

Abstract
Mycobactins are small-molecule iron chelators (siderophores) produced by Mycobacterium tuberculosis (Mtb) for iron mobilization. The bifunctional salicylate synthase MbtI catalyzes the first step of mycobactin biosynthesis through the conversion of the primary metabolite chorismate into salicylic acid via isochorismate. We report the design, synthesis and biochemical evaluation of an inhibitor based on the putative transition-state (TS) for the isochorismatase partial reaction of MbtI. The inhibitor mimics the hypothesized charge build-up at C-4 of chorismate in the TS as well as C-O bond-formation at C-6. Another important design element of the inhibitor is replacement of the labile pyruvate side-chain in chorismate with a stable C-linked propionate isostere. We developed a stereocontrolled synthesis of the highly functionalized cyclohexene inhibitor that features an asymmetric aldol reaction using a titanium enolate, diastereoselective Grignard addition to a tert-butanesulfinyl aldimine, and ring closing olefin metathesis as key steps.
INTRODUCTION
Tuberculosis (TB), the leading cause of infectious disease mortality after HIV-AIDS, is due to members of the Mycobacterium tuberculosis (Mtb) complex that includes seven closely related species.1 The emergence of multi-drug and extensively-drug resistant strains of Mtb has renewed focus on the development of anti-tubercular agents with novel modes of action.2 With the exception of pyrazinamide, whose mechanism remains an enigma,3 the antibiotics presently used in TB chemotherapy target a limited set of biochemical pathways in macromolecular and cofactor biosynthesis.4 Iron is a required cofactor for more than 40 enzymes in Mtb, but is highly restricted in a vertebrate host where the concentration of free iron (Fe2+) is estimated as 10−24 M.5 In order to obtain iron Mtb deploys two complementary strategies: synthesis of the mycobactin siderophores,6 which are small-molecule iron chelators that scavenge iron from host tissues and uptake of heme through a specialized heme receptor followed by heme degradation to release the iron.7 The relative contribution of each mechanism for iron mobilization in vivo is unknown and both may play a role during different stages of infection. Mtb mutants unable to produce mycobactins cannot replicate under iron-restricted conditions and are cleared in vivo, thereby confirming the importance of mycobactin-mediated iron acquisition in Mtb.8
The bifunctional salicylate synthase MbtI is responsible for the first committed step in mycobactin biosynthesis through the conversion of chorismate into salicylic acid, which is accomplished in two partial reactions at the same active site.9 In the first reaction, MbtI catalyzes the interconversion of chorismate to isochorismate that has been hypothesized to proceed through a concerted SN2″ reaction mechanism via transition state TS1 (Figure 1).9c, 10 The isochorismatase activity of MbtI requires Lys205, which nucleophilically activates a water molecule for attack on chorismate at C-6 and Glu252 that polarizes the C-4 hydroxyl-leaving group. In the second reaction, pyruvate is eliminated through an intramolecular [3,3]-sigmatropic rearrangement, formerly a retro-Ene reaction, to afford salicylic acid via bicyclic transition state TS2 (Figure 1).11
Figure 1.

Conversion of chorismate to salicylate via isochorismate catalyzed by MbtI.
MbtI represents an appealing target for development of inhibitors of mycobactin biosynthesis since it is structurally and biochemically characterized, has no human orthologs, and is conditionally essential under iron-deficient conditions. Following the foundational studies of Abell et al. on inhibitors of chorismate-utilizing enzymes,12a–e Payne and coworkers were the first to disclose inhibitors of MbtI and their most potent compound is the rationally designed substrate mimic 1 (Figure 2A) containing a 2,3-dihydroxybenzoate scaffold.12f–g This compound is a competitive inhibitor and possesses an inhibition constant (Ki) of approximately 10 μM. In a complementary approach employing high-throughput screening, Vasan and co-workers reported benzimidazole-2-thione 2 as a noncompetitive inhibitor with similar potency to 1 (Figure 2A).13 Based on the modest potency of the described MbtI inhibitors, we chose to investigate potential transition-state (TS) inhibitors of MbtI. TS inhibitors exploit the affinity of an enzyme for the TS relative to the Michaelis complex.14 Capturing even a small percentage of the TS binding energy with an inhibitor, in theory, allows the design of exceptionally potent inhibitors. TS strategies are less successful when the non-catalyzed rate is high since the amount of potential TS stabilization will be smaller. In the case of MbtI, the first transition state TS1 is more desirable to mimic than the second bicyclic transition state TS2 due to ease of the non-catalyzed sigmatropic rearrangment.11, 15
Figure 2.

(A) Reported MbtI inhibitors; (B) Rationale for TS inhibitor design of 4; (C) Oseltamivir (Tamiflu) a neuraminidase TS inhibitor. Ki values are with respect to MbtI.
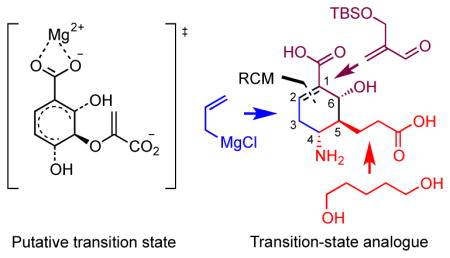
Inspired by the pioneering work of Kozlowski, Bartlett et al., who reported on the synthesis of 3 as a TS inhibitor for several chorismate-utilizing enzymes,16 we report the synthesis and characterization of cyclohexene 4 that mimics putative TS1 through incorporation of an amino group at C-4 to resemble the charged leaving group of TS1 (Figure 2B). Another key design element is replacement of the sigmatropically labile pyruvate side-chain at C-5 with a stable Clinked propionate fragment because MbtI possesses pyruvate lyase activity, unlike the monofunctional chorismate-utilizing enzymes investigated by Kozlowski et al.. Interestingly, inhibitor 4 bears a striking resemblance to Oseltamivir (trade name Tamiflu) 5 (Figure 2C), an antiviral drug developed at Gilead that is a TS inhibitor of viral neuraminidase.17
RESULTS AND DISCUSSION
Retrosynthetic Plan
The challenge of the target molecule 4 resides in the construction of three contiguous stereocenters in the highly functionalized cyclohexene scaffold. Our first retrosynthetic plan involved disconnection of the C-5 propionate side-chain to thionocarbonate 6 through a radical coupling with tert-butyl acrylate. This would allow us to dovetail into the elegant racemic synthesis of 7 reported by Kozlowski and co-workers (Figure 3A).16b We also noted the structural similarity between 4 and Oseltamivir 5 (Figure 2B and 2C), and were inspired by the efficient ring-closing metathesis (RCM) reaction reported by Yao and coworkers18 for construction of the cyclohexene core of Oseltamivir 5. Accordingly, as shown in Figure 3B, RCM disconnection of 8 leads to acyclic diene 9 that can be further simplified to syn aldol adduct 10 via an asymmetric Grignard addition employing Ellman’s N-sulfinamide auxiliary.19 Intermediate 10 can be readily assembled through syn-aldol reaction between oxazolidinone 11 and aldehyde 12. An advantage of this second approach is the ability to control stereochemistry on an acyclic precursor using contemporary methods in asymmetric synthesis.
Figure 3.

Retrosynthetic analysis.
Radical coupling strategy
Silyloxybutadiene 13 was prepared from crotonaldehyde in 78% yield through modification of the reported procedure16b using TBSOTf. The key Diels-Alder reaction between 13 and ethyl propiolate afforded cyclohexadiene in variable yields from 0–61% (from more than a dozen attempts) with the aromatized benzoate derivative as a major byproduct even though the reaction was rigorously degassed and antioxidants were employed. Notwithstanding the inconsistent yields several grams of 14 were secured and elaborated to cyclohexene derivative (±)-7 as described16b in four steps with an overall yield of 24% (average of 70% per step). Conversion to the thionocarbonate (±)-6 mediated by N-methylimidazole (NMI) proceeded in 90% yield at room temperature, whereas 4-dimethylaminopyridine (DMAP), a more conventional acylation catalyst resulted in only low conversion (<10%).20 Several conditions were explored to effect a radical coupling with tert-butyl acrylate and other radical traps21; however, despite considerable effort, only the deoxygenated product (±)-16 was isolated in yields up to 63% yield (84% brsm) likely due to the sterically demanding environment at C-5.
RCM approach
As a result of the capricious yield of the cyclohexadiene 14 coupled with inability to advance intermediate 15, we next focused attention on the complementary RCM route as described below since the propionate side-chain is installed early in the synthesis. Synthesis began with acylation of Evans’ (R)-phenylalanine-derived oxazolidinone with the known carboxylic acid 1722 under mixed anhydride conditions to afford N-acyloxazolidinone 18 in 93% yield. The required aldehyde 19 coupling partner was prepared with a single purification step through Baylis-Hillman reaction23 of tert-butyl acrylate and formaldehyde in aqueous tetrahydrofuran (THF) followed by tert-butyldimethylsilyl (TBS) protection and diisobutylaluminum hydride (DIBAL-H) reduction (Scheme 2, bottom insert). Initially, asymmetric boron-mediated aldol reaction24 of 18 with aldehyde 19 using dibutylboron triflate as a Lewis acid in Et2O provided the syn aldol adduct 20 in high diastereoselectivity (dr > 20:1) but only 36% yield. As an alternative, the titanium enolate of 18 was investigated according to methodology developed by Crimmins and coworkers.25 Treatment of 18 with TiCl4 and 2.5 equiv of (−)-sparteine in CH2Cl2 at −78 °C followed by the addition of aldehyde 19 afforded 20 (dr > 20:1) in an improved 48% yield. Protection as a methoxymethyl (MOM) ether employing CH3OCH2Cl and catalytic tetrabutylammonium iodide (TBAI) furnished 21. The chiral auxiliary was then reductively removed with sodium borohydride26 and Dess-Martin oxidation27 of the resultant alcohol 22 provided aldehyde 23. Condensation with Ellman’s (S)-(−)-tert-butanesulfinamide auxiliary yielded tert-butanesulfinyl aldimine 24. As first reported by Xu and co-workers the combination of pyridinium p-toluenesulfonate (PPTS) and anhydrous CuSO4 dramatically promoted imine formation, which was necessary for the sterically congested aldehyde 23.28 The stereocontrolled installation of the allyl group in 25 was achieved in 87% yield and excellent diastereoselectivity (dr > 20:1) by the addition of allyl magnesium bromide to 24 in CH2Cl2 at −78 °C.19, 29 Ring-closing metathesis of 25 employing the Grubbs second-generation catalyst30 gave cyclohexene derivative 26 in 95% yield.
Scheme 2. Synthesis of cyclohexene 26a.

aReagents and conditions: (a) PivCl, Et3N, THF, 0 °C, 6 h; (b) (R)-4-(benzyl)oxazolidin-2-one, n-BuLi, -78 °C, 5 min, then added to the mixed anhydride, −78 °C for 30 min, 0 °C for 30 min; (c) TiCl4, (−)-sparteine, CH2Cl2, −78 °C, 1.5 h, then 19 was added, −40 °C for 3 h, −20 °C overnight; (d) CH3OCH2Cl, i-Pr2NEt, TBAI, toluene, 90 °C, 7 h; (e) NaBH4, THF/H2O (3:1), 0 °C to rt, overnight; (f) Dess-Martin periodinane, NaHCO3, CH2Cl2, rt, 30 min; (g) (S)-(−)-2-methyl-2-propanesulfinamide, anhydrous CuSO4, PPTS, CH2Cl2, rt, 24 h; (h) allylmagnesium bromide, CH2Cl2, −20 °C, 2.5 h; (i) 14 mol% Grubbs second-generation catalyst, CH2Cl2, reflux, 14 h, then DMSO was added, rt, 16 h; (j) HCHO, DABCO, THF–H2O (1:1), reflux, 16 h; (k) TBSCl, imidazole, CH2Cl2, rt, 16 h; (l) DIBAL-H, CH2Cl2, −78 °C, 1 h.
The remaining synthetic operations involve oxidation of the primary alcohols at C-9 and C-10, removal of the tert-butanesulfinamide auxiliary, and deprotection of the MOM group at C-6 of cyclohexene 26. We initially sought to adjust the oxidation level at C-9 and C-10 and maintain the tert-butanesulfinamide as a nitrogen protecting group. However we discovered the tert-butanesulfinamide moiety is readily oxidized under mild oxidation conditions to a sulfonamide,19 which undergoes facile elimination due to its axial conformation combined with the enhanced acidity of the vicinal allylic proton. Thus, the sulfinamide auxiliary was first removed along with the TBS group by treatment with 4 M HCl in dioxane–MeOH. The resultant amino alcohol was Boc-protected to afford 27 in 89% yield over two steps. Subsequent PMB deprotection with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) gave diol 28. Direct oxidation to the dicarboxylic acid 29 was very low yielding, so we employed a two-stage procedure with tetrapropylammonium perruthenate (TPAP)31 to provide an intermediate dialdehyde that was converted to dicarboxylic acid 29 through Pinnick-Lindgren oxidation.32 Deprotection of 29 with trimethylsilyl bromide33 in CH2Cl2 at −78 °C afforded enantiopure target compound 4 as the acetate salt that was purified by silica gel chromatography employing a quaternary solvent system consisting of CHCl3–MeOH–H2O–AcOH.
The relative configurations of three contiguous stereocenters in 5 were confirmed by the 2D NOESY studies of cyclohexene 28. As shown in Figure 4, NOE correlation of H-4 with H-7 and H-8 indicates an anti relationship between H-4 and H-5. Due to NOE correlation of H-6 with H-7 and H-8 as well as the correlation of H-5 with the CH2 of the MOM group at C-6, we conclude that H-5 and H-6 also have an anti relationship.
Figure 4.
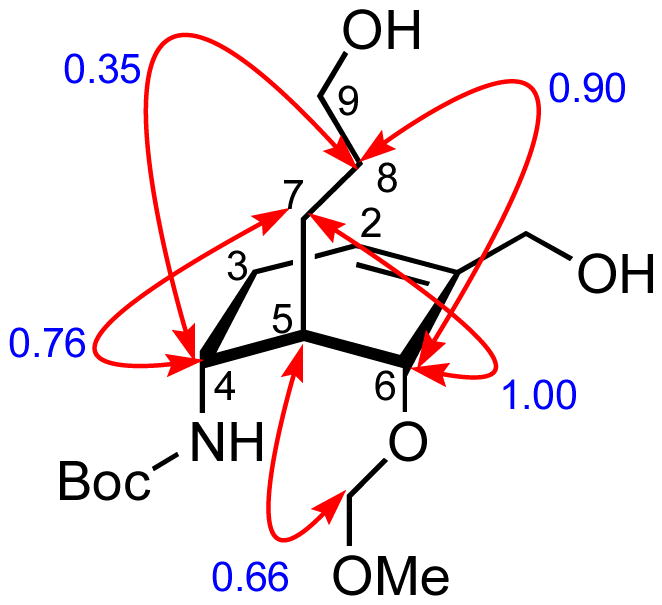
Key NOE correlations of 28. The integral of the H-6/H-9 cross-peak was used as the internal calibrant (its integral was set to 1.00).
Biological Evaluation
The putative transition-state inhibitor 4 was evaluated for enzyme inhibition against recombinant MbtI under initial-velocity conditions as described,13 but showed less than 10% inhibition at 100 μM. The modest potency of 4 clearly indicates it is a poor TS mimic. To rationalize the observed activity, we docked 4 into MbtI using the recently reported co-crystal structure of MbtI with a chorismate analog.34 Introduction of a CH2 moiety in place of the C-5 oxygen atom of chorismate, led to loss of key hydrogen bond with Arg405 while the protonated C-4 amino group made a potentially repulsive electrostatic interaction with Arg405 (Figure S1).
CONCLUSION
We designed and synthesized an inhibitor based on the hypothetical transition state of the isochorismate partial reaction catalyzed by MbtI wherein the C-4 hydroxyl group of chorismate is protonated by Glu252 resulting in bond cleavage and concomitant C-O bond formation at C-6 due to nucleophilic activation of a water molecule by Lys205. MbtI is a bifunctional enzyme and also catalyzes pyruvate elimination via an intramolecular [3,3]-sigmatropic reaction. In order to prevent this potential reaction from occurring in our inhibitor, the pyruvate side-chain was replaced with a stable propionate isostere. Two complementary synthetic routes were explored to the target inhibitor 4. The initial route capitalized on the beautiful chemistry developed by Bartlett and Kozlowski for the preparation of a cyclohexene intermediate (±)-7. Frustrated by our inability to install the propionate side-chain through a radical-mediated process and the fickle yield in the key Diels-Alder reaction, we undertook a novel synthetic route to enantiopure 4. This second approach featured an asymmetric aldol reaction of a titanium enolate, a diastereoselective Grignard addition to a tert-butylsulfinyl aldimine, and ring closing olefin metathesis as key steps. Enzyme inhibition studies revealed 4 is a poor TS mimic and potentially suggests an alternate TS that does not involve charge build-up at C-4 and/or a more prominent role of the C-5 oxygen atom in chorismate for binding. These studies provide a synthetic and mechanistic foundation for future efforts to develop TS-based inhibitors of MbtI.
EXPERIMENTAL SECTION
General Method
All reactions were carried out under a dry Ar atmosphere using oven-dried glassware and magnetic stirring. The solvents were dried before use as follows: THF and Et2O were heated at reflux over sodium benzophenone ketyl; toluene was heated at reflux over sodium; CH2Cl2 was dried over CaH2. Anhydrous N,N-diisopropylethylamine, triethylamine were used directly as purchased. Commercially available reagents were used without further purification unless otherwise noted. Aluminum TLC sheets (silica gel 60 F254) of 0.2-mm thickness were used to monitor the reactions. The spots were visualized with short wavelength UV light or by charring after spraying with a solution prepared from one of the following solutions: phosphomolybdic acid (5.0 g) in 95% EtOH (100 mL); p-anisaldehyde solution (2.5 mL of p-anisaldehyde, 2 mL of AcOH, and 3.5 mL of conc. H2SO4 in 100 mL of 95% EtOH); or ninhydrin solution (0.3g ninhydrin in 100 ml of n-butanol; add 3 ml AcOH). Flash chromatography was carried out with silica gel 60 (230–400 ASTM mesh). NMR spectra were obtained on a 400-MHz or 600-MHz spectrometer. Proton chemical data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad), coupling constant, and integration. Chemical shifts were referenced on residual solvent peaks: CDCl3 (δ = 7.26 ppm for 1H NMR and 77.00 ppm for 13C NMR), CD2Cl2 (δ = 5.32 ppm for 1H NMR and 53.84 ppm for 13C NMR), CD3OD (δ = 3.31 ppm for 1H NMR and 49.00 ppm for 13C NMR), D2O (δ = 4.79 ppm for 1H NMR). Optical rotations were measured at rt in a 1.0-dm cell. High-resolution mass spectrometry was performed on an linear trap quadrupole (LTQ) mass spectrometer with electrospray ionization and a resolution of 60,000 (at m/z=400).
(E)-1-(tert-Butyldimethylsilyloxy)-1,3-butadiene (13)
To a solution of freshly distilled crotonaldehyde (5.00 mL, 60.3 mmol, 1.00 equiv) in CH2Cl2 (70 mL) at −10 °C was added Et3N (12.0 mL, 36.6 mmol, 1.40 equiv) and the reaction stirred 5 min. Next, TBSOTf (15.0 mL, 65.3 mmol, 1.10 equiv) was slowly added and the reaction warmed to 23 °C over 2 h, then stirred an additional 16 h at 23 °C. The reaction was partitioned between CH2Cl2 (75 mL) and saturated aqueous NaHCO3 (75 mL), dried (MgSO4) and concentrated under reduced pressure. The title compound was purified by distillation over a Vigreux column (bp 60–62°C/11 mm Hg) to afford the title compound (8.70 g, 78%) as a colorless liquid. The analytical data (1H, 13C NMR and HRMS) matched the reported data for this compounds prepared by an alternate procedure [reported yield 61% using crotonaldehyde (1.0 equiv) TBSCl (1.2 equiv), Et3N (1.2 equiv), ZnCl2 (0.013 equiv), hydroquinone (0.02 equiv), benzene, 80 °C, 24 h].16b
(±)-(4R,5S,6S) Ethyl 4-(tert-butoxycarbonylamino)-6-(tert-butyldimethylsilyloxy)-5-(phenoxythiocarbonyloxy)cyclohex-1-enecarboxylate (15)
To a solution of alcohol (±)-716b (76.0 mg, 0.180 mmol, 1.00 equiv) in CH2Cl2 (5 mL) at 23 °C was added phenyl thionochloroformate (50.0 μL, 0.36 mmol, 2.00 equiv) followed by N-methylimidazole (50.0 μL, 0.65 mmol, 3.60 equiv). The reaction mixture was stirred for 16 h at 23 °C then quenched with H2O (30 mL) and extracted with CH2Cl2 (3 × 30 mL). The combined organic extracts were washed with saturated aqueous NaCl (30 mL), dried (Na2SO4) and concentrated in vacuo. Purification by flash chromatography (5:1 hexane–EtOAc) afforded the title compound (89 mg, 90%) as white solid: 1H NMR (400 MHz, CDCl3) δ 0.16 (s, 3H), 0.30 (s, 3H), 0.90 (s, 9H), 1.33 (t, J = 7.1 Hz, 3H), 1.42 (s, 9H), 2.44–2.69 (m, 2H), 4.12–4.25 (m, 1H), 4.25–4.38 (m, 2H), 4.83 (s, 1H), 5.49–5.57 (m, 1H), 6.24 (d, J = 9.1 Hz, 1H), 7.04–7.11 (m, 3H), 7.27–7.32 (m, 1H), 7.37–7.44 (m, 2H); 13C NMR (100 MHz, CDCl3) δ −5.1, −4.5, 14.3, 18.0, 25.8, 28.3, 29.6, 43.9, 60.9, 63.7, 79.0, 79.5, 121.7, 126.7, 129.55, 129.64, 138.8, 153.3, 154.9, 165.9, 193.6; HRMS (ESI+) calcd for C27H41NNaO7SSi+ [M + Na]+ 574.2265, found 574.2266 (error 0.2 ppm).
(±)-(4R,6R) Ethyl 4-(tert-butoxycarbonylamino)-6-(tert-butyldimethylsilyloxy)cyclohex-1-enecarboxylate (16)
To a solution of ester (±)-15 (44.0 mg, 0.080 mmol, 1.00 equiv) in refluxing toluene (4 mL) was added dropwise a solution of (Me3Si)3SiH (40 μL, 0.12 mmol, 1.55 equiv), AIBN (7 mg, 0.04 mmol, 0.50 equiv) and tert-butyl acrylate (20 μL, 0.12 mmol, 1.55 equiv) in toluene (2 mL). The reaction mixture was heated at reflux for 2 h. After cooling to room temperature, the solvent was removed in vacuo. Purification by flash chromatography (5:1 hexane–EtOAc) afforded the title compound (20 mg, 63%) as white solid: 1H NMR (400 MHz, CDCl3) δ 0.10 (s, 3H), 0.18 (s, 3H), 0.88 (s, 9H), 1.31 (t, J = 7.1 Hz, 3H), 1.40 (s, 9H), 1.73 (dt, J = 14.3, 3.8 Hz, 1H), 2.01–2.09 (m, 1H), 2.27–2.39 (m, 1H), 2.55 (dd, J = 20.0, 5.1 Hz, 1H), 4.07 (q, J = 4.4 Hz, 1H), 4.10–4.20 (m, 1H), 4.21–4.32 (m, 1H), 4.75–4.81 (m, 1H), 6.55 (d, J = 8.1 Hz, 1H), 6.91–6.98 (m, 1H); 13C NMR (100 MHz, CDCl3) δ −5.0, −4.8, 14.3, 18.0, 25.8, 28.4, 32.6, 33.9, 42.0, 60.6, 62.7, 78.6, 132.0, 138.8, 155.3, 166.3; HRMS (ESI+) calcd for C20H37NNaO5Si+ [M + Na]+ 422.2333, found 422.2337 (error 0.9 ppm).
(R)-3-[5-(4-Methoxybenzyloxy)pentanoyl]-4-benzyloxazolidin-2-one (18)
To a solution of acid 1722 (10.66 g, 44.7 mmol, 1.15 equiv) in THF (500 mL) at −78 °C was added Et3N (10.8 mL, 77.2 mmol, 2.00 equiv) followed by pivaloyl chloride (6.00 mL, 49.2 mmol, 1.15 equiv) and the reaction was warmed to 0 °C. The resulting thick white precipitate was stirred for 6 h at 0 °C and then re-cooled to −78 °C. In a separate flask, n-BuLi (2.5 M in hexane, 17.0 mL, 42.5 mmol, 1.10 equiv) was added by syringe over 5 min to a solution of (R)-4-benzyl-2-oxazolidinone (6.84 g, 38.6 mmol, 1.00 equiv) in THF (100 mL) at −78 °C. The oxazolidinone solution was transferred by cannula to the flask containing the mixed anhydride. The mixture was stirred for 30 min at −78 °C and 30 min at 0 °C, then quenched with saturated aqueous NH4CI (200 mL). THF was removed under reduced pressure and the aqueous layer was extracted with CH2Cl2 (3 × 200 mL). The combined organic layers were washed with 10% aqueous NaOH solution, dried (MgSO4), and concentrated. The crude residue was purified by flash chromatography (3:1→2:1→3:2 hexanes–EtOAc) to afford the title compound (14.2 g, 93%) as a colorless oil: −37.8 (c 3.20, CHCl3); 1H NMR (400 MHz, CDCl3) δ 1.67–1.75 (m, 2H), 1.76–1.85 (m, 2H), 2.75 (dd, J = 13.4, 9.6 Hz, 1H), 2.89–3.05 (m, 2H), 3.29 (dd, J = 13.3, 3.2 Hz, 1H), 3.51 (t, J = 6.2 Hz, 2H), 3.80 (s, 3H), 4.14–4.19 (m, 2H), 4.45 (s, 2H), 4.66 (dddd, J = 13.3, 10.1, 7.1, 3.5 Hz, 1H), 6.87–6.92 (m, 2H), 7.19–7.23 (m, 2H), 7.25–7.37 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 21.0, 29.0, 35.1, 37.8, 55.0, 55.2, 66.1, 69.6, 72.5, 113.7, 127.2, 128.9, 129.2, 129.3, 130.6, 135.3, 153.4, 159.0, 173.0; HRMS (ESI+) calcd for C23H27NNaO5+ [M + Na]+ 420.1781, found 420.1786 (error 1.2 ppm).
2-(tert-Butyldimethylsilyloxymethyl)acrolein (19)
To a solution of tert-butyl 2-(hydroxymethyl)acrylate prepared according to the reported procedure23, 35 (10.0 g, 63.2 mmol, 1.00 equiv) in CH2Cl2 (500 mL) at 0 °C was added imidazole (11.1 g, 165 mmol, 2.61 equiv) followed by tert-butyldimethylsilyl chloride (19.1 g, 126 mmol, 2.00 equiv). The reaction mixture was stirred at 23 °C overnight. After 16 h, the solvent was removed in vacuo to provide a colorless oil, which was dissolved in 10:1 hexane–EtOAc (220 mL). The solution was passed through a short pad of silica gel, which was washed with hexane–EtOAc (10:1). The filtrate was concentrated in vacuo and dried under high vacuum to afford a colorless oil, which was then used directly in the next step without further purification.
To the solution of the crude tert-butyl 2-(tert-butyldimethylsilyloxymethyl)acrylate prepared above in CH2Cl2 (300 mL) at −78 °C was added diisobutylaluminum hydride (100 mL, 1.0 M in hexane, 100 mmol) dropwise. The reaction mixture was stirred for 1 h at −78 °C, then diluted with Et2O (300 mL), and allowed to slowly warm to 23 °C. While the reaction mixture slowly warmed to room temperature, the reaction mixture was quenched by the sequential dropwise addition of H2O (4 mL), 15% aqueous NaOH solution (4 mL), and H2O (10 mL). After warming to 23 °C, the reaction mixture was vigorously stirred for 30 min, then treated with anhydrous MgSO4, and stirred for an additional 15 min. The mixture was filtered through a pad of Celite and the resulting filtrate was concentrated in vacuo to afford the title compound (6.00 g, 50%, two steps) as a colorless oil, whose 1H and 13C NMR agreed with the reported data for 19 prepared by an alternate synthetic route.36
(R)-4-Benzyl-3-[(2R,3R)-4-(tert-butyldimethylsilyloxy)methyl-2-{3-[(4-methoxybenzyl)oxy]propyl}-3-(methoxymethoxy)pent-4-enoyl]oxazolidin-2-one (21)
To a solution of N-acyloxazolidinone 18 (3.00 g, 7.50 mmol, 1.00 equiv) in CH2Cl2 (100 mL) at −78 °C was added a 1.0 M solution of TiCl4 in CH2Cl2 (8.50 mL, 8.50 mmol, 1.13 equiv) dropwise and the solution was stirred for 10 min. (−)-Sparteine (4.30 mL, 18.8 mmol, 2.50 equiv) was added dropwise to the mixture and the solution stirred at −78 °C for 1.5 h. Freshly distilled aldehyde 19 (3.00 g, 15.0 mmol, 2.00 equiv) was then added dropwise. The reaction was stirred for 1.5 h at −78 °C, 3 h at −40 °C, and 12 h at −20 °C. The reaction was quenched at −40 °C by addition of half-saturated aqueous NH4Cl (100 mL) and quickly transferred to a separatory funnel. The organic layer was separated and the aqueous layer extracted with CH2Cl2 (2 × 100 mL). The combined organic layers were dried (MgSO4), filtered and concentrated. Purification of the crude residue by flash chromatography (10:1→8:1→7:1→6:1 PhMe–EtOAc) provided aldol adduct 20 (2.13 g, 48%) as a colorless oil, which was directly carried onto the next step.
Chloromethyl methyl ether (1.69 g, 21.0 mmol, 5.90 equiv) was added to a solution of aldol adduct 20 (2.13 g, 3.56 mmol, 1.0 equiv), i-Pr2NEt (3.66 mL, 21.0 mmol, 5.90 equiv), and Bu4NI (131 mg, 0.36 mmol, 0.10 equiv) in PhMe (50 mL) and the mixture stirred at 90 °C for 7 h. The reaction mixture was cooled to rt and treated with saturated aqueous NaHCO3 (30 mL). The layers were separated and the aqueous layer was extracted with EtOAc (3 × 50 mL). The combined organic extracts were washed with saturated aqueous NaCl (30 mL), dried (Na2SO4) and concentrated. Purification by flash chromatography (4:1→3:1 hexane–EtOAc) afforded the title compound (1.88 g, 82%) as colorless oil: +13.5 (c 1.10, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.00 (s, 6H), 0.84 (s, 9H), 1.57–1.67 (m, 2H), 1.82–1.92 (m, 2H), 2.55 (dd, J = 13.2, 10.2 Hz, 1H), 3.22 (dd, J = 13.2, 3.0 Hz, 1H), 3.27 (s, 3H), 3.37–3.42 (m, 2H), 3.70 (s, 3H), 3.97–4.13 (m, 4H), 4.16–4.23 (m, 1H), 4.26 (d, J = 7.9 Hz, 1H), 4.35 (s, 2H), 4.41 (d, J = 6.8 Hz, 1H), 4.47 (dddd, J = 12.9, 9.8, 6.4, 3.0 Hz, 1H), 4.55 (d, J = 6.8 Hz, 1H), 5.09 (s, 1H), 5.29 (s, 1H), 6.76–6.81 (m, 2H), 7.10–7.15 (m, 2H), 7.15–7.27 (m, 5H); 13C NMR (100 MHz, CDCl3) δ −5.6, 18.2, 25.0, 25.7, 27.0, 37.7, 46.4, 55.0, 55.7, 55.8, 62.1, 65.8, 69.7, 72.3, 77.9, 94.2, 112.8, 113.5, 127.1, 128.8, 129.1, 129.2, 130.5, 135.3, 145.8, 152.9, 158.9, 173.3; HRMS (ESI+) calcd for C35H51NNaO8Si+ [M + Na]+ 664.3276, found 664.3283 (error 1.05 ppm).
(2S,3R)-4-{[(tert-Butyldimethylsilyl)oxy]methyl}-2-{3-[(4-methoxybenzyl)oxy]propyl}-3-(methoxymethoxy)pent-4-en-1-ol (22)
Sodium borohydride (450 mg, 11.7 mmol, 4.00 equiv) was added in one portion to a solution of oxazolidinone 21 (1.88 g, 2.93 mmol, 1.00 equiv) in 3:1 THF–H2O (60 mL) at 0 °C. The reaction mixture was allowed to warm to 23 °C and stirred overnight (~16 h). The reaction was quenched by the slow addition of saturated aqueous NH4Cl (60 mL) and extracted with diethyl ether (3 × 50 mL). The combined organic extracts were washed with saturated aqueous NaCl, dried (MgSO4), and concentrated under reduced pressure. Purification by flash chromatography (3:1→2:1→3:2 hexane–EtOAc) afforded the title compound (920 mg, 67%) as a colorless oil: +60.7 (c 0.400, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.00 (s, 6H), 0.84 (s, 9H), 1.22–1.34 (m, 1H), 1.43–1.59 (m, 2H), 1.59–1.71 (m, 2H), 2.67 (br s, 1H), 3.35 (s, 3H), 3.41 (t, J = 6.3 Hz, 2H), 3.58–3.63 (m, 2H), 3.76 (s, 3H), 4.09 (s, 2H), 4.19 (d, J = 6.1 Hz, 1H), 4.39 (s, 2H), 4.48 (d, J = 6.5 Hz, 1H), 4.58 (d, J = 6.5 Hz, 1H), 5.12 (s, 1H), 5.32 (s, 1H), 6.83 (d, J = 8.5 Hz, 2H), 7.22 (d, J = 8.5 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ −5.58, −5.56, 18.2, 22.7, 25.8, 27.3, 43.1, 55.0, 55.8, 62.4, 63.2, 70.2, 72.4, 78.5, 94.6, 112.9, 113.6, 129.0, 130.5, 146.1, 159.0; HRMS (ESI+) calcd for C25H44NaO6Si+ [M + Na]+ 491.2799, found 491.2806 (error 1.4 ppm).
(2S,3R)-4-{[(tert-Butyldimethylsilyl)oxy]methyl}-2-{3-[(4-methoxybenzyl)oxy]propyl}-3-(methoxymethoxy)pent-4-enal (23)
To a solution of alcohol 22 (920 mg, 1.96 mmol, 1.00 equiv) in CH2Cl2 (20 mL) at 23 °C was added solid NaHCO3 (1.65 g, 19.6 mmol, 10.0 equiv) followed by Dess–Martin periodinane (1.25 g, 2.94 mmol, 1.50 equiv). The reaction was stirred for 30 min at 23 °C then quenched by 1:1 10% aqueous Na2S2O3–saturated aqueous NaHCO3 (20 mL). The layers were separated and the aqueous layer was extracted with CH2Cl2 (3 × 20 ml). The combined organic layers were dried (MgSO4) and concentrated under reduced pressure. The resulting residue was dissolved in a minimum amount of 3:1 hexane–EtOAc and filtered through a short pad of silica gel, which was rinsed with 3:1 hexane–EtOAc (200 mL). The filtrate was concentrated to afford the title compound (821 mg, 90%) as colorless oil: 1H NMR (400 MHz, CDCl3) δ 0.00 (s, 6H), 0.84 (s, 9H), 1.43–1.54 (m, 1H), 1.57–1.79 (m, 3H), 2.50 (dddd, J = 11.5, 8.9, 5.6, 2.7 Hz, 1H), 3.26 (s, 3H), 3.36 (t, J = 6.0 Hz, 2H), 3.73 (s, 3H), 4.05 (s, 2H), 4.34 (s, 2H), 4.41 (d, J = 6.7 Hz, 1H), 4.44 (d, J = 5.9 Hz, 1H), 4.56 (d, J = 6.8 Hz, 1H), 5.08 (s, 1H), 5.27 (s, 1H), 6.80 (d, J = 8.6 Hz, 2H), 7.19 (d, J = 8.6 Hz, 2H), 9.61 (d, J = 2.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ −5.54, −5.52, 18.2, 21.2, 25.8, 27.6, 54.6, 55.1, 55.9, 63.2, 69.7, 72.4, 75.8, 94.1, 113.6, 114.0, 129.1, 130.5, 144.8, 159.0, 203.1; HRMS (ESI+) calcd for C25H42NaO6Si+ [M + Na]+ 489.2643, found 489.2647 (error 0.8 ppm).
(S,E)-N-[(2S,3R)-4-{[(tert-Butyldimethylsilyl)oxy]methyl}-2-{3-[(4-methoxybenzyl)oxy]propyl}-3-(methoxymethoxy)pent-4-en-1-ylidene]-tert-butylsulfinamide (24)
To a solution of aldehyde 23 (820 mg, 1.76 mmol, 1.00 equiv) in CH2Cl2 (10 mL) was added (S)-(−)-tert-butylsulfinamide (235 mg, 1.94 mmol, 1.10 equiv), anhydrous CuSO4 (562 mg, 3.52 mmol, 2.00 equiv) and pyridinium p-toluenesulfonate (442 mg, 1.76 mmol, 1.00 equiv). The mixture was stirred at 23 °C for 24 h, then filtered through a pad of Celite, and the filter cake was washed with CH2Cl2. The combined organic filtrate was washed with saturated aqueous NaCl, dried (MgSO4), and concentrated. Purification by flash chromatography (3:1→2:1 hexane–EtOAc) afforded the title compound (616 mg, 62%, 75% brsm) as a colorless oil: +165 (c 0.700, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.00 (s, 6H), 0.84 (s, 9H), 1.13 (s, 9H), 1.42–1.69 (m, 3H), 1.80–1.91 (m, 1H), 2.73–2.83 (m, 1H), 3.30 (s, 3H), 3.36 (t, J = 6.1 Hz, 2H), 3.72 (s, 3H), 4.06 (s, 2H), 4.24 (d, J = 7.1 Hz, 1H), 4.34 (s, 2H), 4.42 (d, J = 6.7 Hz, 1H), 4.58 (d, J = 6.7 Hz, 1H), 5.06 (s, 1H), 5.25 (s, 1H), 6.80 (d, J = 8.3 Hz, 2H), 7.17 (d, J = 8.3 Hz, 2H), 7.87 (d, J = 6.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ −5.71, −5.69, 18.0, 22.2, 24.2, 25.7, 27.2, 48.7, 54.9, 55.8, 56.6, 62.3, 69.4, 72.2, 77.8, 93.7, 113.5, 114.1, 128.9, 130.4, 144.6, 158.9, 169.8; HRMS (ESI+) calcd for C29H51NNaO6SSi+ [M + Na]+ 592.3099, found 592.3105 (error 1.0 ppm).
(S)-N-[(4R,5S,6R)-7-{[(tert-Butyldimethylsilyl)oxy]methyl}-5-{3-[(4-methoxybenzyl)oxy]propyl}-6-(methoxymethoxy)octa-1,7-dien-4-yl]-tert-butylsulfinamide (25)
To a solution of tert-butanesulfinyl imine 24 (600 mg, 1.05 mmol, 1.00 equiv) in CH2Cl2 (20 mL) was added allylmagnesium bromide (1.0 M in Et2O, 3.15 mL, 3.15 mmol, 3.00 equiv) at −78 °C. The reaction was warmed to −20 °C and stirred for 2.5 h at this temperature then quenched with saturated aqueous NH4Cl (20 mL). The layers were separated and the aqueous layer was extracted with CH2Cl2 (3 × 20 ml). The combined organic layers were dried (MgSO4), filtered, and concentrated. The resulting residue was dissolved in a minimum amount of Et2O and filtered through a short pad of silica gel, which was rinsed with Et2O. The filtrate was concentrated to provide the title compound (560 mg, 87%) as colorless oil: +80.1 (c 0.600, CHCl3); 1H NMR (400 MHz, CDCl3) δ 0.02 (s, 6H), 0.87 (s, 9H), 1.16 (s, 9H), 1.37–1.51 (m, 2H), 1.55–1.79 (m, 3H), 2.35–2.45 (m, 1H), 2.49–2.59 (m, 1H), 3.22 (d, J = 8.3 Hz, 1H), 3.28 (s, 3H), 3.35–3.38 (m, 3H), 3.74 (s, 3H), 3.99 (d, J = 15.0 Hz, 1H), 4.05 (d, J = 14.6 Hz, 1H), 4.16 (d, J = 7.3 Hz, 1H), 4.36 (s, 2H), 4.39 (d, J = 6.7 Hz, 1H), 4.56 (d, J = 6.7 Hz, 1H), 5.05–5.10 (m, 3H), 5.34 (s, 1H), 5.66–5.80 (m, 1H), 6.81 (d, J = 8.2 Hz, 1H), 7.19 (d, J = 8.2 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ −5.55, −5.52, 18.2, 22.6, 22.8, 25.7, 29.5, 38.5, 42.7, 55.0, 55.9, 56.0, 56.5, 62.1, 70.0, 72.3, 79.0, 93.9, 113.5, 113.9, 118.6, 129.0, 130.5, 134.5, 145.3, 158.9; HRMS (ESI+) calcd for C32H58NO6SSi+ [M + H]+ 612.3749, found 612.3754 (error 0.8 ppm).
(S)-N-[(1R,5R,6S)-4-{[(tert-Butyldimethylsilyl)oxy]methyl}-6-{3-[(4-methoxybenzyl)oxy]propyl}-5-(methoxymethoxy)cyclohex-3-en-1-yl]-tert-butylsulfinamide (26)
[1,3-bis-(2,4,6-Trimethylphenyl)-2-imidazolidinylidene]dichloro(phenylmethylene)(tricyclohexylphosphine)ruthenium (13.8 mg, 0.016 mmol, 0.14 equiv) was added to a solution of diene 25 (70.0 mg, 0.114 mmol, 1.0 equiv) in CH2Cl2 (20 mL) and the reaction was heated at reflux for 14 h. The reaction was cooled to 23 °C, then treated with DMSO (1.5 mL),37 and stirred 16 h at 23 °C. The reaction mixture was washed with saturated aqueous NaCl (50 mL), and the organic layer was dried (MgSO4), filtered and concentrated. Purification by flash chromatography (2:1→3:2→1:1 hexane–EtOAc) afforded the title compound (63 mg, 95%) as a light brown oil: 1H NMR (400 MHz, CDCl3) δ −0.01 (s, 3H), 0.00 (s, 3H), 0.85 (s, 9H), 1.11 (s, 9H), 1.16–1.29 (m, 2H), 1.54–1.70 (m, 2H), 1.99–2.07 (m, 1H), 2.23–2.34 (m, 1H), 2.48–2.59 (m, 1H), 3.26–3.46 (m, 6H), 3.73 (s, 3H), 3.92 (s, 1H), 4.02 (d, J = 12.8 Hz, 1H), 4.14 (d, J = 12.8 Hz, 1H), 4.35 (s, 2H), 4.54 (d, J = 6.6 Hz, 1H), 4.60–4.69 (m, 2H), 5.65 (s, 1H), 6.80 (d, J = 8.2 Hz, 2H), 7.17 (d, J = 8.2 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ −5.6, −5.4, 18.0, 22.5, 25.7, 26.5, 27.5, 29.2, 40.9, 53.5, 55.0, 55.5, 55.6, 64.3, 69.5, 71.6, 72.4, 95.3, 113.5, 121.5, 129.0, 130.3, 135.0, 158.9; HRMS (ESI+) calcd for C30H53NNaO6SSi+ [M + Na]+ 606.3255, found 606.3252 (error 0.5 ppm).
tert-Butyl [(1R,5R,6S)-4-(hydroxymethyl)-6-{3-[(4-methoxybenzyl)oxy]propyl}-5-(methoxymethoxy)cyclohex-3-en-1-yl]carbamate (27)
To a solution of 26 (55 mg, 0.094 mmol, 1.0 equiv) in MeOH (2.0 mL) was added dropwise 4 M HCl in 1,4-dioxane (190 μL, 0.75 mmol, 8.0 equiv) at 0 °C. The resulting mixture was stirred at 0 °C for 1 h then quenched with saturated aqueous NaHCO3 (3 mL). Methanol was removed under reduced pressure and the aqueous layer was treated with CH2Cl2 (2 mL) and 6 M aqueous NaOH (2 mL) at 0 °C. After stirring for 10 min at 0 °C, the layers were separated. The aqueous layer was extracted with CH2Cl2 (3 × 3 mL) and the combined organic layers were dried (K2CO3) and concentrated under reduced pressure to afford the crude amino alcohol as a viscous yellow oil, which was used directly in the next step without further purification.
To the crude amino alcohol in 1:2 H2O–1,4-dioxane (3 mL) at 23 °C was added Et3N (26 μL, 0.19 mmol, 2.0 equiv) followed by di-tert-butyl dicarbonate (28 mg, 0.13 mmol, 1.4 equiv) in 1,4-dioxane (0.5 mL). The reaction mixture was stirred for 1 h at 23 °C, then partitioned between H2O (10 mL) and CH2Cl2 (10 mL). The aqueous layer was extracted with CH2Cl2 (3 × 3 mL) and the combined organic layers were dried (MgSO4), filtered, and concentrated. Purification by flash chromatography (1:1 hexane–EtOAc) afforded the title compound (39 mg, 89%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 1.21–1.31 (m, 2H), 1.42 (s, 9H), 1.63–1.74 (m, 2H), 1.96–2.11 (m, 2H), 2.19–2.26 (m, 2H), 3.37–3.46 (m, 5H), 3.80 (s, 3H), 3.82–3.89 (m, 1H), 3.96 (s, 1H), 4.09 (d, J = 12.0 Hz, 1H), 4.18 (d, J = 12.7 Hz, 1H), 4.40 (s, 2H), 4.64 (d, J = 6.7 Hz, 1H), 4.74 (d, J = 6.7 Hz, 1H), 5.76 (br s, 1H), 5.93 (d, J = 8.1 Hz, 1H), 6.87 (d, J = 8.5 Hz, 2H), 7.23 (d, J = 8.5 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 27.0, 27.68, 27.72, 28.4, 40.0, 46.3, 55.2, 56.1, 65.4, 69.7, 72.5, 74.0, 78.8, 96.2, 113.8, 124.6, 129.2, 130.5, 134.8, 155.4, 159.1; HRMS (ESI+) calcd for C25H39NNaO7+ [M + Na]+ 488.2619, found 488.2621 (error 0.4 ppm).
tert-Butyl [(1R,5R,6S)-4-(hydroxymethyl)-6-(3-hydroxypropyl)-5-(methoxymethoxy)cyclohex-3-en-1-yl]carbamate (28)
To a solution of 27 (40 mg, 0.086 mmol, 1.0 equiv) in 20:1 CH2Cl2–H2O (10 mL) was added DDQ (29 mg, 0.13 mmol, 1.5 equiv) at 0 °C. The reaction was stirred for 2 h at 23 °C, then partitioned between saturated aqueous NaHCO3 (10 mL) and CH2Cl2 (10 mL). The aqueous layer was extracted with CH2Cl2 (2 × 10 mL) and the combined organic layers were dried (Na2SO4), filtered, and concentrated. Purification by flash chromatography (Et2O→EtOAc) afforded the title compound (23 mg, 89%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 1.21–1.31 (m, 2H), 1.42 (s, 9H), 1.58–1.72 (m, 2H), 2.04–2.12 (m, 1H), 2.20–2.26 (m, 2H), 2.30–2.40 (m, 2H), 3.43 (s, 3H), 3.55–3.68 (m, 2H), 3.80–3.89 (m, 1H), 3.97–4.01 (m, 1H), 4.07 (d, J = 12.4 Hz, 1H), 4.18 (d, J = 12.6 Hz, 1H), 4.67 (d, J = 6.8 Hz, 1H), 4.77 (d, J = 6.8 Hz, 1H), 5.72–5.79 (m, 1H), 5.93 (d, J = 7.5 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 26.6, 27.8, 28.4, 30.4, 39.9, 46.4, 56.1, 62.4, 65.1, 73.7, 78.9, 96.2, 124.7, 134.9, 155.5; HRMS (ESI+) calcd for C17H31NNaO6+ [M + Na]+ 368.2044, found 368.2046 (error 0.5 ppm).
(4R,5S,6R)-4-tert-(Butylcarbonyl)amino-5-(2-carboxyethyl)-6-(methoxymethoxy)cyclohex-1-enecarboxylic acid (29)
To a solution of diol 28 (23 mg, 0.076 mmol, 1.0 equiv), N-methylmorpholine-N-oxide (53 mg, 0.45 mmol, 6.0 equiv), and powdered 4 Å molecular sieves (35 mg) in CH2Cl2 (1 mL) at 23 °C was added tetrapropylammonium perruthenate (2.0 mg, 0.0056 mmol, 0.075 equiv). The mixture was stirred for 15 min at 23 °C, then filtered through a short pad of silica gel, eluting with EtOAc. The filtrate was concentrated under reduced pressure to afford the crude dialdehyde, which was directly used in the next step without further purification.
To a solution of freshly prepared dialdehyde and 2-methyl-2-butene (0.20 mL, 1.9 mmol, 25 equiv) in 5:1:1 tert-butyl alcohol–THF–H2O (1 mL) at 0 °C was slowly added a solution of sodium chlorite (80% w/w technical grade, 52 mg, 0.46 mmol, 6.0 equiv) and sodium phosphate monobasic monohydrate (63 mg, 0.46 mmol, 6.0 equiv) in H2O (0.3 mL). The resulting suspension was stirred at 23 °C for 2 h. The reaction was quenched with saturated aqueous NaHSO3 (3 mL) at 0 °C and the aqueous layer was extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification by flash chromatography on silica gel38 (210:25:4→180:25:4 CHCl3–MeOH–AcOH) afforded the title compound (13 mg, 46% for two steps) as a colorless oil that was ~90% pure: 1H NMR (400 MHz, CDCl3) δ 1.41 (s, 9H), 1.44–1.54 (m, 2H), 2.18–2.25 (m, 1H), 2.39–2.60 (m, 4H), 3.40 (s, 3H), 3.88–3.95 (m, 1H), 4.31–4.34 (m, 1H), 4.72 (d, J = 6.9 Hz, 1H), 4.86 (d, J = 6.9 Hz, 1H), 6.29 (d, J = 8.1 Hz, 1H), 7.14–7.18 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 25.0, 28.4, 29.0, 31.8, 40.1, 45.4, 56.1, 71.8, 79.3, 97.5, 128.0, 142.4, 155.5, 171.1, 178.2; HRMS (ESI−) calcd for C17H26NO8− [M − H]− 372.1664, found 372.1668 (error 1.1 ppm).
(4R,5S,6R)-4-Amino-5-(2-carboxyethyl)-6-hydroxycyclohex-1-enecarboxylic acid acetate salt (4)
To a solution of dicarboxylic acid 29 (13 mg, 0.035 mmol, 1.0 equiv) in CH2Cl2 (1 mL) was added TMSBr (40 μL, 0.30 mmol, 8.6 equiv) at −78 °C. The reaction mixture was stirred for 1 h at −78 ° then concentrated in vacuo during which time the flask warmed to 23 °C. The residue was washed with Et2O, and purified by flash chromatography on silica gel38 (65:25:4:2→100:56:8:8 CHCl3–MeOH–H2O–AcOH) to afford the acetate salt of the title compound (5.0 mg, 50%) as a white solid: +6.2 (c 0.40, H2O); 1H NMR (600 MHz, D2O) δ 1.57–1.64 (m, 2H), 2.22–2.29 (m, 1H), 2.46 (t, J = 7.4 Hz, 2H), 2.56 (d, J = 13.5 Hz, 1H), 2.78 (d, J = 13.5 Hz, 1H), 3.68–3.74 (m, 1H), 4.55–4.59 (m, 1H), 6.84–6.88 (m, 1H); 13C NMR (150 MHz, D2O) δ 21.1 (AcOH), 24.0, 65.0, 32.7, 40.7, 46.9, 65.1, 131.8*, 134.3, 172.4*, 177.4* (AcOH), 179.6* (Chemical shifts denoted by * are derived from HMBC); HRMS (ESI−) calcd for C10H14NO5− [M − H]− 228.0877, found 228.0883 (error 2.6 ppm).
MbtI Assay
Reactions were performed under initial velocity conditions in a total volume of 50 μL at 37 °C for 30 min and the production of salicylic acid was monitored continuously by following changes in fluorescence at 420 nm with excitation at 305 nm on a microplate reader. Assays were set up in duplicate and contained 0.5 μM MbtI in reaction buffer (100 mM Tris-HCl, pH 8.0, 1 mM MgCl2, 50 μM chorismate, and 0.0025% Igepal CA-630). A three-fold serial dilution of inhibitor in H2O was added to black 384 well plates coated with a non-binding surface (Greiner). A positive control (H2O only) and negative control (10 mM EDTA) were also included. The IC50 values were calculated from the Hill equation (eq 1). In this equation the fractional activity (vi/v0) versus inhibitor concentration was fit by non-linear regression analysis using GraphPad prism version 6.0 where vi is the reaction velocity at a given [I] and v0 is the reaction velocity of the DMSO control, and h is the Hill slope.
| (1) |
Supplementary Material
Scheme 1. Attempted introduction of propionate side-chaina.

aReagents and conditions: (a) TBSOTf, Et3N, CH2Cl2, −20 °C→rt, (b) 2,6-di-tert-butyl-4-methylphenol (BHT) or methylene blue (0.05 equiv), PhCH3, 65–80 °C, 6–8 d; (c) N-methylimidazole (3.5 equiv), CH2Cl2, rt, 24 h; (d) representative conditions: (Me3Si)3Si (1.05 equiv), tert-butyl acrylate (10 equiv), AIBN (0.8 equiv), PhCH3, reflux, 2 h.
Scheme 3. Synthesis of 5a.

aReagents and conditions: (a) 4 M HCl in 1,4-dioxane, MeOH, 0 °C, 1 h; (b) Boc2O, Et3N, 1,4-dioxane/H2O (2:1), rt, 1 h; (c) DDQ, CH2Cl2–H2O (20:1), rt, 2 h; (d) tetrapropylammonium perruthenate, NMO, 4 Å molecular sieves, CH2Cl2, rt, 15 min; (e) NaClO2, 2-methyl-2-butene, t-BuOH–THF–H2O (5:1:1), rt, 2 h; (f) TMSBr, CH2Cl2, −78 °C, 1 h.
Acknowledgments
This work was supported by a grant from the National Institutes of Health Grant (RO1 AI070219 to CCA) and the National Natural Science Foundation of China (21402058 to ZL). We thank Dr. Youlin Xia from NMR center of University of Minnesota for NMR spectroscopic analysis. Mass spectrometric analysis was carried out in the Analytical Biochemistry Shared Resource of the Masonic Cancer Center, University of Minnesota, funded in part by Cancer Center Support Grant CA-77598.
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information: copies of 1H NMR and 13C NMR spectra of all new compounds and Figure illustrating a docked pose of 4 in MbtI. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES AND NOTES
- 1.(a) Homolka S, Niemann S, Russell DG, Rohde KH. PLoS Pathog. 2010;6:e1000988. doi: 10.1371/journal.ppat.1000988. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) WHO. Global Tuberculosis Report 2013. World Health Organization; 2013. [Google Scholar]
- 2.(a) Zumla A, Nahid P, Cole ST. Nat Rev Drug Discov. 2013;12:388. doi: 10.1038/nrd4001. [DOI] [PubMed] [Google Scholar]; (b) Dartois V, Barry CE., 3rd Bioorg Med Chem Lett. 2013;23:4741. doi: 10.1016/j.bmcl.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gopal P, Dick T. Curr Opin Microbiol. 2014;21C:7. doi: 10.1016/j.mib.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 4.(a) Marriner GA, Nayyar A, Uh E, Wong SY, Mukherjee T, Via LE, Carroll M, Edwards RL, Gruber TD, Choi I, Lee J, Arora K, England KD, Boshoff HIM, Barry CE., 3rd Top Med Chem. 2011;7:47. [Google Scholar]; (b) Aldrich CC, Boshoff HI, Remmel RP. Antitubercular Agents. 7. Vol. 7. Wiley; Hoboken, N. J: 2010. p. 713. [Google Scholar]
- 5.Ratledge C, Dover LG. Annu Rev Microbiol. 2000;54:881. doi: 10.1146/annurev.micro.54.1.881. [DOI] [PubMed] [Google Scholar]
- 6.(a) Snow GA. Bacteriol Rev. 1970;34:99. doi: 10.1128/br.34.2.99-125.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Vergne AF, Walz AJ, Miller MJ. Nat Prod Rep. 2000;17:99. doi: 10.1039/a809397k. [DOI] [PubMed] [Google Scholar]; (c) De Voss JJ, Rutter K, Schroeder BG, Barry CE., 3rd J Bacteriol. 1999;181:4443. doi: 10.1128/jb.181.15.4443-4451.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tullius MV, Harmston CA, Owens CP, Chim N, Morse RP, McMath LM, Iniguez A, Kimmey JM, Sawaya MR, Whitelegge JP, Horwitz MA, Goulding CW. Proc Natl Acad Sci USA. 2011;108:5051. doi: 10.1073/pnas.1009516108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) De Voss JJ, Rutter K, Schroeder BG, Su H, Zhu Y, Barry CE., 3rd Proc Natl Acad Sci USA. 2000;97:1252. doi: 10.1073/pnas.97.3.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Reddy PV, Puri RV, Chauhan P, Kar R, Rohilla A, Khera A, Tyagi AK. J Infect Dis. 2013;208:1255. doi: 10.1093/infdis/jit250. [DOI] [PubMed] [Google Scholar]; (c) Reddy PV, Puri RV, Chauhan P, Kar R, Rohilla A, Khera A, Tyagi AK. J Infect Dis. 2014;209:971. doi: 10.1093/infdis/jit605. [DOI] [PubMed] [Google Scholar]
- 9.(a) Quadri LE, Sello J, Keating TA, Weinreb PH, Walsh CT. Chemistry & biology. 1998;5:631. doi: 10.1016/s1074-5521(98)90291-5. [DOI] [PubMed] [Google Scholar]; (b) Harrison AJ, Yu M, Gardenborg T, Middleditch M, Ramsay RJ, Baker EN, Lott JS. J Bacteriol. 2006;188:6081. doi: 10.1128/JB.00338-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zwahlen J, Kolappan S, Zhou R, Kisker C, Tonge PJ. Biochemistry. 2007;46:954. doi: 10.1021/bi060852x. [DOI] [PubMed] [Google Scholar]
- 10.(a) He Z, Stigers Lavoie KD, Bartlett PA, Toney MD. J Am Chem Soc. 2004;126:2378. doi: 10.1021/ja0389927. [DOI] [PubMed] [Google Scholar]; (b) Meneely KM, Luo Q, Dhar P, Lamb AL. Arch Biochem Biophys. 2013;538:49. doi: 10.1016/j.abb.2013.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Wright SK, DeClue MS, Mandal A, Lee L, Wiest O, Cleland WW, Hilvert D. J Am Chem Soc. 2005;127:12957. doi: 10.1021/ja052929v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) DeClue MS, Baldridge KK, Kunzler DE, Kast P, Hilvert D. J Am Chem Soc. 2005;127:15002. doi: 10.1021/ja055871t. [DOI] [PubMed] [Google Scholar]; (c) DeClue MS, Baldridge KK, Kast P, Hilvert D. J Am Chem Soc. 2006;128:2043. doi: 10.1021/ja056714x. [DOI] [PubMed] [Google Scholar]
- 12.(a) Payne RJ, Kerbarh O, Miguel RN, Abell AD, Abell C. Org Biomol Chem. 2005;3:1825. doi: 10.1039/b503800f. [DOI] [PubMed] [Google Scholar]; (b) Payne RJ, Toscano MD, Bulloch EMM, Abell AD, Abell C. Org Biomol Chem. 2005;3:2271. doi: 10.1039/b503802b. [DOI] [PubMed] [Google Scholar]; (c) Payne RJ, Bulloch EMM, Abell AD, Abell C. Org Biomol Chem. 2005;3:3629. doi: 10.1039/b510633h. [DOI] [PubMed] [Google Scholar]; (d) Payne RJ, Bulloch EMM, Toscano MM, Jones MA, Kerbarh O, Abell C. Org Biomol Chem. 2009;7:2421. doi: 10.1039/b901694e. [DOI] [PubMed] [Google Scholar]; (e) Payne RJ, Bulloch EMM, Kerbarh O, Abell C. Org Biomol Chem. 2010;8:3534. doi: 10.1039/c004062b. [DOI] [PubMed] [Google Scholar]; (f) Manos-Turvey A, Bulloch EM, Rutledge PJ, Baker EN, Lott JS, Payne RJ. ChemMedChem. 2010;5:1067. doi: 10.1002/cmdc.201000137. [DOI] [PubMed] [Google Scholar]; (g) Manos-Turvey A, Cergol KM, Salam NK, Bulloch EM, Chi G, Pang A, Britton WJ, West NP, Baker EN, Lott JS, Payne RJ. Org Biomol Chem. 2012;10:9223. doi: 10.1039/c2ob26736e. [DOI] [PubMed] [Google Scholar]
- 13.Vasan M, Neres J, Williams J, Wilson DJ, Teitelbaum AM, Remmel RP, Aldrich CC. ChemMedChem. 2010;5:2079. doi: 10.1002/cmdc.201000275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Schramm VL. Annu Rev Biochem. 1998;67:693. doi: 10.1146/annurev.biochem.67.1.693. [DOI] [PubMed] [Google Scholar]; (b) Wolfenden R. Bioorg Med Chem. 1999;7:647. doi: 10.1016/s0968-0896(98)00247-8. [DOI] [PubMed] [Google Scholar]
- 15.Ferrer S, Marti S, Moliner V, Tunon I, Bertran J. Phys Chem Chem Phys. 2012;14:3482. doi: 10.1039/c2cp23149b. [DOI] [PubMed] [Google Scholar]
- 16.(a) Kozlowski MC, Bartlett PA. J Am Chem Soc. 1991;113:5897. [Google Scholar]; (b) Kozlowski MC, Tom NJ, Seto CT, Sefler AM, Bartlett PA. J Am Chem Soc. 1995;117:2128. [Google Scholar]
- 17.Magano J. Tetrahedron. 2011;67:7875. [Google Scholar]
- 18.Cong X, Yao ZJ. J Org Chem. 2006;71:5365. doi: 10.1021/jo060633h. [DOI] [PubMed] [Google Scholar]
- 19.Robak MT, Herbage MA, Ellman JA. Chem Rev. 2010;110:3600. doi: 10.1021/cr900382t. [DOI] [PubMed] [Google Scholar]
- 20.Sanchez-Rosello M, Puchlopek AL, Morgan AJ, Miller SJ. J Org Chem. 2008;73:1774. doi: 10.1021/jo702334z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keck GE, Enholm EJ, Yates JB, Wiley MR. Tetrahedron. 1985;41:4079. [Google Scholar]
- 22.Wilson MS, Woo JC, Dake GR. J Org Chem. 2006;71:4237. doi: 10.1021/jo0604585. [DOI] [PubMed] [Google Scholar]
- 23.Byan HS, Reddy KC, Bittman R. Tetrahedron Lett. 1994;35:1371. [Google Scholar]
- 24.Evans DA, Bartroli J, Shih TL. J Am Chem Soc. 1981;103:2127. [Google Scholar]
- 25.Crimmins MT, King BW, Tabet EA, Chauhary K. J Org Chem. 2001;66:894. doi: 10.1021/jo001387r. [DOI] [PubMed] [Google Scholar]
- 26.Prashad M, Har D, Kim HY, Repic O. Tetrahedron Lett. 1998;39:7067. [Google Scholar]
- 27.Dess DB, Martin JC. J Org Chem. 1983;48:4155. [Google Scholar]
- 28.Luo YC, Zhang HH, Liu YZ, Xu PF. Tetrahedron Asymm. 2009;20:1174. [Google Scholar]
- 29.Liu G, Cogan DA, Ellman JA. J Am Chem Soc. 1997;119:9913. [Google Scholar]
- 30.(a) Huang J, Stevens ED, Nolan SP, Petersen JL. J Am Chem Soc. 1999;121:2674. [Google Scholar]; (b) Scholl M, Trnka TM, Morgan JP, Grubbs RH. Tetrahedron Lett. 1999;40:2247. [Google Scholar]
- 31.Ley SV, Norman J, Griffith WP, Marsden SP. Synthesis. 1994:639. [Google Scholar]
- 32.(a) Lindgren BO, Nilsson T. Acta Chem Scand. 1973;27:888. [Google Scholar]; (b) Bal BS, Childers WE, Jr, Pinnick HW. Tetrahedron. 1981;37:2091. [Google Scholar]
- 33.Hanessian S, Delorme D, Dufrensne Y. Tetrahedron Lett. 1984;25:2515. [Google Scholar]
- 34.Chi G, Manos-Turvey A, O’Connor PD, Johnston JM, Evans GL, Baker EN, Payne RJ, Lott JS, Bulloch EM. Biochemistry. 2012;51:4868. doi: 10.1021/bi3002067. [DOI] [PubMed] [Google Scholar]
- 35.Turki T, Villieras J, Amri H. Tetrahedron Lett. 2005;46:3071. [Google Scholar]
- 36.Senter TJ, Fadeyi OO, Lindsley CW. Org Lett. 2012;14:1869. doi: 10.1021/ol300466a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahn YM, Yang K, Georg GI. Org Lett. 2001;3:1411. doi: 10.1021/ol010045k. [DOI] [PubMed] [Google Scholar]
- 38.In order to remove the inorganic salts in silica gel, the packed column was washed successively with MeOH followed by CHCl3 prior to loading the crude product.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.