

Abstract
The human 5-hydroxytryptamine receptor subtype 1A (5-HT1A) is highly expressed in the raphe nuclei region and limbic structures; for that reason 5-HT1A had served as a promising target for treating human mood disorders and neurodegenerative diseases. We have developed binary quantitative structure-activity relationship (QSAR) models for 5-HT1A binding using data retrieved from the WOMBAT database and the k-Nearest Neighbor (kNN) machine learning method. A rigorous QSAR modeling and screening workflow had been followed, with extensive internal and external validation processes. The models’ classification accuracies to discriminate 5-HT1A binders from the non-binders were as high as 96% for the external validation. These models were employed further to mine two major natural products screening libraries, i.e. TimTec Natural Product Library (NPL) and Natural Derivatives Library (NDL). In the end five screening hits were tested by radioligand binding assays with a success rate of 40%, and two new compounds were confirmed to be binders at the µM concentration against the human 5-HT1A receptor. The combined application of rigorous QSAR modeling and model-based virtual screening presents a powerful means for profiling natural products compounds with important biomedical activities.
Keywords: 5-HT1A receptor, high-throughput screening, natural products, QSAR, external validation, profiling activities
GRAPHICAL ABSTRACT

INTRODUCTION
The human 5-HT1A receptor is a subtype of 5-HT receptor that binds the endogenous neurotransmitter serotonin (5-hydroxytryptamine, 5-HT)1. It is the most widespread of all the 5-HT receptors, an important family of G protein-coupled receptors (GPCRs). In the central nervous system (CNS), 5-HT1A receptor exists in the cerebral cortex, hippocampus, septum, amygdala and raphe nucleus in high densities, while it is also located at the basal ganglia and thalamus in low amounts2. It had been among the most important molecular targets that were actively being pursued for drug discovery efforts in psychoactive treatment3. Because of its dense concentration on cortical and hippocampal pyramidal neurons, 5-HT1A receptor had been actively studied in recent years for novel strategies to treat the cognitive deficits in schizophrenia4. In fact, atypical antipsychotic drugs modestly enhance cognition and several drugs in this class were found to show 5-HT1A partial agonist activity (e.g. aripiprazole, clozapine, olanzapine, ziprasidone and quetiapine)5,6,7. They can sometimes be used in low doses as augmentation to standard antidepressants, for example, the selective serotonin reuptake inhibitors (SSRIs)8. In addition, 5-HT1A receptor agonists such as buspirone, tandospirone and flesinoxan showed efficacy in relieving anxiety and depression9,10, and some have been approved for these indications in various parts of the world. Other drugs like gepirone, flesinoxan11,12, flibanserin13,14 and PRX-0002315,16 were also investigated, though none of them had been fully developed and approved so far.
The 5-HT1A receptor has recently received considerable attention for its potential treatments to neurodegenerative diseases. 5-HT1A receptor activation had been shown to increase dopamine release in the medial prefrontal cortex, striatum and hippocampus, and may be useful for improving the symptoms of Parkinson's disease17. As mentioned above, some of the atypical antipsychotics are 5-HT1A receptor partial agonists, and this property had been shown to enhance their clinical efficacy. They act by enhancing dopamine release which plays a major role in the antidepressant and anxiolytic effects seen upon postsynaptic activation of the 5-HT1A receptor18. Moreover, 5-HT1A receptor antagonists such as lecozotan have been shown to facilitate certain types of learning and memory in rodents by stimulating the release of glutamate and acetylcholine in various areas of the brain19. As a result, they are now being developed as novel treatments for Alzheimer's disease. Although several drugs including the ones mentioned above are available on market and show the affinities to human 5-HT1A receptor, few were developed specifically for modulating this target. They were either insufficient in term of efficacy for some patients or exhibit various side effects for others. Taken together, there is still a critical need in developing novel 5-HT1A receptor modulators to benefit the aforementioned diseases.
High throughput screening (HTS) has become a popular and efficient approach for the discovery of lead compounds in recent years. Once a quantitative structure-activity relationship (QSAR) model was built, it can be employed to mine large libraries of small molecules with the goal of identifying drug-like, structurally novel hits with the desired biological efficacy20. Recently, we have demonstrated that QSAR models can be used in HTS, also known as database mining, i.e. finding molecular structures that are similar in their activities to the probe molecules or even profiling the activities of compounds in a library21,22,23,24,25. At the same time, the number of chemicals available for HTS has been growing significantly over the past decade. This constant increase in the size of chemicals libraries available highlights the importance of classifying chemical libraries by their characteristics, so that HTS can be performed accordingly in regards to different objectives.
MATERIALS AND METHODS
Datasets for QSAR Model Building
The data on 5-HT1A binders were retrieved from the WOMBAT database while we collected 5-HT1A non-binders from the Psychoactive Drug Screening Program (PDSP) at the National Institute of Mental Health (NIMH). During this study, we employed 10 µM as the threshold value to specify binders vs. non-binders, and only retrieved ligands tested against cloned human species cell lines using the hot ligand of [3H]-8-OH-DPAT. By submitting such queries, we designated 69 unique compounds as 5-HT1A binders with binding affinities stronger than 10 µM. We also retrieved 61 non-binders that were shown to have no binding affinities to 5-HT1A receptor at 10 µM concentration.
Dataset Curation
For the purposes of reliable modeling, our datasets were curated following the protocols published earlier26. In the beginning, we cleaned all compounds with the Wash Molecules module in Molecular Operating Environment (MOE27, version 2009.10). This module processes chemical structures by carrying out several standard operations including 2D depiction layout, hydrogen correction, salt and solvent removal, chirality and bond type normalization (all details can be found in the MOE manual27). Second, ChemAxon Standardizer28 was used to harmonize the representation of aromatic rings. Finally, the structural duplicates were detected by the analysis of the normalized molecular topologies. The functional data for duplicated compounds were verified to be identical, so in each case only a single data entry was retained. The curated subset of the original 5-HT1A dataset used in this work included 130 unique organic compounds including 69 binders and 61 non-binders.
Natural Products Chemical Libraries
TimTec (http://www.timtec.net/) Natural Product Library (NPL) is a chemical library of 720 natural compounds composed of pure natural products as lead identifying materials. It includes primarily known natural compounds that are also available through a number of domestic and international commercial sources. The value of the library design is in the broad diversity of selected natural material available in a screen-ready format. TimTec does not hold any intellectual property rights for compounds in this collection.
TimTec Natural Derivatives Library (NDL) elaborates on structural variety of pure natural compounds and includes synthetic compounds as well as synthetically modified pure natural compounds: alkaloids, natural phenols, nucleoside analogs, carbohydrates, purines, pyrimidines, flavonoids, steroidal compounds and natural amino acids. It is a “natural” extension of the aforementioned NPL, in both design and structural diversity. It should be noted that there is no overlap between NPL and NDL compounds. All NDL compounds comply with screening purity standards and are available as a collection of either 3,040 individual compounds, or smaller subsets.
Selection of Training, Test, and External Validation Sets
As shown in Fig. 1, we followed the rigorous QSAR workflow for model building, validation and screening established earlier29. For this classification QSAR modeling, we have employed five-fold external cross-validation (CV) protocol, i.e. the sample set of 166 compounds was divided randomly into five subsets, with one subset used for external testing and the other four for model training and internal testing. This procedure was repeated five times and a different one-fifth of the dataset was used for external testing each time. The remaining compounds were considered as modeling dataset; they were further partitioned into multiple pairs of chemically diverse and representative training and test sets of different sizes, using the sphere exclusion algorithm adapted to QSAR modeling efforts30,31.
Fig. (1).

The workflow of cheminfomatics models building, validation and virtual screening of natural product-derived hits as applied to the 5-HT1A dataset.
Generation of 2D Molecular Descriptors
The SMILES32 strings of each compound in the 5-HT1A dataset were converted to 2D chemical structures using the MOE package. The Dragon software33 (version 5.5) was used to calculate a wide range of topological indices of molecular structure. These indices include but not limit to the following descriptor types: simple and valence path, cluster, path/cluster and chain molecular connectivity indices, kappa molecular shape indices, topological and electro-topological state indices, differential connectivity indices, graphs’ radius and diameter, Wiener and Platt indices, Shannon and Bonchev-Trinajstić information indices, counts of different vertices, counts of paths and edges between different kinds of vertices33.
Overall, Dragon generated over 2,000 different molecular descriptors. Most of these descriptors characterize chemical structure, but several depend upon the arbitrary numbering of atoms in a molecule and are introduced solely for bookkeeping purposes. In our study, about 880 chemically relevant descriptors were initially calculated and 672 descriptors were eventually employed for this 5-HT1A binder/non-binder dataset after deleting descriptors with zero value or zero variance. All Dragon descriptors were range-scaled prior to distance calculations since the absolute scales for Dragon descriptors can differ by orders of magnitude. Accordingly, our conversion by range-scaling avoided giving descriptors with significantly higher ranges a disproportional weight upon distance calculations in multidimensional Dragon descriptor space.
k-Nearest Neighbor (kNN) Classification Algorithm
In pattern recognition, the kNN classification QSAR method34,35 is a non-parametric method used for classification and regression. In kNN classification, the output is a class membership. An object is classified by a majority vote of its neighbors, with the object being assigned to the class most common among its k nearest neighbors (k is a positive integer, typically small). If k = 1, then the object is simply assigned to the class of that single nearest neighbor. In our cases, the similarity is calculated using only a subset of all descriptors, which is optimized by simulated annealing (SA) technique in order to reach the best Correct Classification Rate (CCR)36:
| (1) |
where and are the number of binders and non-binders in the dataset, and are the number of known binders predicted as binders (i.e. true positives) and the number of non-binders predicted as non-binders (i.e. true negatives). The statistical significance of the predictive models is defined by the leave-one-out cross-validation (LOO CV), i.e. CCRtrain for the training sets and CCRtest for the test sets. Models of high CCRtrain and CCRtest were used as a matter of external validation, i.e. to predict compounds included neither in the training nor in the test set. In theory, any chemicals represented by their corresponding molecular descriptors can be predicted to a class using the kNN classification approach. Formally, a QSAR model can predict the target property for any compound for which chemical descriptors can be calculated. However, if a compound is highly dissimilar from all compounds of the modeling set, reliable prediction of its activity is unlikely to be realized. A concept of similarity threshold (or applicability domain, AD) was developed and used to avoid such an unjustified extrapolation in activity prediction37. In our studies, the AD is defined as the Euclidean distance threshold DT between a query compound and its closest k-nearest neighbors of the training set. It is calculated as follows:
| (2) |
Here, ȳ is the average Euclidean distance between each compound and its k nearest neighbors in the training set which is also optimized in the course of QSAR modeling, σ is its standard deviation of these Euclidean distances, and Z is an arbitrary parameter called the Zcutoff defined by a user. Typically, we set Z to 0.5, which places the boundary for deciding whether a compound is within or outside of the AD at one half of the standard deviation from ȳ. We also define the AD in the entire descriptor space. Thus, if the distance of the external compound from its nearest neighbor in the training set within either the entire descriptor space or the selected descriptor space exceeds these thresholds, the prediction is not made. It is important to note that raising the value of Z would increase the number of molecules in the external set that are by definition inside the AD but could reduce the prediction accuracy because of the inclusion of dissimilar compounds.
Robustness of QSAR Models
The Y-randomization test has been widely used to ensure the robustness of QSAR models38. To establish model robustness, Y-randomization (randomization of the response variable) test should be used. This test consists of repeating all the calculations with scrambled activities of the training set. Ideally, calculations should be repeated at least five (better, more) times. The goal of this procedure is to establish whether models built with real activities of the training set have good statistics not due to overfitting or chance correlation. If predictive power for the training (i.e. LOO CV CCRtrain) or the test set (i.e. CCRtest) of all models built with randomized activities of the training set is significantly lower than that of models built with real activities of the training set, the latter ones are considered reliable. In the end, we applied the one-tail hypothesis to confirm the robustness of our QSAR models.
HTS using Consensus Models
As illustrated in the workflow of Fig. 1, QSAR models that passed both internal and external validation were employed for our HTS purpose39. A global AD defined by the whole body of descriptor types was applied first in order to filter out compounds that are structurally distant from compounds in the modeling set. All 69 known 5-HT1A binders extracted from WOMBAT were used as probes for the AD calculations. Subsequently, the consensus prediction of kNN ensemble models was conducted only to compounds falling into the global AD. The results were deemed to be acceptable only when the compound was found within the AD of more than 50% of all models employed in consensus prediction and the standard deviation of estimated means across all models was small.
All the modeling and virtual screening calculations were carried out at an Intel Xeon X5667 processor-based, 2-socket rack server Dell PowerEdge R410 and multiple Dell Precision M6500N Mobile TAA workstations (Intel i7–840QM Quad Core), at the Laboratory of Cheminfomatics and Drug Design (LCDD) at Howard University, also the Molecular Modeling and Drug Discovery Core for DC D-CFAR. Part of the calculations were conducted at a 352-processor Beowulf Linux cluster of the ITS Research Computing Division of the University of North Carolina at Chapel Hill (UNC-CH). The cluster runs the Red Hat Enterprise Linux 4.0 (32-bit) and the nodes communicate via a Gigabit Ethernet network.
Radioligand Binding Assays
The experimental validation was performed by the NIMH PDSP. Radioligands were purchased by PDSP from Perkin-Elmer or GE Healthcare. Competition binding assays were performed using transfected or stably expressing cell membrane preparations as previously described40,41 and are available online at http://pdsp.med.unc.edu. All experimental details are available at PDSP’s portal site, http://pdsp.med.unc.edu/UNC-CH Protocol Book.pdf.
RESULTS AND DISCUSSION
QSAR Classification Models
The kNN classification method yielded with great success a set of acceptable QSAR models with high accuracy characterized as CCR for both training and test sets. For each of the individual modeling set out of the five-fold splits, we found that there are over seven models of both CCRtrain and CCRtest equal to or higher than 0.80. Notably, most models of CCRtest greater than 0.80 also showed their corresponding CCRtrain greater than 0.80 for the current data sets, though the opposite was not always true. According to our prior workflow, only models with prediction accuracy of both CCRtrain and CCRtest greater than 0.80 are deemed acceptable and selected for consensus prediction. In current cases, their CCRtrain and CCRtest were actually 0.91 and 0.99 respectively, suggesting that those models can distinguish precisely the majority of true-binders from non-binders for both training and test sets. These kinds of remarkably high prediction accuracies as well as the large number of eligible models indicate that our kNN modeling method was capable of telling apart true-binders versus non-binders with great success using Dragon topological descriptors.
QSAR Model Validations
In recent years, we applied Y-randomization and external validation to be critical steps of our whole QSAR/VS workflow in addition to the internal validation of kNN models by test sets. We exploited only those models that had been validated by the prior two steps for external prediction and database mining37. Furthermore, we had another dataset of 105 5-HT1A binders from PDSP used as the independent external validation, to increase the confidence of our QSAR models. In the Y-randomization test conducted, the binary annotations of 5-HT1A binders and non-binders in training sets were randomly shuffled, and kNN classification models were built with the same parameter settings. The test was performed once for each training/test set split and all runs of Y-randomization test showed that almost all models had both CCRtrain and CCRtest less than 0.50. Moreover, the one-tail hypothesis testing was applied and confirmed that the difference of CCRtrain before and after Y-randomization was significant.
In this study, we employed the five-fold CV approach for external prediction, i.e. the 26 compounds are randomly excluded from the original set for each fold and each compound is assigned to the external set for once. Consensus predictions were carried out using those predictive models with CCRtrain and CCRtest greater than 0.80 under different Z value cutoffs (Zcutoff = 0.5 ~ 3.0). Because of the AD inherent to individual kNN QSAR models, the consensus prediction sometimes cannot cover whole compounds in the single fold. Table 1 shows the consensus scores for each of the five-fold external sets. The consensus score (or kNN score), in term of the average value of class number (1 for binders while 2 for non-binders) in classification QSAR, was calculated by the fraction of models that predict a compound as non-binder over the total number of models used for prediction plus 1. Under Zcutoff = 0.5, most external validation sets achieved a rather high prediction accuracy. For the fourth external set split, the validation attained 100% for binders and 92% for non-binders, leading to CCRevs = 0.96. The only falsely predicted binder (average class number > 1.5) was within the AD of small portion of all models, i.e., the model coverage was fairly low and the prediction value is not greater than 1.7. In general, the prediction with such a low coverage is considered to be of low confidence level. The large value of Zcutoff significantly raised the model coverage for binder/non-binder predictions because of the expanded AD for individual models. However, such kind of AD for consensus models also decreases confidence level of external prediction. Generally speaking, in order to have reliable and accurate prediction, the QSAR model has to have broad model coverage and a small Zcutoff value. In summary, only kNN models with CCRtrain, CCRtest and CCRevs equal to or greater than 0.80 were applied to consensus prediction and virtual screening. In addition, the eligible models chosen for the external prediction had relatively small Zcutoff (= 0.5) and relatively broad coverage for compounds in external datasets (>= 50%).
Table 1.
The validation results for the five-fold external sets as well as the additional independent external set from PDSP using the k-Nearest Neighbor method.
|
aExternal Sets |
Prediction CCR |
Confusion Matrix | Statistics | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| N(1)a | N(2)a | TP | TN | FP | FN | SE | SP | EN(1) | EN(2) | ||
| 1 | 0.93 | 15 | 12 | 14 | 11 | 1 | 0 | 0.93 | 0.92 | 1.93 | 1.86 |
| 2 | 0.92 | 13 | 12 | 12 | 11 | 2 | 1 | 0.92 | 0.91 | 1.69 | 1.85 |
| 3 | 0.91 | 15 | 11 | 15 | 9 | 2 | 0 | 1.00 | 0.82 | 1.69 | 2.00 |
| 4 | 0.96 | 13 | 13 | 13 | 12 | 1 | 0 | 1.00 | 0.92 | 1.86 | 2.00 |
| 5 | 0.96 | 13 | 13 | 13 | 12 | 1 | 0 | 1.00 | 0.92 | 1.86 | 2.00 |
| PDSP | 0.89 | 70b | 0 | 62 | N/A | N/A | 8 | 0.89 | N/A | N/A | N/A |
N(1) = number of binders, N(2) = number of non-binders, TP = true positive (binders predicted as binders), FP = false positives (non-binders predicted as binders), FN = false negatives (binders predicted as non-binders), TN = true negative (non-binders predicted as non-binders), SE = sensitivity = TP/N(1), SP = specificity = TN/N(2), EN - the normalized enrichment, EN(1) = (2TP * N(2))/(TP * N(2) + FP * N(1)), EN(2) = (2TN * N(1))/(TN * N(1) + FN * N(2)), and CCR = correct classification rate.
Some N(1) binders from PDSP were out of the AD of the consensus models (Zcutoff = 0.5), thus having no prediction. Only data for compounds found within the AD were used for statistical summaries.
Independent External Validation
We employed models built from 130 5-HT1A binder/non-binder dataset to verify 105 new 5-HT1A binders from PDSP. We should emphasize that these recent binders are unique in structure from existing PDSP binders. Among the 105 binders, 70 compounds were within the AD of consensus kNN models using Zcutoff = 0.5; 62 were accurately annotated by consensus prediction (CCRevs = 0.89, cf. Table 1). Thus, the majority of ligands were predicted correctly by our consensus models. Since the falsely predicted 5-HT1A binders by kNN had the prediction values less than 1.7, and were within the AD of limited number of models, the kNN prediction is considered as of low confidence. The success of this independent external validation suggested that our QSAR models were predictive and robust enough to be applied to further virtual screening.
Here we should emphasize that our model validation is a critically inherent feature of our QSAR modeling workflow. The issue of model validation had been given a lot of attention by the cheminformatic research community in recent years 42. Previously, most QSAR practitioners presumed that internally validated models built from available training set data should be externally predictive. It has been demonstrated, however, that conventional validation techniques including LOO CV or even leave-many-out (LMO) CV applied to the training set, internal validation as applied to the test set, are insufficient to ensure the external predictive power of QSAR models37,43. Thus, we utilized five-fold external CV protocol in this study as well as the Y-randomization test to ensure the robustness and predictive power of kNN models. Needless to say, the use of externally validated models and AD is particularly critical when the models are employed in the practice of virtual screening.
Model-based HTS
Instead of applying only one and the best model to HTS, we followed the consensus prediction approach in our workflow by taking the average of all predictions from qualified models, i.e. seven models of both Internal and External CCRtrain and CCRtest equal to or greater than 0.80. We only kept the set of models from one particular split of the five-fold CV, the forth split in this case, which yields the highest CCRevs. To narrow the hit list and obtain high confidence level for each prediction, we took both the consensus score and model coverage into consideration. In particular, only the screening hits of a consensus score between 1.0 and 1.1 and the model coverage over 50% were chosen. While these hits are predicted to be 5-HT1A binders with high confidence by our consensus kNN models, it would be interesting to test them experimentally in hope of revealing new scaffolds of 5-HT1A binders. For these screening hits chosen by kNN models, five chemicals from NDL library were further selected for experimental validation based on the following criteria: 1) high confidence of consensus prediction by our models; 2) low structural similarity between hits and known 5-HT1A binders; 3) convenient commercial availability.
Experimental Validation
Using the competition binding assays with the radioligand of [3H]-8-OH-DPAT, five hit compounds were tested in vitro for experimental validation. The selection was based on high predicted activity, availability and structural uniqueness. Two tested compounds, CMPD#19546 and CMPD#19548, showed percent of inhibition of 84.3% and 70.9%, respectively (cf. Table 2). The further secondary experiments confirmed that their IC50 are 2.86 µM for CMPD#19546 and 2.99 µM for CMPD#19548, as can be seen from the Fig. (2). We also employed the positive control of WAY100635, and its measured Ki value of 0.75 nM was consistent with the literature data. The comparison between predicted and experimental data is shown in Table 2. Apparently, our kNN prediction of CMPD#19546 is 1.04 ± 0.20 by an ensemble of 201 models (1: Active/2: Inactive), while for CMPD#19548 the prediction is 1.15 ± 0.36 out of an ensemble of 213 models. After searching chemical structure records in Chemical Abstracts Service (CAS) and PubChem databases, we did not find any prior reports of these two natural product-derived compounds’ action on the human 5HT-1A receptor. It is once again proved that by taking advantage of various predictive tools, such as QSAR modeling, more novel compounds can be revealed with unreported biological activities.
Table 2.
The prediction scores by kNN consensus models and experimental data for five hits identified by virtual screening as putative 5-HT1A binders.
| Structure | No. | CMPD | Source library |
Library ID |
kNN score |
SD | Percent of inhibition |
Exp. IC50 (µM) |
|---|---|---|---|---|---|---|---|---|
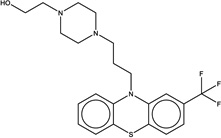
 |
1 | 19546 | NDL | ST076806 | 1.04 | 0.20 | 84.3 | 2.86 |
 |
2 | 19547 | NDL | ST026798 | 1.12 | 0.32 | −6.7 | ND |
 |
3 | 19548 | NDL | ST074727 | 1.15 | 0.36 | 70.9 | 2.99 |
 |
4 | 19549 | NDL | ST049891 | 1.16 | 0.36 | 30.4 | ND |
 |
5 | 19550 | NDL | ST016609 | 1.16 | 0.36 | 32.1 | ND |
The full IC50 curve was generated in further experiment and the Ki value was determined.
Fig. (2).

The full dose response curve for NDL hit compounds ST076806 (CMPD#19546; arrow-down triangles in Upper Panel; Ki = 2,860 nM), ST074727 (CMPD#19548; arrow-up triangles in Lower Panel; Ki = 2,999 nM), and the positive control, WAY100635 (solid squares in both Panels, Ki = 0.75 nM) measured by human 5-HT1A receptor radioligand binding assay. The radioligand is [3H]-8-OH-DPAT at the concentration of 0.5 nM with the standard binding buffer.
CONCLUSIONS
The work reported above shows that our binary kNN classification models built with Dragon topological descriptors can characterize true 5-HT1A binders from non-binders with high accuracy. We applied our prior QSAR modeling workflow to these well-curated data sets and the models were validated in a rigorous manner, internally using multiple divisions of training/test set as well as Y-randomization, and externally using five-fold external CV sets. We have demonstrated that this strategy afforded a set of eligible QSAR models with supreme internal and external predictive power. Those qualified predictors (models) were further validated independently on a new set of PDSP binders, as part of our QSAR modeling workflow. It should be noted that our validation outcomes were highly consistent with the experimental activities of these compounds as 5-HT1A binders. In addition, we exploited these models in a highly conservative manner, i.e. in a consensus fashion and the strictest AD criteria, to screening two major screening libraries of natural products, i.e. NPL and NDL. Five of the screening hits were tested experimentally and two showed strong inhibition activities (>50%) at the single concentration of 10.0 µM. Further secondary assays confirmed that their binding constants of Ki are 2.86 µM and 2.99 µM, respectively. In summary, our current outcome indicates that when a sufficient amount of information on true binders versus non-binder is available, the QSAR modeling techniques can be favorably applied not only to discriminating binders versus non-binders but most importantly, to profiling natural products-derived chemicals with unreported biomedical activities.
ACKNOWLEDGEMENTS
The human 5-HT1A receptor radioligand binding assay and the Ki determinations were generously provided by the National Institute of Mental Health's Psychoactive Drug Screening Program (NIMH PDSP), Contract #HHSN-271-2008-00025-C. The NIMH PDSP is directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscol at NIMH, Bethesda MD, USA. This work was supported in part by District of Columbia Developmental Center for AIDS Research (P30AI087714), National Institutes of Health Administrative Supplements for U.S.-China Biomedical Collaborative Research (5P30A10877714-02), the National Institute on Minority Health and Health Disparities of the National Institutes of Health under Award Number G12MD007597. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
List of Abbreviations
- 5-HT1A
5-Hydroxy Tryptamine receptor subtype 1A
- AD
Applicability Domain
- CCR
Correct Classification Rate
- CCRtrain
Correct Classification Rate for training set
- CCRtest
Correct Classification Rate for test set
- CCRevs
Correct Classification Rate for external validation set
- CNS
central nervous system
- CV
Cross-Validation
- FN
False Negative
- FP
False Positive
- GPCRs
G-protein Coupled Receptors
- HTS
High Throughput Screening
- kNN
k Nearest Neighbor
- LOO
Leave-One-Out
- LMO
Leave-Many-Out
- LOO CV
Leave-One-Out Cross-Validation
- MOE
Molecular Operating Environment
- NIMH
National Institute of Mental Health
- PDSP
Psychoactive Drug Screening Program
- QSAR
Quantitative Structure-Activity Relationship
- SA
Simulated Annealing
- SAR
Structure-Activity Relationship
- SE
Sensitivity
- SP
Specificity
- SSRIs
Selective Serotonin Reuptake Inhibitors
- TN
True Negative
- TP
True Positive
- WDI
World Drug Index
- WOMBAT
World of Molecular Bioactivity
Footnotes
CONFLICT OF INTEREST
The authors confirm that they do not have any conflicts of interest.
REFERENCES
- 1.Gilliam TC, Freimer NB, Kaufmann CA, Powchik PP, Bassett AS, Bengtsson U, Wasmuth JJ. Deletion mapping of DNA markers to a region of chromosome 5 that cosegregates with schizophrenia. Genomics. 1989;5(4):940–944. doi: 10.1016/0888-7543(89)90138-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Ito H, Halldin C, Farde L. Localization of 5-HT1A receptors in the living human brain using [carbonyl-11C]WAY-100635: PET with anatomic standardization technique. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 1999;40(1):102–109. [PubMed] [Google Scholar]; (b) de Almeida J, Mengod G. Serotonin 1A receptors in human and monkey prefrontal cortex are mainly expressed in pyramidal neurons and in a GABAergic interneuron subpopulation: implications for schizophrenia and its treatment. Journal of neurochemistry. 2008;107(2):488–496. doi: 10.1111/j.1471-4159.2008.05649.x. [DOI] [PubMed] [Google Scholar]
- 3.(a) Ramage AG. The mechanism of the sympathoinhibitory action of urapidil: role of 5-HT1A receptors. British journal of pharmacology. 1991;102(4):998–1002. doi: 10.1111/j.1476-5381.1991.tb12290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kolassa N, Beller KD, Sanders KH. Involvement of brain 5-HT1A receptors in the hypotensive response to urapidil. The American journal of cardiology. 1989;64(7):7D–10D. doi: 10.1016/0002-9149(89)90688-7. [DOI] [PubMed] [Google Scholar]; (c) Dabire H. Central 5-hydroxytryptamine (5-HT) receptors in blood pressure regulation. Therapie. 1991;46(6):421–429. [PubMed] [Google Scholar]
- 4.Schechter LE, Dawson LA, Harder JA. The potential utility of 5-HT1A receptor antagonists in the treatment of cognitive dysfunction associated with Alzheimer s disease. Curr. Pharm. Des. 2002;8(2):139–145. doi: 10.2174/1381612023396483. [DOI] [PubMed] [Google Scholar]
- 5.Stark AD, Jordan S, Allers KA, Bertekap RL, Chen R, Mistry Kannan T, Molski TF, Yocca FD, Sharp T, Kikuchi T, Burris KD. Interaction of the novel antipsychotic aripiprazole with 5-HT1A and 5-HT 2A receptors: functional receptor-binding and in vivo electrophysiological studies. Psychopharmacology. 2007;190(3):373–382. doi: 10.1007/s00213-006-0621-y. [DOI] [PubMed] [Google Scholar]
- 6.Rollema H, Lu Y, Schmidt AW, Zorn SH. Clozapine increases dopamine release in prefrontal cortex by 5-HT1A receptor activation. European journal of pharmacology. 1997;338(2):R3–R5. doi: 10.1016/s0014-2999(97)81951-6. [DOI] [PubMed] [Google Scholar]
- 7.Rollema H, Lu Y, Schmidt AW, Sprouse JS, Zorn SH. 5-HT(1A) receptor activation contributes to ziprasidone-induced dopamine release in the rat prefrontal cortex. Biological psychiatry. 2000;48(3):229–237. doi: 10.1016/s0006-3223(00)00850-7. [DOI] [PubMed] [Google Scholar]
- 8.Wheeler Vega JA, Mortimer AM, Tyson PJ. Conventional antipsychotic prescription in unipolar depression, I: an audit and recommendations for practice. J Clin Psychiatry. 2003;64(5):568–574. doi: 10.4088/jcp.v64n0512. [DOI] [PubMed] [Google Scholar]
- 9.Cohn JB, Rickels K. A pooled, double-blind comparison of the effects of buspirone, diazepam and placebo in women with chronic anxiety. Curr Med Res Opin. 1989;11(5):304–320. doi: 10.1185/03007998909115213. [DOI] [PubMed] [Google Scholar]
- 10.Cryan JF, Redmond AM, Kelly JP, Leonard BE. The effects of the 5-HT1A agonist flesinoxan, in three paradigms for assessing antidepressant potential in the rat. European neuropsychopharmacology : the journal of the European College of Neuropsychopharmacology. 1997;7(2):109–114. doi: 10.1016/s0924-977x(96)00391-4. [DOI] [PubMed] [Google Scholar]
- 11.Hadrava V, Blier P, Dennis T, Ortemann C, de Montigny C. Characterization of 5-hydroxytryptamine1A properties of flesinoxan: in vivo electrophysiology and hypothermia study. Neuropharmacology. 1995;34(10):1311–1326. doi: 10.1016/0028-3908(95)00098-q. [DOI] [PubMed] [Google Scholar]
- 12.(a) Schoeffter P, Hoyer D. Centrally acting hypotensive agents with affinity for 5-HT1A binding sites inhibit forskolin-stimulated adenylate cyclase activity in calf hippocampus. British journal of pharmacology. 1988;95(3):975–985. doi: 10.1111/j.1476-5381.1988.tb11728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pitchot W, Wauthy J, Legros JJ, Ansseau M. Hormonal and temperature responses to flesinoxan in normal volunteers: an antagonist study. European neuropsychopharmacology : the journal of the European College of Neuropsychopharmacology. 2004;14(2):151–155. doi: 10.1016/S0924-977X(03)00108-1. [DOI] [PubMed] [Google Scholar]
- 13.D'Aquila P, Monleon S, Borsini F, Brain P, Willner P. Anti-anhedonic actions of the novel serotonergic agent flibanserin, a potential rapidly-acting antidepressant. European journal of pharmacology. 1997;340(2–3):121–132. doi: 10.1016/s0014-2999(97)01412-x. [DOI] [PubMed] [Google Scholar]
- 14.Borsini F, Evans K, Jason K, Rohde F, Alexander B, Pollentier S. Pharmacology of flibanserin. CNS drug reviews. 2002;8(2):117–142. doi: 10.1111/j.1527-3458.2002.tb00219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rickels K, Mathew S, Banov MD, Zimbroff DL, Oshana S, Parsons EC, Jr, Donahue SR, Kauffman M, Iyer GR, Reinhard JF., Jr Effects of PRX-00023, a novel, selective serotonin 1A receptor agonist on measures of anxiety and depression in generalized anxiety disorder: results of a double-blind, placebo-controlled trial. Journal of clinical psychopharmacology. 2008;28(2):235–239. doi: 10.1097/JCP.0b013e31816774de. [DOI] [PubMed] [Google Scholar]
- 16.de Paulis T. Drug evaluation: PRX-00023, a selective 5-HT1A receptor agonist for depression. Current opinion in investigational drugs. 2007;8(1):78–86. [PubMed] [Google Scholar]
- 17.Li Z, Ichikawa J, Dai J, Meltzer HY. Aripiprazole, a novel antipsychotic drug, preferentially increases dopamine release in the prefrontal cortex and hippocampus in rat brain. Eur. J. Pharmacol. 2004;493(1–3):75–83. doi: 10.1016/j.ejphar.2004.04.028. [DOI] [PubMed] [Google Scholar]
- 18.Bantick RA, De Vries MH, Grasby PM. The effect of a 5-HT1A receptor agonist on striatal dopamine release. Synapse. 2005;57(2):67–75. doi: 10.1002/syn.20156. [DOI] [PubMed] [Google Scholar]
- 19.Schechter LE, Smith DL, Rosenzweig-Lipson S, Sukoff SJ, Dawson LA, Marquis K, Jones D, Piesla M, Andree T, Nawoschik S, Harder JA, Womack MD, Buccafusco J, Terry AV, Hoebel B, Rada P, Kelly M, Abou-Gharbia M, Barrett JE, Childers W. Lecozotan (SRA-333): a selective serotonin 1A receptor antagonist that enhances the stimulated release of glutamate and acetylcholine in the hippocampus and possesses cognitive-enhancing properties. The Journal of pharmacology and experimental therapeutics. 2005;314(3):1274–1289. doi: 10.1124/jpet.105.086363. [DOI] [PubMed] [Google Scholar]
- 20.Wang JX, Dipasquale AJ, Bray AM, Maeji NJ, Spellmeyer DC, Geysen HM. Systematic study of substance P analogs. II. Rapid screening of 512 substance P stereoisomers for binding to NK1 receptor. Int J Pept Protein Res. 1993;42(4):392–399. [PubMed] [Google Scholar]
- 21.Hsieh JH, Wang XS, Teotico D, Golbraikh A, Tropsha A. Differentiation of AmpC beta-lactamase binders vs. decoys using classification kNN QSAR modeling and application of the QSAR classifier to virtual screening. J. Comput. Aided Mol. Des. 2008;22(9):593–609. doi: 10.1007/s10822-008-9199-2. [DOI] [PubMed] [Google Scholar]
- 22.Peterson YK, Wang XS, Casey PJ, Tropsha A. Discovery of geranylgeranyltransferase-I inhibitors with novel scaffolds by the means of quantitative structure-activity relationship modeling, virtual screening, and experimental validation. Journal of medicinal chemistry. 2009;52(14):4210–4220. doi: 10.1021/jm8013772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang H, Wang XS, Huang XP, Roth BL, Butler KV, Kozikowski AP, Jung M, Tropsha A. Novel inhibitors of human histone deacetylase (HDAC) identified by QSAR modeling of known inhibitors, virtual screening, and experimental validation. Journal of chemical information and modeling. 2009;49(2):461–476. doi: 10.1021/ci800366f. [DOI] [PubMed] [Google Scholar]
- 24.Hoffman B, Cho SJ, Zheng W, Wyrick S, Nichols DE, Mailman RB, Tropsha A. Quantitative structure-activity relationship modeling of dopamine D(1) antagonists using comparative molecular field analysis, genetic algorithms-partial least-squares, and K nearest neighbor methods. J. Med. Chem. 1999;42(17):3217–3226. doi: 10.1021/jm980415j. [DOI] [PubMed] [Google Scholar]
- 25.Tropsha A, Zheng W. Identification of the descriptor pharmacophores using variable selection QSAR: applications to database mining. Curr. Pharm. Des. 2001;7(7):599–612. doi: 10.2174/1381612013397834. [DOI] [PubMed] [Google Scholar]
- 26.Fourches D, Muratov E, Tropsha A. Trust, but verify: on the importance of chemical structure curation in cheminformatics and QSAR modeling research. J.Chem.Inf.Model. 2010;50(7):1189–1204. doi: 10.1021/ci100176x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Molecular Operating Environment (MOE) Chemical Computing Group Inc.,1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7. 2012 [Online] [Google Scholar]
- 28.Chem Axon. J Chem. 2010 [Google Scholar]
- 29.Hajjo R, Grulke CM, Golbraikh A, Setola V, Huang XP, Roth BL, Tropsha A. Development, validation, and use of quantitative structure-activity relationship models of 5-hydroxytryptamine (2B) receptor ligands to identify novel receptor binders and putative valvulopathic compounds among common drugs. J. Med. Chem. 2010;53(21):7573–7586. doi: 10.1021/jm100600y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Golbraikh A, Shen M, Xiao Z, Xiao YD, Lee KH, Tropsha A. Rational selection of training and test sets for the development of validated QSAR models. J. Comput. Aided Mol. Des. 2003;17(2–4):241–253. doi: 10.1023/a:1025386326946. [DOI] [PubMed] [Google Scholar]
- 31.Golbraikh A, Tropsha A. Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. Mol. Divers. 2002;5(4):231–243. doi: 10.1023/a:1021372108686. [DOI] [PubMed] [Google Scholar]
- 32.D W. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. Journal of Chemical Information and Computer Sciences. 1988;28(1):31–36. [Google Scholar]
- 33.Talete. DRAGON for Windows and Linux. 2007 (Available at http://wwwtaletemiit/help/dragon_help) [Google Scholar]
- 34.Itskowitz P, Tropsha A. kappa Nearest neighbors QSAR modeling as a variational problem: theory and applications. J.Chem.Inf.Model. 2005;45(3):777–785. doi: 10.1021/ci049628+. [DOI] [PubMed] [Google Scholar]
- 35.Zheng W, Tropsha A. Novel variable selection quantitative structure--property relationship approach based on the k-nearest-neighbor principle. J. Chem. Inf. Comput. Sci. 2000;40(1):185–194. doi: 10.1021/ci980033m. [DOI] [PubMed] [Google Scholar]
- 36.de Cerqueira LP, Golbraikh A, Oloff S, Xiao Y, Tropsha A. Combinatorial QSAR modeling of P-glycoprotein substrates. J. Chem. Inf. Model. 2006;46(3):1245–1254. doi: 10.1021/ci0504317. [DOI] [PubMed] [Google Scholar]
- 37.Tropsha A, Gramatica P, Gombar VK. The importance of being earnest: Validation is the absolute essential for successful application and interpretation of QSPR models. Qsar & Combinatorial Science. 2003;22(1):69–77. [Google Scholar]
- 38.Wold S, Eriksson L. Statistical Validation of QSAR Results. In: Waterbeemd Hvd., editor. Chemometrics Methods in Molecular Design (Methods and Principles in Medicinal Chemistry, Vol 2) Weinheim (Germany): Wiley-VCH Verlag GmbH; 1995. pp. 309–318. [Google Scholar]
- 39.Wang XS, Tang H, Golbraikh A, Tropsha A. Combinatorial QSAR modeling of specificity and subtype selectivity of ligands binding to serotonin receptors 5HT1E and 5HT1F. Journal of chemical information and modeling. 2008;48(5):997–1013. doi: 10.1021/ci700404c. [DOI] [PubMed] [Google Scholar]
- 40.Shapiro DA, Renock S, Arrington E, Chiodo LA, Liu LX, Sibley DR, Roth BL, Mailman R. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2003;28(8):1400–1411. doi: 10.1038/sj.npp.1300203. [DOI] [PubMed] [Google Scholar]
- 41.Butterweck V, Nahrstedt A, Evans J, Hufeisen S, Rauser L, Savage J, Popadak B, Ernsberger P, Roth BL. In vitro receptor screening of pure constituents of St. John's wort reveals novel interactions with a number of GPCRs. Psychopharmacology. 2002;162(2):193–202. doi: 10.1007/s00213-002-1073-7. [DOI] [PubMed] [Google Scholar]
- 42.Jorgensen WL, Tirado-Rives J. QSAR/QSPR and Proprietary Data. J Chem. Inf. Model. 2006;46:937–937. [Google Scholar]
- 43.Golbraikh A, Tropsha A. Beware of q(2)! Journal of Molecular Graphics & Modelling. 2002;20(4):269–276. doi: 10.1016/s1093-3263(01)00123-1. [DOI] [PubMed] [Google Scholar]