

Abstract
The family of interferon regulatory factors (IRFs) consists of nine members (IRF1–IRF9) in mammals. They act as transcription factors for the interferons and thus exert essential regulatory functions in the immune system and in oncogenesis. Recent clinical and experimental studies have identified critically important roles of the IRFs in cardiovascular diseases, arising from their participation in divergent and overlapping molecular programmes beyond the immune response. Here we review the current knowledge of the regulatory effects and mechanisms of IRFs on the immune system. The role of IRFs and their potential molecular mechanisms as novel stress sensors and mediators of cardiovascular diseases are highlighted.
Linked Articles
This article is part of a themed section on Chinese Innovation in Cardiovascular Drug Discovery. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2015.172.issue-23
Abbreviations
- ATF3
activating transcription factor 3
- CBP
CREB‐binding protein
- cDCs
conventional dendritic cells
- CLL
chronic lymphocytic leukaemia
- CMPs
common myeloid progenitor cells
- CREB
cAMP‐responsive element‐binding protein
- Dock2
dedicator of cytokinesis 2
- dsRNA
double‐stranded RNA
- ECM
extracellular matrix
- ET‐1
endothelin‐1
- GSK3β
glycogen synthase kinase 3β
- GWAS
genome‐wide association studies
- HATs
histone acetyltransferases
- HDACs
histone deacetylases
- I/R
ischaemia/reperfusion
- IAD
IRF‐associated domain
- IFN
interferon
- IGF‐I
insulin‐like growth factor I
- iNOS
inducible NOS
- IRFs
interferon regulatory factors
- MAVS
mitochondrial antiviral signalling protein
- MDA5
melanoma differentiation‐associated gene 5
- MyD88
myeloid differentiation primary‐response protein 88
- PCAF
p300/CBP‐associated factor
- pDCs
plasma cytoid dendritic cells
- PRR
pattern recognition receptor
- RIG‐I
retinoic acid‐inducible gene I
- SLE
systemic lupus erythematosus
- SRF
serum response factor
- TAD
transcription activation domain
- TBK1
TANK‐binding kinase 1
- TG
transgene
- TIRAP
TIR domain‐containing adaptor protein
- TLRs
Toll‐like receptors
- TRAF
TNF receptor‐associated factor
- TRAM
TRIF‐related adaptor molecule
- TRIF
TIRAP‐inducing IFN‐β
- Tyk2
tyrosine kinase 2
- VSMCs
vascular smooth muscle cells
Tables of Links
| TARGETS | |
|---|---|
| Catalytic receptors a | Enzymes b |
| IFN‐α/β receptor | HAT, histone acetyltransferase |
| IFN‐γ receptor | HDAC, histone deacetylase |
| NLR, NOD‐like receptors | TBK1, TANK binding kinase 1 |
| PRR, pattern recognition receptors | GPCRs c |
| TLR3 | Angiotensin AT2 receptors |
| TLR4 | |
| TLR7 | |
| TLR9 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,cAlexander et al., 2013a, 2013b, 2013c).
Introduction and background
The interferon regulatory factor (IRF) family was first described as transcriptional regulators of the type I interferon (IFN) system. Since 1988, when the first IRF was identified (Miyamoto et al., 1988), most studies of IRFs have focused on their functions in immunity. Abnormalities in IRFs are associated with changes in both innate and adaptive immune responses to cellular and environmental stimuli in a wide variety of pathways. Now, more recent evidence has revealed the remarkable contributions of IRFs to other diseases, such as cardiovascular diseases (Guo et al., 2014; Jiang et al., 2014d; Zhang et al., 2014a), metabolic diseases (Eguchi et al., 2008; 2011; Wang et al., 2013c, 2013d; 2014) and oncogenesis (Yanai et al., 2012). In this review, as documented elsewhere, the roles of IRFs in immune responses are only briefly summarized and discussed. Instead, we focus on the essential and distinct functions of IRFs, as novel stress sensors and regulators in cardiovascular diseases to provide a systematic understanding of their therapeutic promise for cardiac remodelling, stroke and vascular injury. A summary of IRFs and their functions in the immune and cardiovascular systems are shown in Table 1.
Table 1.
A summary of IRF family members and their functions in the immune system and the CVS
| IRF | Expression | Related diseases | Functions | Typical targets | References |
|---|---|---|---|---|---|
| IRF1 | Ubiquitously in most cell types and organs | Chronic hepatitis B and C, heart failure, ischaemic stroke, AS, AAA | Antiviral and bacterial responses; induction of cell cycle arrest and apoptosis; required for NK cells development and the differentiation of CD8+ T‐cells; promotes Th1 differentiation; suppresses Th2 differentiation; accelerates cardiac remodelling; exacerbates ischaemic stroke; protects against neointima formation | IFN‐inducible genes, iNOS, IL‐4, IL‐12, IL‐15, CD40, PPARγ | Liu et al., 2000; Taniguchi et al., 2001; Watford et al., 2003; Wessely et al., 2003; Mellor and Munn, 2004; Passioura et al., 2005; Tamura et al., 2005; Caso et al., 2007; Schmitz et al., 2007; Li et al., 2009; Korachi et al., 2013; Jiang et al., 2014a |
| IRF2 | Widely in various cells and organs | Unknown | Accentuates type I IFN response; essential for CD4+ DC differentiation and NK cells development; promotes Th1 differentiation; suppresses Th2 differentiation | IRF1, IRF9, IL‐4, IL‐12 | Taniguchi et al., 2001; Ichikawa et al., 2004; Jia and Guo, 2008 |
| IRF3 | Most cell types and organs | Dilated cardiomyopathy, hypertrophic cardiomyopathy, stroke, AS, AAA | Induces type I IFNs; reduces cardiac hypertrophy; indispensable for TLR ligands pretreatment‐induced tolerance to stroke; exacerbates outcomes of stroke; inhibits neointimal formation | IFN‐α, IFN‐β, PPARγ, ERK2 | Holland et al., 2008; Stevens et al., 2011; Lu et al., 2013a; Sasai and Yamamoto, 2013; Zhang et al., 2014b |
| IRF4 | Constitutive in cardiocytes, neurons, macrophages and DCs, abundant in lymphocytes | Recurrent bronchitis, atopy, CLL, AS, AAA | Retards interactions of IRF5 with MyD88 and reduces TLR‐dependent inflammation; required for differentiation of CD4+ DCs, pDCs and macrophages; exacerbates cardiac remodelling; improves stroke outcomes; required for B‐lymphocytes development | IRF5, IL‐4, CREB, SRF | Pernis, 2002; Schiavoni et al., 2002; Lehtonen et al., 2003; Kanno et al., 2005; Negishi et al., 2005; Tamura et al., 2005; Di Bernardo et al., 2008; Jiang et al., 2013; Pinto et al., 2013; Guo et al., 2014 |
| IRF5 | Mainly in B‐cells and DCs; expressed in the heart | SLE, cardiac hypertrophy | Stimulates type I IFNs and immune responses upon virus infection | IFN‐α, IFN‐β, IL‐6, TNF‐α | Brown et al., 2011 |
| IRF6 | Constitutive in skin | Unknown | Required for keratinocyte differentiation | Unknown | Restivo et al., 2011; Biggs et al., 2014 |
| IRF7 | Most cell types and organs | Influenza infection, SLE, heart failure, stroke, AS, AAA | Positively regulates TLR‐dependent proinflammatory responses; suppresses cardiac remodelling; required for TLR ligands pretreatment‐elicited neuroprotection; alleviates ischaemic stroke; inhibits neointimal formation | IFN‐α, IFN‐β, IKKβ, ATF3 | Erickson and Gale, 2008; Holland et al., 2008; Stevens et al., 2011; Sasai and Yamamoto, 2013; Jiang et al., 2014b |
| IRF8 | Expressed in cardiocytes, neurons, and lymphocytes | Heart failure, ischaemic stroke, AS, AAA | Promotes type I and II IFNs; required for CD8+ DCs and pDCs differentiation; promotes macrophage differentiation; promotes myeloid apoptosis; essential for B‐cells development; alleviates cardiac hypertrophy; reduces stroke‐induced infarct lesions; accelerates vascular hyperplasia | IFN‐α, IFN‐β, Bcl2l1, Nf1, Bax, iNOS, IL‐12, NFATc1, SRF | Lehtonen et al., 2003; Kanno et al., 2005; Tamura et al., 2005; Huang et al., 2007; Yang et al., 2011; Scheller et al., 2013; Jiang et al., 2014d; Xiang et al., 2014; Zhang et al., 2014a |
| IRF9 | Broadly in various cells and organs | Heart failure, MI, stroke, AS, AAA | Forms ISGF3 to promote type I IFN‐inducible genes; anti‐hypertrophy; promotes myocardial I/R responses; aggravates stroke responses; augments intimal hyperplasia | IFN‐α, IFN‐β, SRF, Sirt1 | Chen et al., 2014; Jiang et al., 2014c; Zhang et al., 2014c |
AAA, abdominal aortic aneurysm; AS, atherosclerosis; MI, myocardial infarction.
Structural basis of IRFs
The first IRF, IRF1, was originally identified as a protein binding to the virus‐inducible elements of the human IFN‐β gene (Miyamoto et al., 1988). IRF2 was recognized in the following year through cross‐hybridization with IRF1 cDNA (Harada et al., 1989). As shown in Figure 1, IRF1 and IRF2 have 62% homology located at their amino‐terminal regions, which comprise a DNA‐binding domain (DBD) with five highly conserved tryptophan repeats (Harada et al., 1989). Using three of the five tryptophan repeats, the DBDs of IRF1 and IRF2 form a helix‐turn‐helix motif to recognize and bind a consensus DNA sequence known as the IRF‐element (IRF‐E) (Tanaka et al., 1993; Fujii et al., 1999). In the carboxyl‐terminal region, IRF1 contains many acidic amino acids and serine‐threonine residues, whereas IRF2 is relatively rich in basic amino acids (Harada et al., 1989), suggesting that these factors have distinct functions.
Figure 1.
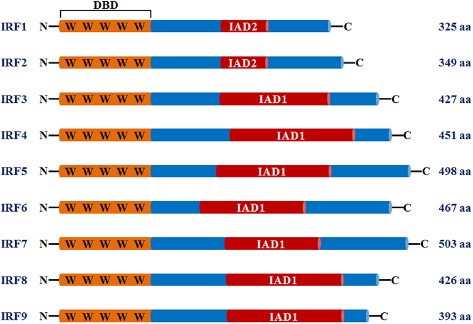
Schematic illustration of mammalian IRFs (IRF1–IRF9). All of the IRFs possess a homology spanning 115 amino acids in their N‐terminal regions, including a DBD characterized by five highly conserved tryptophan repeats. In the C‐terminal region, the IRFs, except for IRF1 and IRF2, contain IAD1, while IRF1 and IRF2 share IAD2.
Following the identification of these two structurally related factors, numerous approaches have been taken to explore additional IRF factors, and seven other mammalian IRFs have been identified: IRF3, IRF4 (LSIRF, PIP, MUMI or ICSAT), IRF5, IRF6, IRF7, IRF8 (ICSBP) and IRF9 (p48 or ISGF3γ) (Taniguchi et al., 2001; Honda and Taniguchi, 2006b). Like IRF1 and IRF2, these members of the IRF family showed a homology of over 115 amino acids in their amino‐terminal regions with five well‐conserved tryptophan repeats and the helix‐turn‐helix DNA‐binding motif, suggesting similar, if not identical, DNA recognition sequences, for example, IRF‐E (Lallemand et al., 2007), IFN‐stimulated response element (ISRE) (Guillot et al., 2005), IFN consensus sequence (Ko et al., 2002), and positive regulatory domain (PRD) (Hiscott, 2007). The carboxyl‐terminal regions of the IRF family, which contain transcriptional activation domains, might constitute regulatory regions. Except for IRF1 and IRF2, IRFs exhibit homology to the C‐terminal domains of the SMAD family and contain an IRF‐associated domain 1 (IAD1), while IRF1 and IRF2 share IAD2 (Eroshkin and Mushegian, 1999; Yanai et al., 2012). The protein–protein interactions that are dependent on these domains may define the binding sites and functions of the protein complex, as a transcriptional activator or repressor.
Cellular events of IRFs
A given transcription factor mediates the associated genetic regulatory network by activating, suppressing and/or interacting with its target genes. The specific regions of IRF members allow them to bind to corresponding motifs of their modifiers, co‐activators and co‐repressors, leading to diverse and specific molecular events, depending on the cell types and variations in stimuli. Accumulating studies have indicated that, following various extracellular and intracellular stimuli, IRFs activate and expand immune responses through various molecular events, especially the IFN and pattern recognition receptor (PRR) signalling pathways (Taniguchi et al., 2001; Honda and Taniguchi, 2006b). These molecular events are summarized in Figure 2.
Figure 2.
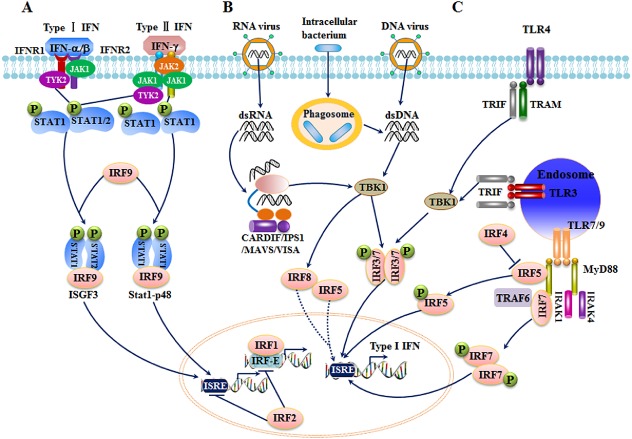
Schematic representation of molecular events involving IRFs in immune responses. (A) Role of IRFs in IFN signalling. In type I IFN responses, IFN‐α/β binds to its receptor and activates JAK1/Tyk2, leading to the recruitment and phosphorylation of STAT2/STAT1. IRF9 forms a complex with the phosphorylated STAT factors, translocates to the nucleus, and activates IFN‐inducible genes. In type II IFN signalling, the STAT1/STAT2/IRF9 and STAT1/STAT1/IRF9 complexes form in a similar pathway. (B) IRFs in cytosolic PRRs. In response to viral infection, RIG‐I or MDA5 recognizes viral RNA and binds to CARDIF/IPS1/MAVS/VISA, leading to the phosphorylation of IRF3 and IRF7 through TBK1. The phosphorylated IRF3 and IRF7 undergo homo‐ or hetero‐dimerization and nuclear translocation, thereby binding to ISRE DNA sequences and activating IFNs and other target genes. In some cases, IRF5 and IRF8 also participate in PRR signalling. (C) IRFs in TLR signalling. Depending on adaptor types, TLR signalling is divided into MyD88‐dependent and TRIF‐dependent pathways. TLR3 and TLR4 respond to stimuli and activate IRF kinase TBK1 mainly through a MyD88‐independent pathway. TBK1 subsequently phosphorylates IRF3 and IRF7, resulting in their dimerization and translocation to the nucleus. In contrast to IRF3 and IRF4, TLR7 and TLR9 trigger IRF activation mainly through a MyD88‐dependent pathway. IRF5 and IRF7 directly bind to MyD88 or TRAF6, dimerize, and translocate to the nucleus. IRF4 inhibits the interaction of IRF5 with MyD88.
IRFs in IFN signalling
IFNs are the central mediators of innate immunity. Two types of IFNs, type I IFNs (IFN‐α and IFN‐β) and type II IFNs (IFN‐γ), have been identified. The type I IFNs are crucial mediators of antiviral responses in a variety of cells following viral infection and are principally regulated by IRFs, whereas type II IFNs are mainly produced by activated T‐cells and NK cells (Taniguchi et al., 2001). In the type I IFN system, the binding of ligands with the IFN‐α/β receptor activates JAK1 and tyrosine kinase 2 (Tyk2). Following tyrosine phosphorylation of residue 455 (Y455) of the intracellular domain of IFN‐α/β receptor 1 through JAK1 and Tyk2, STAT2 is recruited to this Y455 via its SH2 domain, whereas STAT1 is recruited by IFN‐α/β receptor 1 through its interaction with phosphorylated STAT2. These phosphorylated STAT factors, in combination with IRF9, form a heterotrimeric complex, ISGF3, that subsequently translocates to the nucleus and binds to ISRE or IRF‐E, leading to the activation of several IFN‐inducible genes (Taniguchi et al., 2001; Honda and Taniguchi, 2006b).
As in the IFN‐α/β receptor signalling pathway, stimuli on the IFN‐γ receptor may induce formation of the STAT1/STAT2/IRF9 and STAT1/STAT1/IRF9 heterotrimeric complex. Additionally, the dimerization of phosphorylated STAT1 undergoes conversion into the transcriptionally active form, IFN‐γ‐activated factor/IFN‐α‐activated factor, and binds to the IFN‐γ‐activated site (GAS) in the IRF1 promoter, inducing transcription of the IRF1 gene (Takaoka and Yanai, 2006). The IRF family members are thus pivotally involved in IFN signalling from both the α/β and the γ receptors.
IRF1 and IRF2 are ubiquitous and expressed at low levels in various cell types in normal physiological conditions. However, after stimulation with viral infection and immune factors, for example TNF‐α, IL‐1β, IL‐6 and IFNs, the expression of IRFs, particularly IRF1, is dramatically up‐regulated concomitant with posttranslational modification, for example phosphorylation, allowing functional changes from steady‐state levels (Taniguchi et al., 2001). The serine‐phosphorylation of IRF1 by PKA, PKC, and casein kinase II is one of the most common phenomena during IRF‐regulated molecular events (Taniguchi et al., 2001). Phosphorylated IRF1 activates IFN‐α/β promoters and induces endogenous IFN‐α and IFN‐β genes through the mediation of STAT and NF‐κB transcription factors (Sato et al., 2000; Taniguchi and Takaoka, 2002; Honda et al., 2006a). In contrast to IRF1, phosphorylated IRF2 represses both ISRE‐ and IRF‐E‐regulated transcriptional activation, thereby weakening IRF1‐induced IFN‐α/β signalling (Hida et al., 2000; Jia and Guo, 2008). Thus, IRF1 and IRF2 function as a transcriptional activator and attenuator of IFNs respectively.
IRF3 and IRF7 are ubiquitously expressed in a variety of cells and have very similar structures. These IRFs are best known for their critical roles as transcriptional regulators promoting activation of type I IFN by binding to IRF‐E and ISRE after homo‐ or hetero‐dimerization (Holland et al., 2008). Under normal conditions, these factors reside in the cytoplasm; however, upon viral infection, IRF3 and IRF7 are activated, phosphorylated, polymerized and then translocate to the nucleus to regulate IFN production (Taniguchi et al., 2001). After treatment with IFNs, IRF3 expression is constitutive and remains essentially unaffected, whereas the level of IRF7 is dramatically enhanced in IFN‐α/β signalling, indicating that IRF7 could be considered as an IFN‐stimulated gene (ISG) triggering and augmenting IFN expressions to further activate systemic IFN‐induced antiviral defences (Taniguchi et al., 2001; Prakash et al., 2005; Erickson and Gale, 2008; Chen et al., 2013a). The dominant effect of IRF7 in activating IFN‐α/β was demonstrated by the evidence that the induced expression of IFNs in wild‐type mice is similar to that of IRF3 −/− following viral infection, whereas in the IRF7 −/− and IRF3 −/− IRF7 −/− mice, dramatically lower levels of IFN‐α/β expression were obtained (Chen et al., 2013a). In IRF7 −/− mouse embryonic fibroblasts (MEFs), the induction of IFN‐β mRNA was markedly inhibited and the residual mRNA induction was blocked with an additional deficiency of IRF3, suggesting synergistic effects of IRF3 and IRF7 in the induction of IFNs (Honda et al., 2005). In vivo studies demonstrated that the combined absence of IRF3 and IRF7 resulted in uncontrolled viral burdens, higher inflammatory responses, and more rapid death of IRF3 −/− IRF7 −/− mice compared with the individual single IRF knockout (KO) mice, directly confirming the mutually enhanced effects of IRF3 and IRF7 (Daffis et al., 2007; 2008; 2009; 2011; Murphy et al., 2013).
In contrast to the ubiquitous expressions of other IRF members, IRF4 and IRF8 are preferentially expressed in cells responsible for immune responses (Lehtonen et al., 2003; Kanno et al., 2005). In the presence of a high level of IFN‐α, the expression of IRF4 is strongly up‐regulated, whereas the expression of IRF8 is induced by IFN‐γ, rather than by IFN‐α/β (Tamura and Ozato, 2002; Wang and Morse, 2009). These two IRFs regulate cellular immunity by directly binding to IRF‐E or ISRE motifs in the target gene promoters or via their indirect interaction with composite motifs that can be recognized by the Ets transcription factors PU.1 and Spi‐B (Honma et al., 2005; Lu, 2008). The phosphorylated serine residue in the PEST region of PU.1 provides a binding site that is required for the PU.1–IRF4/IRF8 interaction (Escalante et al., 2002). Similarly, IRF4/IRF8 forms a complex with Spi‐B, a factor that is closely associated with PU.1, on an Ets–IRF composite element. Moreover, several other factors regulate the functions of IRF4 and IRF8 in IFN signalling. STAT4, an essential signalling protein for Th1 responses, binds to the GAS‐like element in the promoter region of IRF4. In a yeast two‐hybrid analysis, the immunophilin FK506‐binding protein 52 (FKBP52) post‐translationally regulated and interacted with IRF4, thereby inhibiting the DNA‐binding activity of IRF4 (Mamane et al., 2000).
As mentioned earlier, IRF9 was originally discovered as a DNA‐binding subunit of the transcription factor ISGF3, whose formation is triggered by activation of IFN‐α/β receptors, resulting in the induction of various IFN‐inducible genes. The details of the molecular events involving IRF9 in this pathway have yet to be investigated.
IRFs in PRR signalling
Since the discovery of PRRs, a family that links innate and adaptive immunity, IRFs have received much more attention as regulators of immune responses (Honda and Taniguchi, 2006b). Two classes of PRRs, the cytosolic PRRs and the transmembrane Toll‐like receptors (TLRs), have been defined (Savitsky et al., 2010).
IRFs in cytosolic PRR signalling
Cytosolic PRRs comprise three major types: the retinoic acid‐inducible gene I (RIG‐I) family, IFN‐inducible double‐stranded RNA (dsRNA)‐dependent PKR, and nucleotide‐binding oligomerization domain (NOD) proteins (Janeway and Medzhitov, 2002; Akira et al., 2006), among which the RNA helicase enzymes in the RIG‐1 family, RIG‐I and melanoma differentiation‐associated gene 5 (MDA5), are essential receptors for the recognition of viral RNA and the activation of downstream signalling pathways (Savitsky et al., 2010). In the early phase of viral infection, RIG‐I or MDA5 interacts with the mitochondrial antiviral signalling protein MAVS (also known as IPS‐1, VISA or CARDIF) via the physical binding of their N‐terminal caspase‐recruitment and activation domains (Seth et al., 2005). The signals are then relayed to TANK‐binding kinase 1 (TBK1) and induce the phosphorylation of IRF3 and IRF7 on serine residues, resulting in their homo‐ or hetero‐dimerization and nuclear translocation (Kawai et al., 2004). After binding to the target ISRE DNA sequences, this complex transcriptionally activates type I IFNs and other target genes, such as the chemokine CXCL10. The secreted IFNs bind to IFN receptors in an autocrine or paracrine manner and positively activate the TBK1‐IRF3/7‐IFN loop, leading to the expression of large amounts of IFNα/β proteins in a positive feedback mechanism (Honda et al., 2006a). Four IRF members, IRF3, IRF5, IRF7 and IRF8, are clearly involved in the RIG‐I/MDA5‐regulated PRR signalling pathways.
The IRF3 and IRF7‐related cytosolic PRR events are mediated by various molecules. lgp2, a gene that belongs to the DExD/H‐box‐containing RNA helicase family together with RIG‐1 and MDA5, might compete with RIG‐1 and MDA5 for engagement with viral RNA, thereby negatively regulating the RIG‐I‐ and MDA5‐signalling pathways (Yoneyama et al., 2005). Cyclophilin B, TNF receptor‐associated factor 3 (TRAF3) and the ubiquitin ligase mind bomb, positively regulate TBK1‐activated IRFs, whereas the suppressor of IκB‐kinase ε might function as a negative regulator (Huang et al., 2005; Heineke and Molkentin, 2006; Oganesyan et al., 2006; Li et al., 2011b). Additionally, cAMP‐responsive element‐binding protein (CREB)‐binding protein (CBP) or p300, as co‐activator, promotes the phosphorylation process and forms a holocomplex with IRF3 and IRF7 (Mori et al., 2004; Panne et al., 2007). In contrast, the HCV NS3/4A serine protease blocks the phosphorylation and functions of IRF3 (Foy et al., 2003). The prolyl isomerase, PIN1 (peptidylprolyl cis–trans isomerase, NIMA‐interacting 1), could also blunt or even terminate the action of IRF3 by binding to phosphorylated IRF3 in the nucleus, facilitating the polyubiquitin‐dependent degradation of IRF3 (Saitoh et al., 2006). After the elimination of virus, the IFN response should subside before it becomes deleterious to the host. This attenuation may occur partly through the translational‐suppressive functions of 4E‐BPs on IRF7 mRNA translation, thereby decreasing IFN production and the innate immune response (Erickson and Gale, 2008).
Although IRF3 and IRF7 exert significant effects on IFN production, sustained production of type I IFN was still found with the combined absence of IRF3 and IRF7 under the control of MAVS signalling. However, the simultaneous loss of IRF3, IRF5 and IRF7 (IRF3/5/7‐TKO) in mice yields immune responses relatively equivalent to those seen in MAVS −/− mice (Lazear et al., 2013). Like IRF3 and IRF7, IRF5 can be phosphorylated by TBK1 and translocated into the nucleus to induce IFN activation following viral infection (Savitsky et al., 2010). Lazear et al. (2012) indicated that the RIG‐I like receptor adaptor protein MAVS could induce ISGs through an IRF5‐dependent but IRF3‐ and IRF7‐independent pathway. However, gene‐disruption studies of IRF5 have reached disparate conclusions in terms of IFN production and antibody responses. The identified spontaneous genomic duplication and frameshift mutation in the guanine exchange factor, dedicator of cytokinesis 2 (Dock2), might explain the discrepancies among the published results. Dock2 has emerged in a subset of circulating IRF5 −/− mice and inadvertently to homozygosity. IRF5 −/− mice lacking the mutation in Dock2 exhibited largely intact type I IFN responses and relatively normal antibody responses to viral infection (Purtha et al., 2012). IRF8 is also essential for IFN‐α/β induction in virus stimulated immune cells by directly binding to promoters of type I IFN genes or via participation in the IRF7‐mediated amplifying phase of IFN transcription (Tamura et al., 2008).
IRFs in TLR signalling
The TLR family, which consists of 10 functional members in humans (13 in mice), plays a pivotal role in the activation of both the innate and adaptive immune responses. All TLRs contain extracellular leucine‐rich repeats that recognize pathogen‐associated molecular patterns, whereas the transmembrane and cytoplasmic Toll/IL‐1 receptor (TIR) domains transmit intracellular signals by recruiting TIR‐containing adaptor proteins (Takeda and Akira, 2004; Kawai and Akira, 2006). Depending on their adaptor types, that is myeloid differentiation primary‐response protein 88 (MyD88), TIR domain‐containing adaptor protein (TIRAP), TIRAP‐inducing IFNβ (TRIF) or TRIF‐related adaptor molecule (TRAM), the TLR signalling pathways can be divided into a MyD88‐dependent pathway, which is shared by all TLRs, and a TRIF‐dependent pathway (MyD88‐independent pathway), which is specific to TLR3 and TLR4 (Tamura et al., 2008). TLR3, TLR4, TLR7 and TLR9 are the major TLRs responsible for the activation of IRF‐engaged molecular events through both the MyD88‐ and TRIF‐dependent pathways.
TLR3, which is expressed at the cell surface of endothelial and NK cells or in the membrane of endosomes and phagosomes, employs a TRIF‐dependent pathway to induce type I IFN (Akira et al., 2006). In response to stimuli from dsRNA or other pathogens, TLR3 interacts directly with TRIF to trigger the IRF kinase TBK1, which subsequently phosphorylates IRF3 and IRF7, the essential factors in IFN production mediated by TLR pathways (Sasai and Yamamoto, 2013). Similarly to TLR3, TLR4 can induce IFN gene expression in a MyD88‐independent and TRIF‐ and TBK1‐dependent pathway; however, this induction is limited to the gene encoding IFN‐β and excludes IFN‐α (Piao et al., 2013). Using the intracellular adaptor protein, IRF4 interacts with TRAM‐TRIF, which recruits TBK1 through NF‐κB activating kinase‐associated protein 1 and TRAF3 (Clark et al., 2011). The activated TBK1 phosphorylates IRF3 and IRF7, facilitating their dimerization and translocation to the nucleus, inducing the genes that encode IFN‐β. Gene deletion studies have indicated that IRF3, rather than IRF7, is required for the MyD88‐independent TLR4 signalling pathways (Shaik‐Dasthagirisaheb et al., 2014).
In contrast to TLR3 and TLR4, TLR7 and TLR9 regulate IRFs mainly via MyD88‐dependent signalling. With the stimulation of TLR7/9 ligands, IRF7 directly interacts with the death domain of MyD88 or is activated by a kinase cascade involving IRAK4, IRAK1 and IKKα (O'Neill and Bowie, 2007). Utilizing the TLR7/9–MyD88–IRF7 signalling pathway, a positive feedback loop can be activated, thereby stimulating robust type I IFN production (Colonna, 2007). Similarly to IRF7, IRF5 could bind to and is activated by MyD88 and TRAF6. Following its interaction with an intermediary and part of the TIR domain of MyD88, activated IRF5 dimerised and then translocated to the nucleus (Brown et al., 2011). The binding of IRF5 to MyD88 can be retarded by IRF4, because IRF4 binds to MyD88 in the region overlapping that of IRF5 (Negishi et al., 2005). IRF8 is also involved in TLR9 signalling, amplifying the type I IFN response by interacting with TRAF6 or directly activating the transcription of pro‐inflammatory cytokine genes (Lande and Gilliet, 2010).
Ubiquitination of IRFs
During molecular events involving IRFs, ubiquitination, like phosphorylation, is an efficient cellular process that is responsible for many signal transductions and drives various immunity‐related regulatory events. These two protein‐modification events can occur rapidly through dedicated enzymes, and often act cooperatively in mobilizing particular cellular pathways in response to extracellular signals (Ben‐Neriah, 2002; Wang and Maldonado, 2006). For IRF1, under certain stress conditions, the C‐terminus of Hsc70‐interacting protein binds to and forms a stable complex with IRF1, allowing an increase in IRF1 ubiquitination and a decrease in IRF1 steady‐state levels (Narayan et al., 2011). Following the removal of the C‐terminal 70 amino acids from IRF1, which could be recognized by an E3 ligase, its degradation and polyubiquitination can be inhibited, suggesting that the C‐terminal of IRF1 promotes ubiquitination. Conversely, the enhancer domain in trans inhibits endogenous E3 ligase‐regulated IRF1 ubiquitination (Pion et al., 2009). For IRF2, two binding sites on IRF2 have been identified for the interaction of IRF2 and Mdm2, leading to the ubiquitin‐induced degradation of IRF2 and the retardation of IRF2‐attenuated IFNs activity (Pettersson et al., 2009).
To protect the host from excessive or even uncontrollable immune responses after viral infection, the activations of IFNs will be limited directly or indirectly through polyubiquitination and subsequent proteasomal degradation, especially of IRF3 and IRF7. Viral proteins have been considered as E3 ubiquitin ligases and target IRFs for proteasomal degradation under the regulation of various factors. Bauhofer and Hilton research groups (Hilton et al., 2006; Bauhofer et al., 2007) demonstrated that the pestiviruses, classical swine fever virus and bovine viral diarrhoea virus, could interact with IRF3 through the protein Npro, prior to its phosphorylation‐induced activation, resulting in polyubiquitination and subsequent proteasomal degradation of IRF3, which could be abolished through thermal inactivation of the E1 ubiquitin activating enzyme (Chen et al., 2007). The non‐structural protein 1 and bICP0 (BHV1‐ICP0) could directly mediate the degradation of IRF3 in a proteasome‐dependent manner, thus inhibiting the IFN‐β promoter (Barro and Patton, 2005; Graff et al., 2007; Saira et al., 2007). Additionally, several other proteins, for example viral protein R, viral infectivity factor, RBCC protein interacting with PKC1, and caspase‐8 have been found to catalyse, maybe not directly promote, the ubiquitination and degradation of IRF3 (Melroe et al., 2007; Okumura et al., 2008; Zhang et al., 2008; Sears et al., 2011). For the regulation of IRF7 degradation, N‐Myc and STATs interactor, a negative regulator of the virus‐triggered type I IFNs, was found to promote K48‐linked IRF7 ubiquitination (Wang et al., 2013a). Notably, among those factors responsible for the ubiquitination and degradation of IRFs, Ro52 (TRIM 21) has been highlighted not only for its direct effects on diminishing the transcriptional activities of IRFs, but also for dual functions in their activation (Higgs et al., 2008; 2010). In certain cell types and conditions, Ro52 may sustain IRF3 and IRF8 activity by facilitating interactions with their co‐activators, whereas under other conditions, Ro52 promotes the inactivation of IRF3, IRF5, IRF7 and IRF8 (Kong et al., 2007; Yang et al., 2009; Oke and Wahren‐Herlenius, 2012). Further evidence to substantiate that the protein degradation is not the only outcome of ubiquitination can be found in ubiquitin‐mediated IRF7 activity. For instance, following Lys63‐linked ubiquitination stimulated by TRAF6 and the viral oncoprotein latent membrane protein 1, the activation of IRF7 is enhanced (Kawai et al., 2004; Huye et al., 2007).
The ubiquitination‐controlled activities of other IRFs have remained poorly understood until recently. Among the cellular events regulating IRF5 activity, TRAF6‐mediated K63‐linked polyubiquitination also occurred and is required for nuclear translocation and activation of IRF5 (Balkhi et al., 2008). For IRF8, Cb1 is likely to be the ubiquitin–protein isopeptide ligase and could form a complex with IRF8, contributing to its degradation (Xiong et al., 2005). As ubiquitination plays multiple roles in IRF activations, related factors and mechanisms of this molecular programme require further investigation.
Acetylation of IRFs
Acetylation has pivotal roles in activating cytokine receptors and their downstream signalling pathways. Acetylation of diverse domains of IRF members leads to their conformational change and thus influences their DNA‐binding activities (Tang et al., 2007b). Generally, acetylation in DBD represses DNA binding, whereas acetylation adjacent to DBD enhances DNA binding (Masumi, 2011). Therefore, it is not surprising that both transcriptional activating and silencing can be mediated through the orchestra of acetylation‐associated enzymes and IRFs.
Modification of reversible histone acetylation and deacetylation processes is governed by the opposing activities of histone acetyltransferases (HATs) and histone deacetylases (HDACs). These enzymes have emerged as major regulators of the expression and acetylation activation of genes (Feng et al., 2010). Both IRF1 and IRF2 can be acetylated by forming a multiprotein complex with HATs such as p300/CBP and PCAF (p300/CBP‐associated factor), enabling the crosstalk between IRFs and cell apoptosis or growth‐regulated promoters (Masumi et al., 2003; Marsili et al., 2004). Lys78 of helix α3 within DBD is the major acetylation site for both IRFs, while Lys75 is specific for IRF2 (Qi et al., 2012). Acetylated IRF2, but not IRF1, inhibits p300‐mediated acetylation of core histones; IRF2 thus possibly acts as a substrate for histone acetylases, competitively inhibiting histone acetylation (Masumi, 2011).
For IRF3 and IRF7, the HATs (including p300, PCAF and GCN5) and the corresponding acetylation are crucial for the DNA binding of their homo‐ or hetero‐complex, which are responsible for IFN transcriptional activation (Masumi, 2011). Like IRF1 and IRF2, the single lysine‐residue target of IRF7 for acetylation is a helix‐α3 lysine, Lys92, which is located in the DBD and conserved throughout the entire IRF family. Acetylated IRF7 exhibits an impaired DNA‐binding capacity (Caillaud et al., 2002; Masumi et al., 2003).
The HAT‐regulated acetylations of other IRFs, including IRF4, IRF5 and IRF9, are inducible upon viral infection or IFN treatment. Under the indicated conditions, IRF4 binds to and could be acetylated by GCN5 (Kikuchi et al., 2014), while IRF5 is reversibly acetylated by p300 and CBP (Feng et al., 2010). In response to IFN stimulus, IFN‐α/β receptor 2 and IRF9 undergo CBP‐mediated acetylation and activate ISGF3. All of the acetylated lysine sites of IRF9 are highly conserved and reside within the N‐terminal domain (Tang et al., 2007b). These results collectively demonstrate that the activation and functions of the IRF family are intricately regulated by diverse and/or overlapping molecular programmes, the in‐depth investigation of which will greatly expand our understanding of their broad functions in related diseases.
Implication of IRFs in immunity
Benefiting from emerging molecular biotechnological approaches, genetic techniques and network analysis, the responses of IRFs involved in classic molecular responses to internal and external stimuli have been expanded to their roles in the development and functioning of several cell lines of immunological importance, including dendritic cells (DCs), myeloid cells, NK cells, T‐cells and B‐cells.
DCs, comprising conventional DCs [cDCs, including CD4+ DCs, CD8α+ DCs and CD4−CD8α− (double‐negative, DN) DCs] and pDCs, are crucial for mounting both the innate and adaptive immune systems (Geissmann et al., 2010; Mildner et al., 2013). The development of DCs is mediated by several IRFs, among which IRF4 and IRF8 initially emerged as key players. Studies on IRF4 −/−, IRF8 −/− and IRF4 −/− IRF8 −/− mice have demonstrated that IRF4 and IRF8 are essential for the generation of CD4+ and CD8α+ DCs, respectively; while, in conventional DN DCs and pDCs, overlapping effects of IRF4 and IRF8 regulate cell development (Schiavoni et al., 2002; Tamura et al., 2005; Hambleton et al., 2011). Also, IRF4 and IRF8 are functionally separated in DC differentiation, which occurs in a subset‐selective manner in response to two different growth factors, Flt3 ligand (Flt3L) and GM‐CSF (Gilliet et al., 2002; Tamura et al., 2005). IRF1 and IRF2 also regulate DCs development and differentiation. IRF1‐KO mice exhibit reduced numbers of CD8α+ DCs (Gabriele et al., 2006), whereas IRF2−/− DCs display a significant defect in CD4+CD8α− DCs (Ichikawa et al., 2004). Additionally, IRF1 may inhibit the immunosuppressive features of DCs, as indicated by the failure of IRF1−/− splenic DCs to mature in response to viral or bacterial stimuli (Mellor and Munn, 2004).
Common myeloid progenitor cells (CMPs) give rise to granulocytes (neutrophils) and macrophages. Clinical and experimental studies have shown that IRF4 and IRF8 function as notable effectors in the development, growth and apoptosis of myeloid cells. Although IRF4 is expressed at a much lower level than IRF8 in granulocyte‐macrophage progenitors, both of these IRFs strongly promote the differentiation of CMPs to macrophages. However, neutrophilic differentiation is inhibited by the ectopic expression of IRF4 or IRF8, possibly as a result of altered PU.1 expression (Yamamoto et al., 2011; Becker et al., 2012). Presumably because of the lower expression level, IRF4 −/− mice, but not IRF8 −/− mice, exhibit no obvious abnormality in the development of myeloid cells compared with their wild‐type controls (Yamamoto et al., 2011). Additionally, IRF8 shows a potent ability to inhibit myeloid cell growth and to promote apoptosis potentially by regulating several key genes, for example Bcl2l1, Nf1 and Bax (Huang et al., 2007; Yang et al., 2011; Scheller et al., 2013). It was also reported that IRF1 stimulates cell differentiation and mediates the N‐ras‐induced growth suppression of myeloid cells (Passioura et al., 2005; Schmitz et al., 2007).
Lymphocytes are a diverse group of cells that are central to the development of immunologic memory and tolerance. NK cells, originally identified as effector lymphocytes of innate immunity with constitutive cytolytic functions, are now known to have much more sophisticated biological functions, such as the early control of viral infection and the secretion of cytokines to shape T‐cell responses (Vivier et al., 2011). In vivo experiments have demonstrated that IRF1 robustly regulates the development and functions of NK cells, as suggested by the finding that IRF1 deficiency not only reduced the number of NK cells in the spleen and liver, but also abolished NK cell‐mediated cytolytic activity. The impaired transcriptional induction of genes encoding IL‐15 and IL‐12, cytokines responsible for the development and functions of NK cells, might largely, if not entirely, account for the potent effects of IRF1 on NK cells (Liu et al., 2000; Watford et al., 2003). Interestingly, in contrast to the results seen with the reverse functions in IFN signalling, IRF2 −/− mice carry similar defects in the development and functions of NK cells compared with IRF1 −/− mice, whereas the number of NK cells was maintained at a normal level in IRF2 −/− mice (Taki et al., 2005).
Mammalian adaptive immunity is regulated by two additional types of lymphocytes: B‐ and T‐cells. After encountering antigens, naive B‐ and T‐cells undergo cell division and maturation before exerting their effector functions (Vivier et al., 2011). Among the discovered IRFs, most research on the functions of B‐ and T‐cells has focused on IRF4 and IRF8, whose expression is largely restricted to lymphocytes. Molecular analysis indicated that IRF4 and IRF8 are required for the down‐regulation of surrogate light‐chain gene expression and its sequential DNA rearrangements during the development of B‐lymphocytes (Lu et al., 2003; Lazorchak et al., 2006). Genetic studies also demonstrated that IRF4 is critical to mounting the antibody, cytotoxic or anti‐tumour responses of mature B‐ and T‐cell lineages, while IRF8 is partly involved in the transcriptional network governing B‐cell specification, commitment, and differentiation (Pernis, 2002; Wang et al., 2008). In addition to their involvement in B‐cell development and functions, IRF8 and IRF4 promote the differentiation of Th1 and Th2 cells by regulating their promoting cytokines IL‐12 and IL‐4 respectively (Masumi et al., 2002; Rengarajan et al., 2002). In addition, IRF4 is an indispensable effector of IL‐21‐mediated induction, amplification and stabilization of Th17 phenotypes (Huber et al., 2008; Zhou and Littman, 2009). Like IRF8, IRF1 is critical for IL‐12‐promoted Th1 cell differentiation, while IRF2 facilitates IL‐12 production in cooperation with IRF1 (Elser et al., 2002; Kano et al., 2008). In addition to the extensively investigated IRFs, IRF7 governs the induction of CD8+ T‐cell responses through the MyD88‐IRF7 pathway (Honda et al., 2005).
Given the diverse and essential molecular roles of IRFs in controlling the growth, differentiation and death of various cell types, it is reasonable that IRFs exert pivotal and versatile effects on human diseases, especially on immune diseases and oncogenesis, which have been well documented (Tamura et al., 2008; Savitsky et al., 2010; Yanai et al., 2012). Interestingly, our recent studies and others have demonstrated that IRFs play intricate roles in cardiovascular diseases, especially cardiac remodelling, stroke and vascular injury. In this review, we summarize and discuss the regulatory effects of IRFs as critical novel stress sensors and regulators of the cardiovascular system, and the ways in which they exert their pathophysiological roles. Figure 3 summarizes the functions and potential mechanisms of IRFs in cardiovascular stress.
Figure 3.
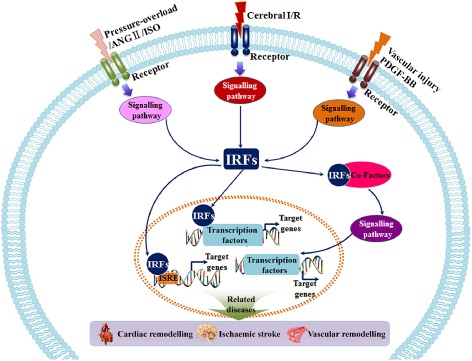
IRFs act as sensors and regulators of cardiovascular stress. Under physiological conditions, IRFs stay in a latent or stable form in the cytoplasm. After cardiovascular stress, such as cardiac pressure‐overload, cerebral I/R and wire‐induced vascular injury, IRF expression, structure and function are dramatically altered. IRFs convert upstream signalling to their co‐activators, co‐repressors or modifiers, triggering diverse and specific molecular events that further regulate the expression and activation of target genes. The dysfunction of these molecular programmes cause various cardiovascular diseases, for example cardiac remodelling, ischaemic stroke and vascular hyperplasia. Ang II, angiotensin II; ISO, isoprenaline.
Functions of IRFs in cardiovascular diseases
IRFs in cardiac remodelling
Cardiac hypertrophy, typically initiated by ill‐defined biomechanical stretch stress or neuroendocrine factors, is characterized by increased cardiomyocyte size accompanied by altered cardiac pump function and reduced ventricular wall tension (Sharif‐Naeini et al., 2010; Li et al., 2014). Pathological cardiac hypertrophy is now recognized as the strongest predictor of the development of heart failure, arrhythmia and sudden death. Initially, compensatory hypertrophic growth allows the heart to cope with various pathogenic stimuli, but continuous pathological stimuli terminally convert this change into a maladaptive response with perturbed contractiles performance and typical heart remodelling (van Berlo et al., 2013).
In addition to ventricular and cellular remodelling, pathological cardiac hypertrophy involves molecular alterations such as the activation of neuroendocrine factors, the re‐expression of fetal genes and changes in proteins involved in excitation–contraction coupling (Kehat and Molkentin, 2010). Angiotensin II, endothelin‐1 (ET‐1) and insulin‐like growth factor I (IGF‐I) are well‐established neurohumoural and endocrine hormones that promote cardiac hypertrophy, while glycogen synthase kinase 3β (GSK3β), regulator of G‐protein signalling 5, class II HDACs, PLA2 and the cyclic GMP‐dependent PKGI have distinct anti‐hypertrophic properties (Fiedler et al., 2002; Sanbe et al., 2003; Li et al., 2010a, 2010b; Yan et al., 2011). Based on these critical molecules, numerous dominant signalling pathways regulating cardiac remodelling have been revealed, particularly the GPCR–calcineurin–NFAT pathway, Ras–MAPK signalling, and the IGF‐I–PI3K–Akt/PKB–mammalian target of rapamycin/GSK3β axis (Heineke and Molkentin, 2006; Huang et al., 2010; Chen et al., 2013b; Jiang et al., 2014e; Liu et al., 2014; Zhang et al., 2014d). Experimental evidence from our laboratory and others' indicated the involvement of IRF1, IRF3, IRF4, IRF5, IRF7, IRF8 and IRF9 in the initiation and development of cardiac remodelling.
IRF1 is expressed ubiquitously, including in cardiomyocytes. In a well‐established mouse model of cardiac hypertrophy induced by chronic aortic banding, IRF1 protein expression increases progressively during the early phase, and then dramatically decreases to below baseline levels during the later stage both in the animal model and in patients with dilated and hypertrophic cardiomyopathy. Gene‐intervention studies have validated the profoundly detrimental effects of IRF1 in cardiac remodelling, as demonstrated by the accelerated ventricular hypertrophy and fibrosis in aortic banding‐stimulated, cardiac‐specific IRF1‐transgene (TG) mice, with reverse phenotypes in IRF1 −/− mice and rats. However, neither IRF1 overexpression nor its deletion impair basal cardiac functions (Jiang et al., 2014a). IRF1 functionally regulates inducible NOS (iNOS) expression by interacting with the binding site in the promoter region of the iNOS gene, which is significantly up‐regulated in patients with heart failure (Huang et al., 2011). The deletion of iNOS in IRF1‐TG mice prevents the exacerbated heart remodelling and dysfunction (Jiang et al., 2014a).
In contrast to IRF1, IRF3 and IRF7 function as negative regulators of pathological cardiac remodelling. In vivo chronic pressure overload and, in vitro isoprenaline stimulation could promote IRF3 phosphorylation and nuclear accumulation. Intriguingly, while sharing similar structures and phenotypes with IRF3, the IRF7 protein levels were markedly reduced in response to hypertrophic stimuli, possibly as a result of their disparate signalling pathways (Lu et al., 2013a; Jiang et al., 2014b). IRF3 directly interacts with ERK2 in cardiomyocytes, thereby inactivating the ERK1/2 signalling pathway (Lu et al., 2013a). Alternatively, IRF7 directly interacts with IKKβ by masking its kinase domain and then blocking IKKβ–IκB–NF‐κB‐regulated cardiac remodelling and dysfunction (Jiang et al., 2014b). Additionally, the functions of IRF3 in cardiac remodelling are disputed. Tsushima et al. (2011) reported that IRF3 regulates angiotensin II‐induced cardiac fibrosis, but not hypertrophy. Different hypertrophic stresses and discrepant gene technologies might contribute to these seemingly paradoxical results. In Tsushima's study, ERK1/2 was also linked to angiotensin II‐stimulated IRF3 activation, but located upstream of IRF3. Using a myxoma virus‐infected non‐permissive primary MEF model, Wang et al. (2004) also demonstrated the upstream regulatory effect of ERK1/2 on IRF3 activation. A conservative interpretation of these results suggests that, a negative feedback loop might exist in IRF3‐to‐ERK1/2 signalling for specific factors involved in cardiac remodelling. Collectively, the regulatory effects of IRF3 on cardiac remodelling in response to diverse stimuli are undefined, and the relationship between IRF3 and ERK1/2 remains to be rigorously clarified.
Although IRF4 and IRF8 were previously reported to be restricted to lymphocytes, our research group found that both of these transcription factors are highly expressed in human and animal hearts. Gain‐ and loss‐of‐function approaches have elucidated the exacerbating and alleviating effects of IRF4 and IRF8, respectively, on cardiac remodelling (Jiang et al., 2013; 2014d). Previous experimental approaches have confirmed that the transcription factor CREB was necessary for activation of the fetal gene programme in cardiac hypertrophy (Kong et al., 2006). In our recent studies, the CREB promoter was demonstrated to contain three IRF4 binding sites and to be responsible for the regulatory effects of IRF4 (Jiang et al., 2013). Studies on the mechanisms of IRF8, however, highlighted a critical modulator of hypertrophic process, NFATc1, located downstream of IRF8. IRF8 shows a potent capacity to directly interact with NFATc1 depending on its IAD1 domain to prevent NFATc1 translocation to the nucleus and leading to at least partial inhibition of cardiac remodelling (Jiang et al., 2014d).
As in the immune response, IRF5 is activated and translocated into the nucleus in the heart from tight skin (Tsk −/+) mice with spontaneous heart failure. While total and phosphorylated IRF5 are comparable with their levels in the heart from WT controls, the total ubiquitin and K63 ubiquitin levels of IRF5 are dramatically altered in the Tsk −/+ heart, suggesting that the structural modification of IRF5 might influence its function in pathological cardiac remodelling and heart failure. The treatment of apoAI mimetic 4F, a protein that could reduce inflammation by inhibiting IRF5 activation, delays the onset of heart failure (Xu et al., 2012). Although the protective effect in heart remodelling might result from reduction of the inflammatory responses by IRF5, nuclear translocation of IRF5 did take place in the myocardium, indicating that IRF5 decrease‐induced cardiac protection could arise through an immune‐independent pathway. Nonetheless, the involvement of IRF5 in cardiac remodelling merits further exploration.
As noted earlier, in cardiac remodelling, crucial transcription factors, including NFAT, myocyte enhancer factor 2 (MEF2) and SRF, recruit co‐activators or co‐repressors to mediate the hypertrophic responses. IRF9 binds to the transcription activation domain (TAD) of myocardin, a co‐activator of SRF, in response to aortic banding or to isoprenaline infusion, thereby strongly inhibiting CArG box‐dependent reporters. Intriguingly, this anti‐hypertrophic property of IRF9 is countered by p300, which also binds directly to TAD to activate myocardin. IRF9 protein expression is dramatically elevated in remodelled human and mouse hearts. Accordingly, IRF9‐KO mice exhibited aggravated cardiac hypertrophy and fibrosis after hypertrophic stimuli, whereas the up‐regulation of IRF9 expression alleviated these responses. Thus, IRF9 functions as a previously unidentified negative regulator of cardiac remodelling (Jiang et al., 2014c).
In addition to biomechanical and neuroendocrine stresses, myocardial ischaemia/reperfusion (I/R) injury also contributes to maladaptive cardiac damage (Timmers et al., 2008; Xiao et al., 2012; Frohlich et al., 2013). Unexpectedly, our recent data indicate that, in contrast to the protective effect on the hypertrophic response, IRF9 functions as a promoter of myocardial I/R‐derived cardiac dysfunction and remodelling (Zhang et al., 2014c). Unlike in cardiac hypertrophy, cardiomyocyte death and the inflammatory response are the dominant pathological factors during I/R injury (Hausenloy and Yellon, 2013). Thus the molecular mechanism by which IRF9 regulates myocardial I/R is likely to be different from that of cardiac hypertrophy. Our investigation indicated that the detrimental effect of IRF9 might depend on its modulation of Sirt1 expression and the corresponding apoptotic signalling. Further in‐depth studies are needed to elucidate the underlying molecular events in IRF9‐dependent myocardial I/R injury.
Functions of IRFs in stroke
Stroke usually arises from either ischaemia or haemorrhage, which interrupt blood flow to the brain. Ischaemic strokes account for approximately 87% of all of such cases (Candelario‐Jalil, 2009; Go et al., 2014). Once cerebral I/R occurs, a series of intricate and highly interconnected cellular and molecular events are initiated and ultimately disturb central autonomic control (Soros and Hachinski, 2012). In the early stage following ischaemic injury, metabolic compromise, calcium overload and oxidative stress rapidly lead to neuron death in the infarct core (Wang et al., 2012). Compared with other organs, the brain is much more susceptible to oxidative attack as a result of its high content of polyunsaturated fatty acids, oxidative metabolic activity and low antioxidant capacity (Halliwell, 2006). In later events, the overly abundant oxygen radicals participate in signalling pathways of neuroinflammation and apoptosis, exacerbating the initial ischaemic damage and yielding penumbral tissue death (Candelario‐Jalil, 2009; Lu et al., 2013b; Wang et al., 2013b). Stroke‐induced cell death occurs preferentially via an apoptotic mechanism rather than necrosis. An alteration in genes encoding caspases and the Bcl family is involved in both the early and late phases of stroke (Stankowski and Gupta, 2011). In the inflammatory response, the TLR family exerts key functions and has attracted much more attention in recent years for its potential therapeutic roles in treating stroke. Generally, following the activation of TLRs, downstream signalling elicits the production and secretion of cytokines and chemokines, which initiate a severely detrimental inflammatory response to cerebral ischaemia (Tang et al., 2007a; Ziegler et al., 2007; Marsh and Stenzel‐Poore, 2008). Paradoxically, emerging studies have suggested that the preconditioning by exogenous TLR ligands might confer robust neuroprotection against subsequent I/R injury (Marsh et al., 2009b; Stevens et al., 2011; Chamorro et al., 2012). All of these phenomena appear to protect against or promote stroke at least partly by reprogramming the genomic response. The IRF family has been identified as one of these genetic factors.
The strong regulatory functions of IRFs in cerebral I/R were originally described in studies demonstrating that IRF1 expression was dramatically up‐regulated in the ischaemic regions of a middle cerebral artery occlusion‐induced mouse stroke model (Nguyen et al., 1997; Iadecola et al., 1999; Raghavendra Rao et al., 2002). Iadecola and subsequent researchers (Alexander et al., 2003) indicated that IRF1 deletion was beneficial to the post‐ischaemic brain, although the underlying mechanisms were largely unknown. Using a loss‐of‐function approach, Caso and colleagues (Caso et al., 2007) observed that TLR4‐deficient mice exhibit minor infarction and inflammation after an experimental stroke. In these mice, IRF1 expression was significantly reduced compared with that of their wild‐type littermates, explaining at least in part the neuroprotective effect of the TLR4 deletion. IRF2 also showed potential involvement in the nervous system, given that IRF2 physically interacts with NF‐κB in neural cells (Drew et al., 1995). However, the effects of IRF2 on stroke are obscure. Additional studies on the functions and underlying molecular mechanisms are required to clarify the modulatory effects of IRF1 and IRF2 on stroke.
Early studies with IRF3 and IRF7 found no direct evidence to indicate the involvement of these IRFs in stroke, but were focused on their essential roles in TLR ligand pretreatment‐induced tolerance to ischaemic brain injury. Systemic preconditioning by LPS or the unmethylated CpG oligonucleotides provides neuroprotection against cerebral I/R injury by activating their receptors, TLR4 and TLR9 respectively (Marsh et al., 2009a; Stevens et al., 2011; Vartanian et al., 2011). An analysis of the reprogrammed genes located downstream of TLRs indicated that IRFs are closely related to the beneficial effects on stroke of preconditioning by TLR ligands. A subsequent gene‐disruption approach validated the indispensable roles of IRF3 and IRF7 in these cases (Stevens et al., 2011). Most recently, Cui et al. (2013) demonstrated that pretreatment with chloroquine markedly suppressed the TLR3/IRF3–IFN‐β signalling pathway, which was activated after global cerebral ischaemia in rats, thereby improving their spatial memory capacity. Additionally, TLR7 preconditioning also provided robust neuroprotection in response to stroke, an effect that was IRF7‐dependent (Leung et al., 2012). These protective effects seem to involve the IRFs that are related to immune/inflammatory responses. Notably, our laboratory recently generated a novel rat strain, IRF3‐KO, and provided direct evidence that IRF3 ablation attenuates stroke in rats, as indicated by blunted apoptosis (H. Li, unpubl. data). The direct effects of IRF3 on stroke in different experimental studies and the underlying mechanisms require further research.
In addition to the heart, IRF4 and IRF8 are also expressed constitutively in neurons and can be induced and suppressed, respectively, by cerebral ischaemia. Our recent investigations have demonstrated that neuron‐specific IRF4‐TG and IRF8‐TG mice exhibited improved stroke outcomes and reduced infarct lesions, whereas these protective effects were reversed in IRF4‐KO and IRF8‐KO mice, respectively (Guo et al., 2014; Xiang et al., 2014). Mechanistically, SRF, a transcription factor that is essential for neuron survival, is required for the beneficial functions of IRF4, as indicated by the complete absence of phenotypes of IRF4‐TG mice following SRF ablation (Guo et al., 2014).
The influence of IRF9 on stroke has been reported recently by our research group. Promisingly, our studies indicated that IRF9 is a positive regulator of stroke by activating neuronal death‐associated signalling pathways. After stimulation with cerebral I/R, IRF9 −/− mice showed markedly decreased neuronal death and neurological deficits, whereas IRF9 overexpression sensitized the neurons to death. Using a bioinformatic approach and genetic manipulation, the IRF9/Sirt1/p53 axis was identified as a crucial signalling pathway in stroke events. Our recent data suggest that, in response to acute I/R stress, IRF9 suppresses Sirt1 expression, followed by the activation of p53‐regulated cell death signalling (Chen et al., 2014).
IRFs involved in regulating vascular injury
Intimal hyperplasia, a prevalent form of severe arterial remodelling after vascular injury, is almost inevitable during various vascular diseases and their treatments, such as atherosclerosis, stenting, angioplasty and bypass surgery (Dzau et al., 2002). This pathological condition consists of multifaceted interactions among vascular smooth muscle cells (VSMCs) and other cell types, as well as complex intracellular and intercellular regulatory mechanisms. In response to biomechanical injury or other stimuli, endothelial cells trigger and amplify inflammatory responses by recruiting inflammatory cells. Although this inflammation is initially a reparative mechanism, persistent inflammatory processes stimulate VSMC proliferation, migration, phenotypic switching and extracellular matrix (ECM) deposition, the most important pathological bases of neointimal thickening (Davis et al., 2003; Inoue et al., 2011). The deregulation of the VSMC is the central core of neointimal formation. Immediately after injury, VSMCs undergo phenotypic change, switching from a differentiated (or contractile) state to a de‐differentiated (also termed ‘synthetic’) phenotype (Davis‐Dusenbery et al., 2011). The de‐differentiated VSMCs, which are characterized by the reduced expression of contractile genes, are particularly susceptible to proliferation, migration, and ECM production (Alexander and Owens, 2012). These changes in VSMCs are regulated by a range of molecular pathways, especially growth factor/cytokine signalling (Dzau et al., 2002; Li et al., 2011a). Additionally, the involvement of immune mechanisms has also been demonstrated under these pathological conditions (Schiffrin, 2014). Several IRFs serve as crucial factors in vascular injury, not only through their participation in immune responses.
In the effort to explore the effects of IRF members on vascular injury, IRF1 has been studied more often than other IRFs and is deeply involved in vascular remodelling. IRF1 can be induced after IFN‐γ stimulation both in vitro and in vivo. Horiuchi et al. (2000) indicated that IRF1, along with the JAK−STAT pathway, participates in the response to activation, by exogenous IFN–γ, of angiotensin AT2 receptors, which are closely associated with cell growth, differentiation and injury. Increased level of IRF1 also partly mediates endogenous IFN–γ‐promoted intimal thickening in immune‐deficient Rag‐1‐KO mice (Dimayuga et al., 2005; Kusaba et al., 2007). In addition to IFNs, ET‐1, a potent vasoconstrictor and growth‐promoting mediator, can induce IRF1 activation in human VSMCs (Woods et al., 2003). However, the beneficial or deleterious effects of IRF1 activation on intimal hyperplasia are less clearly defined. Experimental results from the Wessely and Li research groups (Wessely et al., 2003; Li et al., 2009) have provided direct evidence about the effects of IRF1 on vascular remodelling, indicated by the fact that vascular injury‐stimulated neointimal formation was significantly aggravated in IRF1‐deficient mice compared with wild‐type mice. From a mechanistic standpoint, IRF1 exhibits pleiotropic anti‐hyperplastic activities by regulating the expressions of various factors, such as AT2 receptors, iNOS, mitogens, CD40 and PPARγ, to attenuate endothelial dysfunction, cellular migration and VSMC proliferation (Wagner et al., 2002; Wessely et al., 2003; Lin et al., 2004; Li et al., 2009)
Consistent with the results from IRF1, our recent studies have indicated that both IRF3 and IRF7 act as inhibitors of neointimal formation, based on gain‐ and loss‐of‐function approaches in rodents and in vitro (Zhang et al., 2014b). A significant down‐regulation of IRF3 and IRF7 expression in VSMC was observed after carotid wire injury in vivo and stimulation by PDGF‐BB in vitro. IRF3 binds to the AB domain of PPARγ, a negative regulator of intimal hyperplasia (Lim et al., 2006; Duan et al., 2008), facilitating PPARγ transactivation and the subsequent down‐regulation of the expression of PCNA to ultimately suppress VSMC proliferation (Zhang et al., 2014b). IRF7 is important for neointimal formation because of its ability to directly interact with the transcription factor ATF3 and to inhibit the ATF3‐mediated induction of PCNA (L. Huang, submitted).
Several studies of the underlying mechanisms of VSMC phenotypic modulation have focused on another transcription factor, SRF. In addition to participating in cardiac hypertrophy and neuron survival, SRF and its co‐activator, myocardin, play essential roles in SMC differentiation and phenotypic switching (Camoretti‐Mercado et al., 2003; Miano, 2003; Yoshida et al., 2003). Recent studies from our group have demonstrated that IRF8 can act as an accelerated regulator of neointimal formation through a direct interaction with the SRF/myocardin complex, thereby regulating SRF transactivation to inhibit the expression of SMC‐marker genes (Zhang et al., 2014a).
Most recently, IRF9 was investigated in our research laboratory because of its regulatory effects on vascular remodelling in response to both in vivo and in vitro stimuli. Compared with controls, IRF9 protein expression is significantly higher in human femoral artery specimens with in‐stent restenosis, a wire injury‐induced neointimal formation mouse model, and PDGF‐BB‐stimulated VSMC. Furthermore, IRF9 ablation attenuates VSMC proliferation and intimal thickening in response to injury, whereas IRF9 up‐regulation results in the opposite phenotype, suggesting that IRF9 is an activator of intimal hyperplasia. Detailed mechanistic studies have indicated that IRF9 negatively regulates the transcription of a vascular protective factor, Sirt1, after vascular injury, and that IRF9‐controlled intimal remodelling is dependent on Sirt1 deacetylase activity (S. M. Zhang, submitted).
Among vascular diseases that typically exhibt vascular remodelling, atherosclerosis and abdominal aortic aneurysm (AAA) present high‐risk factors for other severe cardiovascular diseases. In our recent investigations, the regulatory functions of IRFs in these two diseases have been assessed. Several IRFs, including IRF1, IRF3, IRF4, IRF7, IRF8 and IRF9, are critically involved in the development of atherosclerosis and AAA (H. Li, unpubl. data). Further, more extensive studies of their underlying mechanisms are needed.
IRFs, the sensors and regulators of cardiovascular stress
As stress sensors and regulators, the involvement of IRFs in immunity, metabolism, and related diseases has been well documented (Zhao et al., 2014). In the results summarized in this review, IRFs involved in cardiovascular diseases are expressed in a latent or stable form in the cytoplasm under normal conditions, but are dramatically altered in response to pathological cardiovascular stress or neuroendocrine factors, other than viral infection, suggesting that IRFs might act as cardiovascular stress‐inducible factors. Furthermore, IRFs respond to various intracellular or extracellular forms of stress, undergo structural/functional variation, and initiate corresponding signalling pathways to regulate a series of phenotypic alterations. From the published studies described in this review and unpublished data from our group, it is clear that the IRF family is intrinsically associated with and dramatically regulates the aetiologies of cardiac, neural and vascular diseases through its potent effects on cell dilation, proliferation, apoptosis, phenotype switching and inflammatory responses. Thus, IRFs have a much more complex role in these cardiovascular diseases and do not simply act through their mediation of the immune system. Interestingly, under basal conditions, neither the artificial up‐regulation nor down‐regulation of IRFs negatively affected the physiological functions of mice or cells. Rather, they exert significant influence only on pathological stimuli. These results suggest that the contributions of dysregulated IRFs might depend on their responses to detrimental cardiovascular states. Thus, in addition to their effects in the immune system, IRFs might also act as prominent stress sensors and regulators of cardiovascular disease.
Although the effects and mechanisms of IRFs have been systematically investigated, several important questions require further elucidation. For instance, other IRFs expressed in the cardiovascular system might have critical roles under pathophysiological conditions. Moreover, the timing and relative IRF protein levels and their relationships with functions should be determined. Additionally, many non‐parenchymal cells in tissues exert important regulatory functions in response to environmental stimuli. The expression profiles and effects of IRFs in these non‐parenchymal cells merit in‐depth investigation. Our ongoing studies focus on these unresolved issues, the results of which will provide promising therapeutic targets for cardiovascular diseases.
Potential clinical applications of IRFs
Given the versatile and potent effects of IRFs, it is not surprising that this transcription factor family has a profound role in various diseases, especially in the related diseases in the immune and cardiovascular systems. In the recent years, genome‐wide association studies (GWAS) have greatly facilitated wide screening and deep exploration of the latent associations between human diseases and specific genes. Utilizing this advanced technology, the preliminary clinical relevance of IRFs has been established. A recent study observed that IRF1, at positions ‐388 and ‐410, might be a candidate gene marker for chronic hepatitis B and C (Korachi et al., 2013). Applying genome‐wide SNP association study, Di Bernardo et al. (2008) demonstrated that IRF4 is strongly associated with susceptibility to chronic lymphocytic leukaemia (CLL). The CLL risk loci were identified at 6p25.3 (rs872071, IRF4). Additionally, the SNP rs9378805 in IRF4 is associated with recurrent bronchitis, as supported by the GWAS data set (Pinto et al., 2013). In studies of the immune system, although cellular mechanisms that initiate pathogen recognition and IFN production are critical to protection against viral infection response, over‐activation of these molecular events can contribute to adverse pathogenic effects characteristic of autoimmune disorders. One representative example is systemic lupus erythematosus (SLE), a genetic and environmental factors‐triggered disease marked by high levels of type I IFNs in serum (Banchereau and Pascual, 2006; Yang et al., 2012). Several large‐scale genetic association studies have shown that, the major alleles, rs2004640 and rs1131665 (412Q) in IRF5 and IRF7, respectively, predispose the host to the development of SLE in several ethnic groups, cumulatively providing direct genetic evidence that IRFs are closely linked to human autoimmune diseases (Sigurdsson et al., 2005; Graham et al., 2006; Cunninghame Graham et al., 2007; Fu et al., 2011).
In terms of the relevance of IRFs to cardiovascular diseases, previous and ongoing studies have suggested the involvement of IRFs in cardiac remodelling, stroke and vascular injury, through the markedly altered expression levels of IRFs in response to the corresponding pathological cardiovascular conditions. For instance, IRF1, IRF4 and IRF8 are significantly down‐regulated in failing human hearts compared with healthy controls (Jiang et al., 2013, 2014a, 2014d), whereas IRF3 is profoundly up‐regulated in the hearts of patients with dilated or hypertrophic cardiomyopathy (Lu et al., 2013a). All of these alterations in patients are consistent with those seen in animal models of cardiac remodelling in the maladaptive stage. However, the alterations of IRF expression are not always consistent in the compensatory and maladaptive hypertrophy. For example the progressively increasing level of IRF1 in the early stage and markedly reduced IRF1 expression in the late stage of cardiac hypertrophy have not been observed and merit further investigation in human hearts. Furthermore, improved understanding of the timing of changes and the switches to turn on or turn off the expression and activation of IRFs in particular clinical diseases will provide promising bases for novel therapies.
Conclusions and perspectives
A growing number of studies have demonstrated that intricate signalling cascades converge on certain transcription factors that critically participate in the development of various diseases. Accordingly, an understanding of the regulatory mechanisms and counterbalancing molecules that selectively affect these specific transcriptional factors could be of great interest for preventing and treating these diseases. IRFs are a family of such transcription factors. The involvement of IRFs in the immune response has been known for a long time. Intriguingly, recent studies provide novel insights into the regulatory effects of IRFs on diseases of the heart, brain and vasculature. As cardiovascular stress‐responsive regulators, IRFs undergo a series of structural and functional modifications to adapt or mediate environmental or intracellular stimuli‐triggered pathological conditions, as described in this review. However, the functions of IRFs are much more extensive than earlier estimates. Our recent studies have also demonstrated potential functions of IRFs in metabolic diseases (Wang et al., 2013c, 2013d; 2014), and these studies are still ongoing. Future studies should examine the influences of the IRF family on other diseases, for example respiratory diseases, tuberculosis and diarrhoea. Although the growth of genetic approaches, bioinformatic methods and high‐throughput techniques facilitate our experimental studies, the molecular events underlying the IRF‐regulated cellular responses are only partly understood and remain one of the most understudied areas in clinical and experimental research. Studies on the expression and function of IRFs raise a particularly interesting question: why are the trends in the altered expression of IRFs not consistent with their protective or deleterious effects? Among the divergent and overlapping molecular programmes of the IRF family, priorities for investigation should include the interplay of IRF members, the functional and physical interactions with their co‐factors, and the bridges for the association of upstream or downstream events to IRFs. In addition, given that IRFs are shared by different signalling pathways under common pathological conditions, the governing effects of IRFs on these pathways are worthy of further validation and investigation. Most importantly, a deeper understanding of the precise regulatory effects and molecular mechanisms of these regulators will be crucial not only for disease treatment, but also for a global understanding of gene networks and cell behaviour.
Conflict of interest
None.
Acknowledgements
We apologize to the colleagues whose work could not be cited because of space constraints. We thank Wen‐Lin Cheng for assistance with the figures. This work was supported by grants from the National Natural Science Foundation of China (Nos. 81100230, 81070089, 81200071, 81270306, 81270184 and 81370365), the National Science and Technology Support Project (Nos. 2011BAI15B02, 2012BAI39B05, 2013YQ030923‐05 and 2014BAI02B01), the Key Project of the National Natural Science Foundation (No. 81330005) and the National Basic Research Program China (No. 2011CB503902).
References
- Akira S, Uematsu S, Takeuchi O (2006). Pathogen recognition and innate immunity. Cell 124: 783–801. [DOI] [PubMed] [Google Scholar]
- Alexander M, Forster C, Sugimoto K, Clark HB, Vogel S, Ross ME et al (2003). Interferon regulatory factor‐1 immunoreactivity in neurons and inflammatory cells following ischemic stroke in rodents and humans. Acta Neuropathol 105: 420–424. [DOI] [PubMed] [Google Scholar]
- Alexander MR, Owens GK (2012). Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol 74: 13–40. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013a). The Concise Guide to PHARMACOLOGY 2013/14: Catalytic Receptors. Br J Pharmacol 170: 1676–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013b). The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013c). The Concise Guide to PHARMACOLOGY 2013/14: G Protein‐Coupled Receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkhi MY, Fitzgerald KA, Pitha PM (2008). Functional regulation of MyD88‐activated interferon regulatory factor 5 by K63‐linked polyubiquitination. Mol Cell Biol 28: 7296–7308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau J, Pascual V (2006). Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity 25: 383–392. [DOI] [PubMed] [Google Scholar]
- Barro M, Patton JT (2005). Rotavirus nonstructural protein 1 subverts innate immune response by inducing degradation of IFN regulatory factor 3. Proc Natl Acad Sci U S A 102: 4114–4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauhofer O, Summerfield A, Sakoda Y, Tratschin JD, Hofmann MA, Ruggli N (2007). Classical swine fever virus Npro interacts with interferon regulatory factor 3 and induces its proteasomal degradation. J Virol 81: 3087–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker AM, Michael DG, Satpathy AT, Sciammas R, Singh H, Bhattacharya D (2012). IRF‐8 extinguishes neutrophil production and promotes dendritic cell lineage commitment in both myeloid and lymphoid mouse progenitors. Blood 119: 2003–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐Neriah Y (2002). Regulatory functions of ubiquitination in the immune system. Nat Immunol 3: 20–26. [DOI] [PubMed] [Google Scholar]
- van Berlo JH, Maillet M, Molkentin JD (2013). Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest 123: 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs LC, Naridze RL, DeMali KA, Lusche DF, Kuhl S, Soll DR et al (2014). Interferon regulatory factor 6 regulates keratinocyte migration. J Cell Sci 127 (Pt 13): 2840–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J, Wang H, Hajishengallis GN, Martin M (2011). TLR‐signaling networks: an integration of adaptor molecules, kinases, and cross‐talk. J Dent Res 90: 417–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillaud A, Prakash A, Smith E, Masumi A, Hovanessian AG, Levy DE et al (2002). Acetylation of interferon regulatory factor‐7 by p300/CREB‐binding protein (CBP)‐associated factor (PCAF) impairs its DNA binding. J Biol Chem 277: 49417–49421. [DOI] [PubMed] [Google Scholar]
- Camoretti‐Mercado B, Dulin NO, Solway J (2003). Serum response factor function and dysfunction in smooth muscle. Respir Physiol Neurobiol 137: 223–235. [DOI] [PubMed] [Google Scholar]
- Candelario‐Jalil E (2009). Injury and repair mechanisms in ischemic stroke: considerations for the development of novel neurotherapeutics. Curr Opin Investig Drugs 10: 644–654. [PubMed] [Google Scholar]
- Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I (2007). Toll‐like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation 115: 1599–1608. [DOI] [PubMed] [Google Scholar]
- Chamorro A, Meisel A, Planas AM, Urra X, van de Beek D, Veltkamp R (2012). The immunology of acute stroke. Nat Rev Neurol 8: 401–410. [DOI] [PubMed] [Google Scholar]
- Chen HW, King K, Tu J, Sanchez M, Luster AD, Shresta S (2013a). The roles of IRF‐3 and IRF‐7 in innate antiviral immunity against dengue virus. J Immunol 191: 4194–4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HZ, Guo S, Li ZZ, Lu Y, Jiang DS, Zhang R et al (2014). A critical role for interferon regulatory factor 9 in cerebral ischemic stroke. J Neurosci 34: 11897–11912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Gao L, Liu Y, Zhang Y, Jiang DS, Wei X et al (2013b). Vinexin‐beta protects against cardiac hypertrophy by blocking the Akt‐dependent signalling pathway. Basic Res Cardiol 108: 338. [DOI] [PubMed] [Google Scholar]
- Chen Z, Rijnbrand R, Jangra RK, Devaraj SG, Qu L, Ma Y et al (2007). Ubiquitination and proteasomal degradation of interferon regulatory factor‐3 induced by Npro from a cytopathic bovine viral diarrhea virus. Virology 366: 277–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K, Takeuchi O, Akira S, Cohen P (2011). The TRAF‐associated protein TANK facilitates cross‐talk within the IkappaB kinase family during Toll‐like receptor signaling. Proc Natl Acad Sci U S A 108: 17093–17098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M (2007). TLR pathways and IFN‐regulatory factors: to each its own. Eur J Immunol 37: 306–309. [DOI] [PubMed] [Google Scholar]
- Cui G, Ye X, Zuo T, Zhao H, Zhao Q, Chen W et al (2013). Chloroquine pretreatment inhibits Toll‐like receptor 3 signaling after stroke. Neurosci Lett 548: 101–104. [DOI] [PubMed] [Google Scholar]
- Cunninghame Graham DS, Manku H, Wagner S, Reid J, Timms K, Gutin A et al (2007). Association of IRF5 in UK SLE families identifies a variant involved in polyadenylation. Hum Mol Genet 16: 579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daffis S, Samuel MA, Keller BC, Gale M Jr, Diamond MS (2007). Cell‐specific IRF‐3 responses protect against West Nile virus infection by interferon‐dependent and ‐independent mechanisms. PLoS Pathog 3: e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daffis S, Samuel MA, Suthar MS, Keller BC, Gale M Jr, Diamond MS (2008). Interferon regulatory factor IRF‐7 induces the antiviral alpha interferon response and protects against lethal West Nile virus infection. J Virol 82: 8465–8475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daffis S, Suthar MS, Szretter KJ, Gale M Jr, Diamond MS (2009). Induction of IFN‐beta and the innate antiviral response in myeloid cells occurs through an IPS‐1‐dependent signal that does not require IRF‐3 and IRF‐7. PLoS Pathog 5: e1000607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daffis S, Lazear HM, Liu WJ, Audsley M, Engle M, Khromykh AA et al (2011). The naturally attenuated Kunjin strain of West Nile virus shows enhanced sensitivity to the host type I interferon response. J Virol 85: 5664–5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis C, Fischer J, Ley K, Sarembock IJ (2003). The role of inflammation in vascular injury and repair. J Thromb Haemost 1: 1699–1709. [DOI] [PubMed] [Google Scholar]
- Davis‐Dusenbery BN, Wu C, Hata A (2011). Micromanaging vascular smooth muscle cell differentiation and phenotypic modulation. Arterioscler Thromb Vasc Biol 31: 2370–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bernardo MC, Crowther‐Swanepoel D, Broderick P, Webb E, Sellick G, Wild R et al (2008). A genome‐wide association study identifies six susceptibility loci for chronic lymphocytic leukemia. Nat Genet 40: 1204–1210. [DOI] [PubMed] [Google Scholar]
- Dimayuga PC, Li H, Chyu KY, Fredrikson GN, Nilsson J, Fishbein MC et al (2005). T‐cell modulation of intimal thickening after vascular injury: the bimodal role of IFN‐gamma in immune deficiency. Arterioscler Thromb Vasc Biol 25: 2528–2534. [DOI] [PubMed] [Google Scholar]
- Drew PD, Franzoso G, Carlson LM, Biddison WE, Siebenlist U, Ozato K (1995). Interferon regulatory factor‐2 physically interacts with NF‐kappa B in vitro and inhibits NF‐kappa B induction of major histocompatibility class I and beta 2‐microglobulin gene expression in transfected human neuroblastoma cells. J Neuroimmunol 63: 157–162. [DOI] [PubMed] [Google Scholar]
- Duan SZ, Usher MG, Mortensen RM (2008). Peroxisome proliferator‐activated receptor‐gamma‐mediated effects in the vasculature. Circ Res 102: 283–294. [DOI] [PubMed] [Google Scholar]
- Dzau VJ, Braun‐Dullaeus RC, Sedding DG (2002). Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat Med 8: 1249–1256. [DOI] [PubMed] [Google Scholar]
- Eguchi J, Yan QW, Schones DE, Kamal M, Hsu CH, Zhang MQ et al (2008). Interferon regulatory factors are transcriptional regulators of adipogenesis. Cell Metab 7: 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi J, Wang X, Yu S, Kershaw EE, Chiu PC, Dushay J et al (2011). Transcriptional control of adipose lipid handling by IRF4. Cell Metab 13: 249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elser B, Lohoff M, Kock S, Giaisi M, Kirchhoff S, Krammer PH et al (2002). IFN‐gamma represses IL‐4 expression via IRF‐1 and IRF‐2. Immunity 17: 703–712. [DOI] [PubMed] [Google Scholar]
- Erickson AK, Gale M Jr (2008). Regulation of interferon production and innate antiviral immunity through translational control of IRF‐7. Cell Res 18: 433–435. [DOI] [PubMed] [Google Scholar]
- Eroshkin A, Mushegian A (1999). Conserved transactivation domain shared by interferon regulatory factors and SMAD morphogens. J Mol Med (Berl) 77: 403–405. [DOI] [PubMed] [Google Scholar]
- Escalante CR, Brass AL, Pongubala JM, Shatova E, Shen L, Singh H et al (2002). Crystal structure of PU.1/IRF‐4/DNA ternary complex. Mol Cell 10: 1097–1105. [DOI] [PubMed] [Google Scholar]
- Feng D, Sangster‐Guity N, Stone R, Korczeniewska J, Mancl ME, Fitzgerald‐Bocarsly P et al (2010). Differential requirement of histone acetylase and deacetylase activities for IRF5‐mediated proinflammatory cytokine expression. J Immunol 185: 6003–6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedler B, Lohmann SM, Smolenski A, Linnemuller S, Pieske B, Schroder F et al (2002). Inhibition of calcineurin‐NFAT hypertrophy signaling by cGMP‐dependent protein kinase type I in cardiac myocytes. Proc Natl Acad Sci U S A 99: 11363–11368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foy E, Li K, Wang C, Sumpter R Jr, Ikeda M, Lemon SM et al (2003). Regulation of interferon regulatory factor‐3 by the hepatitis C virus serine protease. Science 300: 1145–1148. [DOI] [PubMed] [Google Scholar]
- Frohlich GM, Meier P, White SK, Yellon DM, Hausenloy DJ (2013). Myocardial reperfusion injury: looking beyond primary PCI. Eur Heart J 34: 1714–1722. [DOI] [PubMed] [Google Scholar]
- Fu Q, Zhao J, Qian X, Wong JL, Kaufman KM, Yu CY et al (2011). Association of a functional IRF7 variant with systemic lupus erythematosus. Arthritis Rheum 63: 749–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii Y, Shimizu T, Kusumoto M, Kyogoku Y, Taniguchi T, Hakoshima T (1999). Crystal structure of an IRF‐DNA complex reveals novel DNA recognition and cooperative binding to a tandem repeat of core sequences. EMBO J 18: 5028–5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriele L, Fragale A, Borghi P, Sestili P, Stellacci E, Venditti M et al (2006). IRF‐1 deficiency skews the differentiation of dendritic cells toward plasmacytoid and tolerogenic features. J Leukoc Biol 80: 1500–1511. [DOI] [PubMed] [Google Scholar]
- Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K (2010). Development of monocytes, macrophages, and dendritic cells. Science 327: 656–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliet M, Boonstra A, Paturel C, Antonenko S, Xu XL, Trinchieri G et al (2002). The development of murine plasmacytoid dendritic cell precursors is differentially regulated by FLT3‐ligand and granulocyte/macrophage colony‐stimulating factor. J Exp Med 195: 953–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ et al (2014). Executive summary: heart disease and stroke statistics – 2014 update: a report from the American Heart Association. Circulation 129: 399–410. [DOI] [PubMed] [Google Scholar]
- Graff JW, Ewen J, Ettayebi K, Hardy ME (2007). Zinc‐binding domain of rotavirus NSP1 is required for proteasome‐dependent degradation of IRF3 and autoregulatory NSP1 stability. J Gen Virol 88 (Pt 2): 613–620. [DOI] [PubMed] [Google Scholar]
- Graham RR, Kozyrev SV, Baechler EC, Reddy MV, Plenge RM, Bauer JW et al (2006). A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet 38: 550–555. [DOI] [PubMed] [Google Scholar]
- Guillot L, Le Goffic R, Bloch S, Escriou N, Akira S, Chignard M et al (2005). Involvement of Toll‐like receptor 3 in the immune response of lung epithelial cells to double‐stranded RNA and influenza A virus. J Biol Chem 280: 5571–5580. [DOI] [PubMed] [Google Scholar]
- Guo S, Li ZZ, Jiang DS, Lu YY, Liu Y, Gao L et al (2014). IRF4 is a novel mediator for neuronal survival in ischaemic stroke. Cell Death Differ 21: 888–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B (2006). Oxidative stress and neurodegeneration: where are we now? J Neurochem 97: 1634–1658. [DOI] [PubMed] [Google Scholar]
- Hambleton S, Salem S, Bustamante J, Bigley V, Boisson‐Dupuis S, Azevedo J et al (2011). IRF8 mutations and human dendritic‐cell immunodeficiency. N Engl J Med 365: 127–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada H, Fujita T, Miyamoto M, Kimura Y, Maruyama M, Furia A et al (1989). Structurally similar but functionally distinct factors, IRF‐1 and IRF‐2, bind to the same regulatory elements of IFN and IFN‐inducible genes. Cell 58: 729–739. [DOI] [PubMed] [Google Scholar]
- Hausenloy DJ, Yellon DM (2013). Myocardial ischemia‐reperfusion injury: a neglected therapeutic target. J Clin Invest 123: 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heineke J, Molkentin JD (2006). Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol 7: 589–600. [DOI] [PubMed] [Google Scholar]
- Hida S, Ogasawara K, Sato K, Abe M, Takayanagi H, Yokochi T et al (2000). CD8(+) T‐cell‐mediated skin disease in mice lacking IRF‐2, the transcriptional attenuator of interferon‐alpha/beta signaling. Immunity 13: 643–655. [DOI] [PubMed] [Google Scholar]
- Higgs R, Ni Gabhann J, Ben Larbi N, Breen EP, Fitzgerald KA, Jefferies CA (2008). The E3 ubiquitin ligase Ro52 negatively regulates IFN‐beta production post‐pathogen recognition by polyubiquitin‐mediated degradation of IRF3. J Immunol 181: 1780–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs R, Lazzari E, Wynne C, Ni Gabhann J, Espinosa A, Wahren‐Herlenius M et al (2010). Self protection from anti‐viral responses – Ro52 promotes degradation of the transcription factor IRF7 downstream of the viral Toll‐like receptors. PLoS ONE 5: e11776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton L, Moganeradj K, Zhang G, Chen YH, Randall RE, McCauley JW et al (2006). The NPro product of bovine viral diarrhea virus inhibits DNA binding by interferon regulatory factor 3 and targets it for proteasomal degradation. J Virol 80: 11723–11732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiscott J (2007). Triggering the innate antiviral response through IRF‐3 activation. J Biol Chem 282: 15325–15329. [DOI] [PubMed] [Google Scholar]
- Holland JW, Bird S, Williamson B, Woudstra C, Mustafa A, Wang T et al (2008). Molecular characterization of IRF3 and IRF7 in rainbow trout, Oncorhynchus mykiss: functional analysis and transcriptional modulation. Mol Immunol 46: 269–285. [DOI] [PubMed] [Google Scholar]
- Honda K, Taniguchi T (2006b). IRFs: master regulators of signalling by Toll‐like receptors and cytosolic pattern‐recognition receptors. Nat Rev Immunol 6: 644–658. [DOI] [PubMed] [Google Scholar]
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T et al (2005). IRF‐7 is the master regulator of type‐I interferon‐dependent immune responses. Nature 434: 772–777. [DOI] [PubMed] [Google Scholar]
- Honda K, Takaoka A, Taniguchi T (2006a). Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 25: 349–360. [DOI] [PubMed] [Google Scholar]
- Honma K, Udono H, Kohno T, Yamamoto K, Ogawa A, Takemori T et al (2005). Interferon regulatory factor 4 negatively regulates the production of proinflammatory cytokines by macrophages in response to LPS. Proc Natl Acad Sci U S A 102: 16001–16006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi M, Hayashida W, Akishita M, Yamada S, Lehtonen JY, Tamura K et al (2000). Interferon‐gamma induces AT(2) receptor expression in fibroblasts by JAK/STAT pathway and interferon regulatory factor‐1. Circ Res 86: 233–240. [DOI] [PubMed] [Google Scholar]
- Huang H, Tang QZ, Wang AB, Chen M, Yan L, Liu C et al (2010). Tumor suppressor A20 protects against cardiac hypertrophy and fibrosis by blocking transforming growth factor‐beta‐activated kinase 1‐dependent signaling. Hypertension 56: 232–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Liu T, Xu LG, Chen D, Zhai Z, Shu HB (2005). SIKE is an IKK epsilon/TBK1‐associated suppressor of TLR3‐ and virus‐triggered IRF‐3 activation pathways. EMBO J 24: 4018–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Xiao A, Zhou M, Zhu Z, Lin S, Zhang B (2011). Heritable gene targeting in zebrafish using customized TALENs. Nat Biotechnol 29: 699–700. [DOI] [PubMed] [Google Scholar]
- Huang W, Horvath E, Eklund EA (2007). PU.1, interferon regulatory factor (IRF) 2, and the interferon consensus sequence‐binding protein (ICSBP/IRF8) cooperate to activate NF1 transcription in differentiating myeloid cells. J Biol Chem 282: 6629–6643. [DOI] [PubMed] [Google Scholar]
- Huber M, Brustle A, Reinhard K, Guralnik A, Walter G, Mahiny A et al (2008). IRF4 is essential for IL‐21‐mediated induction, amplification, and stabilization of the Th17 phenotype. Proc Natl Acad Sci U S A 105: 20846–20851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huye LE, Ning S, Kelliher M, Pagano JS (2007). Interferon regulatory factor 7 is activated by a viral oncoprotein through RIP‐dependent ubiquitination. Mol Cell Biol 27: 2910–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Salkowski CA, Zhang F, Aber T, Nagayama M, Vogel SN et al (1999). The transcription factor interferon regulatory factor 1 is expressed after cerebral ischemia and contributes to ischemic brain injury. J Exp Med 189: 719–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa E, Hida S, Omatsu Y, Shimoyama S, Takahara K, Miyagawa S et al (2004). Defective development of splenic and epidermal CD4+ dendritic cells in mice deficient for IFN regulatory factor‐2. Proc Natl Acad Sci U S A 101: 3909–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Croce K, Morooka T, Sakuma M, Node K, Simon DI (2011). Vascular inflammation and repair: implications for re‐endothelialization, restenosis, and stent thrombosis. JACC Cardiovasc Interv 4: 1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway CA Jr, Medzhitov R (2002). Innate immune recognition. Annu Rev Immunol 20: 197–216. [DOI] [PubMed] [Google Scholar]
- Jia W, Guo Q (2008). Gene structures and promoter characteristics of interferon regulatory factor 1 (IRF‐1), IRF‐2 and IRF‐7 from snakehead Channa argus . Mol Immunol 45: 2419–2428. [DOI] [PubMed] [Google Scholar]
- Jiang DS, Bian ZY, Zhang Y, Zhang SM, Liu Y, Zhang R et al (2013). Role of interferon regulatory factor 4 in the regulation of pathological cardiac hypertrophy. Hypertension 61: 1193–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang DS, Li L, Huang L, Gong J, Xia H, Liu X et al (2014a). Interferon regulatory factor 1 is required for cardiac remodeling in response to pressure overload. Hypertension 64: 77–86. [DOI] [PubMed] [Google Scholar]
- Jiang DS, Liu Y, Zhou H, Zhang Y, Zhang XD, Zhang XF et al (2014b). Interferon regulatory factor 7 functions as a novel negative regulator of pathological cardiac hypertrophy. Hypertension 63: 713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang DS, Luo YX, Zhang R, Zhang XD, Chen HZ, Zhang Y et al (2014c). Interferon regulatory factor 9 protects against cardiac hypertrophy by targeting myocardin. Hypertension 63: 119–127. [DOI] [PubMed] [Google Scholar]
- Jiang DS, Wei X, Zhang XF, Liu Y, Zhang Y, Chen K et al (2014d). IRF8 suppresses pathological cardiac remodelling by inhibiting calcineurin signalling. Nat Commun 5: 3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang DS, Zhang XF, Gao L, Zong J, Zhou H, Liu Y et al (2014e). Signal regulatory protein‐alpha protects against cardiac hypertrophy via the disruption of toll‐like receptor 4 signaling. Hypertension 63: 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno Y, Levi BZ, Tamura T, Ozato K (2005). Immune cell‐specific amplification of interferon signaling by the IRF‐4/8‐PU.1 complex. J Interferon Cytokine Res 25: 770–779. [DOI] [PubMed] [Google Scholar]
- Kano S, Sato K, Morishita Y, Vollstedt S, Kim S, Bishop K et al (2008). The contribution of transcription factor IRF1 to the interferon‐gamma‐interleukin 12 signaling axis and TH1 versus TH‐17 differentiation of CD4+ T‐cells. Nat Immunol 9: 34–41. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S (2006). TLR signaling. Cell Death Differ 13: 816–825. [DOI] [PubMed] [Google Scholar]
- Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M et al (2004). Interferon‐alpha induction through Toll‐like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol 5: 1061–1068. [DOI] [PubMed] [Google Scholar]
- Kehat I, Molkentin JD (2010). Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation 122: 2727–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi H, Nakayama M, Kuribayashi F, Imajoh‐Ohmi S, Nishitoh H, Takami Y et al (2014). GCN5 is essential for IRF‐4 gene expression followed by transcriptional activation of Blimp‐1 in immature B cells. J Leukoc Biol 95: 399–404. [DOI] [PubMed] [Google Scholar]
- Ko J, Gendron‐Fitzpatrick A, Splitter GA (2002). Susceptibility of IFN regulatory factor‐1 and IFN consensus sequence binding protein‐deficient mice to brucellosis. J Immunol 168: 2433–2440. [DOI] [PubMed] [Google Scholar]
- Kong HJ, Anderson DE, Lee CH, Jang MK, Tamura T, Tailor P et al (2007). Cutting edge: autoantigen Ro52 is an interferon inducible E3 ligase that ubiquitinates IRF‐8 and enhances cytokine expression in macrophages. J Immunol 179: 26–30. [DOI] [PubMed] [Google Scholar]
- Kong Y, Tannous P, Lu G, Berenji K, Rothermel BA, Olson EN et al (2006). Suppression of class I and II histone deacetylases blunts pressure‐overload cardiac hypertrophy. Circulation 113: 2579–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korachi M, Ceran N, Adaleti R, Nigdelioglu A, Sokmen M (2013). An association study of functional polymorphic genes IRF‐1, IFNGR‐1, and IFN‐gamma with disease progression, aspartate aminotransferase, alanine aminotransferase, and viral load in chronic hepatitis B and C. Int J Infect Dis 17: e44–e49. [DOI] [PubMed] [Google Scholar]
- Kusaba K, Kai H, Koga M, Takayama N, Ikeda A, Yasukawa H et al (2007). Inhibition of intrinsic interferon‐gamma function prevents neointima formation after balloon injury. Hypertension 49: 909–915. [DOI] [PubMed] [Google Scholar]
- Lallemand C, Blanchard B, Palmieri M, Lebon P, May E, Tovey MG (2007). Single‐stranded RNA viruses inactivate the transcriptional activity of p53 but induce NOXA‐dependent apoptosis via post‐translational modifications of IRF‐1, IRF‐3 and CREB. Oncogene 26: 328–338. [DOI] [PubMed] [Google Scholar]
- Lande R, Gilliet M (2010). Plasmacytoid dendritic cells: key players in the initiation and regulation of immune responses. Ann N Y Acad Sci 1183: 89–103. [DOI] [PubMed] [Google Scholar]
- Lazear HM, Lancaster A, Wilkins C, Suthar MS, Huang A, Vick SC et al (2012). IRF‐3, IRF‐5, and IRF‐7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog 9: e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear HM, Lancaster A, Wilkins C, Suthar MS, Huang A, Vick SC et al (2013). IRF‐3, IRF‐5, and IRF‐7 coordinately regulate the type I IFN response in myeloid dendritic cells downstream of MAVS signaling. PLoS Pathog 9: e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazorchak AS, Schlissel MS, Zhuang Y (2006). E2A and IRF‐4/Pip promote chromatin modification and transcription of the immunoglobulin kappa locus in pre‐B cells. Mol Cell Biol 26: 810–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtonen A, Lund R, Lahesmaa R, Julkunen I, Sareneva T, Matikainen S (2003). IFN‐alpha and IL‐12 activate IFN regulatory factor 1 (IRF‐1), IRF‐4, and IRF‐8 gene expression in human NK and T‐cells. Cytokine 24: 81–90. [DOI] [PubMed] [Google Scholar]
- Leung PY, Stevens SL, Packard AE, Lessov NS, Yang T, Conrad VK et al (2012). Toll‐like receptor 7 preconditioning induces robust neuroprotection against stroke by a novel type I interferon‐mediated mechanism. Stroke 43: 1383–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, He C, Feng J, Zhang Y, Tang Q, Bian Z et al (2010a). Regulator of G protein signaling 5 protects against cardiac hypertrophy and fibrosis during biomechanical stress of pressure overload. Proc Natl Acad Sci U S A 107: 13818–13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Tang QZ, Liu C, Moon M, Chen M, Yan L et al (2010b). Cellular FLICE‐inhibitory protein protects against cardiac remodeling induced by angiotensin II in mice. Hypertension 56: 1109–1117. [DOI] [PubMed] [Google Scholar]
- Li H, Li H, Bao Y, Zhang X, Yu Y (2011a). Free fatty acids induce endothelial dysfunction and activate protein kinase C and nuclear factor‐kappaB pathway in rat aorta. Int J Cardiol 152: 218–224. [DOI] [PubMed] [Google Scholar]
- Li L, Chen W, Zhu Y, Wang X, Jiang DS, Huang F et al (2014). Caspase recruitment domain 6 protects against cardiac hypertrophy in response to pressure overload. Hypertension 64: 94–102. [DOI] [PubMed] [Google Scholar]
- Li S, Wang L, Berman M, Kong YY, Dorf ME (2011b). Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity 35: 426–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Wang ZG, Bian C, Chen XD, Li JW, Chen X et al (2009). Interferon regulatory factor‐1 exerts inhibitory effect on neointimal formation after vascular injury. Chin Med Sci J 24: 91–96. [DOI] [PubMed] [Google Scholar]
- Lim S, Jin CJ, Kim M, Chung SS, Park HS, Lee IK et al (2006). PPARgamma gene transfer sustains apoptosis, inhibits vascular smooth muscle cell proliferation, and reduces neointima formation after balloon injury in rats. Arterioscler Thromb Vasc Biol 26: 808–813. [DOI] [PubMed] [Google Scholar]
- Lin Y, Zhu X, McLntee FL, Xiao H, Zhang J, Fu M et al (2004). Interferon regulatory factor‐1 mediates PPARgamma‐induced apoptosis in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 24: 257–263. [DOI] [PubMed] [Google Scholar]
- Liu CC, Perussia B, Young JD (2000). The emerging role of IL‐15 in NK‐cell development. Immunol Today 21: 113–116. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jiang XL, Liu Y, Jiang DS, Zhang Y, Zhang R et al (2014). Toll‐interacting protein (Tollip) negatively regulates pressure overload‐induced ventricular hypertrophy in mice. Cardiovasc Res 101: 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Bian ZY, Zhang R, Zhang Y, Liu C, Yan L et al (2013a). Interferon regulatory factor 3 is a negative regulator of pathological cardiac hypertrophy. Basic Res Cardiol 108: 326. [DOI] [PubMed] [Google Scholar]
- Lu R (2008). Interferon regulatory factor 4 and 8 in B‐cell development. Trends Immunol 29: 487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Medina KL, Lancki DW, Singh H (2003). IRF‐4,8 orchestrate the pre‐B‐to‐B transition in lymphocyte development. Genes Dev 17: 1703–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YY, Li ZZ, Jiang DS, Wang L, Zhang Y, Chen K et al (2013b). TRAF1 is a critical regulator of cerebral ischaemia‐reperfusion injury and neuronal death. Nat Commun 4: 2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamane Y, Sharma S, Petropoulos L, Lin R, Hiscott J (2000). Posttranslational regulation of IRF‐4 activity by the immunophilin FKBP52. Immunity 12: 129–140. [DOI] [PubMed] [Google Scholar]
- Marsh B, Stevens SL, Packard AE, Gopalan B, Hunter B, Leung PY et al (2009a). Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: a critical role for IRF3. J Neurosci 29: 9839–9849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh BJ, Stenzel‐Poore MP (2008). Toll‐like receptors: novel pharmacological targets for the treatment of neurological diseases. Curr Opin Pharmacol 8: 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh BJ, Williams‐Karnesky RL, Stenzel‐Poore MP (2009b). Toll‐like receptor signaling in endogenous neuroprotection and stroke. Neuroscience 158: 1007–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsili G, Remoli AL, Sgarbanti M, Battistini A (2004). Role of acetylases and deacetylase inhibitors in IRF‐1‐mediated HIV‐1 long terminal repeat transcription. Ann N Y Acad Sci 1030: 636–643. [DOI] [PubMed] [Google Scholar]
- Masumi A (2011). Histone acetyltransferases as regulators of nonhistone proteins: the role of interferon regulatory factor acetylation on gene transcription. J Biomed Biotechnol 2011: 640610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masumi A, Tamaoki S, Wang IM, Ozato K, Komuro K (2002). IRF‐8/ICSBP and IRF‐1 cooperatively stimulate mouse IL‐12 promoter activity in macrophages. FEBS Lett 531: 348–353. [DOI] [PubMed] [Google Scholar]
- Masumi A, Yamakawa Y, Fukazawa H, Ozato K, Komuro K (2003). Interferon regulatory factor‐2 regulates cell growth through its acetylation. J Biol Chem 278: 25401–25407. [DOI] [PubMed] [Google Scholar]
- Mellor AL, Munn DH (2004). IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol 4: 762–774. [DOI] [PubMed] [Google Scholar]
- Melroe GT, Silva L, Schaffer PA, Knipe DM (2007). Recruitment of activated IRF‐3 and CBP/p300 to herpes simplex virus ICP0 nuclear foci: Potential role in blocking IFN‐beta induction. Virology 360: 305–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miano JM (2003). Serum response factor: toggling between disparate programs of gene expression. J Mol Cell Cardiol 35: 577–593. [DOI] [PubMed] [Google Scholar]
- Mildner A, Chapnik E, Manor O, Yona S, Kim KW, Aychek T et al (2013). Mononuclear phagocyte miRNome analysis identifies miR‐142 as critical regulator of murine dendritic cell homeostasis. Blood 121: 1016–1027. [DOI] [PubMed] [Google Scholar]
- Miyamoto M, Fujita T, Kimura Y, Maruyama M, Harada H, Sudo Y et al (1988). Regulated expression of a gene encoding a nuclear factor, IRF‐1, that specifically binds to IFN‐beta gene regulatory elements. Cell 54: 903–913. [DOI] [PubMed] [Google Scholar]
- Mori M, Yoneyama M, Ito T, Takahashi K, Inagaki F, Fujita T (2004). Identification of Ser‐386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. J Biol Chem 279: 9698–9702. [DOI] [PubMed] [Google Scholar]
- Murphy AA, Rosato PC, Parker ZM, Khalenkov A, Leib DA (2013). Synergistic control of herpes simplex virus pathogenesis by IRF‐3, and IRF‐7 revealed through non‐invasive bioluminescence imaging. Virology 444: 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan V, Pion E, Landre V, Muller P, Ball KL (2011). Docking‐dependent ubiquitination of the interferon regulatory factor‐1 tumor suppressor protein by the ubiquitin ligase CHIP. J Biol Chem 286: 607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negishi H, Ohba Y, Yanai H, Takaoka A, Honma K, Yui K et al (2005). Negative regulation of Toll‐like‐receptor signaling by IRF‐4. Proc Natl Acad Sci U S A 102: 15989–15994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H, Hiscott J, Pitha PM (1997). The growing family of interferon regulatory factors. Cytokine Growth Factor Rev 8: 293–312. [DOI] [PubMed] [Google Scholar]
- Oganesyan G, Saha SK, Guo B, He JQ, Shahangian A, Zarnegar B et al (2006). Critical role of TRAF3 in the Toll‐like receptor‐dependent and ‐independent antiviral response. Nature 439: 208–211. [DOI] [PubMed] [Google Scholar]
- Oke V, Wahren‐Herlenius M (2012). The immunobiology of Ro52 (TRIM21) in autoimmunity: a critical review. J Autoimmun 39: 77–82. [DOI] [PubMed] [Google Scholar]
- Okumura A, Alce T, Lubyova B, Ezelle H, Strebel K, Pitha PM (2008). HIV‐1 accessory proteins VPR and Vif modulate antiviral response by targeting IRF‐3 for degradation. Virology 373: 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill LA, Bowie AG (2007). The family of five: TIR‐domain‐containing adaptors in Toll‐like receptor signalling. Nat Rev Immunol 7: 353–364. [DOI] [PubMed] [Google Scholar]
- Panne D, McWhirter SM, Maniatis T, Harrison SC (2007). Interferon regulatory factor 3 is regulated by a dual phosphorylation‐dependent switch. J Biol Chem 282: 22816–22822. [DOI] [PubMed] [Google Scholar]
- Passioura T, Dolnikov A, Shen S, Symonds G (2005). N‐ras‐induced growth suppression of myeloid cells is mediated by IRF‐1. Cancer Res 65: 797–804. [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al; NC‐IUPHAR (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernis AB (2002). The role of IRF‐4 in B and T‐cell activation and differentiation. J Interferon Cytokine Res 22: 111–120. [DOI] [PubMed] [Google Scholar]
- Pettersson S, Kelleher M, Pion E, Wallace M, Ball KL (2009). Role of Mdm2 acid domain interactions in recognition and ubiquitination of the transcription factor IRF‐2. Biochem J 418: 575–585. [DOI] [PubMed] [Google Scholar]
- Piao W, Ru LW, Piepenbrink KH, Sundberg EJ, Vogel SN, Toshchakov VY (2013). Recruitment of TLR adapter TRIF to TLR4 signaling complex is mediated by the second helical region of TRIF TIR domain. Proc Natl Acad Sci U S A 110: 19036–19041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto LA, Michel S, Klopp N, Vogelberg C, von Berg A, Bufe A et al (2013). Polymorphisms in the IRF‐4 gene, asthma and recurrent bronchitis in children. Clin Exp Allergy 43: 1152–1159. [DOI] [PubMed] [Google Scholar]
- Pion E, Narayan V, Eckert M, Ball KL (2009). Role of the IRF‐1 enhancer domain in signalling polyubiquitination and degradation. Cell Signal 21: 1479–1487. [DOI] [PubMed] [Google Scholar]
- Prakash A, Smith E, Lee CK, Levy DE (2005). Tissue‐specific positive feedback requirements for production of type I interferon following virus infection. J Biol Chem 280: 18651–18657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purtha WE, Swiecki M, Colonna M, Diamond MS, Bhattacharya D (2012). Spontaneous mutation of the Dock2 gene in Irf5−/− mice complicates interpretation of type I interferon production and antibody responses. Proc Natl Acad Sci U S A 109: E898–E904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H, Zhu H, Lou M, Fan Y, Liu H, Shen J et al (2012). Interferon regulatory factor 1 transactivates expression of human DNA polymerase eta in response to carcinogen N‐methyl‐N′‐nitro‐N‐nitrosoguanidine. J Biol Chem 287: 12622–12633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavendra Rao VL, Bowen KK, Dhodda VK, Song G, Franklin JL, Gavva NR et al (2002). Gene expression analysis of spontaneously hypertensive rat cerebral cortex following transient focal cerebral ischemia. J Neurochem 83: 1072–1086. [DOI] [PubMed] [Google Scholar]
- Rengarajan J, Mowen KA, McBride KD, Smith ED, Singh H, Glimcher LH (2002). Interferon regulatory factor 4 (IRF4) interacts with NFATc2 to modulate interleukin 4 gene expression. J Exp Med 195: 1003–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restivo G, Nguyen BC, Dziunycz P, Ristorcelli E, Ryan RJ, Ozuysal OY et al (2011). IRF6 is a mediator of Notch pro‐differentiation and tumour suppressive function in keratinocytes. EMBO J 30: 4571–4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saira K, Zhou Y, Jones C (2007). The infected cell protein 0 encoded by bovine herpesvirus 1 (bICP0) induces degradation of interferon response factor 3 and, consequently, inhibits beta interferon promoter activity. J Virol 81: 3077–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Tun‐Kyi A, Ryo A, Yamamoto M, Finn G, Fujita T et al (2006). Negative regulation of interferon‐regulatory factor 3‐dependent innate antiviral response by the prolyl isomerase Pin1. Nat Immunol 7: 598–605. [DOI] [PubMed] [Google Scholar]
- Sanbe A, Gulick J, Hanks MC, Liang Q, Osinska H, Robbins J (2003). Reengineering inducible cardiac‐specific transgenesis with an attenuated myosin heavy chain promoter. Circ Res 92: 609–616. [DOI] [PubMed] [Google Scholar]
- Sasai M, Yamamoto M (2013). Pathogen recognition receptors: ligands and signaling pathways by Toll‐like receptors. Int Rev Immunol 32: 116–133. [DOI] [PubMed] [Google Scholar]
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K et al (2000). Distinct and essential roles of transcription factors IRF‐3 and IRF‐7 in response to viruses for IFN‐alpha/beta gene induction. Immunity 13: 539–548. [DOI] [PubMed] [Google Scholar]
- Savitsky D, Tamura T, Yanai H, Taniguchi T (2010). Regulation of immunity and oncogenesis by the IRF transcription factor family. Cancer Immunol Immunother 59: 489–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheller M, Schonheit J, Zimmermann K, Leser U, Rosenbauer F, Leutz A (2013). Cross talk between Wnt/beta‐catenin and Irf8 in leukemia progression and drug resistance. J Exp Med 210: 2239–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavoni G, Mattei F, Sestili P, Borghi P, Venditti M, Morse HC 3rd et al (2002). ICSBP is essential for the development of mouse type I interferon‐producing cells and for the generation and activation of CD8alpha(+) dendritic cells. J Exp Med 196: 1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffrin EL (2014). Immune mechanisms in hypertension and vascular injury. Clin Sci (Lond) 126: 267–274. [DOI] [PubMed] [Google Scholar]
- Schmitz F, Heit A, Guggemoos S, Krug A, Mages J, Schiemann M et al (2007). Interferon‐regulatory‐factor 1 controls Toll‐like receptor 9‐mediated IFN‐beta production in myeloid dendritic cells. Eur J Immunol 37: 315–327. [DOI] [PubMed] [Google Scholar]
- Sears N, Sen GC, Stark GR, Chattopadhyay S (2011). Caspase‐8‐mediated cleavage inhibits IRF‐3 protein by facilitating its proteasome‐mediated degradation. J Biol Chem 286: 33037–33044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea C‐K, Chen ZJ (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF‐κB and IRF3. Cell 122: 669–682. [DOI] [PubMed] [Google Scholar]
- Shaik‐Dasthagirisaheb YB, Huang N, Gibson FC 3rd (2014). Inflammatory response to Porphyromonas gingivalis partially requires interferon regulatory factor (IRF) 3. Innate Immun 20: 312–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif‐Naeini R, Folgering JH, Bichet D, Duprat F, Delmas P, Patel A et al (2010). Sensing pressure in the cardiovascular system: Gq‐coupled mechanoreceptors and TRP channels. J Mol Cell Cardiol 48: 83–89. [DOI] [PubMed] [Google Scholar]
- Sigurdsson S, Nordmark G, Goring HH, Lindroos K, Wiman AC, Sturfelt G et al (2005). Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet 76: 528–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soros P, Hachinski V (2012). Cardiovascular and neurological causes of sudden death after ischaemic stroke. Lancet Neurol 11: 179–188. [DOI] [PubMed] [Google Scholar]
- Stankowski JN, Gupta R (2011). Therapeutic targets for neuroprotection in acute ischemic stroke: lost in translation? Antioxid Redox Signal 14: 1841–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens SL, Leung PY, Vartanian KB, Gopalan B, Yang T, Simon RP et al (2011). Multiple preconditioning paradigms converge on interferon regulatory factor‐dependent signaling to promote tolerance to ischemic brain injury. J Neurosci 31: 8456–8463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A, Yanai H (2006). Interferon signalling network in innate defence. Cell Microbiol 8: 907–922. [DOI] [PubMed] [Google Scholar]
- Takeda K, Akira S (2004). TLR signaling pathways. Semin Immunol 16: 3–9. [DOI] [PubMed] [Google Scholar]
- Taki S, Nakajima S, Ichikawa E, Saito T, Hida S (2005). IFN regulatory factor‐2 deficiency revealed a novel checkpoint critical for the generation of peripheral NK cells. J Immunol 174: 6005–6012. [DOI] [PubMed] [Google Scholar]
- Tamura T, Ozato K (2002). ICSBP/IRF‐8: its regulatory roles in the development of myeloid cells. J Interferon Cytokine Res 22: 145–152. [DOI] [PubMed] [Google Scholar]
- Tamura T, Tailor P, Yamaoka K, Kong HJ, Tsujimura H, O'Shea JJ et al (2005). IFN regulatory factor‐4 and ‐8 govern dendritic cell subset development and their functional diversity. J Immunol 174: 2573–2581. [DOI] [PubMed] [Google Scholar]
- Tamura T, Yanai H, Savitsky D, Taniguchi T (2008). The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol 26: 535–584. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Kawakami T, Taniguchi T (1993). Recognition DNA sequences of interferon regulatory factor 1 (IRF‐1) and IRF‐2, regulators of cell growth and the interferon system. Mol Cell Biol 13: 4531–4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SC, Arumugam TV, Xu X, Cheng A, Mughal MR, Jo DG et al (2007a). Pivotal role for neuronal Toll‐like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A 104: 13798–13803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Gao JS, Guan YJ, McLane KE, Yuan ZL, Ramratnam B et al (2007b). Acetylation‐dependent signal transduction for type I interferon receptor. Cell 131: 93–105. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Takaoka A (2002). The interferon‐alpha/beta system in antiviral responses: a multimodal machinery of gene regulation by the IRF family of transcription factors. Curr Opin Immunol 14: 111–116. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Ogasawara K, Takaoka A, Tanaka N (2001). IRF family of transcription factors as regulators of host defense. Annu Rev Immunol 19: 623–655. [DOI] [PubMed] [Google Scholar]
- Timmers L, Sluijter JP, van Keulen JK, Hoefer IE, Nederhoff MG, Goumans MJ et al (2008). Toll‐like receptor 4 mediates maladaptive left ventricular remodeling and impairs cardiac function after myocardial infarction. Circ Res 102: 257–264. [DOI] [PubMed] [Google Scholar]
- Tsushima K, Osawa T, Yanai H, Nakajima A, Takaoka A, Manabe I et al (2011). IRF3 regulates cardiac fibrosis but not hypertrophy in mice during angiotensin II‐induced hypertension. FASEB J 25: 1531–1543. [DOI] [PubMed] [Google Scholar]
- Vartanian KB, Stevens SL, Marsh BJ, Williams‐Karnesky R, Lessov NS, Stenzel‐Poore MP (2011). LPS preconditioning redirects TLR signaling following stroke: TRIF‐IRF3 plays a seminal role in mediating tolerance to ischemic injury. J Neuroinflammation 8: 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL et al (2011). Innate or adaptive immunity? The example of natural killer cells. Science 331: 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner AH, Gebauer M, Pollok‐Kopp B, Hecker M (2002). Cytokine‐inducible CD40 expression in human endothelial cells is mediated by interferon regulatory factor‐1. Blood 99: 520–525. [DOI] [PubMed] [Google Scholar]
- Wang F, Ma Y, Barrett JW, Gao X, Loh J, Barton E et al (2004). Disruption of Erk‐dependent type I interferon induction breaks the myxoma virus species barrier. Nat Immunol 5: 1266–1274. [DOI] [PubMed] [Google Scholar]
- Wang H, Morse HC 3rd (2009). IRF8 regulates myeloid and B lymphoid lineage diversification. Immunol Res 43: 109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Lee CH, Qi C, Tailor P, Feng J, Abbasi S et al (2008). IRF8 regulates B‐cell lineage specification, commitment, and differentiation. Blood 112: 4028–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Maldonado MA (2006). The ubiquitin–proteasome system and its role in inflammatory and autoimmune diseases. Cell Mol Immunol 3: 255–261. [PubMed] [Google Scholar]
- Wang J, Yang B, Hu Y, Zheng Y, Zhou H, Wang Y et al (2013a). Negative regulation of Nmi on virus‐triggered type I IFN production by targeting IRF7. J Immunol 191: 3393–3399. [DOI] [PubMed] [Google Scholar]
- Wang L, Lu Y, Deng S, Zhang Y, Yang L, Guan Y et al (2012). SHPS‐1 deficiency induces robust neuroprotection against experimental stroke by attenuating oxidative stress. J Neurochem 122: 834–843. [DOI] [PubMed] [Google Scholar]
- Wang L, Lu Y, Zhang X, Zhang Y, Jiang D, Dong X et al (2013b). Mindin is a critical mediator of ischemic brain injury in an experimental stroke model. Exp Neurol 247: 506–516. [DOI] [PubMed] [Google Scholar]
- Wang XA, Zhang R, Jiang D, Deng W, Zhang S, Deng S et al (2013c). Interferon regulatory factor 9 protects against hepatic insulin resistance and steatosis in male mice. Hepatology 58: 603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XA, Zhang R, She ZG, Zhang XF, Jiang DS, Wang T et al (2014). Interferon regulatory factor 3 constrains IKKbeta/NF‐kappaB signaling to alleviate hepatic steatosis and insulin resistance. Hepatology 59: 870–885. [DOI] [PubMed] [Google Scholar]
- Wang XA, Zhang R, Zhang S, Deng S, Jiang D, Zhong J et al (2013d). Interferon regulatory factor 7 deficiency prevents diet‐induced obesity and insulin resistance. Am J Physiol Endocrinol Metab 305: E485–E495. [DOI] [PubMed] [Google Scholar]
- Watford WT, Moriguchi M, Morinobu A, O'Shea JJ (2003). The biology of IL‐12: coordinating innate and adaptive immune responses. Cytokine Growth Factor Rev 14: 361–368. [DOI] [PubMed] [Google Scholar]
- Wessely R, Hengst L, Jaschke B, Wegener F, Richter T, Lupetti R et al (2003). A central role of interferon regulatory factor‐1 for the limitation of neointimal hyperplasia. Hum Mol Genet 12: 177–187. [DOI] [PubMed] [Google Scholar]
- Woods M, Wood EG, Bardswell SC, Bishop‐Bailey D, Barker S, Wort SJ et al (2003). Role for nuclear factor‐kappaB and signal transducer and activator of transcription 1/interferon regulatory factor‐1 in cytokine‐induced endothelin‐1 release in human vascular smooth muscle cells. Mol Pharmacol 64: 923–931. [DOI] [PubMed] [Google Scholar]
- Xiang M, Wang L, Guo S, Lu YY, Lei H, Jiang DS et al (2014). Interferon regulatory factor 8 protects against cerebral ischaemic‐reperfusion injury. J Neurochem 129: 988–1001. [DOI] [PubMed] [Google Scholar]
- Xiao J, Moon M, Yan L, Nian M, Zhang Y, Liu C et al (2012). Cellular FLICE‐inhibitory protein protects against cardiac remodelling after myocardial infarction. Basic Res Cardiol 107: 239. [DOI] [PubMed] [Google Scholar]
- Xiong H, Li H, Kong HJ, Chen Y, Zhao J, Xiong S et al (2005). Ubiquitin‐dependent degradation of interferon regulatory factor‐8 mediated by Cbl down‐regulates interleukin‐12 expression. J Biol Chem 280: 23531–23539. [DOI] [PubMed] [Google Scholar]
- Xu H, Krolikowski JG, Jones DW, Ge ZD, Pagel PS, Pritchard KA Jr et al (2012). 4F decreases IRF5 expression and activation in hearts of tight skin mice. PLoS ONE 7: e52046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Kato T, Hotta C, Nishiyama A, Kurotaki D, Yoshinari M et al (2011). Shared and distinct functions of the transcription factors IRF4 and IRF8 in myeloid cell development. PLoS ONE 6: e25812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Wei X, Tang QZ, Feng J, Zhang Y, Liu C et al (2011). Cardiac‐specific mindin overexpression attenuates cardiac hypertrophy via blocking AKT/GSK3beta and TGF‐beta1‐SMAD signalling. Cardiovasc Res 92: 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai H, Negishi H, Taniguchi T (2012). The IRF family of transcription factors: inception, impact and implications in oncogenesis. Oncoimmunology 1: 1376–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Hu X, Zimmerman M, Torres CM, Yang D, Smith SB et al (2011). Cutting edge: IRF8 regulates Bax transcription in vivo in primary myeloid cells. J Immunol 187: 4426–4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Shi HX, Liu XY, Shan YF, Wei B, Chen S et al (2009). TRIM21 is essential to sustain IFN regulatory factor 3 activation during antiviral response. J Immunol 182: 3782–3792. [DOI] [PubMed] [Google Scholar]
- Yang L, Feng D, Bi X, Stone RC, Barnes BJ (2012). Monocytes from Irf5−/− mice have an intrinsic defect in their response to pristane‐induced lupus. J Immunol 189: 3741–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K et al (2005). Shared and unique functions of the DExD/H‐box helicases RIG‐I, MDA5, and LGP2 in antiviral innate immunity. J Immunol 175: 2851–2858. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Sinha S, Dandre F, Wamhoff BR, Hoofnagle MH, Kremer BE et al (2003). Myocardin is a key regulator of CArG‐dependent transcription of multiple smooth muscle marker genes. Circ Res 92: 856–864. [DOI] [PubMed] [Google Scholar]
- Zhang M, Tian Y, Wang RP, Gao D, Zhang Y, Diao FC et al (2008). Negative feedback regulation of cellular antiviral signaling by RBCK1‐mediated degradation of IRF3. Cell Res 18: 1096–1104. [DOI] [PubMed] [Google Scholar]
- Zhang SM, Gao L, Zhang XF, Zhang R, Zhu LH, Wang PX et al (2014a). Interferon regulatory factor 8 modulates phenotypic switching of smooth muscle cells by regulating the activity of myocardin. Mol Cell Biol 34: 400–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SM, Zhu LH, Li ZZ, Wang PX, Chen HZ, Guan HJ et al (2014b). Interferon regulatory factor 3 protects against adverse neo‐intima formation. Cardiovasc Res 102: 469–479. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu X, She ZG, Jiang DS, Wan N, Xia H et al (2014c). Interferon regulatory factor 9 is an essential mediator of heart dysfunction and cell death following myocardial ischemia/reperfusion injury. Basic Res Cardiol 109: 434. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu Y, Zhu XH, Zhang XD, Jiang DS, Bian ZY et al (2014d). Dickkopf‐3 attenuates pressure overload‐induced cardiac remodelling. Cardiovasc Res 102: 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao GN, Jiang DS, Li H (2014). Interferon regulatory factors: at the crossroads of immunity, metabolism, and disease. Biochim Biophys Acta doi: 10.1016/j.bbadis.2014.04.030. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Zhou L, Littman DR (2009). Transcriptional regulatory networks in Th17 cell differentiation. Curr Opin Immunol 21: 146–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler G, Harhausen D, Schepers C, Hoffmann O, Rohr C, Prinz V et al (2007). TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochem Biophys Res Commun 359: 574–579. [DOI] [PubMed] [Google Scholar]