suicidal ideation was confirmed in 41 (23.6%), 47 (26.1%), and 39 (21.3%) patients in the fixed-dose, flexible-dose, and placebo groups, respectively, in the Columbia-Suicide Severity Rating Scale. The proportion of patients in whom suicidal ideation was not confirmed at baseline but occurred after study treatment initiation was 7 (4.0%), 13 (7.3%), and 21 (11.5%) patients in the fixed-dose, flexible-dose, and placebo groups, respectively.
Among the patients enrolled in the open-labeled extension study, 49 (98.0%) patients experienced AEs and SAEs were observed in 3 (6.0%) patients, but there were no deaths during the 52-week study period. Similar to the double-blinded study, frequently reported AEs were nasopharyngitis, headache, dizziness, somnolence, nausea, constipation, and increased blood pressure; most of these were mild or moderate in severity. The only severe AE, which occurred in one patient, was crime (theft and sexual abuse). No new clinically significant safety issues specific to Japanese patients emerged during the long-term study.
Discussion
The present randomized double-blinded placebo-controlled study was carried out in Japan to investigate the efficacy and safety of fixed (75 mg/day) and flexible (75–225 mg/day) doses of venlafaxine ER in patients with MDD. After 8 weeks of double-blinded treatment, the difference in the mean reduction of HAM-D17 as the primary efficacy variable was statistically significant in the fixed-dose, but not the flexible-dose group compared with placebo. However, secondary efficacy parameters such as MADRS, HAM-D6, and CGI-S showed consistent and significantly greater reduction in both venlafaxine groups compared with placebo. In addition, CGI-I showed significantly greater improvement in the flexible-dose group compared with placebo. Thus, it is assumed that MADRS was more sensitive to antidepressant drug effects than HAM-D17 in this study, considering the previous reports (Galinowski and Lehert, 1995; Mulder et al., 2003; Carmody et al., 2006). As both MADRS and HAM-D are recommended as primary measures in clinical trials (Japanese guideline, 2010; EMA guideline, 2013), the treatment difference detected by MADRS in this study was clinically important.
The reason for the inconsistent result for the flexible-dose group among the scales used in the present study may possibly be the characteristics related to sensitivity to the symptoms of depression. In the characteristics of scales for sleep items, HAM-D17 consists of three separate items including early, middle, and late insomnia, whereas MADRS has just one item evaluating the same aspects of insomnia as HAM-D17. As described previously in Fig. 2b, an improving trend was observed in the sleep disturbance scores of HAM-D17 until week 4, following which the flexible-dose group deviated from the improving trend shown by the fixed-dose and placebo groups. The total HAM-D17 score for the flexible-dose group was clearly affected by less improvement in the sleep disturbance scores. Thus, it can be suggested that HAM-D17 was more sensitive in capturing sleep disturbance when the dose was titrated to 225 mg because of venlafaxine’s norepinephrine uptake inhibition. Venlafaxine is known to sequentially engage the mechanisms of serotonin 5-HT at a low dose (75 mg/day) and norepinephrine uptake inhibition at a high dose (225 mg/day) (Harvey et al., 2000; Debonnel et al., 2007). More than 80% of patients in the flexible-dose group took 225 mg/day at week 4, beyond which no improvement in the sleep disturbance score of HAM-D was observed. This could be attributed to the enhancement of norepinephrine signaling, which is a critical component of the arousal pathway (Mitchell and Weinshenker, 2010). For example, reboxetine, an SNRI, was associated with a significantly higher incidence of insomnia than placebo (Tanum, 2000). In addition, almost all patients took the study drug in the evening (data not shown), which might cause lower reduction in the sleep disturbance score of HAM-D in the flexible-dose group because of a noradrenergic effect.
The norepinephrine effect is in line with the suggestion that higher doses, as used in the flexible-dose group of our study (75–225 mg/day), could be more effective for patients with severe disease (Berney, 2005; Debonnel et al., 2007), given that the severity of depression may influence the relationship between SNRI dose and clinical response. This could potentially explain the lack of a statistically significant clinical response in the flexible-dose group compared with placebo for some of the efficacy measures in our study (Table 2). The majority of patients in the present study had moderate depression at baseline, with a mean HAM-D17 score less than 23 (Shelton et al., 2007) and a mean MADRS score less than 35 (Müller et al., 2000); therefore, the relatively lower doses used in the fixed-dose group (75 mg/day) aligned better with the treatment requirements of the patient population, producing better outcomes than in the flexible-dose group. In addition, a mild-to-moderate placebo response, as found in recent placebo-controlled studies of MDD, could have reduced the likelihood of detecting a large treatment difference associated with HAM-D17 response between the venlafaxine groups and placebo in the present study (Papakostas and Fava, 2009b; Khan et al., 2010; Khin et al., 2011). Nevertheless, the minimally effective dose of venlafaxine 75 mg/day was superior to placebo in efficacy parameters investigated in the present study.
The safety profile of venlafaxine in the present study did not identify any new drug-related risks specific to a Japanese population and the most common AEs observed were consistent with the known profile of venlafaxine (Mendels et al., 1993; Cunningham, 1997; Thase 1997; Khan et al., 1998; Rudolph et al., 1998; Rudolph and Feiger, 1999; Silverstone and Ravindran, 1999). Although venlafaxine has a safe cardiovascular profile, an increase in blood pressure/pulse rate is often observed at doses greater than 300 mg (Thase 1998). Consistent with this observation, up to 3% patients showed sustained hypertension in all treatment groups. No other clinically meaningful difference was apparent between venlafaxine and placebo in the benign safety profile, including laboratory measurements and vital signs. Although overall suicidal ideation was comparable across venlafaxine and placebo groups in this study, it should be noted that there were patients who developed suicidal ideation during the course of study treatment (4.0, 7.3, and 11.5% patients in the fixed-dose, flexible-dose, and placebo groups, respectively); therefore, patients should be informed of this risk before commencement of treatment. Nevertheless, only a small proportion of patients (5.2, 5.6, and 1.6% patients in the fixed-dose, flexible-dose, and placebo groups, respectively), comparable across treatment groups, discontinued the study because of AEs, suggesting that both dosing regimens of venlafaxine were well tolerated in Japanese patients with MDD. Furthermore, the well-tolerated safety profile was confirmed by the extension study, with no new AEs arising during this period.
According to the findings from dose–response studies (Khan et al., 1998; Rudolph et al., 1998), improvement in efficacy variables did not necessarily increase monotonically, with increasing doses greater than 75 mg/day. Nevertheless, some patients experienced greater benefit with dose escalation in the flexible-dose group (Fig. 4) without an additional increase in SAEs or discontinuation because of AEs (data not shown) in the present study. On the basis of these findings, a majority of patients could be treated with 75 mg/day venlafaxine, but increasing the dose may perhaps be beneficial for selected patients, in particular, those showing little improvement and tolerability to treatment with lower doses of venlafaxine.
It is considered that the findings of the present study will inform treatment decisions and dosing recommendations for the use of venlafaxine in Japan for treating patients with MDD. However, some limitations might have confounded the study results; the short double-blinded treatment period might not have enabled evaluation of the long-term benefits of venlafaxine and excluded patients, especially those with comorbid conditions, which cannot be considered a manifestation of the real-world practice setting. Despite these limitations, this study should be recognized as the placebo-controlled study of venlafaxine conducted in Japan. Perhaps additional subsequent analyses might be useful to identify specific populations who need not titrate up to 225 mg/day as in the flexible-dose group. The forced titration method used in this study allowed us to gather more conclusive data on efficacy and safety at higher doses, despite it being an uncommon approach in actual clinical practice because physicians adjust the dosage of antidepressants on the basis of both the disease condition and tolerability in each patient. Therefore, further investigations are needed to evaluate the antidepressant efficacy of 225 mg/day dosing of venlafaxine in real-world clinical settings in Japan, in addition to making comparisons with other antidepressant agents. Despite these limitations, this study provides strong evidence supporting the antidepressive efficacy of venlafaxine ER, especially the 75 mg fixed-dose regimen, following 8 weeks of double-blinded treatment in patients with MDD. In addition, venlafaxine ER was found to be associated with an acceptable safety profile that was similar to that established in previous studies. Therefore, venlafaxine ER was efficacious and safe for the treatment of MDD in Japan.
Acknowledgements
The authors gratefully acknowledge the contributions of trial participants, principal investigators, and all the medical personnel in this clinical study. The authors are grateful to the journal reviewers for insightful comments and suggestions that helped improve the content and presentation of this article. Medical writing services were provided by Cactus Communications. All authors retained full control of the manuscript content.
The study received funding from Pfizer, Japan Inc. Editorial support was provided by Lorraine Sweeney of Peloton Advantage and was funded by Pfizer.
Conflicts of interest
Rio Itamura, Yuko Asami, Kazuhiko Kuribayashi, and Takayuki Imaeda are employees of Pfizer Japan Inc., which funded the study. Teruhiko Higuchi received a consulting fee from Sumitomo Dainippon Pharma Co. Ltd, Takeda Pharmaceutical Co. Ltd, Kowa Company Ltd, Chugai Pharmaceutical Co. Ltd, Eli Lilly Japan K.K., Pfizer Japan Inc., Janssen Pharmaceutical K.K., Bristol-Myers Squibb, FP Pharmaceutical Corporation, Nippon Boehringer Ingelheim Co. Ltd, received support for travel to meetings from Pfizer Japan Inc., received grants from AbbVie Inc., Otsuka Pharmaceutical Co. Ltd, Chugai Pharmaceutical Co. Ltd, Mitsubishi Tanabe Pharma Corporation, Janssen Pharmaceutical K.K., Meiji Seika Pharma Co. Ltd, Pfizer Japan Inc., Boehringer Ingelheim Japan Inc., GlaxoSmithKline K.K., and Abbott Japan Co. Ltd; also received payment for lectures from MSD K.K. a subsidiary of Merck & Co. Inc., Asahi Kasei Pharma Corp., Otsuka Pharmaceutical Co. Ltd, Sumitomo Dainippon Pharma Co. Ltd, Shionogi & Co. Ltd, Meiji Seika Pharma Co. Ltd, Pfizer Japan Inc., Janssen Pharmaceutical K.K., Kyowa Hakkou Kirin Co. Ltd, and Novartis Pharma K.K., received payment for manuscript preparation from Eli Lilly Japan K.K., and Kyowa Yakuhin Co Ltd. Kunitoshi Kamijima received consulting fee from Asahi Kasei Pharma Corporation, Pfizer Japan Inc., received support for travel to meetings from Pfizer Japan Inc., received payment for lectures from Pfizer Japan Inc., Astellas Pharma Inc., AbbVie Pharmaceuticals, Eli Lilly Japan, Eisai, Osuka Pharmaceuticals, Kyowa Hakko Kirin, Glaxo Smith Kline, Shionogi, Dainippon Sumitomo Pharma, Tanabe Mitsubishi, Meiji Seika Pharma, and Mochida Pharmaceutical Co. Kazuyuki Nakagome received consulting fee from Pfizer Japan Inc., Otsuka Pharmaceutical Co. Ltd, Takeda Pharmaceutical Co. Ltd, Sumitomo Dainippon Pharma Co. Ltd, Mochida Pharma, Nippon Boehringer Ingelheim Co. Ltd, and Mitsubishi Tanabe Pharma Corporation, received support for travel to meetings from Pfizer Japan Inc., received grants from Mochida Pharma, Tanabe Mitsubishi, Shionogi Pharma, Sumitomo Dainippon Pharma, Otsuka Pharma, Astellas Pharma, and Asahi Kasei Pharma, and received payment for lectures from Otsuka Pharma, Sumitomo Dainippon Pharma, Eli Lilly Pharma, Tanabe Mitsubishi Pharma, Meiji Seika Pharma, GSK, Shionogi Pharma, Janssen Pharma, MSD, and Kyowa Hakko Kirin Co.
References
- American Psychiatric Association (2010). Practice guideline for the treatment of patients with major depressive disorder, 3rd edition. Available at: http://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/mdd.pdf. [Accessed 5 May 2015].
- Bech P, Gram LF, Dein E, Jacobsen O, Vitger J, Bolwig TG. (1975). Quantitative rating of depressive states. Acta Psychiatr Scand 51:161–170. [DOI] [PubMed] [Google Scholar]
- Bech P, Allerup P, Gram LF, Reisby N, Rosenberg R, Jacobsen O, et al. (1981). The Hamilton depression scale: evaluation of objectivity using logistic models. Acta Psychiatr Scand 63:290–299. [DOI] [PubMed] [Google Scholar]
- Berney P. (2005). Dose-response relationship of recent antidepressants in the short-term treatment of depression. Dialogues Clin Neurosci 7:249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromet E, Andrade LH, Hwang I, Sampson NA, Alonso J, de Girolamo G, et al. (2011). Cross-national epidemiology of DSM-IV major depressive episode. BMC Med 9:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmody TJ, Rush AJ, Bernstein I, Warden D, Brannan S, Burnham D, et al. (2006). The Montgomery Asberg and the Hamilton ratings of depression: a comparison of measures. Eur Neuropsychopharmacol 16:601–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary P, Guy W. (1977). Factor analysis of the Hamilton depression scale. Drugs Exp Clin Res 1:115–120. [Google Scholar]
- Council for International Organizations of Medical Sciences (CIOMS) in collaboration with the World Health Organization (WHO) (2002). International Ethical Guidelines for Biomedical Research Involving Human Subjects. http://www.cioms.ch/publications/layout_guide2002.pdf. [Accessed 31 August 2015].
- Cunningham LA. (1997). Once-daily venlafaxine extended release (XR) and venlafaxine immediate release (IR) in outpatients with major depression. Venlafaxine XR 208 Study Group. Ann Clin Psychiatry 9:157–164. [DOI] [PubMed] [Google Scholar]
- Debonnel G, Saint-André E, Hébert C, de Montigny C, Lavoie N, Blier P. (2007). Differential physiological effects of a low dose and high doses of venlafaxine in major depression. Int J Neuropsychopharmacol 10:51–61. [DOI] [PubMed] [Google Scholar]
- EMA guideline (2013). European Medicines Agency publishes guideline on clinical investigation of medicines for depression. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/05/WC500143770.pdf. [Accessed 30 May 2013].
- Galinowski A, Lehert P. (1995). Structural validity of MADRS during antidepressant treatment. Int Clin Psychopharmacol 10:157–161. [DOI] [PubMed] [Google Scholar]
- Harvey AT, Rudolph RL, Preskorn SH. (2000). Evidence of the dual mechanisms of action of venlafaxine. Arch Gen Psychiatry 57:503–509. [DOI] [PubMed] [Google Scholar]
- Japanese guideline (2010). Japanese guideline on clinical evaluation of antidepressant; Ministry of Health, Labour and Welfare. Available at: http://www.pmda.go.jp/files/000157465.pdf. [Accessed 31 August 2015].
- Japanese Society of Mood Disorders (2013). Guideline for treatment of major depressive disorder by the Japanese Society of Mood Disorders. Available at: http://www.secretariat.ne.jp/jsmd/mood_disorder/img/130924.pdf. [Accessed 15 January 2015].
- Keller MB, Trivedi MH, Thase ME, Shelton RC, Kornstein SG, Nemeroff CB, et al. (2007). The Prevention of Recurrent Episodes of Depression with Venlafaxine for Two Years (PREVENT) Study: outcomes from the 2-year and combined maintenance phases. J Clin Psychiatry 68:1246–1256. [DOI] [PubMed] [Google Scholar]
- Khan A, Upton GV, Rudolph RL, Entsuah R, Leventer SM. (1998). The use of venlafaxine in the treatment of major depression and major depression associated with anxiety: a dose-response study. Venlafaxine Investigator Study Group. J Clin Psychopharmacol 18:19–25. [DOI] [PubMed] [Google Scholar]
- Khan A, Bhat A, Kolts R, Thase ME, Brown W. (2010). Why has the antidepressant-placebo difference in antidepressant clinical trials diminished over the past three decades? CNS Neurosci Ther 16:217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khin NA, Chen YF, Yang Y, Yang P, Laughren TP. (2011). Exploratory analyses of efficacy data from major depressive disorder trials submitted to the US Food and Drug Administration in support of new drug applications. J Clin Psychiatry 72:464–472. [DOI] [PubMed] [Google Scholar]
- Kornstein SG, Kocsis JH, Ahmed S, Thase M, Friedman ES, Dunlop BW, et al. (2008). Assessing the efficacy of 2 years of maintenance treatment with venlafaxine extended release 75-225 mg/day in patients with recurrent major depression: a secondary analysis of data from the PREVENT study. Int Clin Psychopharmacol 23:357–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendels J, Johnston R, Mattes J, Riesenberg R. (1993). Efficacy and safety of b.i.d. doses of venlafaxine in a dose-response study. Psychopharmacol Bull 29:169–174. [PubMed] [Google Scholar]
- Mitchell HA, Weinshenker D. (2010). Good night and good luck: norepinephrine in sleep pharmacology. Biochem Pharmacol 79:801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery SA, Entsuah R, Hackett D, Kunz NR, Rudolph RL, Venlafaxine 335 Study Group (2004). Venlafaxine versus placebo in the preventive treatment of recurrent major depression. J Clin Psychiatry 65:328–336. [DOI] [PubMed] [Google Scholar]
- Mulder RT, Joyce PR, Frampton C. (2003). Relationships among measures of treatment outcome in depressed patients. J Affect Disord 76 (1–3):127–135. [DOI] [PubMed] [Google Scholar]
- Müller MJ, Szegedi A, Wetzel H, Benkert O. (2000). Moderate and severe depression. Gradations for the Montgomery–Asberg Depression Rating Scale. J Affect Disord 60:137–140. [DOI] [PubMed] [Google Scholar]
- Nutt DJ, Davidson JR, Gelenberg AJ, Higuchi T, Kanba S, Karamustafalioğlu O, et al. (2010). International consensus statement on major depressive disorder. J Clin Psychiatry 71 (Suppl E1):e08. [DOI] [PubMed] [Google Scholar]
- Papakostas GI. (2009a). Serotonin norepinephrine reuptake inhibitors: spectrum of efficacy in major depressive disorder. Prim psychiatry 16Suppl 4:16–24. [Google Scholar]
- Papakostas GI, Fava M. (2009b). Does the probability of receiving placebo influence clinical trial outcome? A meta-regression of double-blind, randomized clinical trials in MDD. Eur Neuropsychopharmacol 19:34–40. [DOI] [PubMed] [Google Scholar]
- Rudolph RL, Feiger AD. (1999). A double-blind, randomized, placebo-controlled trial of once-daily venlafaxine extended release (XR) and fluoxetine for the treatment of depression. J Affect Disord 56:171–181. [DOI] [PubMed] [Google Scholar]
- Rudolph RL, Fabre LF, Feighner JP, Rickels K, Entsuah R, Derivan AT. (1998). A randomized, placebo-controlled, dose-response trial of venlafaxine hydrochloride in the treatment of major depression. J Clin Psychiatry 59:116–122. [DOI] [PubMed] [Google Scholar]
- Rush AJ, Trivedi MH, Ibrahim HM, Carmody TJ, Arnow B, Klein DN, et al. (2003). The 16-Item Quick Inventory of Depressive Symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biol Psychiatry 54:573–583. [DOI] [PubMed] [Google Scholar]
- Shelton RC, Prakash A, Mallinckrodt CH, Wohlreich MM, Raskin J, Robinson MJ, et al. (2007). Patterns of depressive symptom response in duloxetine-treated outpatients with mild, moderate or more severe depression. Int J Clin Pract 61:1337–1348. [DOI] [PubMed] [Google Scholar]
- Silverstone PH, Ravindran A. (1999). Once-daily venlafaxine extended release (XR) compared with fluoxetine in outpatients with depression and anxiety. Venlafaxine XR 360 Study Group. J Clin Psychiatry 60:22–28. [DOI] [PubMed] [Google Scholar]
- Simon JS, Aguiar LM, Kunz NR, Lei D. (2004). Extended-release venlafaxine in relapse prevention for patients with major depressive disorder. J Psychiatr Res 38:249–257. [DOI] [PubMed] [Google Scholar]
- Tanum L. (2000). Reboxetine: tolerability and safety profile in patients with major depression. Acta Psychiatr Scand Suppl 402:37–40. [DOI] [PubMed] [Google Scholar]
- Thase ME. (1997). Efficacy and tolerability of once-daily venlafaxine extended release (XR) in outpatients with major depression. J Clin Psychiatry 58:393–398. [DOI] [PubMed] [Google Scholar]
- Thase ME. (1998). Effects of venlafaxine on blood pressure: a meta-analysis of original data from 3744 depressed patients. J Clin Psychiatry 59:502–508. [DOI] [PubMed] [Google Scholar]
- Trivedi MH, Daly EJ. (2008). Treatment strategies to improve and sustain remission in major depressive disorder. Dialogues Clin Neurosci 10:377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2012). Depression, fact sheet. Available at: http://www.who.int/mediacentre/factsheets/fs369/en/. [Accessed 5 May 2014].