

Abstract
Decreased expression of cardiac myosin binding protein-C (cMyBP-C) in the myocardium is thought to be a contributing factor to hypertrophic cardiomyopathy in humans, and the initial molecular defect is likely abnormal cross-bridge (XB) function which leads to impaired force generation, decreased contractile performance, and hypertrophy in vivo. The myosin activator omecamtiv mecarbil (OM) is a pharmacological drug that specifically targets the myosin XB and recent evidence suggests that OM induces a significant decrease in vitro motility velocity and an increase in the XB duty cycle. Thus, the molecular effects of OM maybe beneficial in improving contractile function in skinned myocardium lacking cMyBP-C because absence of cMyBP-C in the sarcomere accelerates XB kinetics and enhances XB turnover rate, which presumably reduces contractile efficiency. Therefore, parameters of XB function were measured in skinned myocardium lacking cMyBP-C prior to and following OM incubation. We measured ktr, the rate of force redevelopment as an index of XB transition from both the weakly- to strongly-bound state and from the strongly- to weakly-bound states and performed stretch activation experiments to measure the rates of XB detachment (krel) and XB recruitment (kdf) in detergent-skinned ventricular preparations isolated from hearts of wild-type (WT) and cMyBP-C knockout (KO) mice. Samples from donor human hearts were also used to assess the effects of OM in cardiac muscle expressing a slow β-myosin heavy chain (β-MHC). Incubation of skinned myocardium with OM produced large enhancements in steady-state force generation which were most pronounced at low levels of [Ca2+] activations, suggesting that OM cooperatively recruits additional XB’s into force generating states. Despite a large increase in steady-state force generation following OM incubation, parallel accelerations in XB kinetics as measured by ktr were not observed, and there was a significant OM-induced decrease in krel which was more pronounced in the KO skinned myocardium compared to WT skinned myocardium (58% in WT vs. 76% in KO at pCa 6.1), such that baseline differences in krel between KO and WT skinned myocardium were no longer apparent following OM-incubation. A significant decrease in the kdf was also observed following OM incubation in all groups, which may be related to the increase in the number of cooperatively recruited XB’s at low Ca2+-activations which slows the overall rate of force generation. Our results indicate that OM may be a useful pharmacological approach to normalize hypercontractile XB kinetics in myocardium with decreased cMyBP-C expression due to its molecular effects on XB behavior.
Keywords: Omecamtiv mecarbil, cMyBP-C, contractile function, XB detachment, XB recruitment
INTRODUCTION
Systolic dysfunction is a major cause of heart failure, and is characterized by reduced pumping ability of the heart and a significant reduction in the left ventricular (LV) ejection fraction [1]. The molecular basis of systolic heart failure is impaired myocyte contractile function at the sarcomere level, and specifically, the actomyosin cross-bridge (XB) cycle, which is the fundamental process that determines both the rate and magnitude of force generation in cardiac muscle [2]. Recent efforts to enhance myocardial contractility are directed toward targeting two of the elementary processes that occur during the XB cycle [2]. These efforts at the level of cardiac sarcomere involve: a) promoting the intrinsic rate of the transition of XBs from their weakly- to the strongly-bound, force-generating states, and b) reducing the amount of ADP release during the XB cycle in order to lower the energetic costs associated with the force-generating acto-myosin interactions. Thus, the cardiac sarcomere is considered as a primary target for therapeutic interventions [3–7] to induce improvements in cardiac contractility and in vivo heart function using both pharmacological [8–10] and gene therapy [11–16] approaches.
At the level of the sarcomere, one of the key proteins that regulate the dynamics of XB cycle is the cardiac myosin binding protein-C (cMyBP-C) [17–24]. Previously, studies have shown that skinned murine myocardium lacking cMyBP-C (KO) displayed significantly accelerated rates of XB detachment and recruitment leading to an overall acceleration in the XB cycling kinetics [25]. Accelerated XB cycling kinetics and mechanical dysfunction were also evident in the myocardium expressing even modest reductions in cMyBP-C expression (cMyBP-C+/−) [26, 27]. Furthermore, accelerated XB kinetics have also been reported in skinned myocardium isolated from patients with similar deficits in cMyBP-C expression [28]. At the whole-heart level, the hyper contractile KO hearts displayed aberrant contractile efficiency as indicated by a reduced ejection fraction and an abbreviated ejection time [15, 27, 29]. cMyBP-C+/− hearts displayed more modest systolic dysfunction and hypertrophy manifested as an elevated end-diastolic pressure and decreased peak rate of LV pressure rise [26, 27].
We recently showed that in vivo reconstitution of cMyBP-C by gene transfer in KO hearts improved the in vivo systolic function and reduced cardiac hypertrophy [15]. Improved cardiac performance was primarily due to a normalization of XB behavior in the hypercontractile KO sarcomere due to increased cMyBP-C expression, which markedly slowed the rates of XB cycling [15]. Recently, omecamtiv mecarbil (OM), a cardiac myosin activator, has been shown to improve systolic function in the failing hearts [10, 30], by enhancing XB-mediated force generation via enhancing the rate of transition of XB’s from the weakly-bound to the strongly-bound state [8], and decreases actomyosin in vitro motility velocity thereby increasing the overall XB duty cycle of the myosin motor [31]. Based on these findings, we investigated the utility of OM as a pharmacological approach to correct the molecular defects in the KO sarcomere which result in accelerated XB kinetics. Our findings indicate that incubation of skinned myocardium with OM significant slowed XB kinetics in both wild-type (WT) and KO skinned myocardium. In particular, the acceleration of XB kinetics due to cMyBP-C ablation was largely blunted by OM incubation, suggesting that at the molecular level, OM may normalize the hypercontractile sarcomere.
MATERIALS AND METHODS
Ethical approval, animal incubation protocols, and procurement of donor human cardiac tissue samples
This study was performed as per the protocols given in the Guide for the Care and Use of Laboratory Animals and as per the guidelines of the Institutional Animal Care and Use Committee at Case Western Reserve University. Mice of either sex, aged 3–6 months (SV/129 strain), were used for the experiments. KO mice used in this study were previously generated and well-characterized [32]. WT mice expressing normal, full-length cMyBP-C in the myocardium were used as controls. Left ventricular (LV) human cardiac tissue samples were obtained from the donor hearts and were used as controls that predominantly express a β-myosin heavy chain (β-MHC) along with the controls that predominantly express an α-MHC (murine myocardium). The three donor human cardiac samples used in our experiments were collected from cases of death due to brain injury, cervical fracture, and due to Guillain-Barre syndrome. Samples from the same hearts have been also used in previous studies [28, 33, 34] and are well characterized. Human cardiac samples were collected as per the approved guidelines of the University of Sydney. Immediately after their collection, the donor tissue samples were frozen in liquid nitrogen and stored at −80°C until further use.
Estimation of phosphorylation status of sarcomeric proteins in WT and KO heart samples
Cardiac myofibrils were isolated from frozen mouse ventricles [35]. In brief, a piece of the frozen tissue was thawed in a fresh relaxing solution, homogenized, and the myofibrils were then skinned for 15 minutes with 1% Triton X-100 [26]. Skinned myofibrils were then resuspended in fresh relaxing solution containing protease and phosphatase inhibitors (PhosSTOP and cOmplete ULTRA Tablets; Roche Applied Science, Indianapolis, IN, USA) and stored on ice. To determine the myofilament protein phosphorylation status, ventricular samples were solubilized by adding Laemmli buffer and were heated to 90°C for 5 minutes. For Western blot analysis, 2.5μg of solubilized myofibrils were loaded onto a 4–20% Tris-glycine gel (Lonza Walkersville Inc., Rockland, ME, USA), transferred to PVDF membrane, and incubated overnight with one of the following primary antibodies: total troponin I (TnI), TnI phospho-serine 23 and 24 (detects phosphorylation of Ser23 and Ser24 of TnI), total cMyBP-C, cMyBP-C phospho-serine 273, 282, or 302 (detects phosphorylation of cMyBP-C, Ser273, Ser282, or Ser302), or HSC70. For Pro-Q phosphoprotein analysis, 2.5μg of solubilized cardiac myofibril samples were electrophoretically separated at 180V for 85 minutes, then fixed and stained with Pro-Q diamond phosphoprotein stain (Invitrogen, Carlsbad, CA, USA) to assess the phosphorylation status of sarcomeric proteins. The same procedure was followed for testing the phosphorylation status in samples treated with OM. Densitometric scanning of the stained gels was performed using Image J software (U.S. National Institutes of Health, Bethesda, MD, USA) [35].
Preparation of skinned ventricular myocardial preparations and Ca2+ solutions for experiments
Skinned ventricular myocardial preparations were prepared as described previously [26, 35]. In brief, ventricular tissue was homogenized in a relaxing solution followed by detergent-skinning for 60 minutes using 1% Triton-X 100. Multicellular preparations measuring ~100μm in width and 400μm in length were selected for the experiments. The composition of various Ca2+ activation solutions used for the experiments was calculated using a computer program [36] and using the established stability constants [37]. All solutions contained the following (in mM): 100 N, N-bis (2-hydroxyethyl)-2-aminoethanesulfonic acid (BES), 15 creatine phosphate, 5 dithiothreitol, 1 free Mg2+, and 4 MgATP. The maximal activating solution (pCa 4.5; pCa = −log [Ca2+]free) also contained 7 EGTA and 7.01 CaCl2; while the relaxing solution (pCa 9.0) contained 7 EGTA and 0.02 CaCl2; and the pre-activating solution contained 0.07 EGTA. The pH of the Ca2+ solutions was set to 7.0 with KOH and the ionic strength of the Ca2+ solutions was 180 mM. A range of pCa solutions (pCa 6.6 to 5.5), containing varying amounts of [Ca2+]free, were then prepared by mixing appropriate volumes of pCa 9.0 and 4.5 stock solutions and all the experiments were carried out at 22°C.
Experimental apparatus for the estimation of isometric force generation and force-pCa relationships in the skinned myocardium
Detergent-skinned ventricular preparations were held between a motor arm (312C; Aurora Scientific Inc., Aurora, Ontario, Canada) and a force transducer (403A; Aurora Scientific Inc.) as described previously [15, 26]. Changes in the motor position and signals from the force transducer were sampled at 2.0 kHz using sarcomere length (SL) control software program [38]. For all mechanical measurements, SL of the ventricular preparations was set to 2.1μm [26, 27]. Force-pCa relationships were generated by incubating skinned myocardium in a range of pCa solutions (i.e., 6.6 to 4.5). The apparent cooperativity of force development was estimated from the steepness of a Hill plot transformation of the force-pCa relationships. The force-pCa data were fit using the equation P/Po = [Ca2+]nH/(knH + [Ca2+]nH), where nH is the Hill coefficient and k is the pCa required to produce half-maximal activation (i.e., pCa50) [35].
Preparation of OM solution for incubating the myocardial preparations
OM was procured from Selleckchem (Houston, TX, USA) and was dissolved in DMSO (as per the manufacturer’s instructions). OM stock solution was added to a relaxing solution to prepare a final concentration of 1μM OM. The final concentration of DMSO in our solutions is 0.00625%, which has negligible effects on cardiac contractile function [39]. Basal contractile function was first measured in pCa solutions ranging from 6.6 to 4.5 following which the myocardial preparations were incubated for 2 minutes in a relaxing solution containing 1μM OM. This incubation was followed by repeating the measurement of contractile function in the same myocardial preparation in pCa solutions ranging from 6.6 to 4.5. Measurements for force-pCa relationships were made at various levels of activator [Ca2+] after a 2-minute incubation of the skinned myocardial preparations with OM.
Measurement of the rate of force redevelopment (ktr)
ktr was measured in the myocardial preparations to assess XB transitions from both the weakly- to strongly-bound state and from the strongly- to weakly-bound states [40, 41]. A mechanical slack-restretch protocol was used to measure ktr in the Ca2+-activated myocardial preparations as described earlier [26, 42, 43]. Skinned muscle preparations were transferred from relaxing (pCa 9.0) to activating Ca2+ solutions (pCa ranging from 6.2 to 5.9), and once the myocardial preparations attained a steady-state isometric force, they were rapidly slackened by 20% of their original muscle length and were held for 10ms. The slackening was followed by a brief period of unloaded shortening which causes a rapid decline in force because of the detachment of the strongly-bound XBs. The myocardial preparations were then rapidly restretched back to their original length and the time course of force redevelopment was measured. ktr was estimated by linear transformation of the half-time of force redevelopment, i.e., ktr = 693/t1/2, where t1/2 is the time (in milliseconds) taken to reach the half maximal force of the ktr trace as described previously [15, 26, 43–45]. Baseline force is considered the point on the ktr trace where force begins to redevelop following the slack-restretch maneuver, and peak force development is considered the point in the ktr trace in which force plateaus and reaches a steady-state level.
Stretch activation experiments to measure the rates of XB detachment, XB recruitment, and XB stiffness
Stretch activation experiments were performed as described earlier [26, 35, 46, 47]. Myocardial preparations were placed in Ca2+ solutions and were allowed to attain a steady-state force. Myocardial preparations were then rapidly stretched by 2% of their initial muscle length, held at the new length for 5 seconds and were then returned back to their initial muscle length. The characteristic features of the stretch activation responses in cardiac muscle have been described earlier [48, 49], and the stretch activation parameters measured are shown in Fig. 4A. In brief, a sudden 2% stretch of muscle length causes an instantaneous rise in force (P1) produced by the myocardial preparation, which is due to the strain of elastic elements of the strongly-bound XBs (Phase 1). The force then quickly declines (Phase 2) due to a rapid detachment of the strained XBs which equilibrate into a non-force generating state, with a rate constant krel. In phase 2, XBs are both detaching and re-attaching with the rate of XB detachment greatly exceeding the rate of XB recruitment, and therefore krel measurement is an index of XB detachment. After this phase of quick force decline, the preparations exhibit a gradual force development (Phase 3), with a rate constant kdf, due to stretch-induced recruitment of new XBs into the force-generating state [35, 49]. In phase 3, XBs are both detaching and reattaching with the rate of XB recruitment greatly exceeding the rate of XB detachment, and therefore kdf measurement is an index of XB recruitment. Stretch activation amplitudes were normalized to prestretch Ca2+-activated force and were measured as described previously [27, 35]. krel and kdf were estimated using a linear transformation of the force decay and force redevelopment, respectively. krel was measured by fitting a single exponential to the time course of force decay musing the formula: y=a (1-exp(−k1×x)) a is the amplitude of the single exponential phase and k1 is the rate constant of the force decay as described previously [25, 50, 51].
Figure 4. Effect of OM on the stretch activation responses in WT, KO, and donor human myocardial preparations.
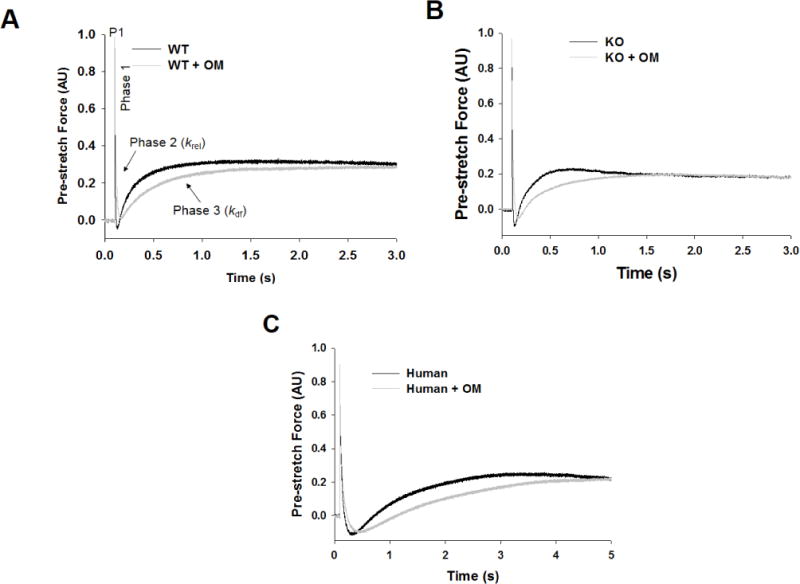
Shown are the representative force responses evoked by a sudden 2% stretch in muscle length (ML) in isometrically-contracting (A) WT, (B) KO, and (C) human myocardial preparations before and following incubation with OM. In panel A, highlighted are the important phases of the force response and various stretch activation parameters that are derived from the response. Phase 1 denotes the immediate increase in force in response to the sudden stretch in ML and represents the XB stiffness. P1 is the magnitude of the immediate force response and is measured from the pre-stretch isometric steady-state force to the peak of phase 1. Phase 2 denotes the rapid decline in the force with a dynamic rate constant krel and is an index of the rate of XB detachment. Phase 3 denotes the delayed force development with a dynamic rate constant kdf and is an index of the rate of XB recruitment. Incubation with OM led to a significant slowing of both krel and kdf in WT, KO, and human myocardial preparations.
kdf was measured by linear transformation of the half-time for force redevelopment using the formula: kdf = −ln0.5×(t1/2)−1 where t1/2 is the time (in milliseconds) taken from the nadir to half the maximal force in phase 3 of the force response shown in Fig. 4A, where maximal force is indicated by a plateau region in phase 3 as described previously [25, 26, 50, 51].
Data Analysis
All data are reported as mean ± SEM. One-way analysis of variance (ANOVA) was used to test whether there are any significant differences in the mean values from multiple groups [52]. Independent t-tests were used to assess whether there are any significant differences between two different groups, and paired t-tests were used to assess whether there are any significant differences pre- and post-OM treatment within the same group [35]. Correlation analysis was performed to substantiate the trends in pCa vs. % decreases in krel and kdf. The criterion for statistical significance was set at P < 0.05 and the asterisks in figures and tables represent statistical significance using t-tests.
RESULTS
Effect of OM on the phosphorylation levels of sarcomeric proteins
To determine if OM treatment alters the phosphorylation status of key regulatory sarcomeric proteins, WT and KO myocardial samples were subjected to Western blot and Pro-Q phospho-analysis prior to and following treatment with OM (Fig. 1). Our Western blot analysis shows that in WT samples, phosphorylation of TnI at residues Ser23/24 and cMyBP-C at Ser273, Ser282, and Ser302 was unaltered by OM treatment (Fig. 1A). TnI phosphorylation was similarly unaltered by OM treatment in KO samples and cMyBP-C phosphorylation and its total protein was not detected in KO samples (Fig. 1A). Ventricular samples from WT and KO hearts were also stained with Pro-Q Diamond stain to assess the effects of OM treatment on the phosphorylation levels of various regulatory myofilament proteins (Fig. 1B). As we previously reported [53] the phosphorylation levels of various sarcomeric proteins such as cardiac TnT, cardiac TnI and regulatory light chain were not significantly different between WT and KO myocardial samples (Fig. 1B). Furthermore, our data also shows that treatment with OM did not affect the phosphorylation status of myofilament proteins in both WT and KO hearts as shown by the absence of significant differences between pre and post OM samples within WT and KO groups (Fig. 1C).
FIGURE 1. Western Blot and Pro-Q analysis to assess the phosphorylation status of myofilament filament proteins in WT and KO heart preparations.
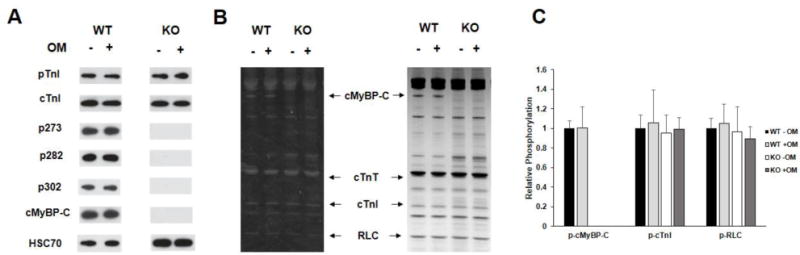
(A) Western blots showing cMyBP-C and TnI phosphorylation before and after treatment with OM in WT and KO heart samples. TnI phosphorylation at residues Ser23/24 was similar between WT and KO samples and was unaffected by OM treatment. cMyBP-C phosphorylation of residues Ser273, Ser282, and Ser302 was absent in KO tissue, and their phosphorylation levels in WT was unaffected by OM treatment. (B) Representative Pro-Q Diamond-stained (left) and Coomassie stained (right) SDS gel showing the phosphorylation status of myofilament proteins before and after treatment with OM in WT and KO heart samples. (C) Quantification of protein phosphorylation as determined by Pro-Q and Coomassie stains from 6 WT and 6 KO hearts. The intensity of the phosphorylation signaling was normalized to the intensity of the total protein signal and the untreated WT myofibril protein phosphorylation was set to 1 as done in our previous study [35]. cMyBP-C phosphorylation was unaffected by OM treatment in WT preparations and was absent in KO preparations. No differences in phosphorylation status of myofilament proteins were observed between WT and KO hearts. Furthermore, treatment with OM for 2 minutes did not induce any significant changes in the phosphorylation status of myofilament proteins in WT and KO hearts. WT, wild-type; KO, knockout; cMyBP-C, cardiac myosin binding protein-C; cTnT, cardiac troponin T; cTnI, cardiac troponin I; RLC, regulatory light chain.
Effect of OM on Ca2+-activated force generation in WT and KO myocardial preparations
Ca2+-activated force production was first measured in skinned ventricular preparations in Ca2+ solutions with increasing amounts of Ca2+ (pCa 6.2, 6.1, 6.0, and 5.9, i.e., ~10 to 40% of maximal force [53]). This was followed by a 2-minute incubation of the preparations in 1.0μM OM and measuring the Ca2+-activated force production in pCa solutions: 6.2, 6.1, 6.0, and 5.9. Our results indicate that there was a significant increase in the force production in both WT and KO preparations after 2-minute incubation with OM (Fig. 2). Furthermore, our results show that the % increase in force from baseline (pre-OM) to post-OM was more pronounced at low levels of activator [Ca2+] (pCa 6.2) and progressively decreased as the level of activator [Ca2+] increased (Fig. 2) in skinned mouse myocardium. Similar trends were observed in preparations isolated from human left ventricular samples which predominantly express the slow β-MHC isoform, although, the force enhancement was less pronounced than what was observed in the predominantly α-MHC background of the mouse myocardium (WT and KO). For human heart preparations, the % increases in force production from the baseline were 60.4±14.6, 49.5±10.8, 27.6±9.9, and 14.0±4.8, respectively at pCa’s 6.2, 6.1, 6.0, and 5.9. Furthermore, incubation with OM did not affect the force generation at maximal Ca2+ activation (pCa 4.5) in any of the groups (Table 1).
FIGURE 2. Effect of OM on force enhancement at various levels of activator [Ca2+].
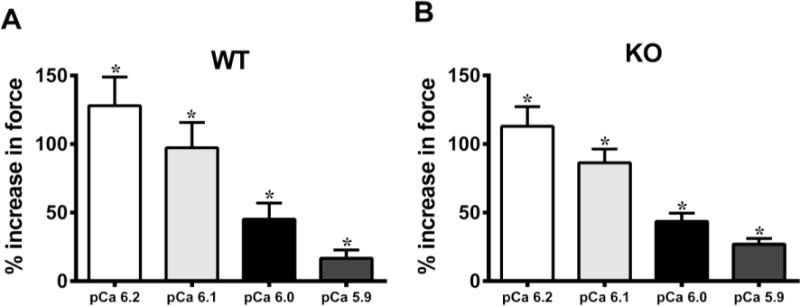
Baseline forces generated by the skinned ventricular preparations were first measured in Ca2+ solutions with pCa ranging from 6.2 to 5.9. Forces were again measured on the same preparations using the same range of pCa solutions following 2-minute incubation with OM. The net increase in force generation following incubation with OM at each level of activator [Ca2+] was calculated and is expressed as % increase in force from baseline in (A) WT and (B) KO preparations. Thus, the % increases shown in panels A and B following OM treatment are over and above the Ca2+-mediated force generation, at each pCa. The ability of OM to enhance force production decreases as the level of activator [Ca2+] in the sarcomere increases. No statistical differences were found between the % increases in forces following OM treatment in WT and KO groups. This indicates that OM enhanced force generation in the WT and KO preparations to the same extent. Values are expressed as mean ± S.E.M. Independent t-tests were used to compare the data between WT and KO groups and paired t-tests were used to compare the data between pre- and post-OM treatment with in the same group. 16 preparations were analyzed from 5 WT hearts and 18 preparations were analyzed from 5 KO hearts, with multiple preparations from each heart. * P < 0.05 when comparing forces generated before incubation with OM vs. forces generated following incubation with OM within each group.
Table 1.
Myofilament Ca2+ sensitivity, Hill coefficient of force production, and maximal force generation in WT, KO, and human myocardial preparations
| Group | pCa50 | nH | Fmax (mN/mm2) |
|---|---|---|---|
| Pre-OM | |||
| WT | 5.83±0.02 | 2.42±0.13 | 24.52±2.67 |
| KO | 5.86±0.01 | 2.33±0.14 | 20.76±2.27 |
| Human | 5.85±0.02 | 2.49±0.12 | 21.35±4.87 |
| Post-OM | |||
| WT | 5.97±0.03* | 1.92±0.17* | 24.04±3.05 |
| KO | 6.01±0.02* | 1.87±0.08* | 22.29±3.78 |
| Human | 5.92±0.02* | 2.07±0.12* | 22.38±4.35 |
pCa50: myofilament Ca2+ sensitivity; nH: cooperativity of force production; Fmax: force generation at maximal Ca2+ activation (pCa 4.5). Independent t-tests were used to compare the pre-OM and post-OM data in WT, KO, and human groups. 14–20 preparations were analyzed from 5 WT hearts, 15 preparations were analyzed from 5 KO hearts, and 9–14 preparations were analyzed from 3 human hearts with multiple preparations from each heart. Values are expressed as mean ± S.E.M.
Significantly different from the corresponding pre-OM group; P < 0.05.
Effect of OM on myofilament Ca2+ sensitivity (pCa50) and cooperativity of force development (nH)
The effect of OM on pCa50 was assessed by plotting normalized force values against a range of pCa and constructing force-pCa relationships at SL 2.1 μm in WT, KO, and human heart preparations. pCa50, the pCa required to generate half-maximal force, was estimated by fitting the Hill equation to the force-pCa relationships (Fig. 3). Our data shows that treatment with OM increased the responsiveness of the cardiac myofilaments to Ca2+ at submaximal Ca2+-activations as indicated by a significant left-ward shift in the force-pCa relationships in all the groups (Figs. 3A, B, and C). pCa50 values for WT, KO, and human heart preparations are shown in Table 1. The effect of OM on nH was assessed by fitting the Hill equation to the force-pCa relationships. nH values decreased post-OM treatment in all the groups indicating that incubation with OM decreased the overall cooperativity of force production (Table 1).
FIGURE 3. Effect of OM on myofilament Ca2+ sensitivity (pCa50).
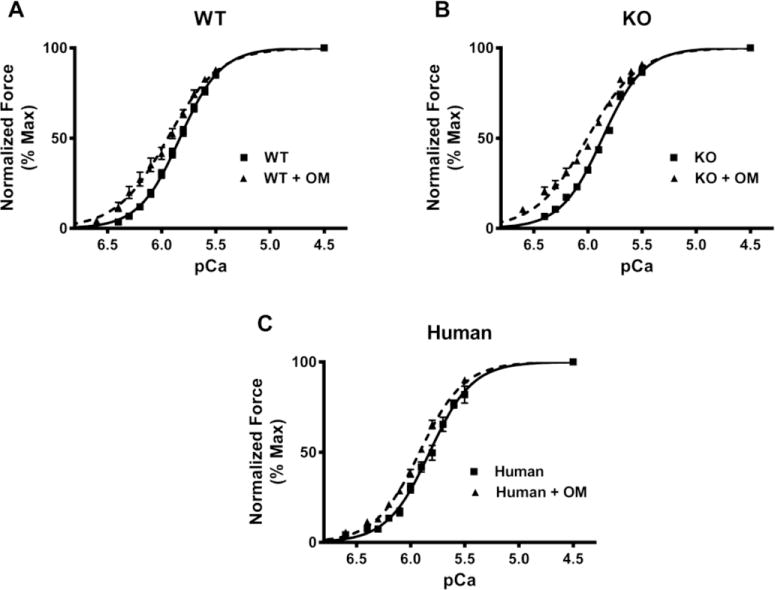
Force-pCa relationships were constructed by plotting normalized forces generated against a range of pCa. (A) Effect of OM on the force-pCa relationships in WT preparations. (B) Effect of OM on the force-pCa relationships in KO preparations. (C) Effect of OM on the force-pCa relationships in donor human heart preparations. Treatment with OM caused a significant left-ward shift in the force-pCa relationships in all the three groups indicating that OM caused a significant increase in pCa50. 14–15 preparations from 5 hearts were analyzed for WT and WT+OM groups. 15 preparations from 5 hearts were analyzed for KO and KO+OM groups. 14 preparations from 3 hearts and 13 preparations from 3 hearts were analyzed for human and human+OM groups, respectively with multiple preparations from each heart.
Effect of OM on the rate of force redevelopment (ktr) in WT and KO myocardial preparations
ktr, is an index of XB transition from both the weakly- to strongly-bound state and from the strongly- to weakly-bound states [40, 41] and OM has been shown to accelerate the transition of XBs to the strongly-bound force-producing state in an in vitro assay [8]. In this study we wanted to test the effects of OM on ktr under conditions where the cardiac sarcomeric lattice structure is intact. To gain better insights regarding the effect of OM on ktr, we measured ktr at same level of activator [Ca2+] (pCa 6.1) before and following incubation with OM. The force produced was significantly higher at the same level of activator [Ca2+] following incubation with OM (Fig. 2). Because ktr is dependent on the level of activation (i.e., force generation, [41, 43]), we also examined the effects of OM on ktr at equivalent levels of force generation (Table 2).
Table 2.
Steady-state and dynamic contractile parameters measured before and following incubation with OM at similar levels of force production
| Group | pCa | P1 | ktr (s−1) | krel (s−1) | kdf (s−1) |
|---|---|---|---|---|---|
| Pre-OM | |||||
| WT | 5.98±0.02 | 0.561±0.015 | 2.90±0.28 | 251.24±23.92 | 4.54±0.30 |
| KO | 5.94±0.02 | 0.482±0.021* | 6.32±0.50* | 409.13±48.34* | 7.17±0.42* |
| Post-OM | |||||
| WT | 6.14±0.02† | 0.672±0.026† | 2.08±0.23† | 99.78±9.24† | 1.58±0.19† |
| KO | 6.12±0.02† | 0.571±0.025*† | 3.61±0.26*† | 89.30±15.69† | 2.79±0.24*† |
P1: XB stiffness; ktr: rate of force redevelopment; krel: rate of XB detachment; kdf: rate of XB recruitment. Independent t-tests were used to compare the data between WT and KO groups and paired t-tests were used to compare the data between pre- and post-OM treatment with in the same group. 13–16 preparations were analyzed from 5 WT hearts, and 17–18 preparations were analyzed from 7 KO hearts, with multiple preparations from each heart. Values are expressed as mean ± S.E.M.
Significantly different from the corresponding WT within each group; P < 0.05.
Significantly different from the corresponding pre-OM group; P < 0.05.
When studied at the same level of Ca2+ activation, we found that ktr was not increased even after incubation with OM in both WT and KO preparations (Fig. 5A). A decreased ktr was also observed following incubation with OM in the control human heart preparations, despite an increase in steady-state force generation (Table 3). We also studied the effects of OM on ktr when the forces were matched pre- and post-OM incubations (Table 2). Our data indicates that ktr was significantly decreased following incubation with OM in WT and KO preparations when force generation was closely matched (Table 2). Similar results were obtained for the control human heart preparations as well (Table 4). Numerous studies have shown that ktr increases in proportion to the level of force generation (i.e., proportional to the number of force generating XB’s) [26, 41, 43, 44, 54]. Therefore, because OM incubation produced a dramatic increase in force generation at pCa 6.1 in both WT and KO preparations (Fig. 2), one would also expect to observe parallel and significant increases in ktr. However, no such increases in ktr were observed at pCa 6.1 (Fig. 5A) even when there were significant increases in force production (Fig. 2) following OM incubation. At slightly higher pCa (pCa = ~6.0), it is clear that OM treatment significantly decreased ktr (Fig. 5B). Thus, our data indicate that the rate of transition of XBs into the force-bearing state is decreased following incubation with OM.
FIGURE 5. Effect of OM on the rate of force redevelopment (ktr).
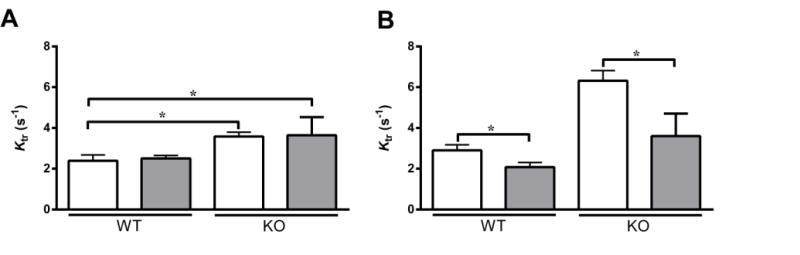
The effects of OM on ktr were assessed both at equivalent pCa (pCa 6.1) (A) and at equivalent levels of activation (see Table 2 for pCa values) (B) in WT and KO groups, using a mechanical slack-restretch maneuver [35], before (white bars) and following (grey bars) incubation with OM. As reported earlier [53], KO preparations exhibited a significant acceleration of ktr when compared to the WT preparations prior to incubation with OM. Similar trend persisted even after incubation with OM. Values are expressed as mean ± S.E.M. Independent t-tests were used to compare the data between WT and KO groups and paired t-tests were used to compare the data between pre- and post-OM treatment with in the same group. 14 preparations were analyzed from 5 WT hearts and 18 preparations from 7 KO hearts, with multiple preparations from each heart. * P < 0.05.
Table 3.
Steady-state and dynamic contractile parameters measured before and following incubation with OM at equivalent levels of activator [Ca2+] (pCa 6.1) in control donor human heart preparations
| Group | Force (mN/mm2) | P1 | ktr (s−1) | krel (s−1) | kdf (s−1) |
|---|---|---|---|---|---|
| Pre-OM | 6.06±1.53 | 0.677±0.050 | 0.76±0.07 | 56.32±4.77 | 0.86±0.07 |
| Post-OM | 8.56±1.87* | 0.724±0.033 | 0.52±0.05* | 38.26±4.32* | 0.49±0.03* |
P1: XB stiffness; ktr: rate of force redevelopment; krel: rate of XB detachment; kdf: rate of XB recruitment. Paired t-tests were used to compare the data between pre- and post-OM treatment groups. 10 preparations were analyzed from 3 hearts, with multiple preparations from each heart. Values are expressed as mean ± S.E.M.
Significantly different from the corresponding pre-OM group; P < 0.05.
Table 4.
Steady-state and dynamic contractile parameters measured before and following incubation with OM at similar level of force production in control donor human heart preparations
| Group | pCa | P1 | ktr (s−1) | krel (s−1) | kdf (s−1) |
|---|---|---|---|---|---|
| Pre-OM | 6.01±0.02 | 0.576±0.041 | 0.87±0.08 | 55.76±5.02 | 0.89±0.07 |
| Post-OM | 6.13±0.02* | 0.698±0.028* | 0.52±0.05* | 37.11±4.16* | 0.47±0.04* |
P1: XB stiffness; ktr: rate of force redevelopment; krel: rate of XB detachment; kdf: rate of XB recruitment. Paired t-tests were used to compare the data between pre- and post-OM treatment groups. 8–10 preparations were analyzed from 3 hearts, with multiple preparations from each heart. Values are expressed as mean ± S.E.M.
Significantly different from the corresponding pre-OM group; P < 0.05.
Effects of OM on the rates of XB detachment (krel) and XB recruitment (kdf) in WT and KO myocardial preparations
Our data show that OM slows the XB turnover rate, ktr in WT and KO preparations (Fig. 5; Table 2). Because ktr is proportional to the sum of f (rate of XB recruitment) + g (rate of XB detachment) according to a simple two-state XB model [40], we tested whether the OM-induced decrease in ktr arises from changes in either the rate of XB detachment or the rate of XB recruitment, or due to both. We used stretch activation experiments (see the Methods section) to measure krel and kdf which are indices of the rates of XB detachment and XB recruitment, respectively [26, 35].
When studied at the same level of activator [Ca2+] (pCa 6.1), we found that krel was significantly slowed following incubation with OM in both WT and KO preparations (Fig. 6A). As shown previously [15, 50, 53] KO preparations exhibited an increased basal krel when compared to WT preparations. However, following incubation with OM, the decrease in krel was more pronounced (at pCa 6.1) in the KO preparations (~76%) when compared to the WT preparations (~58%), such that the differences observed in baseline krel between WT and KO preparations were now no longer apparent (Table 5 and Fig. 6A). A similar trend was observed in the human skinned myocardium as krel decreased by 32% post OM incubation (Table 3).
FIGURE 6. Effect of OM on the rate of XB detachment (krel) and the rate of XB recruitment (kdf).
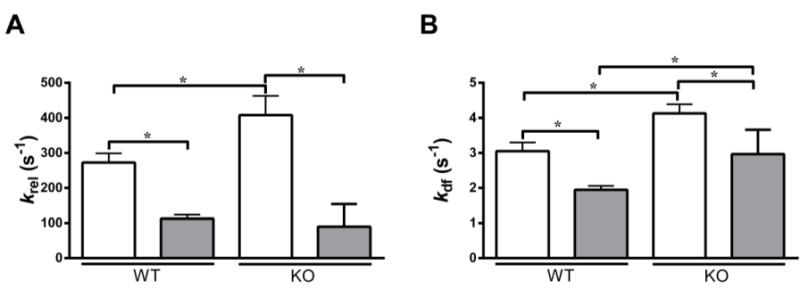
Isometrically-activated ventricular preparations were subjected to a sudden 2% stretch in their muscle length and the elicited force responses were used to estimate (A) krel and (B) kdf in WT and KO preparations before (white bars) and following (grey bars) incubation with OM at pCa 6.1 [26, 35]. As reported earlier [53], KO preparations exhibited a significantly increased krel when compared to the WT preparations before incubation with OM. However, such a trend was abolished after incubation with OM because the krel in the KO decreased to the level observed in the WT preparations following incubation with OM. Furthermore, krel was significantly decreased in WT and KO groups when compared to their respective pre-OM groups–indicating that the rate of XB detachment from actin was significantly slowed upon incubation with OM. As reported earlier [53], KO preparations exhibited a significant acceleration of kdf when compared to the WT preparations before incubation with OM, a trend that persisted even after incubation with OM. Furthermore, kdf was significantly decreased in both WT and KO groups when compared to their respective pre-OM groups–indicating that the rate of XB recruitment into the force-generating state was significantly slowed upon OM incubation. Values are expressed as mean ± S.E.M. Independent t-tests were used to compare the data between WT and KO groups and paired t-tests were used to compare the data between pre- and post-OM treatment with in the same group. 14–16 preparations and 15–18 preparations from 5 and 7 hearts were analyzed for WT and KO groups, respectively with multiple preparations from each heart. * P < 0.05.
Table 5.
Percentage decreases in the rates of XB detachment and recruitment in WT and KO preparations following incubation with OM at different levels of activator [Ca2+].
| pCa | WT | KO | ||
|---|---|---|---|---|
| krel (% decrease) | kdf (% decrease) | krel (% decrease) | kdf (% decrease) | |
| 6.2 | 66.5±3.8 | 55.7±3.5 | 63.2±3.0 | 40.2±4.5* |
| 6.1 | 57.9±2.4 | 30.2±5.7 | 76.4±3.0* | 30.1±3.0 |
| 6.0 | 50.8±1.6 | 26.3±4.8 | 61.7±2.7* | 27.3±2.7 |
| 5.9 | 41.8±2.3 | 18.6±3.3 | 60.9±1.9* | 24.3±3.7 |
krel: rate of XB detachment; kdf: rate of XB recruitment. Values are expressed as mean ± S.E.M. Paired t-tests were used to compare the data between pre- and post-OM treatment groups. 12–16 preparations were analyzed from 5 hearts and 13–18 preparations were analyzed from 7 hearts for WT and KO groups, respectively with multiple preparations from each heart. The decrease in krel was less pronounced as the level of activator [Ca2+] increased in the WT group but such a trend was absent in the KO group. The slowing in kdf was progressively decreased as the level of activator [Ca2+] increased in both the WT and KO groups. Furthermore, the % decreases in krel in the KO group were more pronounced than WT at pCa’s 6.1, 6.0, and 5.9. The % decreases in kdf in the WT group were significantly higher than KO at pCa 6.2. Correlation analysis showed a strong positive correlation between pCa vs. % decreases in krel and kdf in the WT group (correlation coefficient values of 0.99 and 0.93, respectively). In the KO group, there was a strong positive correlation between pCa vs. % decreases for kdf (correlation coefficient value of 0.94), and a moderate positive correlation for pCa vs. % decreases in krel in the KO group (correlation coefficient value of 0.38). This is due to the fact that the % decreases in krel in the KO group are less sensitive to increases in submaximal [Ca2+]. Independent t-tests were used to test the differences between WT and KO groups at each pCa.
Significantly different from the corresponding WT group; P < 0.05.
Akin to our observations at the same level of activator [Ca2+], krel was significantly decreased following incubation with OM in WT and KO preparations when force-matched comparisons were made (Table 2). The differences observed in krel between WT and KO groups before incubation with OM were no longer apparent following incubation with OM (Table 2). Such significant decreases in krel following incubation with OM were also seen in the control human heart preparations (Table 4). Furthermore, the decrease in krel following incubation with OM was progressively diminished as the level of activator [Ca2+] increased in the WT group, but this trend was not observed in the KO group as krel was highly decreased across all levels of activator [Ca2+] (Table 5), indicating that the OM-induced decrease in krel was more pronounced in the absence of cMyBP-C in the myocardium, especially at pCa’s 6.1 to 5.9. Similar to observations in the WT group, the decrease in krel was progressively diminished as the level of activator [Ca2+] increased in the human heart preparations. The percentage decreases in krel were 39.30±4.68, 33.45±3.62, 28.92±3.05, 26.91±2.24, respectively at pCa’s 6.2, 6.1, 6.0 and 5.9 in human heart preparations.
When studied at the same level of activator [Ca2+] (pCa 6.1), kdf was significantly decreased by 36% and 28% following incubation with OM in WT and KO preparations, respectively (Fig. 6B). This result indicates that the rate of XB recruitment is significantly decreased upon incubation with OM. Similarly, kdf decreased by 43% in the control human heart preparations upon incubation with OM (Table 3). When force-matched comparisons were made, the decrease in kdf was even more pronounced following incubation with OM in WT and KO preparations than what was observed at the same level of activator [Ca2+]. kdf decreased by 65% and 61% following incubation with OM in WT and KO preparations, respectively (Table 2). kdf was decreased by 47% in the control human heart preparations upon incubation with OM (Table 4). Furthermore, the decrease in kdf following incubation with OM was progressively diminished as the level of activator [Ca2+] increased in both WT and KO groups (Table 5). Similar trends were observed in the human heart preparations. The percentage decreases in kdf were 55.39±3.73, 41.92±3.10, 23.57±3.67, 17.11±2.62, respectively at pCa’s 6.2, 6.1, 6.0 and 5.9 in human heart preparations. Collectively, our results suggest that incubation with OM slows the overall XB cycling kinetics and prolongs the duty cycle of the myosin heads. In particular, slowed XB detachment rate (krel) would act to maintain thin filament activation for a longer time period which can then increase the force production (Fig. 2) by allowing enhanced XB-mediated cooperative XB recruitment and binding to open actin molecules [55], which contributes to an overall decrease in kdf and ktr [41]. Thus, slowed kdf is likely a secondary effect of the slowed XB detachment-induced prolongation in the XB duty cycle.
Effect of OM on XB stiffness (P1) in WT and KO myocardial preparations
Using stretch activation experiments we tested whether incubation with OM affects the XB stiffness. We imposed a sudden 2% stretch in muscle length in an isometrically-contracting myocardial preparation and measured the magnitude of the elicited instantaneous increase in force (P1 in Fig. 4A). P1 is a result of a rapid distortion of the elastic regions of the strongly-bound XBs and is an index of the XB stiffness [26, 48, 56]. As shown previously [53] our current data shows that KO preparations exhibited reduced XB stiffness when compared to the WT preparations (Table 2). When the level of Ca2+-activation was equivalent, P1 following OM incubation displayed a trend towards increased P1 in both WT and KO preparations, and when pre-stretch force was matched, XB stiffness was significantly increased in both WT and KO skinned preparations as indicated by increases in P1 (Table 2). At equivalent levels of activator Ca2+ (pCa 6.1), P1 was not affected following OM incubation in human myocardial preparations (Table 3), and P1 increased significantly in human myocardial preparations when the pre-stretch forces levels were matched (Table 4).
DISCUSSION
In this study we tested the utility of OM in attenuating the accelerated contractile XB kinetics associated with the absence of cMyBP-C in the cardiac sarcomere. Our data shows that both the rates of XB detachment from the strongly-bound state and XB recruitment into the force-generating state are significantly slowed in the KO skinned myocardium following incubation with OM, suggesting that pharmacological intervention can be considered to slow hypercontractile XB kinetics in myocardium expressing reduced levels of cMyBP-C.
The effect of OM on steady-state force generation depends on the level of Ca2+ activation
It is known that OM increases myocardial contractility by directly activating the cardiac myosin motor, unlike other commonly clinically used inotropic drugs such as β-adrenergic receptor agonists and phosphodiesterase inhibitors which activate the signaling pathways that ultimately enhance the intracellular Ca2+ transients [57]. In particular, OM infusion has been shown to increase LV systolic ejection time in dog model of systolic heart failure [30] and has been shown to increase cardiac contractility as indicated by significant increases in fractional shortening in rat and dog models [8]. However, to date, the effects of OM on cardiac contractility in the presence of varying levels of activator [Ca2+] in skinned myocardium has not been studied. Here we found that incubation of skinned myocardium with OM significantly increased the force generation at low levels of activator [Ca2+] and significantly increased the myofilament Ca2+ sensitivity (Fig. 3) and indicating that more XB’s are being recruited into the force-producing state following incubation with OM (Fig. 2), however, the magnitude of force enhancement was inversely proportional to the levels of activator [Ca2+], and progressively decreased with increasing [Ca2+] (Fig. 2). Our data also shows that the increase in myofilament Ca2+ sensitivity (pCa50) following incubation with OM was also accompanied by an overall decrease in the Hill coefficient of force production (i.e., shallower nH) (Table 1) indicating enhanced cooperative XB recruitment. Post-OM incubation, increases in force production were more pronounced at low levels of activator Ca2+ and progressively decreased with increased levels of activator Ca2+ (Fig. 2). This phenomenon is likely due to the fact that at low Ca2+ activations thin filament activation is more reliant on XB-mediated cooperative XB recruitment because most of the thin filament regulatory units (RU) are in their off state [55]. At high [Ca2+] most of the RU’s will already be in their on state, which reduces the reliance of the thin filament on XB-mediated cooperative XB recruitment. Because OM promotes enhanced force generation through increased cooperative XB recruitment, the effects on force generation are more pronounced at low Ca2+-activations, thereby resulting in a decrease in the steepness of nH (Table 1) [58]. Similar trends were observed in donor human heart preparations but the magnitudes of the force enhancement at various levels of activator [Ca2+] were less pronounced following OM incubation.
Differences in the magnitude of OM-induced force enhancements between murine and human heart preparations may be related to the inherent kinetic differences in the isoform expression of MHC. Human hearts predominantly express the β-MHC isoform which is an inherently slow myosin motor with a longer XB duty ratio [59–62] than the fast α-MHC isoform motor expressed in the mouse heart. Consequently, it is possible that the apparent prolongation of the duty ratio induced by OM may be less prominent in the already slow β-MHC-XBs in the human myocardium compared to the fast α-MHC-XBs in the murine myocardium, thereby, explaining the relatively smaller effects of OM-induced force enhancements observed in human myocardium at various [Ca2+]. Regardless, the effects of OM on force enhancement at low levels of activator [Ca2+] were still highly significant in the human myocardium (Table 3). A greater effect of OM on force generation at lower levels of activator [Ca2+] is consistent with a mechanism that involves cooperative XB-recruitment, which is more pronounced at low levels of Ca2+-activation [41, 54] where relatively few XB’s are in the strongly-bound force-producing states and the recruitment of additional XBs to strongly bound-states can increase force generation significantly. Furthermore, positive feedback effects of the additionally recruited XBs will further help to stabilize the open conformation of the N-terminus of cardiac TnC to sustain the thin filament activation and enhance force generation at low Ca2+ levels [63, 64].
OM-induced slowing of XB kinetics are more pronounced in the hypercontractile KO skinned myocardium
It has been shown that OM allosterically activates the myosin motor by binding to the catalytic domain of the myosin head and promotes the transition of XBs into a strongly-bound, force-generating state [8], and has also been shown to induce a large increase in the average duty cycle of porcine ventricular myosin [31]. Considering that KO skinned myocardium displays a significant acceleration in XB cycling kinetics, OM’s ability to increase the duty cycle of the myosin motor may be a useful pharmacological approach to correct contractile dysfunction in the KO myocardium. Because ktr is an index of rate of transition of XBs from the weakly- to strongly-bound, and strongly- to weakly-bound states [40, 41], we used a slack-restretch maneuver [35, 53] to directly measure the impact of OM on ktr. When studied at the same level of activator [Ca2+], we found that ktr was unaffected following incubation with OM in both WT and KO preparations (Fig. 5A), and when steady-state forces were matched, ktr was significantly decreased following OM incubation (Fig. 5B, Table 2). This result was somewhat surprising because we have previously shown that ktr increases significantly in both WT and KO preparations as the amount of force generation increases [43]. Our finding that ktr was blunted, or even decreased, despite a large increase in the steady-state force generation following incubation with OM in both WT and KO preparations (Fig. 2), suggests that OM inherently decreases ktr. Decreased ktr was also observed following incubation with OM in the human skinned myocardium both at matched Ca2+-activation levels (Table 3) and at matched force generation levels (Table 4), indicating that qualitatively, the effects of OM on ktr were not dependent on the MHC isoform that is expressed in cardiac muscle. Decreased ktr following OM incubation could be due to decreased rates of transition of XBs into the force-bearing state or due to decreased rates of XB detachment, or both.
Therefore, we performed stretch-activation experiments to further probe the effects of OM on the rates of XB detachment (krel) and the XB recruitment (kdf). Our stretch-activation data reveal that both krel and kdf decreased significantly post-OM incubation, whether these rates were measured at same level of activator [Ca2+], or at equivalent levels of pre-stretch force generation (Fig. 6; Table 2). In particular, differences in krel that were observed between WT and KO prior to OM incubation (i.e., KO exhibited significantly accelerated krel at baseline) were abolished following OM incubation (Table 2) indicating the rates of XB detachment in KO preparations were decreased to a greater extent than in the WT preparations, specifically at pCa’s 6.1 to 5.9 (Table 5). Furthermore, the effects of OM on krel were more pronounced in KO skinned myocardium, in that krel was significantly decreased at all levels of activator [Ca2+] studied, whereas the decrease in krel in WT skinned myocardium progressively reduced as the levels of activator [Ca2+] increased (Table 5). Reduced XB detachment rates (i.e., krel) following OM incubation also predicts that ATP consumption during the XB bridge cycle (i.e., tension cost) is significantly reduced because XB detachment rate is highly correlated with tension cost [40, 65–67]. The decrease in krel may also be related to increased XB stiffness that was observed in WT and KO preparations following OM incubation [53] (Table 2). The increased XB stiffness along with the related conformation changes in the myosin heads may have likely increased the recruitment time of the XBs to actin, maintaining the XBs for a longer time period in their force-generating state by resisting strain-induced XB detachment (Fig. 6). Taken together, these effects would be predicted to enhance force generation and concomitantly increasing the efficiency of muscle contraction, as has been shown in previous studies [30] . On the other hand, it may seem counter-intuitive that increased force generation following OM incubation (Fig. 2) resulted in a decreased kdf (Fig. 6). However, decreased kdf may be due to the fact that enhanced XB recruitment involves both Ca2+ and XB-mediated cooperative spread of thin filament activation that aids in the transition of additional XBs from the non-cycling pool or a weakly-bound state to the strongly-bound state. Because cooperative activation is a time consuming process it acts to limit the overall rate of force development [41]. Therefore, it is likely that OM primarily acts as a XB-mediated activator of the thin filament which sustains the already bound XBs in their attached state for a longer period. The potential secondary effect of prolonged XB recruitment time is that thin filament regulatory units remain open for a longer time period, thereby enhancing the access of unbound XBs to bind to open actin sites. In this regard, consistent with ktr findings, a significant decrease in both krel and kdf post-OM incubation was also observed in the human preparations that predominantly express β-MHC isoform (Tables 3 and 4), further confirming that the effects of OM on XB behavior are not cardiac MHC isoform dependent.
Potential in vivo consequences of OM-induced slowing of XB kinetics in the KO myocardium
Deficiency of cMyBP-C accelerates XB cycling kinetics and reduces the time that XBs effectively spend in their force-generating state, thereby, contributing to a premature truncation of the systolic ejection phase in vivo [68, 69]. In this study, we observed a large increase in the force generation at low levels of activator [Ca2+] following OM incubation (Fig. 2) which is due to decreased rates of XB detachment which prolongs XB duty cycle and enhances cooperative XB-mediated XB recruitment. In this regard, a recent report [31] demonstrated a 10-fold increase in the duty cycle of OM-treated isolated porcine cardiac myosin, which would be predicted to significantly enhance force generation. These molecular effects of OM on XB behavior can be predicted to significantly promote the recruitment of XBs into the force-bearing state at the initial phase of isovolumic contraction in vivo when [Ca2+] levels in the sarcomere are still relatively low. It is also possible that the OM-induced slowing of XB detachment may subsequently prolong duty cycle of the force-bearing state of XBs later in systole even at a time point when the cytosolic Ca2+ levels wane off, specifically, extending the period of the late systolic ejection phase, as shown previously [8, 30]. On the other hand, although, prolonged systolic ejection due to delayed XB detachment could significantly augment systolic function and cardiac output, such a mechanism could also have an adverse impact on diastolic function if systole is sufficiently prolonged such that it impinges on the period of diastolic relaxation which would delay diastolic filling, a possibility that requires further investigation.
Limitations of the study
Our experiments at the myofilament level shows that treatment with OM can be a potential pharmacological approach to slow XB kinetics in myocardium lacking cMyBP-C, but extrapolating these findings to the whole organ level is premature. The present study was performed in isolated skinned myocardial preparations where the level of Ca2+ activation and experimental conditions are precisely controlled. However, modulation of cardiac function at the whole organ level is much more complex, involving a complex interplay of the myofilaments with the cellular Ca2+-handling machinery, neurohormonal signaling, electrical activation, etc. Thus, it is also possible that the molecular findings presented here may not completely translate to benefits at the whole heart level in the KO model. Therefore, future experiments will have to be performed to investigate the utility of OM in improving in vivo contractile and hemodynamic function in cMyBP-C deficient hearts under basal conditions, and also in response to increased workload, such as with increased β-adrenergic stimulation. These in vivo experiments will provide vital insights into the effectiveness of OM infusion in improving cMyBP-C-related contractile dysfunction.
Conclusions
Altered XB contractile dynamics is a common feature of animal models and human patients with hypertrophic cardiomyopathy (HCM). Increased ktr and rates of XB detachment have been observed in skinned myocardium isolated from patients expressing HCM-causing mutations in cMyBP-C and MHC [28, 70, 71], and are thought to contribute to increased tension cost of contraction [72]. In this study, we show a significant slowing in the intrinsically faster XB kinetics in the skinned KO murine myocardium following OM incubation. Because hypercontractile XB behavior is thought to be an important feature in the pathogenesis of cMyBP-C-related HCM, the pharmacological use of OM may have utility in normalizing contractile function at the level of sarcomere, and therefore, the effects of OM on improving in vivo function should be investigated.
Highlights.
-
-
Omecamtiv mecarbil (OM) is a novel drug that targets the cardiac XB cycle
-
-
OM enhances force generation in skinned myocardium but slows XB kinetics
-
-
The effects of OM on XB behavior were most pronounced at low Ca2+-activation
-
-
OM significantly slowed rates of XB detachment in cMyBP-C−/− skinned myocardium
-
-
OM can ameliorate contractile dysfunction due to decreased cMyBP-C expression
Acknowledgments
This work was supported by the National Heart, Lung, and Blood Institute Grant (HL-114770).
ABBREVIATIONS
- cMyBP-C
cardiac myosin binding protein-C
- OM
omecamtiv mecarbil
- KO
knockout
- WT
wild-type
- XB
cross-bridge
- ktr
rate of force redevelopment
- krel
rate of XB detachment
- kdf
rate of XB recruitment
- MHC
myosin heavy chain
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST STATEMENT
There are no conflicts of interest.
AUTHOR AND CONTRIBUTIONS
R.M. K.S.G, and J.E.S contributed to the conception and design of the experiments. R.M., K.S.G A.L., C.G.D.R., and J.E.S participated in performing the experiments, data acquisition, data analysis, data interpretation, drafting, and revising the manuscript. All authors approved the final version of the manuscript.
References
- 1.Chatterjee K, Massie B. Systolic and diastolic heart failure: differences and similarities. Journal of cardiac failure. 2007;13:569–76. doi: 10.1016/j.cardfail.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 2.Teerlink JR. A novel approach to improve cardiac performance: cardiac myosin activators. Heart failure reviews. 2009;14:289–98. doi: 10.1007/s10741-009-9135-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feest ER, Steven Korte F, Tu AY, Dai J, Razumova MV, Murry CE, et al. Thin filament incorporation of an engineered cardiac troponin C variant (L48Q) enhances contractility in intact cardiomyocytes from healthy and infarcted hearts. Journal of molecular and cellular cardiology. 2014;72:219–27. doi: 10.1016/j.yjmcc.2014.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lundy SD, Murphy SA, Dupras SK, Dai J, Murry CE, Laflamme MA, et al. Cell-based delivery of dATP via gap junctions enhances cardiac contractility. Journal of molecular and cellular cardiology. 2014;72:350–9. doi: 10.1016/j.yjmcc.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mamidi R, Li J, Gresham KS, Stelzer JE. Cardiac myosin binding protein-C: a novel sarcomeric target for gene therapy. Pflugers Archiv: European journal of physiology. 2014;466:225–30. doi: 10.1007/s00424-013-1412-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nowakowski SG, Kolwicz SC, Korte FS, Luo Z, Robinson-Hamm JN, Page JL, et al. Transgenic overexpression of ribonucleotide reductase improves cardiac performance. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:6187–92. doi: 10.1073/pnas.1220693110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tardiff JC, Carrier L, Bers DM, Poggesi C, Ferrantini C, Coppini R, et al. Targets for Therapy in Sarcomeric Cardiomyopathies. Cardiovascular research. 2015 doi: 10.1093/cvr/cvv023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malik FI, Hartman JJ, Elias KA, Morgan BP, Rodriguez H, Brejc K, et al. Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science. 2011;331:1439–43. doi: 10.1126/science.1200113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Solaro RJ. CK-1827452, a sarcomere-directed cardiac myosin activator for acute and chronic heart disease. IDrugs: the investigational drugs journal. 2009;12:243–51. [PubMed] [Google Scholar]
- 10.Teerlink JR, Clarke CP, Saikali KG, Lee JH, Chen MM, Escandon RD, et al. Dose-dependent augmentation of cardiac systolic function with the selective cardiac myosin activator, omecamtiv mecarbil: a first-in-man study. Lancet. 2011;378:667–75. doi: 10.1016/S0140-6736(11)61219-1. [DOI] [PubMed] [Google Scholar]
- 11.Davis J, Westfall MV, Townsend D, Blankinship M, Herron TJ, Guerrero-Serna G, et al. Designing heart performance by gene transfer. Physiological reviews. 2008;88:1567–651. doi: 10.1152/physrev.00039.2007. [DOI] [PubMed] [Google Scholar]
- 12.Day SM, Westfall MV, Fomicheva EV, Hoyer K, Yasuda S, La Cross NC, et al. Histidine button engineered into cardiac troponin I protects the ischemic and failing heart. Nature medicine. 2006;12:181–9. doi: 10.1038/nm1346. [DOI] [PubMed] [Google Scholar]
- 13.Herron TJ, Devaney E, Mundada L, Arden E, Day S, Guerrero-Serna G, et al. Ca2+-independent positive molecular inotropy for failing rabbit and human cardiac muscle by alpha-myosin motor gene transfer. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2010;24:415–24. doi: 10.1096/fj.09-140566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mearini G, Stimpel D, Geertz B, Weinberger F, Kramer E, Schlossarek S, et al. Mybpc3 gene therapy for neonatal cardiomyopathy enables long-term disease prevention in mice. Nature communications. 2014;5:5515. doi: 10.1038/ncomms6515. [DOI] [PubMed] [Google Scholar]
- 15.Merkulov S, Chen X, Chandler MP, Stelzer JE. In vivo cardiac myosin binding protein C gene transfer rescues myofilament contractile dysfunction in cardiac myosin binding protein C null mice. Circulation Heart failure. 2012;5:635–44. doi: 10.1161/CIRCHEARTFAILURE.112.968941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pena JR, Szkudlarek AC, Warren CM, Heinrich LS, Gaffin RD, Jagatheesan G, et al. Neonatal gene transfer of Serca2a delays onset of hypertrophic remodeling and improves function in familial hypertrophic cardiomyopathy. Journal of molecular and cellular cardiology. 2010;49:993–1002. doi: 10.1016/j.yjmcc.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coulton AT, Stelzer JE. Cardiac myosin binding protein C and its phosphorylation regulate multiple steps in the cross-bridge cycle of muscle contraction. Biochemistry. 2012;51:3292–301. doi: 10.1021/bi300085x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuster DW, Sequeira V, Najafi A, Boontje NM, Wijnker PJ, Witjas-Paalberends ER, et al. GSK3beta phosphorylates newly identified site in the proline-alanine-rich region of cardiac myosin-binding protein C and alters cross-bridge cycling kinetics in human: short communication. Circulation research. 2013;112:633–9. doi: 10.1161/CIRCRESAHA.112.275602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palmer BM, Sadayappan S, Wang Y, Weith AE, Previs MJ, Bekyarova T, et al. Roles for cardiac MyBP-C in maintaining myofilament lattice rigidity and prolonging myosin cross-bridge lifetime. Biophysical journal. 2011;101:1661–9. doi: 10.1016/j.bpj.2011.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Previs MJ, Beck Previs S, Gulick J, Robbins J, Warshaw DM. Molecular mechanics of cardiac myosin-binding protein C in native thick filaments. Science. 2012;337:1215–8. doi: 10.1126/science.1223602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Razumova MV, Bezold KL, Tu AY, Regnier M, Harris SP. Contribution of the myosin binding protein C motif to functional effects in permeabilized rat trabeculae. The Journal of general physiology. 2008;132:575–85. doi: 10.1085/jgp.200810013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Razumova MV, Shaffer JF, Tu AY, Flint GV, Regnier M, Harris SP. Effects of the N-terminal domains of myosin binding protein-C in an in vitro motility assay: Evidence for long-lived cross-bridges. The Journal of biological chemistry. 2006;281:35846–54. doi: 10.1074/jbc.M606949200. [DOI] [PubMed] [Google Scholar]
- 23.Rosas PC, Liu Y, Abdalla MI, Thomas CM, Kidwell DT, Dusio GF, et al. Phosphorylation of Cardiac Myosin Binding Protein-C is a Critical Mediator of Diastolic Function. Circulation Heart failure. 2015 doi: 10.1161/CIRCHEARTFAILURE.114.001550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L, Sadayappan S, Kawai M. Cardiac Myosin Binding Protein C Phosphorylation Affects Cross-Bridge Cycle’s Elementary Steps in a Site-Specific Manner. PloS one. 2014;9:e113417. doi: 10.1371/journal.pone.0113417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stelzer JE, Patel JR, Moss RL. Protein kinase A-mediated acceleration of the stretch activation response in murine skinned myocardium is eliminated by ablation of cMyBP-C. Circulation research. 2006;99:884–90. doi: 10.1161/01.RES.0000245191.34690.66. [DOI] [PubMed] [Google Scholar]
- 26.Cheng Y, Wan X, McElfresh TA, Chen X, Gresham KS, Rosenbaum DS, et al. Impaired contractile function due to decreased cardiac myosin binding protein C content in the sarcomere. American journal of physiology Heart and circulatory physiology. 2013;305:H52–65. doi: 10.1152/ajpheart.00929.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Desjardins CL, Chen Y, Coulton AT, Hoit BD, Yu X, Stelzer JE. Cardiac myosin binding protein C insufficiency leads to early onset of mechanical dysfunction. Circulation Cardiovascular imaging. 2012;5:127–36. doi: 10.1161/CIRCIMAGING.111.965772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoskins AC, Jacques A, Bardswell SC, McKenna WJ, Tsang V, dos Remedios CG, et al. Normal passive viscoelasticity but abnormal myofibrillar force generation in human hypertrophic cardiomyopathy. Journal of molecular and cellular cardiology. 2010;49:737–45. doi: 10.1016/j.yjmcc.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tong CW, Stelzer JE, Greaser ML, Powers PA, Moss RL. Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function. Circulation research. 2008;103:974–82. doi: 10.1161/CIRCRESAHA.108.177683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen YT, Malik FI, Zhao X, Depre C, Dhar SK, Abarzua P, et al. Improvement of cardiac function by a cardiac Myosin activator in conscious dogs with systolic heart failure. Circulation Heart failure. 2010;3:522–7. doi: 10.1161/CIRCHEARTFAILURE.109.930321. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Ajtai K, Burghardt TP. Analytical comparison of natural and pharmaceutical ventricular myosin activators. Biochemistry. 2014;53:5298–306. doi: 10.1021/bi500730t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harris SP, Bartley CR, Hacker TA, McDonald KS, Douglas PS, Greaser ML, et al. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circulation research. 2002;90:594–601. doi: 10.1161/01.res.0000012222.70819.64. [DOI] [PubMed] [Google Scholar]
- 33.Kooij V, Saes M, Jaquet K, Zaremba R, Foster DB, Murphy AM, et al. Effect of troponin I Ser23/24 phosphorylation on Ca2+-sensitivity in human myocardium depends on the phosphorylation background. Journal of molecular and cellular cardiology. 2010;48:954–63. doi: 10.1016/j.yjmcc.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mollova M, Bersell K, Walsh S, Savla J, Das LT, Park SY, et al. Cardiomyocyte proliferation contributes to heart growth in young humans. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:1446–51. doi: 10.1073/pnas.1214608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gresham KS, Mamidi R, Stelzer JE. The contribution of cardiac myosin binding protein-c Ser282 phosphorylation to the rate of force generation and in vivo cardiac contractility. The Journal of physiology. 2014;592:3747–65. doi: 10.1113/jphysiol.2014.276022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fabiato A. Computer programs for calculating total from specified free or free from specified total ionic concentrations in aqueous solutions containing multiple metals and ligands. Methods in enzymology. 1988;157:378–417. doi: 10.1016/0076-6879(88)57093-3. [DOI] [PubMed] [Google Scholar]
- 37.Godt RE, Lindley BD. Influence of temperature upon contractile activation and isometric force production in mechanically skinned muscle fibers of the frog. The Journal of general physiology. 1982;80:279–97. doi: 10.1085/jgp.80.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Campbell KS, Moss RL. SLControl: PC-based data acquisition and analysis for muscle mechanics. American journal of physiology Heart and circulatory physiology. 2003;285:H2857–64. doi: 10.1152/ajpheart.00295.2003. [DOI] [PubMed] [Google Scholar]
- 39.Takahashi R, Talukder MA, Endoh M. Inotropic effects of OR-1896, an active metabolite of levosimendan, on canine ventricular myocardium. European journal of pharmacology. 2000;400:103–12. doi: 10.1016/s0014-2999(00)00385-x. [DOI] [PubMed] [Google Scholar]
- 40.Brenner B. Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: implications for regulation of muscle contraction. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:3265–9. doi: 10.1073/pnas.85.9.3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Campbell K. Rate constant of muscle force redevelopment reflects cooperative activation as well as cross-bridge kinetics. Biophysical journal. 1997;72:254–62. doi: 10.1016/S0006-3495(97)78664-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen PP, Patel JR, Rybakova IN, Walker JW, Moss RL. Protein kinase A-induced myofilament desensitization to Ca(2+) as a result of phosphorylation of cardiac myosin-binding protein C. The Journal of general physiology. 2010;136:615–27. doi: 10.1085/jgp.201010448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stelzer JE, Fitzsimons DP, Moss RL. Ablation of myosin-binding protein-C accelerates force development in mouse myocardium. Biophysical journal. 2006;90:4119–27. doi: 10.1529/biophysj.105.078147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fitzsimons DP, Patel JR, Campbell KS, Moss RL. Cooperative mechanisms in the activation dependence of the rate of force development in rabbit skinned skeletal muscle fibers. The Journal of general physiology. 2001;117:133–48. doi: 10.1085/jgp.117.2.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Regnier M, Martyn DA, Chase PB. Calcium regulation of tension redevelopment kinetics with 2-deoxy-ATP or low [ATP] in rabbit skeletal muscle. Biophysical journal. 1998;74:2005–15. doi: 10.1016/S0006-3495(98)77907-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ford SJ, Mamidi R, Jimenez J, Tardiff JC, Chandra M. Effects of R92 mutations in mouse cardiac troponin T are influenced by changes in myosin heavy chain isoform. Journal of molecular and cellular cardiology. 2012;53:542–51. doi: 10.1016/j.yjmcc.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Michael JJ, Gollapudi SK, Chandra M. Effects of pseudo-phosphorylated rat cardiac troponin T are differently modulated by alpha- and beta-myosin heavy chain isoforms. Basic research in cardiology. 2014;109:442. doi: 10.1007/s00395-014-0442-9. [DOI] [PubMed] [Google Scholar]
- 48.Ford SJ, Chandra M, Mamidi R, Dong W, Campbell KB. Model representation of the nonlinear step response in cardiac muscle. The Journal of general physiology. 2010;136:159–77. doi: 10.1085/jgp.201010467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stelzer JE, Patel JR, Moss RL. Acceleration of stretch activation in murine myocardium due to phosphorylation of myosin regulatory light chain. The Journal of general physiology. 2006;128:261–72. doi: 10.1085/jgp.200609547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stelzer JE, Dunning SB, Moss RL. Ablation of cardiac myosin-binding protein-C accelerates stretch activation in murine skinned myocardium. Circulation research. 2006;98:1212–8. doi: 10.1161/01.RES.0000219863.94390.ce. [DOI] [PubMed] [Google Scholar]
- 51.Stelzer JE, Larsson L, Fitzsimons DP, Moss RL. Activation dependence of stretch activation in mouse skinned myocardium: implications for ventricular function. The Journal of general physiology. 2006;127:95–107. doi: 10.1085/jgp.200509432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mamidi R, Gollapudi SK, Mallampalli SL, Chandra M. Alanine or aspartic acid substitutions at serine23/24 of cardiac troponin I decrease thin filament activation, with no effect on crossbridge detachment kinetics. Archives of biochemistry and biophysics. 2012;525:1–8. doi: 10.1016/j.abb.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mamidi R, Gresham KS, Stelzer JE. Length-dependent changes in contractile dynamics are blunted due to cardiac myosin binding protein-C ablation. Frontiers in physiology. 2014;5:461. doi: 10.3389/fphys.2014.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moss RL, Razumova M, Fitzsimons DP. Myosin crossbridge activation of cardiac thin filaments: implications for myocardial function in health and disease. Circulation research. 2004;94:1290–300. doi: 10.1161/01.RES.0000127125.61647.4F. [DOI] [PubMed] [Google Scholar]
- 55.Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiological reviews. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 56.Stelzer JE, Moss RL. Contributions of stretch activation to length-dependent contraction in murine myocardium. The Journal of general physiology. 2006;128:461–71. doi: 10.1085/jgp.200609634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Malik FI, Morgan BP. Cardiac myosin activation part 1: from concept to clinic. Journal of molecular and cellular cardiology. 2011;51:454–61. doi: 10.1016/j.yjmcc.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 58.Razumova MV, Bukatina AE, Campbell KB. Different myofilament nearest-neighbor interactions have distinctive effects on contractile behavior. Biophysical journal. 2000;78:3120–37. doi: 10.1016/S0006-3495(00)76849-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Locher MR, Razumova MV, Stelzer JE, Norman HS, Patel JR, Moss RL. Determination of rate constants for turnover of myosin isoforms in rat myocardium: implications for in vivo contractile kinetics. American journal of physiology Heart and circulatory physiology. 2009;297:H247–56. doi: 10.1152/ajpheart.00922.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mamidi R, Chandra M. Divergent effects of alpha- and beta-myosin heavy chain isoforms on the N terminus of rat cardiac troponin T. The Journal of general physiology. 2013;142:413–23. doi: 10.1085/jgp.201310971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Palmiter KA, Tyska MJ, Dupuis DE, Alpert NR, Warshaw DM. Kinetic differences at the single molecule level account for the functional diversity of rabbit cardiac myosin isoforms. The Journal of physiology. 1999;519(Pt 3):669–78. doi: 10.1111/j.1469-7793.1999.0669n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rundell VL, Manaves V, Martin AF, de Tombe PP. Impact of beta-myosin heavy chain isoform expression on cross-bridge cycling kinetics. American journal of physiology Heart and circulatory physiology. 2005;288:H896–903. doi: 10.1152/ajpheart.00407.2004. [DOI] [PubMed] [Google Scholar]
- 63.Li KL, Rieck D, Solaro RJ, Dong W. In situ time-resolved FRET reveals effects of sarcomere length on cardiac thin-filament activation. Biophysical journal. 2014;107:682–93. doi: 10.1016/j.bpj.2014.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rieck DC, Li KL, Ouyang Y, Solaro RJ, Dong WJ. Structural basis for the in situ Ca(2+) sensitization of cardiac troponin C by positive feedback from force-generating myosin cross-bridges. Archives of biochemistry and biophysics. 2013;537:198–209. doi: 10.1016/j.abb.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chandra M, Rundell VL, Tardiff JC, Leinwand LA, De Tombe PP, Solaro RJ. Ca(2+) activation of myofilaments from transgenic mouse hearts expressing R92Q mutant cardiac troponin T. American journal of physiology Heart and circulatory physiology. 2001;280:H705–13. doi: 10.1152/ajpheart.2001.280.2.H705. [DOI] [PubMed] [Google Scholar]
- 66.de Tombe PP, Stienen GJ. Protein kinase A does not alter economy of force maintenance in skinned rat cardiac trabeculae. Circulation research. 1995;76:734–41. doi: 10.1161/01.res.76.5.734. [DOI] [PubMed] [Google Scholar]
- 67.Mamidi R, Michael JJ, Muthuchamy M, Chandra M. Interplay between the overlapping ends of tropomyosin and the N terminus of cardiac troponin T affects tropomyosin states on actin. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2013;27:3848–59. doi: 10.1096/fj.13-232363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nagayama T, Takimoto E, Sadayappan S, Mudd JO, Seidman JG, Robbins J, et al. Control of in vivo left ventricular [correction] contraction/relaxation kinetics by myosin binding protein C: protein kinase A phosphorylation dependent and independent regulation. Circulation. 2007;116:2399–408. doi: 10.1161/CIRCULATIONAHA.107.706523. [DOI] [PubMed] [Google Scholar]
- 69.Palmer BM, Georgakopoulos D, Janssen PM, Wang Y, Alpert NR, Belardi DF, et al. Role of cardiac myosin binding protein C in sustaining left ventricular systolic stiffening. Circulation research. 2004;94:1249–55. doi: 10.1161/01.RES.0000126898.95550.31. [DOI] [PubMed] [Google Scholar]
- 70.Belus A, Piroddi N, Scellini B, Tesi C, D’Amati G, Girolami F, et al. The familial hypertrophic cardiomyopathy-associated myosin mutation R403Q accelerates tension generation and relaxation of human cardiac myofibrils. The Journal of physiology. 2008;586:3639–44. doi: 10.1113/jphysiol.2008.155952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Witjas-Paalberends ER, Ferrara C, Scellini B, Piroddi N, Montag J, Tesi C, et al. Faster cross-bridge detachment and increased tension cost in human hypertrophic cardiomyopathy with the R403Q MYH7 mutation. The Journal of physiology. 2014;592:3257–72. doi: 10.1113/jphysiol.2014.274571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Witjas-Paalberends ER, Guclu A, Germans T, Knaapen P, Harms HJ, Vermeer AM, et al. Gene-specific increase in the energetic cost of contraction in hypertrophic cardiomyopathy caused by thick filament mutations. Cardiovascular research. 2014;103:248–57. doi: 10.1093/cvr/cvu127. [DOI] [PubMed] [Google Scholar]