

Abstract
The association between pulmonary inflammation and lung cancer is well-established. However, currently there are no appropriate models that recapitulate inflammation-related lung cancer in humans. In the present study, we examined, in two tumor bioassays, enhancement by bacterial lipopolysaccharide (LPS) of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-induced lung tumorigenesis in A/J mice. Mice that were treated with NNK alone developed 29.6 ± 9.8 and 36.2 ± 4.1 lung tumors/mouse in experiment 1 and 2, respectively. Chronic intranasal instillation of LPS to NNK-treated mice increased the multiplicity of lung tumors to 47.3 ± 16.1 and 51.2 ± 4.8 lung tumors/mouse in experiment 1 and 2, corresponding to a significant increase by 60% and 41%, respectively. Moreover, administration of LPS to NNK-pretreated mice significantly increased the multiplicity of larger tumors and histopathologically more advanced lesions (adenoma with dysplasia and adenocarcinoma), macrophage recruitment to the peritumoral area and expression of inflammation-, cell proliferation- and survival-related proteins. Overall, our findings demonstrated the promise of the NNK-LPS-A/J mice model to better understand inflammation-driven lung cancer, dissect the molecular pathways involved and identify more effective preventive and therapeutic agents against lung cancer.
Keywords: 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, animal model, lipopolysaccharide, lung tumor, macrophage
Introduction
Lung cancer is the leading cause of cancer-related mortality in the United States and worldwide.15 Although about 90% of lung cancer cases are attributed to the habit of tobacco smoking, only 10–15% of tobacco smokers develop lung cancer, which suggests the existence of other risk factors independent of smoking. One of these factors is chronic pulmonary inflammation. Indeed, smokers with chronic obstructive pulmonary disease (COPD), the main form of pulmonary inflammatory disease, have up to 5-fold increased risk for lung cancer as compared to smokers without COPD.21 The association between chronic pulmonary inflammation and lung cancer has been ascribed to the co-localization of genes that determine responsiveness to pro-inflammatory toxicants and neoplasia.2
Endogenous as well as exogenous inflammatory stimuli defining the intrinsic and extrinsic inflammatory pathways, respectively, play important roles in the promotion and progression of lung tumorigenesis.1,22 Endogenous inflammatory stimuli are initiated by activation of oncogenes such as K-Ras1 which result in the generation of inflammatory microenvironment in the tumors. The principal exogenous inflammatory stimulus in the lung is tobacco smoke and this inflammatory condition persists even after cessation of smoking,32,35 most likely due to colonization of the lung with respiratory pathogens as a consequence of tobacco smoke-induced disruption of the innate immunity of the lung.30
Although substantial epidemiological evidence indicates an association between pulmonary inflammation and lung cancer, the molecular events underlying inflammation-related lung tumorigenesis are not well known. To this end, a novel animal model is required in which murine lung cancer develops with a background of pulmonary inflammation mimicking human COPD. Such a model would lead not only to a better understanding of how inflammation contributes to lung tumorigenesis but also to the development of preventive and therapeutic drugs against pulmonary inflammation and inflammation-driven lung cancer.
In the present study, we examined whether chronic intranasal administration of lipopolysaccharide (LPS), a major proinflammatory glycolipid component of the gram-negative bacterial outer membrane, enhances 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-induced lung tumorigenesis in mice. Moreover we present preliminary studies on the mechanisms responsible for these effects of LPS. An earlier study has shown that LPS-induced inflammatory and pathologic changes in mouse lung mimic changes observed in human subjects with COPD,33 suggesting the importance of this model to study the role of inflammation in lung tumorigenesis. LPS is ubiquitously present in the environment, including soil, plant, water, dust, and ambient air. In particular, its level in cigarette smoke is high,11 indicating that LPS could have a significant role in tobacco smoke-induced pulmonary inflammation.
Materials and Methods
Lung Tumor Bioassay
Female A/J mice, 5–6 weeks of age, were obtained from The Jackson Laboratory (Bar Harbor, ME). Upon arrival (Week 0), the mice were housed in the specific pathogen-free animal quarters of Research Animal Resources (RAR), University of Minnesota Academic Health Center, randomized into different groups and maintained on AIN-93 pelleted diet. All studies were approved by the Institutional Animal Care and Use Committee. Beginning one week after arrival, mice in groups 3 and 4 (15 mice/group) received intraperitoneal injections of NNK (four injections, twice a week, at a dose of 50 mg/kg in 0.2 ml physiological solution), whereas mice in groups 1 and 2 (10 mice/group) were given physiological saline solution. NNK was synthesized as described elsewhere.12 Beginning one week after the last dose of NNK, mice in groups 2 and 4 received weekly intranasal instillation of LPS (from E. coli O55:B5, Sigma, St Louis, MO) at a dose of 5 µg/mouse in 50 µl phosphate buffered saline. LPS was administered under isofluorane anesthesia for a total of 22 weeks. Mice in group 1 were administered 50 µl PBS in a similar manner. At week 27, the mice were euthanized with carbon dioxide. The lungs were harvested and tumors on the surface of the lung counted and the size of the tumors determined under a dissecting microscope. The left lobes of the lungs were preserved in 10% neutral buffered formalin and used for histopathological studies. Tumors on the right lobes of the lung were microdissected, kept at −80°C and used for subsequent protein and RNA studies. In order to examine the reproducibility of the results, the tumor bioassay was performed twice.
Histopathological Analysis of Lung Tumors
Formalin-fixed lung tissues were processed through a series of graded alcohols, embedded in paraffin and three step sections (each 200 µm apart) having a thickness of 4 µm were cut and stained with hematoxylin and eosin. Proliferative lesions were counted in each step section, and care was taken not to recount large lesions that were present at more than one level.
Proliferative lesions in the lungs were classified as hyperplasia, adenoma, adenoma with dysplasia or adenocarcinoma based on our previous reports7,28 and the recommendations published by the Mouse Models of Human Cancers Consortium25.
Immunohistochemistry for F4/80
F4/80 is a specific macrophage marker. Four µm thick formalin-fixed, paraffin-embedded sections of lung were deparaffinized and rehydrated, followed by treatment with trypsin (Biocare). Immunohistochemistry was performed on a Dako Autostainer using a rat anti-mouse F4/80 antibody (Cedarlane) as primary antibody (after blocking endogenous peroxidase with hydrogen peroxide and applying a protein block), with detection by a Rat on Mouse HRP (horse radish peroxidase)-Polymer kit (Biocare) using diaminobenzidine (Dako) as the chromagen. Mayer’s Hematoxylin (Dako) was used as the counterstain. Mouse spleen was used as a positive control tissue, and for negative control slides the primary antibody was substituted with Super Sensitive Rat Negative Serum (Biogenex). Immunolabelled slides were evaluated using light microscopy.
Macrophage numbers were indirectly assessed by quantifying the extent of F4/80 immunohistochemistry labeling of cells in tissue sections. Digital images of uniform size were taken of randomly selected macrophage-rich areas adjacent to tumors with a Spot Insight 4 MP CCD Scientific Color Digital camera (Diagnostic Instruments) mounted on a Nikon E-800 microscope (Nikon Plan Apo 40×/0.95 lens). A threshold for F4/80 positive labeling was established using Image-Pro Plus version 6.2 (Media Cybernetics), and used to analyze all images. Data were generated from 3 mice/group and 4 microscopic fields/mouse and expressed as the number of positive pixels present per image field.
Western Immunoblotting
Protein samples from mouse lung tissues were prepared as follows. About 30 mg of normal lung tissue (pooled samples from vehicle- or LPS-treated mice, 3 mice each) or microdissected lung tumors (pooled samples from NNK- or NNK plus LPS-treated mice, 3 mice each) were ground using a mortar and pestle and the resulting lung tissue powder homogenized in an ice-cold lysis buffer containing the following constituents: 50 mmol/L Tris-HCl, 150 mmol/L NaCl, 1 mmol/L EGTA, 1 mmol/L EDTA, 20 mmol/L, 1% Triton X-100, pH 7.4, and protease inhibitors [aprotinin (1 µg/mL), leupeptin (1 µg/mL), pepstatin (1 µmol/L), and phenylmethylsulfonyl fluoride (0.1 mmol/L)] and phosphatase inhibitors Na3VO4 (1 mmol/L) and NaF (1 mmol/L). The preparations were centrifuged (14,000 × g for 20 min) and supernatants stored at −80°C.
For Western immunoblotting, 60 µg of protein were loaded onto a 4% to 12% Novex Tris-glycine gel (Invitrogen) and run for 60 min at 200 V. The proteins were then transferred onto a nitrocellulose membrane (Bio-Rad) for 1 h at 30 V. Protein transfer was confirmed by staining membranes with BLOT-FastStain (Chemicon). Subsequently, membranes were blocked in 5% Blotto nonfat dry milk in Tris buffer containing 1% Tween 20 for 1 h and probed overnight with the following primary antibodies obtained from Cell Signaling Technology (Beverly, MA): anti-phospho-Akt, anti-PTEN, anti-phospho-Stat3, anti-phospho-NF-κB, and anti-beta-actin. All primary antibodies were used at a dilution of 1:1,000. After incubating the membranes with a secondary antibody (goat anti-rabbit IgG; 1:5,000) for 1 h, chemiluminescent immunodetection was used. Signal was visualized by exposing membranes to HyBolt CL autoradiography film. All membranes were stripped and reprobed with anti-β-actin (1:1000) to check for differences in the amount of protein loaded in each lane. Three independent Western immunoblotting assays were performed, and each time three different pooled samples from three mice. Blots were quantified by scanning densitometry using UN-SCAN-IT (Silk Scientific Corporation, Orem, UT) normalizing protein expression to the β-actin loading control.
Statistical Analysis
Wilcoxon rank sum test was used for pairwise comparisons of the number of tumors on the surface of the lung (groups treated with NNK plus LPS versus the group treated with NNK only). Since the frequency of the different histopathological lesions (hyperplasia, adenoma, adenoma with progression and adenocarcinoma) was approximately normally distributed, two-sample-t-test was employed for the pairwise comparison of this variable between NNK plus LPS group versus the group treated with NNK only. Two-sided P-values ≤ 0.05 were considered statistically significant. For the analysis of tumor infiltrating macrophages, we used linear mixed model (LMM) after log transformation of pixel counts. Within-animal correlation was handled with exchangeable correlation assumption in the LMM. Restricted maximum likelihood (REML) method was used for model fitting. For the analyses of results from the Western immunoblotting assay, the two-sided Student’s t-test was used. Data are reported as mean±SD of triplicate determinations. P values of < 0.05 were considered statistically significant.
Results
Effects of LPS on NNK-induced Lung Tumorigenesis
In order to determine if LPS enhances lung tumorigenesis, we compared the multiplicity, size and progression of lung tumors in A/J mice treated with NNK or NNK plus LPS in two sets of tumor bioassays. In experiment 1 (Table 1), treatment of A/J mice with NNK alone induced 29.6 ± 9.8 tumors/mouse, whereas chronic administration of LPS to NNK pretreated mice significantly increased the lung tumor multiplicity by 60% to 47.3 ± 16.1 (P < 0.05). Similarly, in experiment 2, NNK alone induced 36.2 ± 4.1 tumors/mouse, whereas chronic administration of LPS to NNK pretreated mice significantly increased the lung tumor multiplicity by 41% to 51.2 ± 4.8 (P < 0.05). The various size classes of lung tumors induced by NNK alone or NNK plus LPS are indicated in Figs. 1a and 1b. Generally, smaller tumors (< 1 mm) were more abundant in the NNK group than in the NNK plus LPS group, whereas larger tumors (> 2 mm) were relatively fewer or absent in the NNK group as compared to the NNK plus LPS group. Representative gross and microscopic images of NNK- and NNK plus LPS-induced lung tumors are depicted in Figs. 2 and 3, respectively. Since the size of human lung tumors has been shown to be related to the degree of malignant progression,34,37 we examined if mice treated with NNK plus LPS harbored more advanced lung tumors than mice treated with NNK alone. As shown in Table 2, multiplicities of more advanced lesions (adenoma with dysplasia and adenocarcinoma) were significantly increased in the NNK plus LPS group as compared to that of the NNK group. In contrast, multiplicities of less advanced lesions (hyperplastic foci and adenoma) were similar in the two groups. A photomicrograph of an adenocarcinoma from the NNK plus LPS group that has invaded the airways is shown in Fig. 4.
Table 1.
Effects of chronic LPS administration on NNK-induced lung tumors in A/J micea
| Group | Treatment | No. of mice |
Mean body weight at termination |
Lung tumors | |||
|---|---|---|---|---|---|---|---|
| Tumor incidence (%) |
Tumors/mouse (Mean ± SD) |
Increase in tumor multiplicity (%) |
Pb | ||||
| Experiment 1 | |||||||
| 1 | Vehicle control | 10 | 24.2 ±.1.4 | 20 | 0.2 ± 0.4 | ||
| 2 | LPS | 10 | 21.7± 1.8 | 20 | 0.2 ± 0.4 | ||
| 3 | NNK | 15 | 23.5 ± 1.2 | 100 | 29.6 ± 9.8 | ||
| 4 | NNK plus LPS | 15 | 22.2 ± 1.6 | 100 | 47.3 ± 16.1 | 160b | < 0.05 |
| Experiment 2 | |||||||
| 1 | Vehicle control | 10 | 23.9 ± 2.7 | 10 | 0.1 ± 0.3 | ||
| 2 | LPS | 10 | 23.4 ± 1.8 | 20 | 0.2 ± 0.4 | ||
| 3 | NNK | 15 | 24.2 ± 2.1 | 100 | 36.2 ± 4.1 | ||
| 4 | NNK + LPS | 15 | 23.7 ± 1.6 | 100 | 51.2 ± 4.8 | 141b | < 0.05 |
Beginning at age 6–7 weeks, groups of female A/J mice received intraperitoneal injection of NNK (four doses of 50 mg/kg, twice a week in 0.2 ml physiological saline solution). LPS was administered weekly by intranasal instillation (5 µg/mouse in 50 µl physiological saline solution, 25 µl in each nostril) beginning one week after the last dose of NNK until termination of the study at week 27.
Compared to the tumor multiplicity in the NNK group, group 3.
Figure 1.

Number of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)- or NNK plus lipopolysaccharide (LPS)-induced lung tumors in each size range. 1a, Experiment 1; 1b, Experiment 2. * p < 0.05, compared to the group treated with NNK plus LPS (for tumors < 1 mm) or compared to the group treated with NNK (for tumors ≥ 1mm).
Figure 2.

Lung; mouse No. 4 (a), mouse No. 1 (b), mouse No. 6 (c). Representative gross images from vehicle-treated (a), NNK-treated (b), and NNK plus LPS-treated (c) mice showing a lower number and size of tumors in NNK-treated mice as compared to that of NNK + LPS-treated mice.
Figure 3.

Cross-section of the lung; mouse No. 1 (a), mouse No. 6 (b). The number and size of lung tumors was lower in the NNK group (a) relative to the NNK plus LPS group (b). Hematoxylin and eosin. * is an adenocarcinoma.
Table 2.
Effect of LPS administration on NNK-induced microscopic lung lesionsa
| Group | Treatment | Hyperplastic foci/mouse |
Adenoma/ mouse |
Adenoma with cellular pleomorphism/mouse |
Adenocarcinoma/ mouse |
|---|---|---|---|---|---|
| 1 | NNK | 3.0 ± 1.2 | 7.1 ± 3.6 | 1.3 ± 1.1 | 0.1 ± 0.4 |
| 2 | NNK plus LPS | 3.1 ± 1.8 | 11.8 ± 5.2 | 3.9 ± 1.8b | 0.6 ± 0.7b |
Groups of female A/J mice received NNK or NNK plus LPS as described in Table 1. Upon termination of the study, the left lobe of the lung was preserved in 10% buffered formalin and multiplicities of the different pulmonary lesions induced by NNK or NNK plus LPS were determined as described in the Materials and Methods section.
P < 0.05, compared to group 1.
Figure 4.
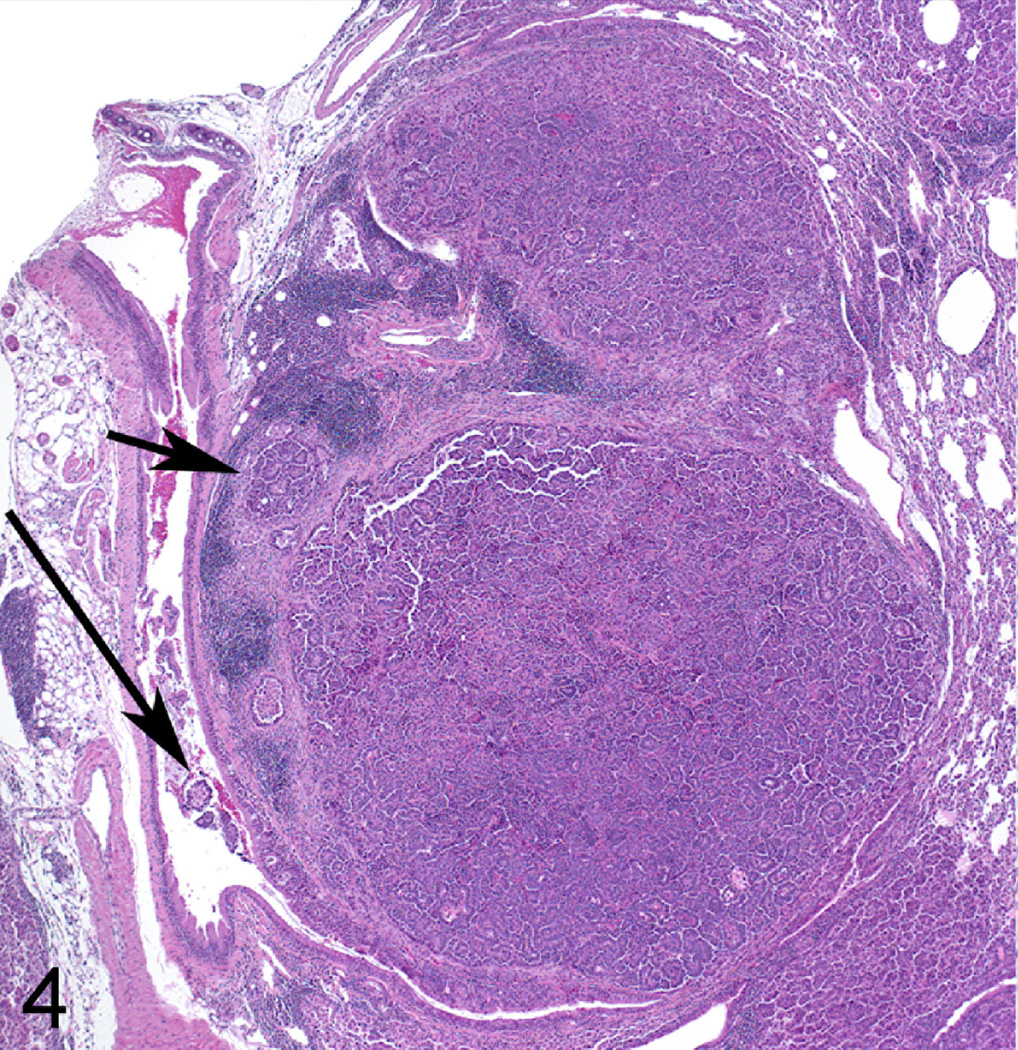
Lung adenocarcinoma; mouse No. 6. Tumor labeled * in Figure 3, showing invasion of airway and surrounding tissue (long and short arrows, respectively). Hematoxylin and Eosin.
The multiplicity of lung tumors in mice treated with the vehicle was in the range of historical data of lung tumor multiplicity in this strain (a maximum of 1–2 lung tumors/mouse). A/J mice are among the most sensitive to develop both spontaneous and chemically-induced lung tumors, due to a polymorphism in intron 2 of Kras, which influences mutational activation of K-ras oncogene and susceptibility to lung cancer.20 In spontaneously developing lung tumors in A/J mice, the tumors were always adenomas. The lung tumor multiplicity of mice treated with LPS alone was similar to that of the vehicle control group (Table 1), indicating that in this model, LPS alone is not tumorigenic. The percentage of mice that developed lung tumors in NNK and NNK plus LPS groups was similar. This result was not unexpected since the most sensitive indicator in the A/J mouse lung tumorigenesis model is tumor multiplicity.
Effects of LPS on Macrophage Recruitment
Since macrophages play a critical role in promoting lung tumorigenesis37 and LPS is a potent inducer of macrophage activation,23 we sought to examine the possibility of increased recruitment of macrophages to the lung tumors. As assessed both qualitatively and quantitatively and shown in Figs. 5 and 6, respectively, LPS treatment dramatically increased the recruitment of macrophages to NNK-induced lung tumors, as determined by immunolabelling for F4/80, a macrophage-specific marker in the mouse. Interestingly, in both groups of mice, macrophages were located predominantly at the periphery of the tumors, and did not infiltrate the tumor parenchyma much. Also, lung tissues from LPS-treated mice exhibited a higher number of macrophages as compared to lungs from vehicle-treated mice (data not shown).
Figure 5.

Lung; mouse No. 1 (a), mouse No. 6 (b). Tumors from NNK-treated mice (a) exhibited a lower level of macrophage recruitment to tumors, as revealed by decreased F4/80 detection, a specific mouse macrophage marker, than tumors from mice treated with NNK plus LPS (b). Diaminobenzidine (chromagen) and Mayer’s Hematoxylin (counterstain).
Figure 6.

Comparative macrophage frequency in lung tumors induced by NNK or NNK plus LPS. Data were generated by counting the number of positive pixel counts (number of pixel columns multiplied by the number of pixel rows), present per image field. * p < 0.05, compared to the group treated with NNK.
Enhanced Activation of Inflammation-, Cell Proliferation- and Survival-related Proteins by LPS
Engagement of TLR4, a LPS sensing receptor, has been shown to phosphorylate various intracellular kinases, including PI3K and a variety of transcription factors such as NF-κB, and STAT3 in macrophages, monocytes and epithelial cells.4,9,36 To shed some light on the potential mechanisms through which LPS enhanced NNK-induced lung tumorigenesis, we determined the relative activation of Akt, NF-κB and STAT3 and expression of PTEN in lung tissues of vehicle-, LPS-, NNK-, or NNK plus LPS-treated mice. These results are depicted in Figs. 7a and 7b. In both NNK- and LPS- treated mice, levels of activated Akt, NF-κB and STAT3 were significantly higher as compared to the expression of these proteins in vehicle-treated mice; PTEN expression was significantly altered only in the in NNK-treated group. Chronic administration of LPS to NNK-pretreated mice further enhanced levels of activated Akt, NF-κB, and STAT3 expression but decreased PTEN expression.
Figure 7.

a. Immunoblots showing modulation of phospho-Akt (P-Akt), phospho-nuclear factor-KappaB (P-NF-κB), phospho-signal transducer and activator of transcription 3 (P-STAT3) and phosphatase and tensin homolog (PTEN) expression levels in lung tissues of mice treated with LPS, NNK or NNK plus LPS. The blot was stripped and re-probed with anti β-Actin antibody to correct for differences in protein loading. Figure 7b. Quantitative data showing P-Akt, P-NF-κB, P-STAT3 and PTEN levels in the different groups. Data were generated from three independent experiments. * p < 0.05, compared to the vehicle group; ** p < 0.05 compared to the NNK group.
Discussion
The principal aim of the present study was to develop a facile mouse model of inflammation-driven lung tumorigenesis. Development of such models could contribute towards better understanding of the association between pulmonary inflammation and lung cancer, and lead to the identification of more effective chemopreventive and therapeutic agents for pulmonary inflammation and lung cancer. We found that chronic exposure of NNK-pretreated A/J mice to LPS enhanced lung tumorigenesis, as evidenced by increased lung tumor multiplicity, size and tumor progression, with enhanced recruitment of macrophages, and activation of inflammation-and cell proliferation and survival-related proteins.
Although inflammation is an integral component of body defense, non-regulated inflammation is detrimental causing, among other effects, genetic and epigenetic alterations leading to cancer.6,14,24,38 Indeed, according to epidemiological studies, about 25% of human cancers are attributed to chronic inflammation.6 Among human cancers linked to chronic inflammation are liver, colon, prostate, gastric, esophageal, lung and cervical cancers, which are associated, respectively, with viral hepatitis, inflammatory bowel disease, prostatitis, helicobacter pylori infection, gastroesophageal reflux disease, cigarette smoking, and human papilloma virus.6,14,21
The establishment of animal models of inflammation-driven cancers has helped to recapitulate features of these diseases in humans. For instance, the immune dysregulated model or the azoxymethane-dextran sulfate sodium model for colon cancer have shown great promise to study the molecular events taking place during human inflammatory bowel disease (IBD) and IBD-associated colon cancer.26 Currently, there are no such well-established models for inflammation-driven lung cancer. Moghaddam et al. showed that exposure of mice to aerosolized Haemophilus influenzae lysate elicited airway inflammation with a cellular and cytokine profile similar to that in COPD and promoted lung cancer.24 Also, Keohavong and colleagues exposed NNK-pretreated FVB/N mice to LPS to examine if the latter enhances lung tumorigenesis.17 However, whereas the Haemophilus influenzae lysate model is technically complex, the FVB/N model is not ideal for the development of chemopreventive/therapeutic agents since these mice poorly sensitive (less than 50% of carcinogen-treated mice develop tumors) to develop lung tumors even when using extremely high doses of carcinogens. Moreover, since FVB/N mice are highly susceptible to asthma-like airway responsiveness with significant generation of antigen-specific IgE, it would be difficult to discern the effect of LPS-induced inflammation.
Among critical players of inflammation-driven tumorigenesis are PI3K/Akt, NF-κB and STAT3 signaling pathways.3,6,14,19 These signaling pathways positively regulate cell proliferation, survival, invasion, and angiogenesis, thereby enhancing tumorigenesis. In the present study, all three pathways were more activated in NNK plus LPS-induced lung tumors as compared to the level in NNK-induced lung tumors, which was in line with the higher lung tumor size and multiplicity in NNK plus LPS-induced lung tumors. Both NNK and LPS, individually, are known to positively regulate Akt, NF-κB and STAT3 activation4,5,,8–10,13,27,31 and therefore the dramatic activation of these proteins in NNK plus LPS-induced lung tumors could be due to additive/synergistic effects of the agents. Activation of the PI3K/Akt signaling pathway is negatively regulated by the tumor suppressor protein PTEN, which, in the preset study, was markedly down-regulated in NNK plus LPS-induced lung tumors, which may have contributed to increased activation of Akt, a well-established positive regulator of the NF-κB pathway.9,10 There is also a close interplay between NF-κB and STAT3 since maintenance of constitutively elevated NF-κB activity requires STAT3,19 and activation of NF-κB controls activation of STAT3 through upregulation of IL-6 expression.3 Overall, PI3K/Akt, NF-κB and STAT3 signaling pathways could play critical roles in the enhancement by LPS of NNK-induced lung tumorigenesis.
LPS is ubiquitously present in the environment. In humans, it is an important cause of airway inflammation among agricultural and textile workers.28 However, the most important source of human exposure to LPS is cigarette smoke. Smoking one pack of cigarettes/day exposes to about 2.5 µg LPS11,18, a dose of respirable LPS that is comparable to the levels of LPS associated with adverse health effects in cotton textile workers.16 Thus LPS could be one of the major contributors of inflammatory conditions induced by cigarette smoke. In the present study, the dose of LPS used to induce pulmonary inflammation (5 µg/mouse) was equivalent to the amount of LPS smokers receive upon smoking 2 packs of cigarettes/day.11 Given the similarity between LPS-induced inflammatory and pathologic changes in mice human COPD in smokers32 and that COPD is an important risk factor for lung cancer, our model could be an ideal model for inflammation-driven lung cancer in humans.
In conclusion, in the present study, we clearly showed enhancement of lung tumorigenesis by LPS-induced pulmonary inflammation. Since tobacco smoke contains several inflammatory compounds in addition to carcinogens, the existing carcinogen only-based animal models of lung cancer may not fully reflect the situation in smokers. Therefore, further development of this model could better reflect the exposure scenario in smokers and lead to the development of more effective chemopreventive and chemotherapeutic agents against lung cancer.
Acknowledgements
Funding: NIH grant (8K01OD010970) to TM and faculty start up fund to FK from Masonic Cancer Center and College of Veterinary Medicine, University of Minnesota.
Footnotes
Conflict of Interest Statement:
None declared.
References
- 1.Basseres DS, Ebbs A, Levantini E, Baldwin AS. Requirement of the NF-kappaB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res. 2010;70(9):3537–3546. doi: 10.1158/0008-5472.CAN-09-4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bauer AK, Malkinson AM, Kleeberger SR. Susceptibility to neoplastic and non-neoplastic pulmonary diseases in mice: genetic similarities. Am J Physiol Lung Cell Mol Physiol. 2004;287(4):L685–L703. doi: 10.1152/ajplung.00223.2003. [DOI] [PubMed] [Google Scholar]
- 3.Bollrath J, Greten FR. IKK/NF-kappaB and STAT3 pathways: central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep. 2009;2009;10(12):1314–1319. doi: 10.1038/embor.2009.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown J, Wang H, Hajishengallis GN, Martin M. TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. J Dent Res. 2011;90(4):417–427. doi: 10.1177/0022034510381264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen ZB, Liu C, Chen FQ, Li SY, Liang Q, Liu LY. Effects of tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) on the activation of ERK1/2 MAP kinases and the proliferation of human mammary epithelial cells. Environ Toxicol Pharmacol. 2006;22(3):283–291. doi: 10.1016/j.etap.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;2009;30(7):1073–1081. doi: 10.1093/carcin/bgp127. [DOI] [PubMed] [Google Scholar]
- 7.Dagne A, Melkamu T, Schutten MM, Qian X, Upadhyaya P, Luo X, Kassie F. Enhanced inhibition of lung adenocarcinoma by combinatorial treatment with indole-3-carbinol and silibinin in A/J mice. Carcinogenesis. 2011;32(4):561–567. doi: 10.1093/carcin/bgr010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ehlting C, Ronkina N, Böhmer O, Albrecht U, Bode KA, Lang KS, et al. Distinct functions of the mitogen-activated protein kinase-activated protein (MAPKAP) kinases MK2 and MK3: MK2 mediates lipopolysaccharide-induced signal transducers and activators of transcription 3 (STAT3) activation by preventing negative regulatory effects of MK3. J Biol Chem. 2011;286(52):24113–24124. doi: 10.1074/jbc.M111.235275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell Signal. 2001;13(2):85–94. doi: 10.1016/s0898-6568(00)00149-2. [DOI] [PubMed] [Google Scholar]
- 10.Halappanavar S, Russell M, Stampfli MR, Williams A, Yauk CL. Induction of the interleukin 6/ signal transducer and activator of transcription pathway in the lungs of mice sub-chronically exposed to mainstream tobacco smoke. BMC Med Genomics. 2009;2:56. doi: 10.1186/1755-8794-2-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hasday JD, Bascom R, Costa JJ, Fitzgerald T, Dubin W. Bacterial endotoxin is an active component of cigarette smoke. Chest. 1999;115(3):829–835. doi: 10.1378/chest.115.3.829. [DOI] [PubMed] [Google Scholar]
- 12.Hecht SS, Lin D, Castonguay A. Effects of α-deuterium substitution on the mutagenicity of 4-(methyl-nitrosamino)-1-(3-pyridyl)-1-butanone (NNK) Carcinogenesis. 1983;4:305–310. doi: 10.1093/carcin/4.3.305. [DOI] [PubMed] [Google Scholar]
- 13.Ho YS, Chen CH, Wang YJ, Pestell RG, Albanese C, Chen RJ, et al. Tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) induces cell proliferation in normal human bronchial epithelial cells through NFkappaB activation and cyclin D1 up-regulation. Toxicol Appl Pharmacol. 2005;205(2):133–148. doi: 10.1016/j.taap.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 14.Hussain SP, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer. 2007;121(11):2373–2380. doi: 10.1002/ijc.23173. [DOI] [PubMed] [Google Scholar]
- 15.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 16.Kennedy SM, Christiani DC, Eisen EA, Wegman DH, Greaves IA, Olenchock SA, Ye TT, Lu PL. Cotton dust and endotoxin exposure-response relationships in cotton textile workers. Am Rev Respir Dis. 1987;135(1):194–200. doi: 10.1164/arrd.1987.135.1.194. [DOI] [PubMed] [Google Scholar]
- 17.Keohavong P, Kahkonen B, Kinchington E, Yin J, Jin J, Liu X, Siegfried JM, DI YP. K-ras mutations in lung tumors from NNK-treated mice with lipopolysaccharide-elicited lung inflammation. Anticancer Res. 2011;31(9):2877–2882. [PubMed] [Google Scholar]
- 18.Larsson L, Pehrson C, Dechen T, Crane-Godreau M. Microbiological components in mainstream and sidestream cigarette smoke. Tob Induc Dis. 2012;10(1):13. doi: 10.1186/1617-9625-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee H, Herrmann A, Deng JH, Kujawski M, Niu G, Li Z, Forman S, Jove R, Pardoll DM, Yu H. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell. 2009;15(4):283–293. doi: 10.1016/j.ccr.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malkinson AM, You M. The intronic structure of cancer-related genes regulates susceptibility to cancer. Mol. Carcinog. 1994;10(2):61–65. doi: 10.1002/mc.2940100202. [DOI] [PubMed] [Google Scholar]
- 21.Mannino DM, Aguayo SM, Petty TL, Redd SC. Low lung function and incident lung cancer in the United States: data from the First National Health and Nutrition Examination Survey follow-up. Arch Intern Med. 2003;163(12):1475–1480. doi: 10.1001/archinte.163.12.1475. [DOI] [PubMed] [Google Scholar]
- 22.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 23.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 24.Moghaddam SJ, Ochoa CE, Sethi S, Dickey BF. Nontypeable Haemophilus influenzae in chronic obstructive pulmonary disease and lung cancer. Int J Chron Obstruct Pulmon Dis. 2011;6:113–123. doi: 10.2147/COPD.S15417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nikitin AY, Alcaraz A, Anver MR, Bronson RT, Cardiff RD, Dixon D, et al. Classification of proliferative pulmonary lesions of the mouse: recommendations of the mouse models of human cancers consortium. Cancer Res. 2004;64(7):2307–2316. doi: 10.1158/0008-5472.can-03-3376. [DOI] [PubMed] [Google Scholar]
- 26.Ock CY, Kim EH, Hong H, Hong KS, Han YM, Choi KS, Hahm KB, Chung MH. Prevention of colitis-associated colorectal cancer with 8-hydroxydeoxyguanosine. Cancer Prev Res. 2011;4(9):1507–1521. doi: 10.1158/1940-6207.CAPR-11-0161. [DOI] [PubMed] [Google Scholar]
- 27.Ojaniemi M, Glumoff V, Harju K, Liljeroos M, Vuori K, Hallman M. Phosphatidylinositol 3-kinase is involved in Toll-like receptor 4-mediated cytokine expression in mouse macrophages. Eur J Immunol. 2003;33(3):597–605. doi: 10.1002/eji.200323376. [DOI] [PubMed] [Google Scholar]
- 28.Qian X, Melkamu T, Upadhyaya P, Kassie F. Indole-3-carbinol inhibited tobacco smoke carcinogen-induced lung adenocarcinoma in A/J mice when administered during the postinitiation or progression phase of lung tumorigenesis. Cancer Lett. 2011;311(1):57–65. doi: 10.1016/j.canlet.2011.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwartz DA, Thorne PS, Yagla SJ, Burmeister LF, Olenchock SA, Watt JL, Quinn TJ. The role of endotoxin in grain dust-induced lung disease. Am. J Respir Crit Care Med. 1995;152(2):603–608. doi: 10.1164/ajrccm.152.2.7633714. [DOI] [PubMed] [Google Scholar]
- 30.Sethi S, Maloney J, Grove L, Wrona C, Berenson CS. Airway Inflammation and bronchial bacterial colonization in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173(9):991–998. doi: 10.1164/rccm.200509-1525OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsurutani J, Castillo SS, Brognard J, Granville CA, Zhang C, Gills JJ, Sayyah J, Dennis PA. Tobacco components stimulate Akt-dependent proliferation and NFkappaB-dependent survival in lung cancer cells. Carcinogenesis. 2005;26(7):1182–1195. doi: 10.1093/carcin/bgi072. [DOI] [PubMed] [Google Scholar]
- 32.Turato G, Di Stefano A, Maestrelli P, Mapp CE, Ruggieri MP, Roggeri A, Fabbri LM, Saetta M. Effect of smoking cessation on airway inflammation in chronic bronchitis. Am J Respir Crit Care Med. 1995;152(4 Pt 1):1262–1267. doi: 10.1164/ajrccm.152.4.7551380. [DOI] [PubMed] [Google Scholar]
- 33.Vernooy JH, Dentener MA, van Suylen RJ, Buurman WA, Wouters EF. Long term intratracheal lipopolysaccharide exposure in mice results in chronic lung inflammation and persistent pathology. Am J Respir Cell Mol Biol. 2002;26(1):152–159. doi: 10.1165/ajrcmb.26.1.4652. [DOI] [PubMed] [Google Scholar]
- 34.Wisnivesky JP, Yankelevitz D, Henschke CI. Stage of lung cancer in relation to its size: part 2. Evidence. Chest. 2005;127(4):1136–1139. doi: 10.1378/chest.127.4.1136. [DOI] [PubMed] [Google Scholar]
- 35.Wright JL, Lawson LM, Pare PD, Wiggs BR, Kennedy S, Hogg JC. Morphology of peripheral airways in current smokers and ex-smokers. Am Rev Respir Dis. 1983;127(4):474–477. doi: 10.1164/arrd.1983.127.4.474. [DOI] [PubMed] [Google Scholar]
- 36.Wu YL, Kou YR, Ou HL, Chien HY, Chuang KH, Liu HH, Lee TS, Tsai CY, Lu ML. Glucosamine regulation of LPS-mediated inflammation in human bronchial epithelial cells. Eur J Pharmacol. 2010;635(1–3):219–226. doi: 10.1016/j.ejphar.2010.02.044. [DOI] [PubMed] [Google Scholar]
- 37.Yang F, Chen H, Xiang J, Zhang Y, Zhou J, Hu H, Zhang J, Luo X. Relationship between tumor size and disease stage in non-small cell lung cancer. BMC Cancer. 2010;10:474. doi: 10.1186/1471-2407-10-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zaynagetdinov R, Sherrill TP, Polosukhin VV, Han W, Ausborn JA, McLoed AG, McMahon FB, Gleaves LA, Degryse AL, Stathopoulos GT, Yull FE, Blackwell TS. A critical role for macrophages in promotion of urethane-induced lung carcinogenesis. J Immunol. 2011;187(11):5703–5711. doi: 10.4049/jimmunol.1100558. [DOI] [PMC free article] [PubMed] [Google Scholar]