

Abstract
Hydrogen sulfide (H2S) has traditionally been viewed as a highly toxic gas; however, recent studies have implicated H2S as a third member of the gasotransmitter family, exhibiting properties similar to NO and carbon monoxide. Accumulating evidence has suggested that H2S influences a wide range of physiological and pathological processes, among which blood vessel relaxation, cardioprotection and atherosclerosis have been particularly studied. In the cardiovascular system, H2S production is predominantly catalyzed by cystathionine γ‐lyase (CSE). Decreased endogenous H2S levels have been found in hypertensive patients and animals, and CSE −/− mice develop hypertension with age, suggesting that a deficiency in H2S contributes importantly to BP regulation. H2S supplementation attenuates hypertension in different hypertensive animal models. The mechanism by which H2S was originally proposed to attenuate hypertension was by virtue of its action on vascular tone, which may be related to effects on different ion channels. Both H2S and NO cause vasodilatation and there is cross‐talk between these two molecules to regulate BP. Suppression of oxidative stress may also contribute to antihypertensive effects of H2S. This review also summarizes the state of research on H2S and hypertension in China. A better understanding of the role of H2S in hypertension and related cardiovascular diseases will allow novel strategies to be devised for their treatment.
Linked Articles
This article is part of a themed section on Chinese Innovation in Cardiovascular Drug Discovery. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2015.172.issue-23
Abbreviation
- 2K1C
two‐kidney‐one‐clip
- Ang II
angiotensin II
- AOA
aminooxyacetic acid
- AT1 receptor
angiotensin II type 1 receptor
- CBS
cystathionine‐β‐synthase
- CO
carbon monoxide
- CSE
cystathionine‐γ‐lyase
- DBP
diastolic BP
- eNOS
endothelial NOS
- H2S
hydrogen sulfide
- I/R
ischaemia/reperfusion
- L‐NAME
NG‐nitro‐l‐arginine methyl ester
- MAP
mean arterial pressure
- MPST
3‐mercaptopyruvate sulfurtransferase
- MWT
medial wall thickness
- PAAT
pulmonary arterial acceleration time
- PAG
DL‐propargylglycine
- PHT
pulmonary hypertension
- RVET
right ventricular ejection time
- RVH
right ventricular hypertrophy
- SBP
systolic BP
- SHR
spontaneously hypertensive rat
- SMCs
smooth muscle cells
- VEGFR‐1
soluble fms‐like tyrosine kinase 1
- VD
vas deferens
Tables of Links
| TARGETS | |
|---|---|
| GPCR a | Catalytic receptors d |
| AT1 receptor | VEGFR‐1 |
| Muscarinic receptors | Enzymes e |
| Thromboxane A2 receptor | CBS |
| Ligand‐gated ion channels b | CSE |
| Epithelial sodium channels (ENaC) | eNOS |
| Ion channels c | ERK1/2 |
| BKCa channels | HO1 |
| CaV channels | p38MAPK |
| CaV1.1‐1.4 (L‐type Ca) channels | PKG |
| KATP channels | PTEN |
| Kir channels | MPST |
| KV channels | |
| KV7.x (KCNQ) channels |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,c,d,eAlexander et al., 2013a, 2013b, 2013c, 2013d, 2013e).
Introduction
In recent years, the ‘gasotransmitters’ NO, carbon monoxide (CO) and hydrogen sulfide (H2S) have been the object of intense research (Papapetropoulos et al., 2014). In the last two decades, NO has been extensively studied; and in more recent times, the importance of H2S in cardiovascular regulation has become increasingly apparent (Polhemus and Lefer, 2014). Until the H2S content within the brain was first measured in postmortem studies in 1989, H2S was traditionally viewed as a highly toxic gas devoid of beneficial biological or physiological functions (Goodwin et al., 1989). Subsequently, H2S quickly emerged as an important signalling molecule with widespread physiological functions.
Three enzymatic pathways have been identified which produce H2S in mammals: cystathionine‐β‐synthase (CBS), cystathionine‐γ‐lyase (CSE) and 3‐mercaptopyruvate sulfurtransferase (MPST). CBS and CSE are believed to be the critical enzymes for H2S generation in the modulation of neurological and cardiovascular functions, respectively, while expression of MPST has also been found in vascular endothelium (Wang, 2012). Accumulating evidence has confirmed that a wide range of physiological and pathological processes can be mediated by H2S, especially in the cardiovascular system, including blood vessel relaxation, cardioprotection and atherosclerosis (Wang, 2011; Tang et al., 2013; Yang et al., 2013; Bos et al., 2014; King et al., 2014; Mani et al., 2014). This review will focus on the roles of H2S in hypertension and related cardiovascular diseases.
Deficiency of H2S production and hypertension development
Several studies have shown that H2S may play important roles in the pathogenesis and development of hypertension in humans. In patients with grade 2 and 3 hypertension, plasma H2S concentrations were found to be lower than in subjects with normal BP (Sun et al., 2007). In patients with portal hypertension, endogenous H2S levels have been reported to be lower than in healthy controls; and an inverse relationship was seen between disease severity and lower plasma H2S levels, the latter being inversely correlated with portal vein diameters and Child‐Pugh score (Wang et al., 2014). In patients with pulmonary hypertension (PHT), levels of H2S and expression of CSE have been found to be reduced; based on evaluation of the receiver‐operating characteristic curve, CSE had the most significant sensitivity and specificity to predict dynamic PHT (Sun et al., 2014). H2S deficiency might be mainly ascribed to reduce CSE activity or expression, although little is known about transcriptional regulation or post‐translational modification of CSE.
Genetic modulations of CSE, CBS or MPST levels are effective means to experimentally investigate the cardiovascular actions of H2S. However, complete genetic deficiency of CBS (as in a homozygote knockout mouse) exhibits a neonatally lethal phenotype because of liver dysfunction (Watanabe et al., 1995). It is possible to circumvent this lethality problem by insertion of a transgene that expresses a cDNA encoding for a human CBS protein that contains the I278T mutation under control of a zinc‐inducible metallothionein promoter (Wang et al., 2005). Indeed, expression of this mutant CBS protein was able to prevent the neonatally lethal phenotype, but failed to normalize the elevated total homocysteine levels (Gupta et al., 2014). Some evidence suggests that, in the context of hyperhomocysteinemia, homocysteine can inactivate CSE by homocysteinylation, a phenomenon seen in hypertension and other cardiovascular diseases (Sen et al., 2010). On the other hand, MPST knockout is not lethal to mice, but there exists a paucity of information regarding any possible effect of genetic deficiency of MPST on cardiovascular disease (Nagahara, 2013). It is worth noting that CSE mutant mice with a C57BL/6J × 129SvEv mixed genetic background develop hypertension with age. Systolic BP (SBP) in the mutant mice has been reported to increase at more than 135 mmHg at 12 weeks of age, which was almost 18 mmHg higher than in control mice. The BP of CSE−/− mice was about 10 mmHg higher than that of CSE−/+ mice after 10 weeks of age, all of which suggests that a deficiency of CSE/H2S may contribute to BP augmentation (Yang et al., 2008). However, it has been controversially reported that C57BL/6J mice with CSE deficiency actually exhibit normal BP (Ishii et al., 2010). This discrepancy may be due to differences in their genetic background. However, it should also be noted that, in the earlier studies, BP was measured using a standard tail‐cuff non‐invasive measurement system or by intra‐arterial catheterization method rather than by telemetric BP recordings, and the latter is much less prone to error than either of the former methods. An augmented level of less than 20 mmHg in CSE−/− mice might also be subjected to error and possible bias. An electronic BP monitor is the only reliable method for analysing systemic BP nowadays. Additionally, there is a limitation that all of the phenotypes are observed in mice with an overall CSE deficiency, not just a conditional knockout in the cardiovascular system. Therefore, the true effect of CSE deficiency on BP remains unclear at present. Lucock et al. found that both genetic variants of CSE‐G1364T and CBS‐844ins68 are biological determinants of H2S synthesis and are aetiologically important in the regulation of BP (Lucock et al., 2013). In addition, it has been observed that mean arterial pressure (MAP) in rats is not enhanced after treatment with either the CSE inhibitor DL‐propargylglycine (PAG) or the CBS inhibitor aminooxyacetic acid (AOA) alone for 4 weeks; however, MAP increased gradually from 99 ± 2 to 130 ± 1 mmHg during 4 weeks of treatment with the combination of PAG and AOA (Roy et al., 2012). The balance of evidence therefore suggests that H2S plays an important role in regulating BP.
H2S supplementation attenuates hypertension
It has been reported in many studies that H2S supplementation decreases BP in different hypertensive models. The protective effects of H2S in different conditions are discussed in the succeeding text.
Role of H2S in the spontaneously hypertensive rat (SHR)
Many of the cardiovascular changes in the SHR are similar to those in hypertensive human subjects. Levels of H2S in plasma, urinary and gene expression and activity of CSE in thoracic aorta are all suppressed in SHR (Yan et al., 2004; Ahmad et al., 2014). A profound antihypertensive effect of H2S in SHR has been identified by several groups. Yan et al. and Zhao et al. found that exogenous administration of NaHS (56 μmol·kg−1 day−1) for 5 weeks attenuated the elevation of pressure and lessened aortic structural remodelling and collagen accumulation during the development of hypertension (Yan et al., 2004; Zhao et al., 2008). Shi et al. reported that NaHS treatment for 3 months at doses of 30 and 90 μmol·kg−1 day−1 reduced SBP, diastolic BP (DBP) and MAP to similar extents in SHR. Moreover, NaHS at 10 μmol·kg−1 day−1 reduced DBP and MAP, indicating that lower doses of NaHS can reduce BP if treatment if given for a long enough period (Shi et al., 2007). These findings indicate that NaHS at doses between 10 and 90 μmol·kg−1 day−1 reduce BP in SHR. On the other hand, it is well established that NaHS promotes apoptotic cell death of cultured fibroblasts and smooth muscle cells (SMCs; Baskar et al., 2007) and additionally releases copious amounts of H2S over a short time frame (s), which does not effectively mimic physiological concentrations of H2S in vivo and might be harmful. The studies have not evaluated specifically for possible toxicity of NaHS; therefore, whether the antihypertensive effect was simply a manifestation of H2S toxicity remains to be clarified. Because NaHS is not an ideal H2S donor, several other H2S donors, including GYY4137 (a slow‐releasing H2S donor), has been synthesized to evaluate the true physiological role of H2S. GYY4137 did not cause detectable cytotoxicity or alter the cell‐cycle profile or p53 expression of cultured rat vascular SMCs; additionally, it releases low amounts of H2S slowly and persistently and does not trigger signalling pathways leading to cell death. Chronic treatment of conscious animals with GYY4137 at 133 μmol·kg−1 reduced SBP in SHR, the fall in BP being apparent after 2 days and persisting after 14 days of treatment. On cessation of drug therapy, the BP of Wistar‐Kyoto (WKY) rats returned to pre‐injection values within 7 days; while for SHR, the BP was still well controlled at this time point, with BP of all animals returning to pretreatment levels 14 days after cessation of treatment. Despite such lack of toxicity, GYY4137 similarly gave rise to a BP reduction (Li et al., 2008a). In a word, an H2S supplement is more or less beneficial in reducing BP in SHR.
Role of H2S in PHT
In tissue bath preparations of small peripheral airways (<5 mm in diameter) from porcine lungs precontracted with the muscarinic ACh receptor agonist carbachol, both H2S donor NaHS and the precursor L‐cysteine causes a large relaxation of the airways (Rashid et al., 2013). Ariyaratnam et al. found that 500 μM NaHS causes a reduction in both pulmonary artery and bronchial airway pressures (Ariyaratnam et al., 2013). Moreover, Na2S, GYY4137 and L‐cysteine also cause relaxation of airways (Parkinson et al., 1988; Castro‐Piedras and Perez‐Zoghbi, 2013).
Collectively, these data suggest that H2S is a potent dilator of human pulmonary arteries in vitro. However, the influence of H2S on pulmonary blood flow in vivo is extremely complex. The notion that H2S has powerful therapeutic potential for PHT, therefore, needs further confirmation in vivo. Rats exposed to 21 days of hypoxia exhibit decreased plasma H2S concentration and H2S production in their lungs (Wei et al., 2008). Administration of NaHS (10 μmol·kg−1·day−1) has been found to reduce the mean pulmonary artery pressure by 31.2%, but this protective effect was largely abolished by PAG, indicating that H2S might be involved in the development of hypoxia‐induced PHT. H2S supplementation can also attenuate hypoxia‐induced hypertension in broilers (Yang et al., 2012). Up‐regulating the endogenous H2S pathway also reduces pulmonary arterial pressure in rats with PHT induced by high pulmonary blood flow (Luo et al., 2013). After aorta‐veno cava shunting for 11 weeks, rats exhibit PHT and pulmonary artery collagen remodelling in association with a decrease in lung tissue H2S content, suggesting that a reduced level of H2S may contribute to the detrimental effect of shunting (Li et al., 2008b). PHT is also considered as a complication of severe bronchopulmonary dysplasia. Neonatal rats exposed to chronic hyperoxia develop PHT, as demonstrated by a significant decrease in the pulmonary arterial acceleration time/right ventricular ejection time (PAAT/RVET) and an increase in medial wall thickness (MWT) of small pulmonary arteries and right ventricular hypertrophy (RVH); GYY4137 attenuates these functional and structural features of PHT with an increase in mean PAAT/RVET, a decrease in MWT and a reduction in RVH (Vadivel et al., 2014). In human subjects, basal exhaled H2S is higher than the ambient concentration of H2S in room air, indicative of endogenous H2S production in humans; after i.v. administration of Na2S, a rapid elevation of exhaled H2S concentration was observed, the amount of exhaled H2S rapidly decreases after discontinuation of the Na2S infusion, suggesting that exogenously administered H2S diffuses to the bronchial tissue (Toombs et al., 2010). Collectively, these findings suggest that H2S may offer a novel therapeutic target for PHT. However, disagreement exists in H2S concentration and the possibility of lung injury. Inhalation of 80 ppm H2S has been reported to ameliorate lung pathology in lipopolysaccharide‐induced (Faller et al., 2012) and in ventilator‐induced (Faller et al., 2010) lung injury. On the other hand, Francis et al. observed that 1 or 5 ppm H2S did not alter ventilation‐induced lung injury, while 60 ppm H2S worsened it (Francis et al., 2011). It is clear that non‐specific toxicity may in itself evoke a decrease in BP. The equipotent inhalation concentration of different H2S donors by injection has not been evaluated. In other words, further work needs to be done to investigate possible H2S lung toxicity in relation to dose.
Role of H2S in other types of hypertension
Pre‐eclampsia is a hypertensive syndrome that affects 4–7% of all pregnancies and is a major contributor to maternal and fetal morbidity and mortality worldwide. Plasma H2S levels and CSE mRNA expression in the pre‐eclamptic placenta have been found to be reduced in pre‐eclampsia compared with normotensive controls (Wang et al., 2013). After PAG treatment of pregnant C57Bl6/J mice from E8.5 to E17.5, a dose‐dependent decrease in circulating H2S levels was observed; importantly, MAP increased in response to PAG in a dose‐dependent manner, an effect which was attenuated by co‐administration of 0.25 mg·kg−1 GYY4137 (Wang et al., 2013). On the other hand, Holwerda et al. observed that placental CBS mRNA expression decreased in the early‐onset pre‐eclampsia, whereas CSE mRNA in placenta was unchanged in severe pre‐eclampsia (Holwerda et al., 2012). Cindrova‐Davies et al. reported CSE level to be reduced in placentas from pregnancies with severe early‐onset growth restriction and pre‐eclampsia displaying abnormal umbilical artery Doppler waveforms, compared with both pre‐eclamptic placentas with normal waveforms and controls (Cindrova‐Davies et al., 2013). These contradictory findings regarding placental expression of H2S‐synthesizing enzymes may be explained by a lack of significant results because of the small sample sizes in these studies (Patel et al., 2009; Holwerda et al., 2012; Cindrova‐Davies et al., 2013) and highlight the need for larger studies to be performed. Soluble fms‐related tyrosine kinase 1 (VEGFR‐1), a circulating anti‐angiogenic protein, contributes to the development of pre‐eclampsia; treatment with NaHS (50 μmol·kg−1, twice daily) for 8 days reduced VEGFR‐1‐induced hypertension by up‐regulating VEGF expression in rats, although whether this effect is sustained at later time points is presently unknown (Holwerda et al., 2014). Collectively, these data suggest that endogenous H2S is required for healthy placental vascular function and that a decrease in CSE/H2S activity may contribute to the pathogenesis of pre‐eclampsia.
Following induction of hypertension in Wistar rats by oral administration of the L‐arginine analogue NG‐nitro‐l‐arginine methyl ester (L‐NAME) in drinking water for 6 weeks, NaHS treatment decreased in SBP by 19%; furthermore, the observed inhibition of H2S generation and CSE activity in these rats was also greatly attenuated by NaHS treatment (Zhong et al., 2003). Li et al. also found that acute i.v. pre‐injection of GYY4137 (133 μmol·kg−1), but not of NaHS (2.5 μmol·kg−1) or saline, reduced the L‐NAME‐mediated hypertension (Li et al., 2008a). This suggests that there is likely to be an optimal balance between H2S and NO to maintain a dynamic equilibrium and that, if the balance is disturbed for example by L‐NAME administration, H2S supplementation can redress the disturbance in BP.
Tan et al. reported that CCl4 reduces serum H2S levels, hepatic H2S production and CSE expression in rats; exogenous NaHS was found to attenuate CCl4‐induced hepatotoxicity, liver cirrhosis and portal hypertension, indicating that targeting H2S may present a promising approach, particularly in relation to prevention, against portal hypertension (Tan et al., 2011). Rats with cirrhosis induced by bile duct ligation for 4 weeks were treated daily with NaHS for 5 days, then isolated livers were perfused first with NaHS for 20 min followed by noradrenaline (NA). It was found that bile duct ligation resulted in down‐regulation of CSE mRNA/protein levels and activity, indicating that a reduction of CSE expression in the liver with cirrhosis contributes to the development of increased intrahepatic resistance and portal hypertension. NA administration resulted in a dose‐dependent increase of portal pressure and this effect was restored by H2S treatment (Fiorucci et al., 2005). However, inhibition of CSE prevents acute inflammatory liver failure by augmenting thiosulfate levels and up‐regulating antioxidant and anti‐apoptotic defence in the liver (Shirozu et al., 2014). It is possible that excess or even an exogenous supplement of H2S might result in liver injury. That is, there is a possibility that the attenuating effect of H2S on portal hypertension is not due to its pharmacological characteristics but is due merely to non‐specific toxic effects. A more accurate conclusion can only be made on the precondition that the H2S supplement is not harmful to the liver.
To date, few data are available on the effect of H2S on renal hypertension, a type of secondary hypertension. Renal hypertension can be induced with two‐kidney‐one‐clip (2K1C, a clip constricting one renal artery) in animals. One group found that NaHS treatment (5.6 mg·kg−1·day−1) over 4 weeks reversed the BP elevation in 2K1C rats but not in one‐kidney‐one‐clip rats, suggesting that the antihypertensive effect of H2S may be greater in hypertension associated with higher plasma renin activity (Lu et al., 2010). Zhang et al. found that H2S also prevented H2O2‐induced activation of epithelial sodium channels, through which sodium can be reabsorbed in the distal renal tubules to regulate salt‐sensitive hypertension, through a phosphatase and tensins homologue (PTEN; previously known as phosphatidylinositol 3,4,5‐trisphosphate‐dependent) pathway (Zhang et al., 2013). Further investigation of the effect of H2S on epithelial sodium channel activity in animal models may therefore be relevant to the clinical management of salt‐sensitive hypertension. Recently, it has been reported that exogenous administration of an H2S donor attenuates angiotensin II (Ang II)‐induced hypertension (Snijder et al., 2014). Although Zhao et al. found that NaHS decreases the binding affinity of the angiotensin II type 1 (AT1) receptor and attenuates AT1 receptor activation (Zhao et al., 2008), the mechanism by which H2S regulates Ang II‐induced hypertension is not clear because of the involvement of complex interacting networks including renal sympathetic nerve activity and the cardiac sympathetic afferent reflex.
Principal possible mechanisms of the antihypertensive effect of H2S
Relaxation of vascular smooth muscle
One of the earliest proposed beneficial physiological effects of H2S was its action on vascular tone. The endothelium‐dependent vasorelaxation induced by H2S shares many common mechanistic traits with that of endothelium‐derived hyperpolarizing factor (Edwards et al., 2012). Deficiency in CSE expression diminishes endothelium‐dependent relaxation of resistance arteries (Yang et al., 2008). Tang et al. also found that CSE‐knockout mice exhibit elevated resting membrane potential of SMCs, and lack a methacholine‐induced endothelium‐dependent relaxation of mesenteric arteries, whereas that of aorta is preserved; methacholine caused hyperpolarization of SMC in endothelium‐intact mesenteric arteries from wild‐type mice, but this effect was abolished in CSE‐knockout mice, and treatment with exogenous H2S hyperpolarized vascular SMCs and endothelial cells from both wild‐type and CSE‐knockout mice, suggesting that H2S is indeed an endothelium‐derived hyperpolarizing factor (Tang et al., 2013). Loss of endothelium attenuates the relaxation of rat aortic tissues induced by H2S and shifts the H2S concentration‐response curve to the right (Zhao and Wang, 2002). The endothelium dependence of the H2S effect is more pronounced in isolated and perfused rat mesenteric artery bed, such that removal of functional endothelium reduced H2S‐induced relaxation of rat mesenteric artery bed by about sevenfold, with an increase in EC50 of H2S from 25 to 161 μM (Cheng et al., 2004). This tissue‐selective endothelium‐dependent effect of H2S is similar to that of endothelium‐derived hyperpolarizing factor. However, NaHS (0.1–3.0 mM) has been found to elicit concentration‐dependent relaxation of rat middle cerebral arteries, which is unaffected by endothelium removal (Streeter et al., 2012). NaHS relaxes coronary arteries precontracted by U46619 (a thromboxane A2 agonist), this relaxation is similarly unaffected by endothelium removal (Cheang et al., 2010). Expression of CSE and CBS protein has been observed in vascular endothelial cells (Wen et al., 2013). However, it remains unclear why the endothelium dependence of H2S‐mediated relaxation appears to vary from one blood vessel type to another. Specific targets in the endothelium are still not available nowadays.
A large part of H2S‐induced vasorelaxation appears to be dependent on the activation of ATP‐sensitive K+ channel (KATP) in vascular smooth muscle (Liu et al., 2011; Wang, 2011) by increasing whole‐cell KATP currents to hyperpolarize membrane potentials and improving single‐channel activity by enhancing permeability of single KATP channels (Tang et al., 2005). Using the whole‐cell and single‐channel patch‐clamp technique, direct evidence was obtained that exogenous H2S activates KATP channels and hyperpolarizes cell membrane of rat aorta and mesenteric artery SMCs, and that inhibition of endogenous H2S production with PAG reduces whole‐cell KATP currents (Zhao et al., 2001). In the concentration range 100 nM–100 μM, GYY4137 elicits a concentration‐dependent relaxation of phenylephrine‐induced contraction in isolated posterior ciliary arteries, which is attenuated by the KATP channel blocker glibenclamide, suggesting that vascular smooth muscle relaxation induced by H2S is mediated, at least in part, by KATP channels (Chitnis et al., 2013). Reduced expression of CSE and increased miR‐21 in placentas are also associated with increased vascular resistance. Perfusion of normal placentas with NaHS, after preconstriction with a thromboxane mimetic, results in a dose‐dependent vasorelaxation, which can be partially blocked by glibenclamide (Cindrova‐Davies et al., 2013). Nevertheless, the detailed mechanism by which H2S activates KATP channels remains to be elucidated. After treatment of mouse aortic rings with NaHS, cGMP‐dependent PKG activation and NaHS‐stimulated relaxation were evoked in a time‐dependent manner, which could be attenuated by DT‐2 (a PKG1 inhibitor), although interestingly, vasodilator responses to a slow‐releasing H2S donor (GYY4137) were unaffected by DT‐2, suggesting that this donor dilates mouse aorta through PKG‐independent pathways. Dilator responses to NaHS were reduced in vessels of PKG−/− mice, and moreover, glibenclamide inhibited NaHS‐induced vasorelaxation in vessels from wild‐type animals, but not PKG‐I−/− mice, suggesting that there is cross‐talk between KATP and PKG (Bucci et al., 2012). Besides PKG, there may also be other as yet undiscovered H2S‐induced signal‐transduction pathways mediating KATP channel activation.
Several other studies have found that H2S may also induce vasodilatation by affecting other ion channels besides KATP. Suppression of L‐type calcium channels with nifedipine or inhibiting potassium conductance with 50 mM K+ reduce the maximum relaxation elicited by NaHS in rat middle cerebral arteries; however, selective blockers of KATP, calcium‐sensitive (KCa), voltage‐dependent (KV) or inward rectifier (Kir) potassium channels alone or in combination did not affect the response to NaHS, indicating that H2S‐mediated relaxation is partly mediated by inhibition of CaV1.1‐1.4 (L‐type) calcium channels, with an additional contribution by K+ channels which are not of the KATP, KCa, KV or Kir subtypes (Streeter et al., 2012). NaHS elicits concentration‐dependent vasorelaxation in mesenteric arteries and aortas, which can be blocked by the KV7.x channel (KCNQ) inhibitor XE991, and the vasodilator capacity of the KCNQ channel opener retigabine is preserved following inhibition of H2S generation (Schleifenbaum et al., 2010). Li et al. reported that CBS and CSE are functionally expressed in vas deferens (VD) and that H2S mediates VD smooth muscle relaxation; transient receptor potential and KATP channels do not appear to contribute to the NaHS‐induced relaxant effect, whereas the large‐conductance Ca2+‐activated potassium (BKCa) channel blockers iberiotoxin or tetraethylammonium largely reverse the relaxant effect, suggesting that H2S may target BKCa channels in VD smooth muscle (Li et al., 2012). NaHS‐induced relaxation and membrane hyperpolarization in coronary arteries were found to be reduced by 4‐aminopyridine but unaffected by glibenclamide (Cheang et al., 2010). NaHS has been reported to dilate cerebral arteries from Sprague‐Dawley rats with the same potency following precontraction by either 5‐hydroxytryptamine or 60 mmol·L−1 KCl, which were unaffected by several K+ channel blockers. Patch clamp recordings showed that NaHS reduced the amplitude of L‐type Ca2+ currents in single myocytes isolated enzymatically from the cerebral artery, indicating that NaHS relaxes cerebral arteries primarily through inhibiting Ca2+ influx via Ca2+ channels (Tian et al., 2012). These data suggest that different ion channels may be responsible for mediating vasorelaxation in different vascular beds. How they may interact with each other, and whether they play different roles in the vasodilator response, needs further investigation in future studies.
Taken together, the results show that the effects of H2S on ion channels varies in different physiological or pathological situations. KATP channels and other ion channels play different roles in the relaxation of vascular smooth muscle. But is there any specific contributions caused by H2S? If this is true, which ion channel plays the crucial role? Is the relaxant effect on vascular smooth muscle just an artifact or a complicated response? More precise mechanisms need to be studied in the future.
Interaction with NO and CO
Although H2S and NO exhibit independent signalling, and H2S but not NO targets KATP channels, both of these gasotransmitters mediate vasodilatation. Previous studies suggest that a cross‐talk exists between these two molecules. H2S content in lung tissue is increased by L‐arginine treatment after aorta‐veno cava shunting, whereas mean pulmonary artery pressure and relative median area of pulmonary arteries are attenuated (Yanfei et al., 2006). Several lines of evidence have reported that H2S therapy results in cardioprotection following transverse aortic constriction via up‐regulation of endothelial NOS (eNOS) and NO bioavailability (Kondo et al., 2013; Polhemus et al., 2013). We have found that GYY4137 partially restores aortic endothelium‐dependent relaxation in apoE−/− mice, which may be related to increased phosphorylation of eNOS in aorta (Liu et al., 2013). Altaany et al. reported that incubation of HUVECs with NaHS stimulates the phosphorylation of eNOS and enhanced NO production; blockade of NO production by eNOS‐specific siRNA or L‐NAME is seen to reverse, and eNOS overexpression to potentiate, the proliferative effect of H2S on HUVECs (Altaany et al., 2013). Mice lacking CSE exhibit dysfunctional eNOS, diminished NO levels and exacerbated myocardial ischaemia/reperfusion (I/R) injury; acute H2S therapy restores eNOS function and NO bioavailability and attenuates I/R injury, however, does not protect against I/R injury in eNOS phospho‐mutant mice (King et al., 2014). Coletta et al. showed that inhibition of eNOS attenuates H2S‐stimulated vasorelaxation (Coletta et al., 2012). These data demonstrate that that H2S‐mediated cytoprotective signalling is dependent largely on eNOS activation and NO generation. However, there is no direct evidence that NO is involved in the antihypertensive effect of H2S. In the future, this possibility could be investigated by examining the effects of H2S in an animal model with both NO deficiency and hypertension, for example the eNOS knockout mouse.
The regulation of eNOS phosphorylation is complex. We have previously found that association of globular actin with eNOS plays an essential role in agonist‐induced eNOS activation through enabling its phosphorylation by Akt at serine residue 1177 (Mi et al., 2011). Several agents, such as pyridoxine (Xie et al., 2012) and 17β‐estradiol (Han et al., 2012), regulate eNOS activity through effects on its phosphorylation. Several lines of evidence suggest that H2S activates PI3K/Akt, thereby increasing eNOS phosphorylation at Ser1177 and enhancing vascular endothelial NO production (Predmore et al., 2011; Coletta et al., 2012). However, one study found that NaHS induced phosphorylation of eNOS at the phosphoserine residue Ser1179 in a manner not affected by PI3K/Akt inhibition in endothelial cells (Kida et al., 2013). In addition, silencing of the CSE abolishes NO‐stimulated cGMP accumulation and attenuates the ACh‐induced vasorelaxation, indicating a partial requirement of H2S in the vascular activity of NO (Coletta et al., 2012). Homocysteine also induces inducible NOS, reduces eNOS in endothelial cells and reduces bioavailability of NO through the formation of nitrotyrosine, which may exacerbate hyperhomocysteinemia‐associated hypertension (Sen et al., 2010). Collectively, these results suggest that NO and H2S may play mutually complementary roles in the physiological control of vascular tone. Although sulfhydration of some proteins by H2S appears to be a physiological determinant of transcriptional activity (Sen et al., 2012), no data currently exist on possible sulfhydration of eNOS after H2S treatment.
Treatment with a slow H2S releasing donor (named as ADTOH) has been shown to inhibit oxidative stress in retinal ganglion cells and increase haem oxygenase 1 (HO1), the main enzyme responsible for endogenous CO generation (Majid et al., 2013; Polhemus and Lefer, 2014). Constitutively, produced CO inhibits CBS physiologically (Kajimura et al., 2010) and hypoxia diminishes CO generation and thereby leads to increasing H2S which mediate the vasodilatation of precapillary arterioles, suggesting that hypoxic regulation of the cerebral microcirculation is mediated by a CO‐sensitive H2S pathway (Morikawa et al., 2012). Reducing CO levels in Brown‐Norway rats increases H2S generation, restores O2 sensing and prevents hypoxia‐induced pulmonary oedema. Increasing CO levels in SHR has been found to enhance carotid H2S generation, prevent hypersensitivity to hypoxia and control hypertension in SHR (Peng et al., 2014). H2S has also been demonstrated to be an effective and specific novel therapy for acute CO poisoning (Yu et al., 2011). Collectively, these data provide evidence for H2S‐CO cross‐talk. Nevertheless, the vast majority of biologically produced CO is exhaled via the lungs, and inhalation of H2S is very difficult to achieve in practice, therefore, an in‐depth study of their interaction is problematic.
Suppression of oxidative stress
The application of H2S may trigger a number of protective actions through its antioxidative effects. Moreover, redox‐sensitive signalling pathways play an important role in hypertension (Shao et al., 2012). NaHS can elicit vasoprotection by both scavenging O2 − and reducing vascular NADPH oxidase‐derived O2 − production, in an acute oxidative stress model with xanthine oxidase or with the O2 − generator pyrogallol (Al‐Magableh et al., 2014). H2S has also been found to inhibit H2O2‐mediated mitochondrial dysfunction in human endothelial cells by preserving the activities and protein expression levels of the antioxidant enzymes superoxide dismutase, catalase, glutathione peroxidase and glutathione‐S‐transferase (Wen et al., 2013). The protective effect of H2S on endothelial cells in the presence of high glucose may also involve an antioxidative stress mechanism (Guan et al., 2012). In cultured H9C2 myoblasts, exogenous H2S exerts a protective effect against H2O2‐induced or high glucose‐induced cell injury by inhibiting activation of the p38 MAPK and ERK1/2 pathways and preventing oxidative stress (Szabo et al., 2011; Xu et al., 2013). As a strong reducing agent, the above‐mentioned antioxidative effects of H2S in vitro are to a large extent predictable. Additionally, in vivo studies have found that H2S acts as an antioxidant in the context of oxidative stress associated with hypoxic PHT and the mechanism appears to be partly through attenuating cellular content of oxidized glutathione (Wei et al., 2007). Treatment with NaHS decreases BP and oxidative stress in SHR, and combined NaHS and tempol therapy in SHR decreases BP to a greater extent (Ahmad et al., 2014). GYY4137 decreases superoxide generation in aorta of high‐fat‐fed apoE−/− mice, while CSE‐knockout mice fed with atherogenic diet exhibit increased lesional oxidative stress (Liu et al., 2013; Mani et al., 2013). Zhou et al. found that H2S protects against chronic alcohol‐induced left ventricular remodelling via attenuating oxidative stress (Zhou et al., 2013). Another group showed that chronic NaHS treatment for 3 months prevents hypertrophy of intramyocardial arterioles and ventricular fibrosis, as well as decreases myocardial reactive oxygen species in SHR (Shi et al., 2007). These findings collectively suggest that H2S decreases BP and suppresses oxidative stress; however, whether the antioxidative ability of H2S in itself is important in reducing the BP in concert with other BP‐lowering actions needs to be further investigated.
Research on H2S and hypertension in China
Professor Chaoshu Tang in Peking University was the first to study the effects of H2S on hypertension in China. His group were the first to find, in 2003, that a lack of endogenous H2S was involved in the pathogenesis of hypoxic PHT and that exogenous H2S could exert a beneficial effect in this context (Chunyu et al., 2003). Subsequently, other groups found that exogenous H2S effectively prevents the development of different forms of hypertension, such as L‐NAME‐induced hypertension, SHR, high blood flow‐induced PHT and portal hypertension (Zhong et al., 2003; Yan et al., 2004; Yanfei et al., 2006; Shi et al., 2007; Tan et al., 2011). They proposed several possible mechanisms, such as vasorelaxation, induction of apoptosis of pulmonary artery SMCs, attenuation of Ang II‐induced AT1 receptor activation and inhibition of oxidative stress (Shi et al., 2007; Zhao et al., 2008; Fang et al., 2009; Li et al., 2009). An important breakthrough occurred with the finding of Zhang et al. that H2S also prevents H2O2‐induced activation of epithelial sodium channels, through which sodium can be reabsorbed in the distal nephron to regulate salt‐sensitive hypertension through a PTEN(phosphatidylinositol 3,4,5‐trisphosphate)‐dependent pathway (Zhang et al., 2013). Their findings raise the possibility that the effect of H2S on epithelial sodium channel activity seen in animal models may further be exploited clinically for the management of salt‐sensitive hypertension. A major focus of future work on H2S in China will be on discovery of novel drugs targeting the H2S pathway and on subsequent clinical translation.
Concluding remarks and future perspectives
Over the last few decades, significant progress has been achieved in delineating the antihypertensive effect and molecular mechanisms underlying the actions of H2S (Figure 1). However, several questions remain to be answered. The precise mechanisms underpinning H2S‐induced vasodilatation need to be better delineated. Moreover, some studies suggest that H2S can in fact exert vasoconstrictor effects (Koenitzer et al., 2007; Polhemus and Lefer, 2014). This apparent discrepancy needs further investigation to specifically determine whether this depends on factors such as concentration of H2S, vascular bed or the oxygen tension. The precise biological roles of H2S in amelioration of oxidative stress also remain unclear. Moreover, the vasodilator actions of H2S may be a result, at least in part, of eNOS‐generated NO promoted by H2S signalling. However, the exact manner of cross‐talk between H2S and NO is incompletely understood and deserves to be better elucidated; an improved understanding of how these two molecules cooperate will allow better design of clinically useful therapies. Finally, precisely how CSE activity is regulated at the post‐translational level needs to be better defined.
Figure 1.
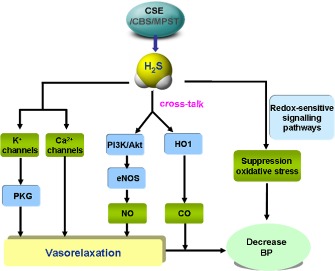
Schematic illustration of possible mechanisms that may underlie H2S‐induced BP lowering. H2S lowers BP via vasodilatation by activation of vascular KATP channels and/or inhibiting Ca2+ influx via Ca2+ channels. NO and H2S share cross‐talk regulatory roles in vasorelaxation via the PI3K/Akt‐eNOS‐NO pathway. H2S increase haem oxygenase 1 (HO1), which is the main enzyme for CO generation. H2S inhibits reactive oxygen species (ROS) production through redox‐sensitive signalling pathways.
Clinically, it remains to be determined whether the research findings described earlier can be translated to practice for the management of hypertension and other related cardiovascular diseases; and most importantly, whether H2S releasing agents and CSE/H2S activators will prevent or treat hypertension and its complications. As well as such efficacy considerations, the safety of such agents needs to be rigorously confirmed, as well as the dose relationship of any toxic effects.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by grants from the National Basic Research Program of China (973) (grant no 2012CB517503), the National Natural Science Foundation of China (grant nos. 81170083, 81330004, 31371156), the Collaborative Innovation Center for Cardiovascular Disease Translational Medicine and PAPD.
References
- Ahmad FU, Sattar MA, Rathore HA, Tan YC, Akhtar S, Jin OH et al (2014). Hydrogen sulphide and tempol treatments improve the blood pressure and renal excretory responses in spontaneously hypertensive rats. Ren Fail 36: 598–605. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013a). The Concise Guide to PHARMACOLOGY 2013/14: G Protein‐Coupled Receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013b). The Concise Guide to PHARMACOLOGY 2013/14: Ligand‐gated ion channels. Br J Pharmacol 170: 1582–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013c). The Concise Guide to PHARMACOLOGY 2013/14: Ion channels. Br J Pharmacol 170: 1607–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013d). The Concise Guide to PHARMACOLOGY 2013/14: G Protein‐Coupled Receptors. Br J Pharmacol 170: 1676–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013e). The Concise Guide to PHARMACOLOGY 2013/14: G Protein‐Coupled Receptors. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Magableh MR, Kemp‐Harper BK, Ng HH, Miller AA, Hart JL (2014). Hydrogen sulfide protects endothelial nitric oxide function under conditions of acute oxidative stress in vitro . Naunyn Schmiedebergs Arch Pharmacol 387: 67–74. [DOI] [PubMed] [Google Scholar]
- Altaany Z, Yang G, Wang R (2013). Crosstalk between hydrogen sulfide and nitric oxide in endothelial cells. J Cell Mol Med 17: 879–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariyaratnam P, Loubani M, Morice AH (2013). Hydrogen sulphide vasodilates human pulmonary arteries: a possible role in pulmonary hypertension? Microvasc Res 90: 135–137. [DOI] [PubMed] [Google Scholar]
- Baskar R, Li L, Moore PK (2007). Hydrogen sulfide‐induces DNA damage and changes in apoptotic gene expression in human lung fibroblast cells. FASEB J 21: 247–255. [DOI] [PubMed] [Google Scholar]
- Bos EM, van Goor H, Joles JA, Whiteman M, Leuvenink HG (2014). Hydrogen sulfide – physiological properties and therapeutic potential in ischaemia. Br J Pharmacol. doi: 10.1111/bph.12869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci M, Papapetropoulos A, Vellecco V, Zhou Z, Zaid A, Giannogonas P et al (2012). cGMP‐dependent protein kinase contributes to hydrogen sulfide‐stimulated vasorelaxation. PLoS ONE 7: e53319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro‐Piedras I, Perez‐Zoghbi JF (2013). Hydrogen sulphide inhibits Ca2+ release through InsP3 receptors and relaxes airway smooth muscle. J Physiol 591 (Pt 23): 5999–6015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheang WS, Wong WT, Shen B, Lau CW, Tian XY, Tsang SY et al (2010). 4‐aminopyridine‐sensitive K+ channels contributes to NaHS‐induced membrane hyperpolarization and relaxation in the rat coronary artery. Vascul Pharmacol 53: 94–98. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Ndisang JF, Tang G, Cao K, Wang R (2004). Hydrogen sulfide‐induced relaxation of resistance mesenteric artery beds of rats. Am J Physiol Heart Circ Physiol 287: H2316–H2323. [DOI] [PubMed] [Google Scholar]
- Chitnis MK, Njie‐Mbye YF, Opere CA, Wood ME, Whiteman M, Ohia SE (2013). Pharmacological actions of the slow release hydrogen sulfide donor GYY4137 on phenylephrine‐induced tone in isolated bovine ciliary artery. Exp Eye Res 116: 350–354. [DOI] [PubMed] [Google Scholar]
- Chunyu Z, Junbao D, Dingfang B, Hui Y, Xiuying T, Chaoshu T (2003). The regulatory effect of hydrogen sulfide on hypoxic pulmonary hypertension in rats. Biochem Biophys Res Commun 302: 810–816. [DOI] [PubMed] [Google Scholar]
- Cindrova‐Davies T, Herrera EA, Niu Y, Kingdom J, Giussani DA, Burton GJ (2013). Reduced cystathionine gamma‐lyase and increased miR‐21 expression are associated with increased vascular resistance in growth‐restricted pregnancies: hydrogen sulfide as a placental vasodilator. Am J Pathol 182: 1448–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Módis K, Panopoulos P et al (2012). Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium‐dependent vasorelaxation. Proc Natl Acad Sci U S A 109: 9161–9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G, Félétou M, Weston AH (2012). Hydrogen sulfide as an endothelium‐derived hyperpolarizing factor in rodent mesenteric arteries. Circ Res 110: e13–e14, author reply e15–e16. [DOI] [PubMed] [Google Scholar]
- Faller S, Ryter SW, Choi AM, Loop T, Schmidt R, Hoetzel A (2010). Inhaled hydrogen sulfide protects against ventilator‐induced lung injury. Anesthesiology 113: 104–115. [DOI] [PubMed] [Google Scholar]
- Faller S, Zimmermann KK, Strosing KM, Engelstaedter H, Buerkle H, Schmidt R et al (2012). Inhaled hydrogen sulfide protects against lipopolysaccharide‐induced acute lung injury in mice. Med Gas Res 2: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Zhao J, Chen Y, Ma T, Xu G, Tang C et al (2009). Hydrogen sulfide derived from periadventitial adipose tissue is a vasodilator. J Hypertens 27: 2174–2185. [DOI] [PubMed] [Google Scholar]
- Fiorucci S, Antonelli E, Mencarelli A, Orlandi S, Renga B, Rizzo G et al (2005). The third gas: H2S regulates perfusion pressure in both the isolated and perfused normal rat liver and in cirrhosis. Hepatology 42: 539–548. [DOI] [PubMed] [Google Scholar]
- Francis RC, Vaporidi K, Bloch KD, Ichinose F, Zapol WM (2011). Protective and detrimental effects of sodium sulfide and hydrogen sulfide in murine ventilator‐induced lung injury. Anesthesiology 115: 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin LR, Francom D, Dieken FP, Taylor JD, Warenycia MW, Reiffenstein RJ et al (1989). Determination of sulfide in brain tissue by gas dialysis/ion chromatography: postmortem studies and two case reports. J Anal Toxicol 13: 105–109. [DOI] [PubMed] [Google Scholar]
- Guan Q, Zhang Y, Yu C, Liu Y, Gao L, Zhao J (2012). Hydrogen sulfide protects against high‐glucose‐induced apoptosis in endothelial cells. J Cardiovasc Pharmacol 59: 188–193. [DOI] [PubMed] [Google Scholar]
- Gupta S, Melnyk SB, Kruger WD (2014). Cystathionine beta‐synthase‐deficient mice thrive on a low‐methionine diet. FASEB J 28: 781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Li X, Zhou S, Meng G, Xiao Y, Zhang W et al (2012). 17β‐estradiol antagonizes the down‐regulation of ERalpha/NOS‐3 signaling in vascular endothelial dysfunction of female diabetic rats. PLoS ONE 7: e50402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holwerda KM, Bos EM, Rajakumar A, Ris‐Stalpers C, van Pampus MG, Timmer A et al (2012). Hydrogen sulfide producing enzymes in pregnancy and preeclampsia. Placenta 33: 518–521. [DOI] [PubMed] [Google Scholar]
- Holwerda KM, Burke SD, Faas MM, Zsengeller Z, Stillman IE, Kang PM et al (2014). Hydrogen sulfide attenuates VEGFR‐1‐induced hypertension and renal damage by upregulating vascular endothelial growth factor. J Am Soc Nephrol 25: 717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii I, Akahoshi N, Yamada H, Nakano S, Izumi T, Suematsu M (2010). Cystathionine gamma‐Lyase‐deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J Biol Chem 285: 26358–26368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura M, Fukuda R, Bateman RM, Yamamoto T, Suematsu M (2010). Interactions of multiple gas‐transducing systems: hallmarks and uncertainties of CO, NO, and H2S gas biology. Antioxid Redox Signal 13: 157–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida M, Sugiyama T, Yoshimoto T, Ogawa Y (2013). Hydrogen sulfide increases nitric oxide production with calcium‐dependent activation of endothelial nitric oxide synthase in endothelial cells. Eur J Pharm Sci 48: 211–215. [DOI] [PubMed] [Google Scholar]
- King AL, Polhemus DJ, Bhushan S, Otsuka H, Kondo K, Nicholson CK et al (2014). Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase‐nitric oxide dependent. Proc Natl Acad Sci U S A 111: 3182–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenitzer JR, Isbell TS, Patel HD, Benavides GA, Dickinson DA, Patel RP et al (2007). Hydrogen sulfide mediates vasoactivity in an O2‐dependent manner. Am J Physiol Heart Circ Physiol 292: H1953–H1960. [DOI] [PubMed] [Google Scholar]
- Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S et al (2013). H(2)S protects against pressure overload‐induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation 127: 1116–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Whiteman M, Guan YY, Neo KL, Cheng Y, Lee SW et al (2008a). Characterization of a novel, water‐soluble hydrogen sulfide‐releasing molecule (GYY4137): new insights into the biology of hydrogen sulfide. Circulation 117: 2351–2360. [DOI] [PubMed] [Google Scholar]
- Li W, Jin HF, Liu D, Sun JH, Jian PJ, Li XH et al (2009). Hydrogen sulfide induces apoptosis of pulmonary artery smooth muscle cell in rats with pulmonary hypertension induced by high pulmonary blood flow. Chin Med J 122: 3032–3038. [PubMed] [Google Scholar]
- Li X, Du J, Jin H, Geng B, Tang C (2008b). Sodium hydrosulfide alleviates pulmonary artery collagen remodeling in rats with high pulmonary blood flow. Heart Vessels 23: 409–419. [DOI] [PubMed] [Google Scholar]
- Li Y, Zang Y, Fu S, Zhang H, Gao L, Li J (2012). H2S relaxes vas deferens smooth muscle by modulating the large conductance Ca2+‐activated K+ (BKCa) channels via a redox mechanism. J Sex Med 9: 2806–2813. [DOI] [PubMed] [Google Scholar]
- Liu YH, Yan CD, Bian JS (2011). Hydrogen sulfide: a novel signaling molecule in the vascular system. J Cardiovasc Pharmacol 58: 560–569. [DOI] [PubMed] [Google Scholar]
- Liu Z, Han Y, Li L, Lu H, Meng G, Li X et al (2013). The hydrogen sulfide donor, GYY4137, exhibits anti‐atherosclerotic activity in high fat fed apolipoprotein E(−/−) mice. Br J Pharmacol 169: 1795–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Liu YH, Goh HS, Wang JJ, Yong QC, Wang R et al (2010). Hydrogen sulfide inhibits plasma renin activity. J Am Soc Nephrol 21: 993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucock M, Yates Z, Martin C, Choi JH, Boyd L, Tang S et al (2013). Hydrogen sulphide‐related thiol metabolism and nutrigenetics in relation to hypertension in an elderly population. Genes Nutr 8: 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Liu D, Tang C, Du J, Liu AD, Holmberg L et al (2013). Sulfur dioxide upregulates the inhibited endogenous hydrogen sulfide pathway in rats with pulmonary hypertension induced by high pulmonary blood flow. Biochem Biophys Res Commun 433: 519–525. [DOI] [PubMed] [Google Scholar]
- Majid AS, Majid AM, Yin ZQ, Ji D (2013). Slow regulated release of H2S inhibits oxidative stress induced cell death by influencing certain key signaling molecules. Neurochem Res 38: 1375–1393. [DOI] [PubMed] [Google Scholar]
- Mani S, Li H, Untereiner A, Wu L, Yang G, Austin RC et al (2013). Decreased endogenous production of hydrogen sulfide accelerates atherosclerosis. Circulation 127: 2523–2534. [DOI] [PubMed] [Google Scholar]
- Mani S, Untereiner A, Wu L, Wang R (2014). Hydrogen sulfide and the pathogenesis of atherosclerosis. Antioxid Redox Signal 20: 805–817. [DOI] [PubMed] [Google Scholar]
- Mi Q, Chen N, Shaifta Y, Xie L, Lu H, Liu Z et al (2011). Activation of endothelial nitric oxide synthase is dependent on its interaction with globular actin in human umbilical vein endothelial cells. J Mol Cell Cardiol 51: 419–427. [DOI] [PubMed] [Google Scholar]
- Morikawa T, Kajimura M, Nakamura T, Hishiki T, Nakanishi T, Yukutake Y et al (2012). Hypoxic regulation of the cerebral microcirculation is mediated by a carbon monoxide‐sensitive hydrogen sulfide pathway. Proc Natl Acad Sci U S A 109: 1293–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara N (2013). Regulation of mercaptopyruvate sulfurtransferase activity via intrasubunit and intersubunit redox‐sensing switches. Antioxid Redox Signal 19: 1792–1802. [DOI] [PubMed] [Google Scholar]
- Papapetropoulos A, Whiteman M, Giuseppe C (2014). Pharmacological tools for hydrogen sulfide research: a brief, introductory guide for beginners. Br J Pharmacol. doi: 10.1111/bph.12806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson NA, Christofi G, Obrenovitch TP, Symon L (1988). Simple staining for heat‐fixed brain sections after topographical pH analysis or autoradiographic studies. J Cereb Blood Flow Metab 8: 883–885. [DOI] [PubMed] [Google Scholar]
- Patel P, Vatish M, Heptinstall J, Wang R, Carson RJ (2009). The endogenous production of hydrogen sulphide in intrauterine tissues. Reprod Biol Endocrinol 7: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al; NC‐IUPHAR (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Makarenko VV, Nanduri J, Vasavda C, Raghuraman G, Yuan G et al (2014). Inherent variations in CO‐H2S‐mediated carotid body O2 sensing mediate hypertension and pulmonary edema. Proc Natl Acad Sci U S A 111: 1174–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polhemus DJ, Lefer DJ (2014). Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ Res 114: 730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polhemus DJ, Kondo K, Bhushan S, Bir SC, Kevil CG, Murohara T et al (2013). Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ Heart Fail 6: 1077–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Predmore BL, Julian D, Cardounel AJ (2011). Hydrogen sulfide increases nitric oxide production from endothelial cells by an akt‐dependent mechanism. Front Physiol 2: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid S, Heer JK, Garle MJ, Alexander SP, Roberts RE (2013). Hydrogen sulphide‐induced relaxation of porcine peripheral bronchioles. Br J Pharmacol 168: 1902–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Khan AH, Islam MT, Prieto MC, Majid DS (2012). Interdependency of cystathione gamma‐lyase and cystathione beta‐synthase in hydrogen sulfide‐induced blood pressure regulation in rats. Am J Hypertens 25: 74–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleifenbaum J, Köhn C, Voblova N, Dubrovska G, Zavarirskaya O, Gloe T et al (2010). Systemic peripheral artery relaxation by KCNQ channel openers and hydrogen sulfide. J Hypertens 28: 1875–1882. [DOI] [PubMed] [Google Scholar]
- Sen N, Paul BD, Gadalla MM, Mustafa AK, Sen T, Xu R et al (2012). Hydrogen sulfide‐linked sulfhydration of NF‐kappaB mediates its antiapoptotic actions. Mol Cell 45: 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen U, Mishra PK, Tyagi N, Tyagi SC (2010). Homocysteine to hydrogen sulfide or hypertension. Cell Biochem Biophys 57: 49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao D, Oka S, Brady CD, Haendeler J, Eaton P, Sadoshima J (2012). Redox modification of cell signaling in the cardiovascular system. J Mol Cell Cardiol 52: 550–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi YX, Chen Y, Zhu YZ, Huang GY, Moore PK, Huang SH et al (2007). Chronic sodium hydrosulfide treatment decreases medial thickening of intramyocardial coronary arterioles, interstitial fibrosis, and ROS production in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 293: H2093–H2100. [DOI] [PubMed] [Google Scholar]
- Shirozu K, Tokuda K, Marutani E, Lefer D, Wang R, Ichinose F (2014). Cystathionine gamma‐lyase deficiency protects mice from galactosamine/lipopolysaccharide‐induced acute liver failure. Antioxid Redox Signal 20: 204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder PM, Frenay AS, de Boer RA, Pasch A, Hillebrands J, Leuvenink HG et al (2014). Exogenous administration of thiosulfate, a donor of hydrogen sulfide, attenuates Angiotensin II‐induced hypertensive heart disease in rats. Br J Pharmacol. doi: 10.1111/bph.12825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streeter E, Hart J, Badoer E (2012). An investigation of the mechanisms of hydrogen sulfide‐induced vasorelaxation in rat middle cerebral arteries. Naunyn Schmiedebergs Arch Pharmacol 385: 991–1002. [DOI] [PubMed] [Google Scholar]
- Sun L, Sun S, Li Y, Pan W, Xie Y, Wang S et al (2014). Potential biomarkers predicting risk of pulmonary hypertension in congenital heart disease: the role of homocysteine and hydrogen sulfide. Chin Med J 127: 893–899. [PubMed] [Google Scholar]
- Sun NL, Xi Y, Yang SN, Ma Z, Tang CS (2007). [Plasma hydrogen sulfide and homocysteine levels in hypertensive patients with different blood pressure levels and complications]. Zhonghua Xin Xue Guan Bing Za Zhi 35: 1145–1148. [PubMed] [Google Scholar]
- Szabo G, Veres G, Radovits T, Gero D, Modis K, Miesel‐Groschel C et al (2011). Cardioprotective effects of hydrogen sulfide. Nitric Oxide 25: 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan G, Pan S, Li J, Dong X, Kang K, Zhao M et al (2011). Hydrogen sulfide attenuates carbon tetrachloride‐induced hepatotoxicity, liver cirrhosis and portal hypertension in rats. PLoS ONE 6: e25943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Wu L, Liang W, Wang R (2005). Direct stimulation of K(ATP) channels by exogenous and endogenous hydrogen sulfide in vascular smooth muscle cells. Mol Pharmacol 68: 1757–1764. [DOI] [PubMed] [Google Scholar]
- Tang G, Yang G, Jiang B, Ju Y, Wu L, Wang R (2013). H2S is an endothelium‐derived hyperpolarizing factor. Antioxid Redox Signal 19: 1634–1646. [DOI] [PubMed] [Google Scholar]
- Tian XY, Wong WT, Sayed N, Luo J, Tsang SY, Bian ZX et al (2012). NaHS relaxes rat cerebral artery in vitro via inhibition of l‐type voltage‐sensitive Ca2+ channel. Pharmacol Res 65: 239–246. [DOI] [PubMed] [Google Scholar]
- Toombs CF, Insko MA, Wintner EA, Deckwerth TL, Usansky H, Jamil K et al (2010). Detection of exhaled hydrogen sulphide gas in healthy human volunteers during intravenous administration of sodium sulphide. Br J Clin Pharmacol 69: 626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vadivel A, Alphonse RS, Ionescu L, Machado DS, O'Reilly M, Eaton F et al (2014). Exogenous hydrogen sulfide (H2S) protects alveolar growth in experimental O2‐induced neonatal lung injury. PLoS ONE 9: e90965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Han J, Xiao L, Jin CE, Li DJ, Yang Z (2014). Role of hydrogen sulfide in portal hypertension and esophagogastric junction vascular disease. World J Gastroenterol 20: 1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Ahmad S, Cai M, Rennie J, Fujisawa T, Crispi F et al (2013). Dysregulation of hydrogen sulfide producing enzyme cystathionine gamma‐lyase contributes to maternal hypertension and placental abnormalities in preeclampsia. Circulation 127: 2514–2522. [DOI] [PubMed] [Google Scholar]
- Wang L, Chen X, Tang B, Hua X, Klein‐Szanto A, Kruger WD (2005). Expression of mutant human cystathionine beta‐synthase rescues neonatal lethality but not homocystinuria in a mouse model. Hum Mol Genet 14: 2201–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R (2011). Signaling pathways for the vascular effects of hydrogen sulfide. Curr Opin Nephrol Hypertens 20: 107–112. [DOI] [PubMed] [Google Scholar]
- Wang R (2012). Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev 92: 791–896. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Osada J, Aratani Y, Kluckman K, Reddick R, Malinow MR et al (1995). Mice deficient in cystathionine beta‐synthase: animal models for mild and severe homocyst(e)inemia. Proc Natl Acad Sci U S A 92: 1585–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei HL, Du JB, Tang CS (2007). Effect of hydrogen sulfide on oxidative stress in hypoxic pulmonary hypertension. Beijing Da Xue Xue Bao 39: 565–569. [PubMed] [Google Scholar]
- Wei HL, Zhang CY, Jin HF, Tang CS, Du JB (2008). Hydrogen sulfide regulates lung tissue‐oxidized glutathione and total antioxidant capacity in hypoxic pulmonary hypertensive rats. Acta Pharmacol Sin 29: 670–679. [DOI] [PubMed] [Google Scholar]
- Wen YD, Wang H, Kho SH, Rinkiko S, Sheng X, Shen HM et al (2013). Hydrogen sulfide protects HUVECs against hydrogen peroxide induced mitochondrial dysfunction and oxidative stress. PLoS ONE 8: e53147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Liu Z, Lu H, Zhang W, Mi Q, Li X et al (2012). Pyridoxine inhibits endothelial NOS uncoupling induced by oxidized low‐density lipoprotein via the PKCalpha signalling pathway in human umbilical vein endothelial cells. Br J Pharmacol 165: 754–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Wu W, Chen J, Guo R, Lin J, Liao X et al (2013). Exogenous hydrogen sulfide protects H9c2 cardiac cells against high glucose‐induced injury by inhibiting the activities of the p38 MAPK and ERK1/2 pathways. Int J Mol Med 32: 917–925. [DOI] [PubMed] [Google Scholar]
- Yan H, Du J, Tang C (2004). The possible role of hydrogen sulfide on the pathogenesis of spontaneous hypertension in rats. Biochem Biophys Res Commun 313: 22–27. [DOI] [PubMed] [Google Scholar]
- Yanfei W, Lin S, Junbao D, Chaoshu T (2006). Impact of L‐arginine on hydrogen sulfide/cystathionine‐gamma‐lyase pathway in rats with high blood flow‐induced pulmonary hypertension. Biochem Biophys Res Commun 345: 851–857. [DOI] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K et al (2008). H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma‐lyase. Science 322: 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Zhao K, Ju Y, Mani S, Cao Q, Puukila S et al (2013). Hydrogen sulfide protects against cellular senescence via S‐sulfhydration of Keap1 and activation of Nrf2. Antioxid Redox Signal 18: 1906–1919. [DOI] [PubMed] [Google Scholar]
- Yang Y, Zhang BK, Liu D, Nie W, Yuan JM, Wang Z et al (2012). Sodium hydrosulfide prevents hypoxia‐induced pulmonary arterial hypertension in broilers. Br Poult Sci 53: 608–615. [DOI] [PubMed] [Google Scholar]
- Yu YP, Li ZG, Wang DZ, Zhan X, Shao JH (2011). Hydrogen sulfide as an effective and specific novel therapy for acute carbon monoxide poisoning. Biochem Biophys Res Commun 404: 6–9. [DOI] [PubMed] [Google Scholar]
- Zhang J, Chen S, Liu H, Zhang B, Zhao Y, Ma K et al (2013). Hydrogen sulfide prevents hydrogen peroxide‐induced activation of epithelial sodium channel through a PTEN/PI(3,4,5)P3 dependent pathway. PLoS ONE 8: e64304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Wang R (2002). H(2)S‐induced vasorelaxation and underlying cellular and molecular mechanisms. Am J Physiol Heart Circ Physiol 283: H474–H480. [DOI] [PubMed] [Google Scholar]
- Zhao W, Zhang J, Lu Y, Wang R (2001). The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J 20: 6008–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Zhang LK, Zhang CY, Zeng XJ, Yan H, Jin HF et al (2008). Regulatory effect of hydrogen sulfide on vascular collagen content in spontaneously hypertensive rats. Hypertens Res 31: 1619–1630. [DOI] [PubMed] [Google Scholar]
- Zhong G, Chen F, Cheng Y, Tang C, Du J (2003). The role of hydrogen sulfide generation in the pathogenesis of hypertension in rats induced by inhibition of nitric oxide synthase. J Hypertens 21: 1879–1885. [DOI] [PubMed] [Google Scholar]
- Zhou X, Lu X, Xu W, Chen J (2013). Protective effects of hydrogen sulfide against chronic alcohol intake‐induced left ventricular remodeling in rats. Cardiovasc Drugs Ther 27: 221–227. [DOI] [PubMed] [Google Scholar]