

Abstract
The biosynthesis of pantothenate, the core of coenzyme A (CoA), has been considered an attractive target for the development of antimicrobial agents since this pathway is essential in prokaryotes, but absent in mammals. Pantothenate synthetase, encoded by the gene panC, catalyzes the final condensation of pantoic acid with β–alanine to afford pantothenate via an intermediate pantoyl adenylate. We describe the synthesis and biochemical characterization of five PanC inhibitors that mimic the intermediate pantoyl adenylate. These inhibitors are competitive inhibitors with respect to pantoic acid and possess submicromolar to micromolar inhibition constants. The observed SAR is rationalized through molecular docking studies based on the reported co-crystal structure of 1a with PanC. Finally, whole cell activity is assessed against wild-type Mtb as well as a PanC knockdown strain where PanC is depleted to less than 5% of wild-type levels.
Keywords: pantothenate synthetase, tuberculosis, bisubstrate inhibitor, adenylation, coenzyme A
Graphical abstract
1. Introduction
Tuberculosis (TB) caused by members of the Mycobacterium tuberculosis (Mtb) complex is an ancient scourge that remains the leading source of morbidity and mortality today with an estimated nine million new cases and 1.4 million deaths in 2011, primarily in the developing world.1 The recommended therapy by the World Health Organization (WHO) for drug sensitive TB is known as directly observed treatment, short course (DOTS) and requires 6 months of treatment with isoniazid, rifampin, ethambutol, and pyrazinamide.2 This extremely long duration of treatment is likely due to the ability of Mtb to switch its metabolism to a nonreplicating state,3 the heterogeneous nature of the bacterial subpopulations residing in different lesions types,3 and the lack of drug penetration into the site of infection.4 In order to combat this global health threat, new drugs are needed to shorten the treatment duration and for drug resistant strains including multidrug-resistant (MDR) TB and extensively drug resistant (XDR) TB.5, 6
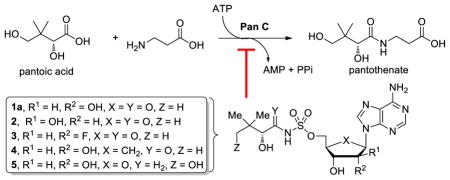
Pantothenate, also known as vitamin B5 is a precursor to coenzyme A (CoA), an essential cofactor required in central and intermediary metabolism where it serves as an acyl group carrier and carbonyl activating group.7, 8 Bioinformatics analysis has identified the de novo biosynthetic pathway to pantothenate as an attractive target for the development of antimicrobial agents since this pathway is absent in mammals, but essential in prokaryotes.9–11 Biosynthesis of pantothenate is accomplished by four enzymes encoded by the genes panB, pancC, panD, and panE. PanB and PanE are responsible for the synthesis of pantoic acid, while PanD, an aspartate α-decarboxylase, produces β-alanine. PanC (Rv3602c, pantothenate synthetase) then catalyzes the condensation of pantoic acid with β-alanine through a two-step adenylation-ligation reaction.12 In the adenylation half-reaction, ATP and pantoic acid react to form a pantoyl-adenylate intermediate (Fig. 1A). Following the release of pyrophosphate, β-alanine binds and PanC catalyzes its ligation with the activated carbonyl of the pantoyl-adenylate to afford pantothenate. The detailed kinetic characterization of PanC from Mtb shows it uses a bi-uni-uni-bi ping pong kinetic mechanism with sequential ordered binding of ATP followed by pantoic acid and sequential ordered release of pantothenate followed by AMP (Fig. 1B).12 The apparent KM values for pantoic acid, ATP, and β-alanine are 0.13, 2.6, and 0.8 mM, respectively. PanC is a 33 kDa protein that exists as a homodimer in solution. The active site is located in the N-terminal domain (residues 1–186) while the C-terminal domain (residues 187–309) covers the active site.13 Substrates must diffuse through a gate comprising residues 75–88, which are flexible and disordered, but becomes ordered upon formation of the pantoyl adenylate. The structures PanC from Mtb in complex with substrates, intermediates, and products have been solved providing a step-by-step view of the PanC reaction.13, 14
Figure 1.

Pantothenate synthetase catalyzed reactions.
Inhibitors of PanC have been identified by high-throughput screening,15–17 fragment-based approaches,18–20 dynamic combinatorial chemistry,21 and through the rationale design of analogues of the pantoyl-adenylate intermediate.22, 23 The pantoyl-adenylate intermediate mimic 1, which is epimeric at the C-2 position of the pantoyl fragment, reported by Ciulli and co-workers is the most potent inhibitor yet reported with a Ki of 0.22 μM with respect to ATP as the varied substrate at saturating concentrations of pantoic acid and β-alanine using a coupled biochemical assay that measures formation of AMP.23 In this paper, we report the design, synthesis, and biochemical evaluation of five inhibitors with the C-2 stereocenter retaining the same configuration (i.e. R) as the reaction intermediate 1a (Fig. 2). To prevent the intramolecular lactonization (Fig. 1C), the terminal C-4 hydroxyl of the pantoyl moiety was removed in 1a–4 while the pantoyl carbonyl group was deleted in 5. All compounds were also evaluated against wild-type Mtb and a PanC depleted Mtb strain.
Figure 2.

Reaction intermediate analogues of pantoyl-adenylate
2. Results and Discussion
2.1. Chemistry
Synthesis of diastereomerically pure 1a was achieved starting from commercially available (R)-2-amino-3,3-dimethylbutanoic acid 6 that was first converted to the corresponding α-hydroxy acid 7 with retention of configuration via an intermediate α-lactone (Scheme 1).24 Protection of the resultant hydroxyl group as a TBS ether provided 8, which was coupled with N-hydroxysuccinimide (NHS) using DCC to afford the common reaction intermediate 9. The pantoyl-adenylate analogue 1a was then prepared by acylation of 2′,3′-isopropylidene-5′-O-(sulfamoyl)adenosine 1025 with 9 employing Cs2CO3 in DMF followed by concomitant deprotection of the TBS and acetonide groups using aqueous trifluoroacetic acid (TFA).26
Scheme 1.

Reagents and conditions: (i) H2SO4, NaNO2, H2O, 0 °C→rt, 16 h, 81%; (ii) TBSCl, imidazole, DMF, 16 h, 81%; (iii) NHS, DCC, DME, 16 h, 60%; (iv) 10, Cs2CO3, DMF, 24 h; (v) 4:1 TFA–H2O, 8 °C, 48 h, 7% from 10.
Analogue 2 that is epimeric at the C-2′ position was synthesized in five steps from vidarabine 11 (Scheme 2). Persilylation with excess TBSCl afforded 12 and regioselective deprotection of the primary 5′-OTBS was accomplished under carefully controlled conditions with TFA in 2:1 H2O–THF to furnish 13. Sulfamoylation with freshly prepared sulfamoyl chloride27 in dioxane yielded 14, which was coupled to 9 and deprotected with 80% aqueous TFA to provide the arabinosyl nucleoside derivative 2.
Scheme 2.

Reagents and conditions: (i) TBSCl (6.0 equiv), imidazole, DMF, 16 h, 70%; (ii) 2:1:1 H2O–THF–TFA, 2 h, 81%; (iii) NaH, H2NSO2Cl, dioxane, 24 h, 60%; (iv) 9, Cs2CO3, DMF, 24 h; (v) 4:1 TFA–H2O, 20 °C, 60 h, 3% from 14.
The 2′-fluoroadenosine analogue 328 was also prepared starting from vidarabine 11 through regioselective protection of the less sterically encumbered 3′ and 5′-hydroxyl groups to yield 15 (Scheme 3). Fluorination of 15 with diethylaminosulfur trifluoride (DAST) afforded 16.29 After selective removal of the 5′-OTBS employing TFA in 2:1 H2O–THF, intermediate 17 was converted to sulfamate 18. The 1H NMR data of 17 and 18 agreed with reported analytical data,30 indicating that the C-2′ configuration of 16 was R. Acylation of 18 with 9 followed by deprotection of the TBS with 80% aqueous TFA provided 3.
Scheme 3.

Reagents and conditions: (i) TBSCl, Et3N, DMF, 16 h, 65%; (ii) DAST, pyridine, CH2Cl2, 6 h, 31%; (iii) 2:1:1 H2O–THF–TFA, 20 min, 92%; (iv) NaH, H2NSO2Cl, dioxane, 16 h, 60%; (v) 9, Cs2CO3, DMF, 20 h; (vi) 4:1 TFA–H2O, 20 °C, 72 h, 4% from 18.
Carbocyclic analogue 4 was prepared from the reported aristeromycin nucleoside analouge 1931 by acylation with 9 followed by deprotection with 80% aqueous TFA (Scheme 4).
Scheme 4.

Reagents and conditions: (i) 9, Cs2CO3, DMF, 20 h; (ii) 4:1 TFA–H2O, 8 °C, 48 h, 7% from 19.
The synthesis of sulfamide 5 began from commercially available (D)-(–)-pantolactone 20 that was first reduced to triol 21 with LiAlH4 as described by Burkart et al., then protected as the 1,3-benzylidene acetal 22.32 Functional group interconversion of primary alcohol 22 to amine 25 was performed by sequential tosylation, azide displacement, and catalytic reduction. Sulfamoylation of 25 with 4-nitrophenyl chlorosulfate afforded 26, which was coupled to nucleoside 2733 followed by global deprotection of the acetonide and benzylidene acetal with 80% aqueous TFA to furnish 5.34
2.2. PanC Inhibition Studies
Pantothenate synthetase (PanC) from Mtb was subcloned from BAC-Rv222 (kindly provided by the Institut Pasteur) into pET28b and expressed in E. coli BL21 (DE3) as described in Materials and Methods to provide an N-terminal His-tagged protein with kinetic parameters commensurate with the native enzyme.12 Kinetic studies to evaluate enzyme inhibition of each compound toward PanC were performed under initial velocity conditions using a continuous coupled assay that measures production of pyrophosphate (see Materials and Methods).35, 36
Since compounds 1a–5 are bisubstrate inhibitors, designed to bind both the pantoic acid and ATP binding pockets, we evaluated inhibition with respect to pantoic at fixed non-saturating concentrations of ATP and saturating concentrations of the third substrate β-alanine. Representative inhibition data for compound 1a are shown in Figure 3. The double-reciprocal plots of initial velocity versus pantoic acid concentration at different inhibitor concentrations of 1a display a pattern of intersecting lines that converge at the y-axis, indicating that the molecule act as a competitive inhibitor towards pantoic acid, in which Km values are increased, but there is no effect on Vmax.37 The data were fitted to Eq. 1 for competitive inhibition yielding a Ki value of 0.27 ± 0.04 μM.
Figure 3.

Inhibition of PanC with inhibitor 1a. Assays were performed at varying concentrations of inhibitor 1 (0–0.8 μM) and pantoic acid, fixed concentrations of ATP (2.6 mM) and β-alanine (2.4 mM).
Inhibitors 2–5 were similarly evaluated and shown to possess identical modalities of inhibition with respect to pantoic acid (see Table 1 for inhibition constants). Inhibitor 1a containing an acyl-sulfamate linkage and the natural ribosyl subunit is the most potent (Ki = 0.27 μM) in the series while inhibitor 5 containing a sulfamide linkage is the least potent (Ki = 3.47 μM), an approximately 13-fold difference in affinity, demonstrating that the linker carbonyl is more important than the terminal hydroxyl group of the pantoyl fragment. Replacement of the ribofuranosyl ring oxygen with a methylene in carbocyclic analogue 3 and substitution of the 2′-hydroxyl with a fluoro group in analogue 4 are reasonably well tolerated resulting in a modest 2–3 fold loss in affinities. Inversion of the 2′-alcohol in analogue 2 results in more pronounced 4–6 fold loss in potency.
Table 1.
Inhibition constants for 1a–5.
| Inhibitor | Ki (μM) |
|---|---|
| 1a | 0.27 ± 0.04 |
| 2 | 1.73 ± 0.24 |
| 3 | 0.99 ± 0.30 |
| 4 | 0.87 ± 0.12 |
| 5 | 3.47 ± 0.48 |
2.3. Computational Modeling
The key interactions between the intermediate analogue (Pan-AMP, Fig. 1A) and PanC involve the secondary hydroxyl and the carbonyl groups of the pantoyl fragment, which form hydrogen bonds with protein residues Gln72 and Thr39, respectively. In the adenylate moiety, the two hydroxyls of ribose form hydrogen bonds with Asp161, Gly158 and Gln 164, while the two nitrogen atoms at the adenine moiety interact with Val187 and Met195. To understand the binding selectivity of our designed PanC inhibitors, we modeled 2–5 into PanC binding site using the reported X-ray structure of the complex of 1 with PanC (Fig. 4A and 4B).17 The removal of the carbonyl group in 5 results in a loss of critical hydrogen bond interaction with Gln 72 and explains the nearly 13-fold loss of affinity relative to 1a. For inhibitor 3, the 2′-fluorine atom maintained hydrogen bond with Asp161. Replacement of the oxygen atom by carbon in inhibitor 4 does not change the compound overall binding mode with the protein and 4 displayed only a slight loss in binding affinity (Ki = 287 nM) compared to 1a.
Figure 4.

Docking results of the inhibitors in the active site of the closed model of E. coli pantothenate synthetase. (A) The detailed binding interactions of inhibitor 1 in the active site of the protein. The inhibitor is shown as sticks with grey carbons. (B) The detailed hydrogen bonds are indicated with dashed lines and H-bond distances are given.
2.4. Evaluation against whole cell M. tuberculosis
Pantoyl-adenylate analogues 1a–5 were evaluated against whole cell Mtb H37RvMA in 7H9 liquid medium; however, none of the compounds displayed any growth inhibition up to 250 μM. Notably, no whole cell activity against wild-type Mtb has yet been observed or disclosed for any previously described PanC inhibitor.15–23 Mizrahi and co-workers recently reported on the preparation of a Mtb panC conditional mutant that expresses less than 5% wild-type PanC levels.38 Depletion of PanC renders this mutant hypersensitive to target-specific inhibitors. In order to provide evidence that 1a–5 possess some target-based activity, the compounds were screened against this panC-depleted strain, but observed no activity was observed with up to 2 mM 1a–5. Although discouraging, it is important to note that the enzyme potency of these bisubstrate inhibitors is relatively modest compared to related bisubstrate inhibitors for other adenylating enzymes in Mtb.39
3. Conclusion
We have reported the synthesis as well as the biochemical and biological characterization of five PanC inhibitors as potential antitubercular agents. These compounds structurally mimic the pantoyl-adenylate reaction intermediate by bioisosteric replacement of the labile acyl-phosphate moiety with either acyl-sulfamate or sulfamide functional groups. We demonstrated that structural variation of the glycosyl domain of these inhibitors is well tolerated and the hydrogen bond interactions between the inhibitors and the active site residues Gln72 and Gln164 are critical for high binding affinity. Surprisingly, despite their close mimicry of the pantoyl-adenylate, none of the compounds were exceptionally potent inhibitors with Ki ranging from ~0.3–3 μM. By contrast, related adenylate inhibitors of the enzymes MbtA and BirA in Mtb possess picomolar dissociation constants.40, 41 Ciulli reported the binding of 1 (as a mixture of C-2 epimers) to PanC by isothermal titration calorimetry is accompanied by a large unfavorable entropic term (-TΔS = +8.8 kcal/mol),23 which may reflect the energetic penalty associated with the disorder-to-order transition of the gate residues (amino acids 75–88) that occurs upon ligand binding.13 Thus, despite an impressive favorable enthalpy of binding PanC (ΔH = −18.2 kcal/mol),23 the overall potency of 1a is limited by the large unfavorable entropy that is likely to restrict the potency of all active-site directed inhibitors of this enzyme. Since the inhibitors have limited potency, we used an Mtb strain wherein PanC is depleted to less than 5% of the wild-type levels in order to provide evidence that the compounds target PanC. However, none of the compounds demonstrated any activity against this knock-down strain suggesting the compounds failed to accumlate intracelluarly. This could be caused by poor cell penetration or active efflux. Alternatively, PanC may not be particularly vulnerable to inhibition and thus may not represent an ideal target for development of new antitubercular agents.
4. Materials and Methods
4.1 General Procedures
All chemicals (reagent grade) used were purchased from Sigma–Aldrich (USA) and Sinopharm Chemical Reagent Co. Ltd. (China).1H NMR spectra were measured on Varian Unity Inova 300 or 400 MHz NMR Spectrometers at 25 °C and referenced to TMS. Chemical shifts are reported in ppm using the residual solvent line as internal standard. Splitting patterns are designed as s, singlet; d, doublet; t, triplet; m, multiplet. HRMS spectra were acquired on an Agilent Technologies 6220 Accurate-Mass TOF LC/MS. Analytical thin-layer chromatography (TLC) was performed on the glass-backed silica gel sheets (silica gel 60 Å GF254). All compounds were detected using UV light (254 or 365 nm). Analytical HPLC was conducted on SHIMADZU LC-20AD. Prior to Ki measurement, all compounds were determined to be >95% pure by HPLC based on the peak area percentage.
4.1.1. (R)-2-Hydroxy-3,3-dimethylbutanoic acid (7)
To a solution of 6 (1.0 g, 7.62 mmol, 1.0 equiv) in 0.5 M H2SO4 at 0 °C was added a solution of NaNO2 (3.15 g, 45.6 mmol, 6.0 equiv) in H2O (10 mL) dropwise over 30 min. After addition was complete the solution was warmed to 23 °C and stirred overnight (~16 h). The reaction mixture was diluted with H2O (30 mL), extracted with diethyl ether (3 × 30 mL) and the combined organic layers were dried (Na2SO4), and concentrated to afford 7 (806 mg, 81%) as a colorless oil, which was used directly in next reaction. 1H NMR (400 MHz, CDCl3) δ 3.90 (s, 1H), 1.02 (s, 9H); MS (ESI–) calcd for C6H11O3 [M–H]– 131.1, found 131.4.
4.1.2. (R)-2-(tert-Butyldimethylsilyl)oxy-3,3-dimethylbutanoic acid (8)
To a solution of 7 (2.06 g, 15.6 mmol, 1.0 equiv) in DMF (15 mL) were added imidazole (5.09 g, 74.8 mmol, 4.8 equiv) and tert-butylchlorodimethylsilane (5.64 g, 37.4 mmol, 2.4 equiv). The mixture was stirred overnight at 23 °C then extracted with 1:1 ethyl acetate–petroleum ether (3 × 30 mL). The organic layer was washed successively with a 10% aqueous citric acid solution, H2O, saturated aqueous NaHCO3, and brine, then dried (Na2SO4), and concentrated. The residue was dissolved in methanol (100 mL), then an aqeuous 0.8 M K2CO3 solution (50 mL) was added. After 4 h, 10% citric acid aqueous solution was added to adjust pH to 4. The mixture was extracted with ethyl acetate (3 × 30 mL) and the combined organic layers were dried (Na2SO4) and concentrated. The residue was purified by flash chromatography (10:1 petroleum ether–ethyl acetate) to afford the title compound (3.1 g, 81%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 9.64 (br s, 1H), 3.83 (s, 1H), 0.97 (s, 9H), 0.93 (s, 9H), 0.08 (s, 3H), 0.06 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 176.6, 80.0, 35.4, 26.0, 25.8, 18.3, −5.2 (2 C); MS (ESI–) calcd for C12H25O3Si [M–H]– 245.2, found 245.2.
4.1.3. (R)-2,5-Dioxopyrrolidin-1-yl-2-(tert-butyldimethylsilyl)oxy-3,3-dimethylbutanoate (9)
To a solution of 8 (1.99 g, 8.1 mmol, 1.0 equiv) in DME (50 mL) at 0 °C, N-hydroxysuccinimide (1.87 g, 16.2 mmol, 2.0 equiv) and DCC (2.34 g, 11.3 mmol, 1.4 equiv) were added and the mixture was stirred overnight at 23 °C. The mixture was filtered through a short pad of Celite washing with ethyl acetate and the filtrate was concentrated. The residue was purified by flash chromatography (1:1 petroleum ether–CH2Cl2) to afford the title compound (1.60 g, 60%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 4.09 (s, 1H), 2.81 (br s, 4H), 1.05 (s, 9H), 0.92 (s, 9H), 0.12 (s, 3H), 0.09 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 169.1, 168.1, 78.5, 36.0, 25.7 (3C), 18.2, −5.2, −5.5; MS (ESI–) calcd for C16H29NO5Si [M]– 343.2, found 342.8.
4.1.4. 5′-O-[N-((R)-2-Hydroxy-3,3-dimethylbutanoyl)sulfamoyl]adenosine (1a)
To a solution of 1025 (100 mg, 0.26 mmol, 1.0 equiv) in DMF (1.0 mL) was added 9 (179 mg, 0.52 mmol, 2.0 equiv) and Cs2CO3 (169 mg, 0.52 mmol, 2.0 equiv) at 23 °C. After stirring 24 h at 23 °C, the solution was concentrated in vacuo. Purification by flash chromatography (500:6:1.5 CH2Cl2–MeOH–Et3N) afforded 5′-O-{N-[(R)-2-(tert-butyldimethylsilyl)oxy-3,3-dimethylbutanoyl]sulfamoyl}-2′,3′-O-isopropylideneadenosine triethylammonium salt (72 mg) as a yellow solid. This compound was dissolved in 80% aqueous TFA (2 mL). After stirring 48 h at 8 °C, the solution was concentrated in vacuo. Purification by flash chromatography (10:1 CH2Cl2–MeOH) afforded the title compound (8 mg, 7% yield from compound 10) as a white solid: 1H NMR (400 MHz, CD3OD) δ 8.57 (s, 1H), 8.33 (s, 1H), 6.38 (s, 1H), 5.05–5.00 (m, 1H), 4.79–4.77 (m, 1H), 4.74–4.70 (m, 1H), 4.40–4.37 (m, 1H), 4.14–4.10 (m, 1H), 3.62 (s, 1H), 0.99 (s, 9H); 13C NMR (100 MHz, CD3OD) δ 179.0, 158.7, 150.3, 140.9, 140.6, 121.2, 95.3, 85.2, 80.3, 77.2, 71.7, 59.7, 36.1, 26.5; HRMS (ESI–) calcd for C16H23N6O8S [M–H]– 459.1304, found 459.1310.
4.1.5. 2′,3′,5′-O-tri(tert-Butyldimethylsilyl)vidarabine (12)
To a solution of vidarabine 11 (200 mg, 0.748 mmol, 1 equiv) in DMF (1.5 mL) were added imidazole (306 mg, 4.49 mmol, 6.0 equiv) and TBSCl (677 mg, 4.49 mmol, 6.0 equiv) and the reaction was stirred overnight at 23 °C The reaction mixture was partitioned between EtOAc and H2O and the organic layer was dried (NaSO4), and concentrated. Purification by flash chromatography (80:1 CH2Cl2–MeOH) afforded the title compound (319 mg, 70%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 1H), 8.03 (s, 1H), 6.48 (d, J = 3.6 Hz, 1H), 5.59 (s, 2H), 4.34–4.32 (m, 1H), 4.19–4.16 (m, 1H), 4.03–3.98 (m, 1H), 3.88–3.84 (m, 2H), 0.94 (s, 9H), 0.92 (s, 9H), 0.72 (s, 9H), 0.15 (s, 6H), 0.09 (s, 3H), 0.08 (s, 3H), −0.08 (s, 3H), −0.45 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 155.5, 153.0, 149.6, 140.8, 119.3, 86.5, 85.9, 78.3, 77.6, 63.0, 26.1, 25.9, 25.7, 18.5, 18.1, 17.9, −4.4, −5.2, −5.5; MS (ESI+) calcd for C28H56N5O4Si3[M+H]+ 610.4, found 610.0.
4.1.6. 2′,3′-O-di(tert-Butyldimethylsilyl)vidarabine (13)
To a solution of 12 (50 mg, 0.082 mmol) in THF (0.5 mL) was added 35% aqueous TFA (2 mL). The reaction mixture was stirred for 2 h at 23 °C., then quenched with saturated aqueous NaHCO3 and extracted with EtOAc. The combined organic layers were dried (NaSO4) and concentrated. Purification by flash chromatography (80:1 CH2Cl2–MeOH) afforded the title compound (33 mg, 81%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 8.30 (s, 1H), 8.05 (s, 1H), 6.44 (d, J = 4.0 Hz, 1H), 5.83 (s, 2H), 4.38–4.34 (m, 1H), 4.31–4.27 (m, 1H), 4.13 (s, 1H), 4.08–4.04 (m, 1H), 3.92–3.88 (m, 2H), 0.94 (s, 9H), 0.67 (s, 9H), 0.16 (s, 6H), −0.06 (s, 3H), −0.41 (s, 3H); 13C NMR (75 MHz, CD3OD) δ 156.8, 153.6, 149.7, 141.9, 119.5, 88.0, 87.1, 79.1, 78.4, 62.6, 26.2, 26.0, 18.6, 18.3, −4.2, −4.4, −4.8, −5.4; MS (ESI+) calcd for C22H42N5O4Si2 [M+H]+ 496.3, found 496.0.
4.1.7. 2′,3′-O-di(tert-Butyldimethylsilyl)-5′-O-(sulfamoyl)vidarabine (14)
To a solution of 13 (100 mg, 0.2 mmol, 1.0 equiv) in 1,4-dioxane (5 mL) was added NaH (60% w/w in mineral oil, 60 mg, 1.5 mmol, 7.5 equiv). The reaction mixture was stirred for 1 h at 23 °C. Next, NH2SO2Cl27 (58 mg, 0.5 mmol, 2.5 equiv) was added and the reaction was stirred for 24 h, then quenched by the slow addition of 5:1 CH2Cl2–MeOH (10 mL). The reaction mixture was filtered through silica gel and the filtrate was concentrated. Purification by flash chromatography (30:1 CH2Cl2–MeOH) afforded the title compound (70 mg, 60%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 8.22 (s, 2H), 6.48 (d, J = 3.2 Hz, 1H), 4.48 (dd, J = 10.4, 7.2 Hz, 1H), 4.40–4.24 (m, 4H), 0.98 (s, 9H), 0.72 (s, 9H), 0.22 (s, 6H), −0.02 (s, 3H), −0.43 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 155.9, 152.7, 149.1, 139.8, 118.3, 84.2, 81.9, 77.3, 76.5, 68.1, 25.7, 25.4, 17.6, 17.3, −4.6, −4.7, −5.4, −5.8; MS (ESI+) calcd for C22H43N6O6SSi2 [M+H]+ 575.2, found 574.8.
4.1.8. 5′-O-[N-((R)-2-Hydroxy-3,3-dimethylbutanoyl)sulfamoyl]vidarabine (2)
To a solution of 14 (420 mg, 0.70 mmol, 1.0 equiv) in DMF (10 mL) was added 9 (497 mg, 1.40 mmol, 2.0 equiv) and Cs2CO3 (476 mg, 1.40 mmol, 2.0 equiv). The reaction was stirred for 24 h then concentrated in vacuo. The residue was purified by flash chromatography (500:6:1.5 CH2Cl2–MeOH–Et3N) to afford 2′,3′-O-di(tert-butyldimethylsilyl)-5′-O-{N-[(R)-2-(tert-butyldimethylsilyloxy)-3,3-dimethylbutanoyl]sul- famoyl}vidarabine triethylammonium salt (80 mg) as a yellow solid. This intermediate was dissolved in 80% aqueous TFA (2 mL). After stirring 60 h at 23 °C, the solution was concentrated in vacuo. Purification by flash chromatography (10:1 CH2Cl2–MeOH) afforded the title compound (10 mg, 3% yield from compound 14) as a white solid: 1H NMR (400 MHz, CDCl3) δ 8.58 (s, 1H), 8.19 (s, 1H), 6.38 (d, J = 5.2 Hz, 1H), 5.01 (d, J = 13.6 Hz, 1H), 4.74 (d, J = 2.4 Hz, 1H), 4.68 (dd, J = 16.0, 2.4 Hz, 1H), 4.52 (t, J = 5.2 Hz, 1H), 3.95 (t, J = 5.2 Hz, 1H), 3.64 (s, 1H), 1.00 (s, 9H); 13C NMR (100 MHz, CD3OD) δ 179.6, 158.7, 150.1, 142.2, 141.5, 121.3, 91.8, 85.7, 81.3, 80.2, 76.9, 59.8, 36.2, 26.5; HRMS (ESI–) calcd for C16H23N6O8S [M–H]– 459.1304, found 459.1318.
4.1.9. 3′,5′-O-di(tert-Butyldimethylsilyl)vidarabine (15)
To a solution of vidarabine 11 (2.0 g, 7.5 mmol, 1.0 equiv) in DMF (40 mL) were added triethylamine (5.2 mL, 37 mmol, 5.0 equiv) and TBSCl (2.82 g, 18.7 mmol, 2.5 equiv). The reaction mixture was stirred for 16 h at 23 °C then partitioned between EtOAc and H2O. The organic layer was dried (NaSO4), and concentrated. Purification by flash chromatography (80:1 CH2Cl2–MeOH) afforded the title compound (2.4 g, 65%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 8.35 (s, 1H), 8.27 (s, 1H), 6.33 (d, J = 2.4 Hz, 1H), 5.65 (s, 2H), 4.88 (d, J = 10.0 Hz, 1H), 4.37–4.34 (m, 1H), 4.14 (d, J = 10.0 Hz, 1H), 4.07 (s, 1H), 3.96 (d, J = 11.2 Hz, 1H), 3.81 (d, J = 11.2 Hz, 1H), 0.93 (s, 9H), 0.93 (s, 9H), 0.15 (s, 6H), 0.14 (s, 6H); MS (ESI+) calcd for C22H42N5O4Si2 [M+H]+ 496.3, found 496.4.
4.1.10. 3′,5′-O-di(tert-Butyldimethylsilyl)-2′-deoxy-2′-fluoroadenosine (16)
To a solution of 15 (100 mg, 0.20 mmol, 1.0 equiv) and pyridine (0.20 mL, 10 mmol, 10 equiv) in CH2Cl2 (3 mL) at 23 °C was added diethylaminosulfur trifluoride (DAST) (50.1 μL, 1.0 mmol, 5.0 equiv). The reaction mixture was stirred for 6 h at 23 °C then quenched with 5% aqueous NaHCO3 (15 mL) and extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were dried (NaSO4) and concentrated. Purification by flash chromatography (50:1 CH2Cl2–MeOH) afforded the title compound (20 mg, 31%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 8.32 (s, 1H), 8.16 (s, 1H), 6.25 (dd, J = 15.6, 1.6 Hz, 1H), 6.05 (s, 2H), 5.32 (dd, J = 52.8, 1.6 Hz, 1H), 4.73–4.64 (m, 1H), 4.16–4.12 (m, 1H), 4.02 (dd, J = 12.0, 2.4 Hz, 1H), 3.79 (dd, J = 12.0, 2.4 Hz, 1H), 0.93 (s, 9H), 0.89 (s, 9H), 0.14 (s, 3H), 0.13 (s, 3H), 0.08 (s, 3H), 0.06 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 155.8, 153.3, 149.4, 139.3, 120.1, 93.0 (d, J = 191.3 Hz), 87.0 (d, J = 33.0 Hz), 84.0, 69.4 (d, J = 15.8 Hz), 61.3, 26.0, 25.8, 18.5, 18.2, −4.6, −4.9, −5.3, −5.4; MS (ESI+) calcd for C22H41FN5O3Si2 [M+H]+ 498.3, found 498.3.
4.1.11. 3′-O-(tert-Butyldimethylsilyl)-2′-deoxy-2′-fluoroadenosine (17)
To a solution of 16 (100 mg, 0.20 mmol) in THF (0.5 mL) was added 35% aqueous 7 TFA (2 mL) at 23 °C. The solution was stirred for 20 min, then quenched with saturated aqueous NaHCO3 solution. The mixture was extracted with EtOAc (3 × 10 mL) and the combined extracts were dried (NaSO4), and concentrated. Purification by flash chromatography (50:1 CH2Cl2–MeOH) afforded the title compound (71 mg, 92%) as a white solid: 1H NMR (400 MHz, CD3OD) δ 8.37 (s, 1H), 8.19 (s, 1H), 6.27 (dd, J = 15.2, 4.0 Hz, 1H), 5.51 (dt, J = 52.8, 4.0 Hz, 1H), 4.79–4.72 (m, 1H), 4.15 (d, J = 2.0 Hz, 1H), 3.90 (d, J = 12.4 Hz, 1H), 3.71 (dd, J = 12.8, 2.4 Hz, 1H), 0.96 (s, 9H), 0.18 (s, 3H), 0.17 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 156.2, 152.6, 148.8, 139.6, 119.2, 92.3 (d, J = 189.0 Hz), 85.7 (d, J = 32.3 Hz), 84.7, 70.0 (d, J = 15.0 Hz), 60.3, 25.6, 17.9, −4.9, −5.1; MS (ESI+) calcd for C16H27FN5O3Si [M+H]+ 384.2, found 384.2.
4.1.12. 3′-O-(tert-Butyldimethylsilyl)-2′-deoxy-2′-fluoro-5′-O-(sulfamoyl)adenosine (18)
To a solution of 17 (100 mg, 0.26 mmol, 1.0 equiv) in 1,4-dioxane (5 mL) was added NaH (60 dispersion w/w in mineral oil, 32 mg, 0.78 mmol, 3.0 equiv) at 23 °C. After 1h, NH2SO2Cl27 (75 mg, 0.65 mmol, 2.5 equiv) was added and the solution was stirred for 16 h at 23 °C. The reaction was quenched by the slow addition of 5:1 CH2Cl2–MeOH (10 mL) then the mixture was filtered through silica gel and the filtrate was concentrated. Purification by flash chromatography (30:1 CH2Cl2–MeOH) afforded the title compound (72 mg, 60%) as a white solid: 1H NMR (300 MHz, CD3OD) δ 8.26 (s, 1H), 8.21 (s, 1H), 6.31 (dd, J = 17.1, 2.7 Hz, 1H), 5.53 (ddd, J = 52.5, 4.5, 2.7 Hz, 1H), 4.94–4.91 (m, 1H), 4.46–4.40 (m, 1H), 4.32–4.26 (m, 2H), 0.97 (s, 9H), 0.21 (s, 3H), 0.19 (s, 3H); 13C NMR (75 MHz, DMSO-d6) δ 156.2, 152.8, 148.8, 139.7, 119.1, 92.2 (d, J = 187.5 Hz), 86.1 (d, J = 33.8 Hz), 80.3, 70.2 (d, J = 15.0 Hz), 67.7, 25.6, 17.8, −4.9, −5.1; MS (ESI+) calcd for C16H28FN6O5SSi [M+H]+ 463.2, found 463.2.
4.1.13. 2′-Deoxy-2′-fluoro-5′-O-[N-((R)-2-Hydroxy-3,3-dimethylbutanoyl)sulfamoyl] adenosine (3)
To a solution of 18 (140 mg, 0.30 mmol, 1.0 equiv) in DMF (8 mL) was added 9 (196 mg, 0.60 mmol, 2.0 equiv) and Cs2CO3 (206 mg, 0.60 mmol, 2.0 equiv) at 23 °C. The reaction mixture was stirred for 20 h at 23 °C, then concentrated in vacuo. The residue was purified by flash chromatography (500:6:1.5 CH2Cl2–MeOH–Et3N) to afford 3′-O(tert-butyldimethylsilyl)-5′-O-{N-[(R)-2-(tert-butyldimethylsilyloxy)-3,3-dimethylbutanoyl] sulfamoyl}-2′-deoxy-2′-fluoroadenosine triethylammonium salt (80 mg) as a yellow solid. The compound was dissolved in 80% aqueous TFA (1 mL) and the solution was stirred at 20 °C for 72 h. The solvent was removed in vacuo. Purification by flash chromatography (10:1 CH2Cl2–MeOH) afforded the title compound (5.0 mg, 4% yield from 18) as a white solid: 1H NMR (400 MHz, CD3OD) δ 8.56 (s, 1H), 8.30 (s, 1H), 6.68 (d, J = 6.8 Hz, 1H), 4.97 (dd, J = 51.6, 4.4 Hz, 1H), 4.86–4.82 (m, 2H), 4.76 (dd, J = 14.0, 2.8 Hz, 1H), 4.50 (dt, J = 18.8, 4.4 Hz, 1H), 3.63 (s, 1H), 0.98 (s, 9H); 13C NMR (100 MHz, CD3OD) δ 179.1, 158.7, 150.4, 141.0, 140.5, 121.3, 94.3 (d, J = 192.0 Hz), 92.4 (d, J = 31.0 Hz), 84.8, 80.2 (d, J = 7.0 Hz), 71.5 (d, J = 16.0 Hz), 59.5, 36.2, 26.5; HRMS (ESI–) calcd for C16H22FN6O7S [M–H]– 461.1260, found 461.1280.
4.1.14. 5′-O-[N-((R)-2-Hydroxy-3,3-dimethylbutanoyl)sulfamoyl]aristeromycin (4)
To a solution of 1931 (89 mg, 0.19 mmol, 1.0 equiv) in DMF (5 mL) was added 9 (128 mg, 0.38 mmol, 2.0 equiv) and Cs2CO3 (122 mg, 0.38 mmol, 2.0 equiv). The reaction mixture was stirred for 20 h at 23 °C then concentrated in vacuo. The residue was purified by flash chromatography (500:8:1.5 CH2Cl2–MeOH–Et3N) to afford 5′-O-{N-[(R)-2-(tert-butyldimethylsilyl)oxy-3,3-dimethylbutanoyl]sulfamoyl}-2′,3′-O-isopropylidenearisteromycin triethylammonium salt (50 mg) as a yellow solid. The compound was dissolved in 80% aqueous TFA (1.0 mL) and the solution was stirred at 8 °C for 48 h. The solvent was removed in vacuo. Purification by flash chromatography (10:1 CH2Cl2–MeOH) afforded the title compound (6 mg, 7% yield from compound 19) as a white solid: 1H NMR (400 MHz, CD3OD) δ 8.56 (s, 1H), 8.27 (s, 1H), 5.04 (d, J = 4.8 Hz, 1H), 4.93 (dd, J = 13.6, 3.6 Hz, 1H), 4.55 (dd, J = 13.6, 2.8 Hz, 1H), 4.11–4.07 (m, 1H), 4.02 (d, J = 4.8 Hz, 1H), 3.69 (s, 1H), 3.05–2.96 (m, 1H), 2.80 (d, J = 10.0 Hz, 1H), 2.04 (d, J = 14.4 Hz, 1H), 1.00 (s, 9H); 13C NMR (100 MHz, CD3OD) δ 179.1, 158.6, 150.5, 143.5, 141.2, 121.7, 80.3, 77.5, 74.4, 67.4, 60.6, 44.0, 36.1, 33.5, 26.5; HRMS (ESI–) calcd for C17H25N6O7S [M–H]– 457.1511, found 457.1510.
4.1.15. (4R)-5,5-Dimethyl-4-hydroxymethyl-2-phenyl-1,3-dioxane (22)
To a mixture of 2132 (371 mg, 2.77 mmol, 1.0 equiv) and benzaldehyde dimethyl acetal (548 mg, 3.04 mmol, 1.1 equiv) in CH2Cl2 (300 mL) was added camphorsulfonic acid (50 mg, 0.28 mmol, 0.1 equiv) at 23 °C. The reaction mixture was refluxed for 24 h, then saturated NaHCO3 aqueous solution was added to adjust the pH to 7. The mixture was extracted with EtOAc, and the combined organic extracts were washed with saturated aqueous NaCl, dried (NaSO4), and concentrated. Purification by flash chromatography (6:1 petroleum ether–EtOAc) afforded the title compound (500 mg, 85%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 7.52 (d, J = 6.8 Hz, 2H), 7.43–7.33 (m, 3H), 5.51 (s, 1H), 3.69–3.60 (m, 5H), 1.14 (s, 3H), 0.84 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 138.4, 129.2, 128.4, 126.4, 102.2, 86.0, 79.0, 61.6, 31.7, 21.5, 19.3; MS (ESI+) calcd for C13H19O3 [M+H]+ 223.1, found 223.0.
4.1.16. (4R)-5,5-Dimethyl-2-phenyl-4-[(trifluoromethanesulfonyl)oxy]methyl-1,3-dioxane (23)
To a solution of 22 (135 mg, 0.61 mmol, 1.0 equiv) and Et3N (0.2 mL) in CH2Cl2 (5 mL) was added TsCl (173 mg, 0.91 mmol, 1.5 equiv) at 0 °C. The mixture was then warmed to 23 °C and stirred for 16 h. The solvent was removed in vacuo and the residue was purified by flash chromatography (10:1 petroleum ether–EtOAc) to afford the title compound (190 mg, 83%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 7.80 (d, J = 8.0 Hz, 2H), 7.48–7.34 (m, 5H), 7.27 (d, J = 8.0 Hz, 2H), 5.43 (s, 1H), 4.28 (dd, J = 10.8, 2.0 Hz, 1H), 4.11–4.03 (m, 1H), 3.91–3.85 (m, 1H), 3.65 (dd, J = 30.0, 11.2 Hz, 2H), 2.43 (s, 3H), 1.10 (s, 3H), 0.87 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 144.8, 138.0, 132.9, 129.8, 129.0, 128.2, 128.0, 126.2, 101.6, 82.5, 78.5, 69.5, 31.8, 21.7, 21.4, 18.8; MS (ESI+) calcd for C20H24O5SNa [M+Na]+ 399.1, found 399.0.
4.1.17. (4R)-4-Azidomethyl-5,5-dimethyl-2-phenyl-1,3-dioxane (24)
To a solution of 23 (185 mg, 0.49 mmol, 1.0 equiv) in DMF (3 mL) was added NaN3 (96 mg, 1.47 mmol, 3.0 equiv) and the mixture was refluxed at 100 °C for 16 h. The mixture was extracted with diethyl ether and the combined organic layers were dried (NaSO4), and concentrated. Purification by flash chromatography (10:1 petroleum ether–EtOAc) afforded the title compound (113 mg, 93%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 7.55 (d, J = 6.8 Hz, 2H), 7.43–7.32 (m, 3H), 5.58 (s, 1H), 3.81 (d, J = 9.2 Hz, 1H), 3.74 (d, J = 11.2 Hz, 1H), 3.65 (d, J = 11.2 Hz, 1H), 3.46 (dd, J = 12.8, 9.2 Hz, 1H), 3.20 (d, J = 13.2 Hz, 1H), 1.16 (s, 3H), 0.84 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 138.1, 128.9, 128.3, 126.1, 101.7, 84.7, 78.8, 50.7, 32.3, 21.5, 18.9; MS (ESI+) calcd for C13H17N3O2Na [M+Na]+ 270.1, found 270.0.
4.1.18. (4R)-4-Aminomethyl-5,5-dimethyl-2-phenyl-1,3-dioxane (25)
To a solution of 24 (100 mg, 0.4 mmol, 1.0 equiv) in MeOH (3 mL) under nitrogen was added 10% w/w Pd/C (20 mg). The nitrogen atmosphere was replaced with an atmosphere of H2 and the mixture was stirred at 23 °C for 2 h. The reaction mixture was then filtered through Celite washing with MeOH and the filtrate was concentrated to afford the title compound (67 mg, 75%) as a white solid: 1H NMR (400 MHz, CDCl3) 1H NMR (300 MHz, CDCl3) δ 7.56–7.28 (m, 5H), 5.47 (s, 1H), 3.89 (br s, 2H), 3.60–3.52 (m, 3H), 2.88–2.67 (m, 2H), 1.08 (s, 3H), 0.78 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 138.6, 128.8, 128.2, 126.1, 101.9, 87.5, 78.8, 41.4, 32.0, 21.4, 19.0; MS (ESI+) calcd for C13H20NO2 [M+H]+ 222.1, found 222.2.
4.1.19. 4-Nitrophenyl-[((4R)-5,5-dimethyl-2-phenyl-1,3-dioxan-4-yl)methyl]sulfamate (26)
To a solution of 25 (60 mg, 0.27 mmol, 1.0 equiv) and 4-nitrophenol (375 mg, 2.7 mmol, 10.0 equiv) in CH2Cl2 (3 mL) was added 4-nitrophenyl chlorosulfate (192 mg, 0.81 mmol, 3.0 equiv) dropwise in CH2Cl2 (3 mL) at −78 °C. The reaction was stirred 2 h at −78 °C, then diluted with CH2Cl2 and warmed to 23 °C followed by consecutive washes with 1 M NaH2PO4 and H2O. The organic layer was dried (Na2SO4) and concentrated. Purification by flash chromatography (50:1 CH2Cl2–MeOH) afforded the title compound (60 mg, 50%) as a white solid: 1H NMR (400 MHz, CDCl3) δ 8.09 (d, J = 9.2 Hz, 2H), 7.50–7.33 (m, 7H), 5.45 (s, 1H), 3.80–3.70 (m, 2H), 3.61 (d, J = 11.2 Hz, 1H), 3.57–3.49 (m, 1H), 3.32–3.21 (m, 1H), 1.20 (s, 3H), 0.89 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 154.3, 145.8, 137.7, 129.5, 128.4, 126.4, 125.4, 122.6, 102.1, 82.8, 78.3, 44.1, 31.9, 21.1, 18.7; MS (ESI+) calcd for C19H22N2O7SNa [M+Na]+ 445.1, found 444.8.
4.1.20. 5′-Amino-5′-deoxy-5′-N-[N-((R)-2,2-dimethylbutane-1,3-diol-4-yl)sulfamoyl] adenosine (5)
To a mixture of 26 (120 mg, 0.28 mmol, 1.0 equiv) and 2733 (167 mg, 0.56 mmol, 2.0 equiv) in CH2Cl2 (10 mL) at 23 °C was added 4Å molecular sieves (80 mg) and Et3N (0.40 mL, 2.80 mmol, 10 equiv). The reaction was stirred for 24 h, then filtered and the filtrate concentrated. Purification by flash chromatography (40:1 CH2Cl2–MeOH) afforded 5′-amino-5′-deoxy-5′-N-(N-{[(2R,4R)-5,5-dimethyl-2-phenyl-1,3-dioxan-4-yl]methylamino} sulfamoyl)-2′,3′-O-isopropylideneadenosine (100 mg) as a white solid. The compound was dissolved in 80% aqueous TFA (2 mL) at −35 °C. After stirring 4 h at −35 °C, the solution was concentrated in vacuo. Purification by flash chromatography (10:1 CH2Cl2–MeOH) afforded the title compound (5 mg, 4% yield from 26) as a white solid: 1H NMR (400 MHz, CD3OD) δ 8.28 (s, 1H), 8.23 (s, 1H), 5.90 (d, J = 6.8 Hz, 1H), 4.91–4.89 (m, 1H), 4.36 (dd, J = 5.2, 2.0 Hz, 1H), 4.32–4.28 (m, 1H), 3.64–3.55 (m, 2H), 3.40–3.34 (m, 3H), 3.16 (dd, J = 12.4, 2.0 Hz, 1H), 2.85 (dd, J = 12.8, 10.0 Hz, 1H), 0.81 (s, 3H), 0.78 (s, 3H); 13C NMR (100 MHz, CD3OD) δ 157.6, 153.9, 150.0, 142.5, 121.2, 91.6, 85.9, 76.3, 74.3, 73.0, 70.2, 46.1, 45.8, 39.6, 21.8, 20.1; HRMS (ESI–) calcd for C16H26N7O7S [M–H]– 460.1620, found 460.1635.
4.2 Pantothenate Synthetase Assay
4.2.1. Materials
Pantoic acid was synthesized as described by Rychlik,42 7-methyl-6-thioguanosine (MesG) was purchased from Berry and Associates (Dexter, MI, USA), E. coli TOPO and BL21(DE3) cells are from Invitrogen (Carlsbad, CA, USA), restriction enzymes and Taq polymerase are from New England Biolabs (Ipswich, MA, USA), the vectorpET28b is from EMD Biosciences (San Diego, CA, USA), the primers for PCR are from Integrated DNA Technologies (Coralville, IA, USA) and the PFU polymerase is from Agilent Technologies (Wilmington, DE, USA). All other chemicals, biological buffers, and the coupling enzymes inorganic pyrophosphatase (I1643) and purine nucleoside phosphorylase (N8264) were purchased from Sigma-Aldrich (St. Louis, MO).
4.2.2. Cloning, Expression and Purification of Recombinant PanC from Mycobacterium tuberculosis
PanC (Rv3602c) was amplified from the H37Rv BAC Rv222 using PFU turbo and the primers GCGAGCAACCACATCGTCAC and CGACTTCAGCATCGTCCGTAAC. The PCR product was treated with Taq using standard procedures to add 3′ overhanging adenosine residues and then cloned into pCR2.1-TOPO. The resulting plasmid (pCDD22) was used as template to amplify the expression construct using the primers GGCATATGACGATTCCTGCGTTCCATCCC and GCTCAAGCTTCAGTTTCTCCAATGTGATTCGAGGATTGCCCGG containing the restriction sites NdeI and HindIII respectively (underlined). The resulting PCR product was cloned into pCR2.1 giving plasmid pCDD23. The construct was digested using NdeI and HindIII and ligated into similarly digested pET28b yielding pCDD24. The recombinant plasmid pCDD24 was transformed into E. coli BL21 (DE3) electrocompetent cells and streaked-out on a Luria Bertani (LB) agar plate with 100 μg mL−1 of kanamycin and incubated overnight at 37 °C. A single colony was selected to inoculate 50 mL of LB with 100 μgmL−1 of kanamycin to be used as a starting culture.
Terrific Broth (TB) cultures (4 L) were supplemented with 100 μg mL−1 kanamycin, and 10 mL of an overnight starting culture added to the media. The cultures were grown at 37 °C until an OD600 of 0.5 and then induced with 0.5 mM of isopropyl β-D-1-thiogalactopyranoside (IPTG) for 4 h at 30 °C. The cells were centrifuged at 6000g for 30 min and the pellets were stored at −80 °C. The frozen pellets were thawed in lysis buffer (50 mM HEPES, 300 mM NaCl, 10 mM imidazole, pH 8.0) and the cells were disrupted by sonication on a Branson Sonifier (5 × 2min, 30% duty cycle, power 8) and centrifuged for 30 min at 50,000g to remove cell debris. To the supernatant was added 1 mL of 50% Ni-NTA agarose resin in 30% ethanol (Qiagen) and the final solution was incubated at 4 °C for 1 h. The sample was then loaded onto a gravity column and the resin was washed with 15 mL of wash buffer (50 mM HEPES, 300 NaCl, 20 mM imidazole, pH 8.0) and eluted with 2.5 mL of elution buffer (50 mM HEPES, 300 NaCl, 250 mM imidazole, pH 8.0). In order to remove the imidazole from the sample, a desalting column (PD-10, GE HealthCare, Piscataway NJ, USA) was used with the storage buffer (10 mM HEPES [pH 8.0], 1 mM EDTA, 5% glycerol). Protein concentrations were determined using ε280 = 15,930 M−1 cm−1 for the native PanC with a hexa-histidine tag and the final yield of this protocol provided approximately 45 mg per liter of cell culture.
4.2.3. In vitro Inhibition Studies
Kinetic studies to evaluate PanC inhibition of each compound were performed under initial velocity conditions using the MesG coupled assay (EnzChek pyrophosphatase assay, Invitrogen) with 400 nM PanC, 100 mM HEPES (pH 8.0), 2.4 mM β-alanine, 10 mM MgCl2, 0.04 unit of pyrophosphatase, 0.1 unit of purine nucleoside phosphorylase, 0.2 mM 7-methyl-6-thioguanosine (MesG) and varying concentrations of inhibitor, pantoic acid, and ATP in a total volume of 100 μL containing up to 5% DMSO. Reactions were initiated by addition of PanC. In this coupled assay, pyrophosphate generated from the PanC reaction is converted to phosphate by pyrophosphatase. Purine nucleoside phosphorylase then catalyzes the phosphorolysis of the substrate 7-methyl-6-thioguanosine (MesG) to the chromogenic product 7-methyl-6-thioguaninethat is measured by an increase in absorbance at 360 nm. Experiments were performed in 96-well plates (UV Half Area plate with Transparent Bottom-Corning) and formation of 7-methyl-6-thioguaninewas measured at 360 nm (ε360 = 11,000 M−1 cm−1) at 25 °C on a microplate reader.
For experiments to determine the apparent inhibition constants with respect to pantoic acid, the initial rates were measured at varying concentrations (50, 100, 130, 180, 240 and 300μM) of pantoic acid with 2.6 mM ATP and 2.4 mM β-alanine at different fixed levels of the inhibitors [inhibitor 1a (0, 0.4, 0.8 and 1 μM), inhibitor 2 (0, 0.08, 0.12 and 0.16 μM), inhibitor 3 (0, 0.2, 0.4 and 0.6 μM), inhibitor 4 (0, 0.2, 0.4 and 0.5 μM) and inhibitor 5 (0, 0.8, 1.6 and 2 μM)]. Inhibition data were fit to equation 1:
| (1) |
where I is the inhibitor concentration, S is the substrate concentration, Km is the Michaelis-Menten constant, Vmax is the maximal velocity, Ki is the competitive inhibition constant.37
4.3. Modeling Studies
The three-dimensional structures of the aforementioned compounds were constructed using Chem. 3D ultra 12.0 software [Chemical Structure Drawing Standard; Cambridge Soft corporation, USA (2010)], which were then energetically minimized by using Tripos force field with 5000 iterations and minimum RMS gradient of 0.05 prior to docking. The crystal structures of pantothenate synthetase (M. tuberculosis. and E. coli) (PDB code: 3IVG.pdb) complex were retrieved from the RCSB Protein Data Bank (http://www.rcsb.org/pdb/home/home.do). All bound waters were eliminated from the protein and hydrogens were added to the protein. Each ligand was docked into the active site of pantothenate synthetase using the Surflex-Dock suite of SYBYL 1.3.
4.4. Mtb whole-cell assays
Whole-cell screening of 1–5 was carried out against wildtype H37RvMA43 and the panC-depleted strain as previously described.38 Briefly, cells were grown to an OD600 of ~0.2 in 7H9 medium prior to 500-fold dilution. In the case of the panC knockdown, the medium was supplemented with Hygromycin B (Hyg; 50 μg/mL), Kanamycin (Km; 25 μg/mL) and Gentamicin (Gm; 5 μg/mL) in order to facilitate maintenance of the regulatory vectors. Using a starting concentration of 4 mM, 2-fold serial dilutions of each inhibitor were performed in 96-well microplates containing 50 μL 7H9 medium, supplemented with Hyg (25 μg/mL), Km (12.5 μg/mL) and Gm (2.5 μg/mL) where appropriate, in both the presence and absence of anhydrotetracycline (ATc; 20 ng/mL) alone or ATc (20 ng/mL) together with pantothenate (50 μg/mL). A volume of 50 μL of the diluted cell suspension was added to each well, yielding a final volume of 100 μL per well. Plates were incubated at 37 °C and growth was observed visually following 7, 10 and 14 days of incubation.
Supplementary Material
Scheme 5.

Reagents and conditions: (i) LiAlH4, THF, 0 °C, 2 h, 58%; (ii) PhCH(OEt)2, CSA, CH2Cl2, reflux, 24 h, 85%; (iii) TsCl, Et3N, CH2Cl2, 16 h, 83%; (iv) NaN3, DMF, reflux, 16 h, 93%; (v) H2, Pd/C, MeOH, 2 h, 75%; (vi) 4-nitrophenyl chlorosulfate, CH2Cl2, −78 °C, 2 h, 50%; (vii) 27, Et3N, CH2Cl2, 4 Å molecular sieves; (viii) 4:1 TFA–H2O, −35 °C, 4 h, 4% from 26.
Acknowledgments
This research was supported by grants from the National Natural Science Foundation of China (Grant No. 81072514, 21002067 to C.Q.), the Ministry of Education Scholarship Fund (The Jiangsu “333” Project to C.Q.) and the National Institutes of Health (Grant No. AI070219 to C.C.A.).
References and notes
- 1.World Health Organization. [Accessed March 2013];Fact sheet on tuberculosis. http://www.who.int/mediacentre/factsheets/fs104/en/index.html.
- 2.Mitchison D, Davies G. Int J Tuberc Lung Dis. 2012;16:724. doi: 10.5588/ijtld.12.0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barry CE, 3rd, Boshoff HI, Dartois V, Dick T, Ehrt S, Flynn J, Schnappinger D, Wilkinson RJ, Young D. Nat Rev Microbiol. 2009;7:845. doi: 10.1038/nrmicro2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kjellsson MC, Via LE, Goh A, Weiner D, Low KM, Kern S, Pillai G, Barry CE, 3rd, Dartois V. Antimicrob Agents Chemother. 2012;56:446. doi: 10.1128/AAC.05208-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barry CE. Curr Top Med Chem. 2011;11:1216. doi: 10.2174/156802611795429158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dartois V, Barry CE., 3rd Bioorg Med Chem Lett. 2013;23:4741. doi: 10.1016/j.bmcl.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Webb ME, Smith AG, Abell C. Nat Prod Rep. 2004;21:695. doi: 10.1039/b316419p. [DOI] [PubMed] [Google Scholar]
- 8.Leonardi R, Zhang YM, Rock CO, Jackowski S. Prog Lipid Res. 2005;44:125. doi: 10.1016/j.plipres.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Spry C, Kirk K, Saliba KJ. FEMS Microbiol Rev. 2008;32:56. doi: 10.1111/j.1574-6976.2007.00093.x. [DOI] [PubMed] [Google Scholar]
- 10.Sambandamurthy VK, Wang X, Chen B, Russell RG, Derrick S, Collins FM, Morris SL, Jacobs WR., Jr Nat Med. 2002;8:1171. doi: 10.1038/nm765. [DOI] [PubMed] [Google Scholar]
- 11.Sambandamurthy VK, Derrick SC, Jalapathy KV, Chen B, Russell RG, Morris SL, Jacobs WR., Jr Infect Immun. 2005;73:1196. doi: 10.1128/IAI.73.2.1196-1203.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng R, Blanchard JS. Biochemistry. 2001;40:12904. doi: 10.1021/bi011522+. [DOI] [PubMed] [Google Scholar]
- 13.Wang S, Eisenberg D. Protein Sci. 2003;12:1097. doi: 10.1110/ps.0241803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang S, Eisenberg D. Biochemistry. 2006;45:1554. doi: 10.1021/bi051873e. [DOI] [PubMed] [Google Scholar]
- 15.Mycobacterium tuberculosis Pantothenate Synthetase Assay - BioAssay Summary. http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=375.
- 16.Velaparthi S, Brunsteiner M, Uddin R, Wan B, Franzblau SG, Petukhov PA. J Med Chem. 2008;51:1999. doi: 10.1021/jm701372r. [DOI] [PubMed] [Google Scholar]
- 17.White EL, Southworth K, Ross L, Cooley S, Gill RB, Sosa MI, Manouvakhova A, Rasmussen L, Goulding C, Eisenberg D, Fletcher TM., 3rd J Biomol Screen. 2007;12:100. doi: 10.1177/1087057106296484. [DOI] [PubMed] [Google Scholar]
- 18.Hung AW, Silvestre HL, Wen S, Ciulli A, Blundell TL, Abell C. Angew Chem Int Ed. 2009;48:8452. doi: 10.1002/anie.200903821. [DOI] [PubMed] [Google Scholar]
- 19.Sledz P, Silvestre HL, Hung AW, Ciulli A, Blundell TL, Abell C. J Am Chem Soc. 2010;132:4544. doi: 10.1021/ja100595u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silvestre HL, Blundell TL, Abell C, Ciulli A. Proc Natl Acad Sci USA. 2013;110:12984. doi: 10.1073/pnas.1304045110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scott DE, Dawes GJ, Ando M, Abell C, Ciulli A. Chembiochem. 2009;10:2772. doi: 10.1002/cbic.200900537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tuck KL, Saldanha SA, Birch LM, Smith AG, Abell C. Org Biomol Chem. 2006;4:3598. doi: 10.1039/b609482a. [DOI] [PubMed] [Google Scholar]
- 23.Ciulli A, Scott DE, Ando M, Reyes F, Saldanha SA, Tuck KL, Chirgadze DY, Blundell TL, Abell C. Chembiochem. 2008;9:2606. doi: 10.1002/cbic.200800437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franz N, Menin L, Klok HA. Eur J Org Chem. 2009;31:5390. doi: 10.1039/b912313j. [DOI] [PubMed] [Google Scholar]
- 25.Ferreras JA, Ryu JS, Di Lello F, Tan DS, Quadri LE. Nat Chem Biol. 2005;1:29. doi: 10.1038/nchembio706. [DOI] [PubMed] [Google Scholar]
- 26.Qiao C, Gupte A, Boshoff HI, Wilson DJ, Bennett EM, Somu RV, Barry CE, 3rd, Aldrich CC. J Med Chem. 2007;50:6080. doi: 10.1021/jm070905o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heacock D, Forsyth CJ, Shiba K, Musier-Forsyth K. Bioorg Chem. 1996;24:273. [Google Scholar]
- 28.Xu ZX, Yin W, Chen JL, Qiao CH. Chin J Org Chem. 2013;33:1578. [Google Scholar]
- 29.Pankiewicz KW, Krzeminski J, Ciszewski LA, Ren WY, Watanabe KA. J Org Chem. 1992;57:553. [Google Scholar]
- 30.Vijver PV, Ostrowski T, Sproat B, Goebels J, Rutgeerts O, Aerschot AV, Wear M, Herdewijin P. J Med Chem. 2008;51:3020. doi: 10.1021/jm8000746. [DOI] [PubMed] [Google Scholar]
- 31.Somu RV, Wilson DJ, Bennett EM, Boshoff HI, Celia L, Beck BJ, Barry CE, 3rd, Aldrich CC. J Med Chem. 2006;49:7623. doi: 10.1021/jm061068d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mandel AL, La Clair JJ, Burkart MD. Org Lett. 2004;6:4801. doi: 10.1021/ol048853+. [DOI] [PubMed] [Google Scholar]
- 33.Liu F, Austin DJ. Tetrahedron Lett. 2001;42:3153. [Google Scholar]
- 34.Fettes KJ, Howard N, Hickman DT, Adah SA, Player MR, Torrence PF, Micklefield J. Chem Commun. 2000:765. [Google Scholar]
- 35.Webb MR. Proc Natl Acad Sci USA. 1992;89:4884. doi: 10.1073/pnas.89.11.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Upson RH, Haugland RP, Malekzadeh MN. Anal Biochem. 1996;243:41. doi: 10.1006/abio.1996.0479. [DOI] [PubMed] [Google Scholar]
- 37.Copeland RA. Evaluation of Enzyme Inhibitors in Drug Discovery: A guide for medicinal chemists and pharmacologists. Hoboken, NJ: Wiley; 2013. [PubMed] [Google Scholar]
- 38.Abrahams GL, Kumar A, Savvi S, Hung AW, Wen S, Abell C, Barry CE, 3rd, Sherman DR, Boshoff HI, Mizrahi V. Chem Biol. 2012;19:844. doi: 10.1016/j.chembiol.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duckworth BP, Nelson KM, Aldrich CC. Curr Top Med Chem. 2012;12:766. doi: 10.2174/156802612799984571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neres J, Labello NP, Somu RV, Boshoff HI, Wilson DJ, Vannada J, Chen L, Barry CE, 3rd, Bennett EM, Aldrich CC. J Med Chem. 2008;51:5349. doi: 10.1021/jm800567v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duckworth BP, Geders TW, Tiwari D, Boshoff HI, Sibbald PA, Barry CE, 3rd, Schnappinger D, Finzel BC, Aldrich CC. Chem Biol. 2011;18:1432. doi: 10.1016/j.chembiol.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rychlik M. J Mass Spectrom. 2001;36:555. doi: 10.1002/jms.157. [DOI] [PubMed] [Google Scholar]
- 43.Ioerger TR, Feng Y, Ganesula K, Chen X, Dobos KM, Fortune S, Jacobs WR, Jr, Mizrahi V, Parish T, Rubin E, Sassetti C, Sacchettini JC. J Bacteriol. 2010;192:3645. doi: 10.1128/JB.00166-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.