

Abstract
Background and Purpose
High‐salt diet induces cardiac remodelling and leads to heart failure, which is closely related to cardiac mitochondrial dysfunction. Transient receptor potential (TRP) channels are implicated in the pathogenesis of cardiac dysfunction. We investigated whether activation of TRP vanilloid (subtype 1) (TRPV1) channels by dietary capsaicin can, by ameliorating cardiac mitochondrial dysfunction, prevent high‐salt diet‐induced cardiac hypertrophy.
Experimental Approach
Male wild‐type (WT) and TRPV1 −/− mice were fed a normal or high‐salt diet with or without capsaicin for 6 months. Their cardiac parameters and endurance capacity were assessed. Mitochondrial respiration and oxygen consumption were measured using high‐resolution respirometry. The expression levels of TRPV1, sirtuin 3 and NDUFA9 were detected in cardiac cells and tissues.
Key Results
Chronic high‐salt diet caused cardiac hypertrophy and reduced physical activity in mice; both effects were ameliorated by capsaicin intake in WT but not in TRPV1 −/− mice. TRPV1 knockout or high‐salt diet significantly jeopardized the proficiency of mitochondrial Complex I oxidative phosphorylation (OXPHOS) and reduced Complex I enzyme activity. Chronic dietary capsaicin increased cardiac mitochondrial sirtuin 3 expression, the proficiency of Complex I OXPHOS, ATP production and Complex I enzyme activity in a TRPV1‐dependent manner.
Conclusions and Implications
TRPV1 activation by dietary capsaicin can antagonize high‐salt diet‐mediated cardiac lesions by ameliorating its deleterious effect on the proficiency of Complex I OXPHOS. TRPV1‐mediated amendment of mitochondrial dysfunction may represent a novel target for management of early cardiac dysfunction.
Linked Articles
This article is part of a themed section on Chinese Innovation in Cardiovascular Drug Discovery. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2015.172.issue-23
Abbreviations
- AngII
angiotensin II
- CI
complex I
- CII
complex II
- CS
citrate synthase
- EF
ejection fraction
- ETC
electronic transport chain
- FS
shortening fraction
- HS
high‐salt diet
- iRTX
5′‐iodoresiniferatoxin
- LVH
left ventricular hypertrophy
- LVID
left ventricular internal diameter
- LVPW
left ventricular posterior wall
- ND
normal‐salt diet
- OXPHOS
oxidative phosphorylation
- RER
respiration exchange ratio
- RES
resveratrol
- ROS
reactive oxygen species
- TRPCs
transient potential receptor channels
- TRPV1
transient receptor potential vanilloid subfamily member 1
- TRPV1−/−
TRPV1 knockout
- WT
wild‐type
Tables of Links
| LIGANDS | |
|---|---|
| 5′‐iodoresiniferatoxin | Capsaicin |
| Angiotensin II | Hydrogen peroxide |
| ATP | NADH |
| Baicalein | Resveratrol |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a, 2013b).
Introduction
Cardiac hypertrophy is the cellular response to pathological stress and is associated with a thickening of ventricular walls (Frey and Olson, 2003). Prolonged cardiac hypertrophy leads to heart failure (Kemp et al., 2012). Cardiac hypertrophy can be caused by hypertension, obesity and type 2 diabetes or an unhealthy lifestyle such as a high salt intake (Palmieri et al., 2003; Burnier et al., 2007; Schmieder et al., 2007). There is plenty of evidence suggesting that cardiac hypertrophy progresses as a result of sympathetic nerve activation and stimulation of the renin‐angiotensin system (Kemp and Conte, 2012). However, the importance of Ca2+ channels in the development of cardiac remodelling and the regulation of cardiac function is also well documented (Zhang and Brown, 2004). In addition to the roles for ion channels, such as the sarcoplasmic reticulum calcium release channels, a non‐selective cation channel, the transient potential receptor channels (TRPCs) have also been implicated in the development of cardiac hypertrophy (Eder and Molkentin, 2011; Beech, 2013). Both TRPC3 and TRPC6 contribute to angiotensin II (Ang II)‐induced cardiac hypertrophy, leading to diacylglycerol production and extracellular calcium influx (Onohara et al., 2006). In contrast, our studies have shown that TRP vanilloid subfamily member 1 (TRPV1) activation lowers high BP by increasing vascular PKA/endothelial NOS phosphorylation and inducing NO production in genetically hypertensive rats as well as reducing the production of vascular reactive oxygen species (ROS) in high‐salt (HS) diet‐induced hypertensive mice (Yang et al., 2010; Hao et al., 2011).
In addition, findings from both clinical and experimental studies suggest that dysfunction of mitochondrial bioenergetics may contribute to the development of cardiac hypertrophy (Rosca and Hoppel, 2010). ATP production through mitochondrial respiration is the major source of cardiac energy (Abel and Doenst, 2011). Damage to mitochondrial respiratory enzymes can cause a malfunction of the electronic transport chain (ETC) and reduce ATP production, which eventually impairs cardiac contractility. There is substantial evidence showing that oxidative stress levels are elevated in both cardiac hypertrophy and failure (Seddon et al., 2007; Takimoto and Kass, 2007; Tsutsui et al., 2009). Sirtuins including sirtuins 1–7 are quite conserved NAD+‐dependent deacetylases and ADP‐ribosyltransferases. Sirtuin 3 catalyzes NAD+‐dependent substrate‐related protein deacetylation (Pillai et al., 2010). Mitochondrial sirtuin 3 can directly increase ROS clearance by the deacetylation of Mn‐SOD (Tao et al., 2014). Sirtuin 3 was also found to be involved in basal ATP maintenance through its regulation of the acetylation and Complex I (CI) activity of the ETC (Ahn et al., 2008). Mitochondrial CI dysfunction leads to the accumulation of NADH and a decrease in the NAD+/NADH ratio thereby inhibiting sirtuin 3 activity (Karamanlidis et al., 2013).
Current strategies for the treatment of cardiac failure include both lifestyle modifications and drug therapies. Patients with the early stages of cardiac failure only have risk factors for structural heart disease but no clinical symptoms (Hunt et al., 2005). Excessive salt intake is closely associated with left ventricular hypertrophy (LVH); furthermore, long‐term moderate sodium restriction decreases LVH in patients with early stage cardiac failure (Jula and Karanko, 1994). Therefore, restricting or ameliorating the effect of a high salt intake on the heart is very important for preventing the development of cardiac failure. Furthermore, it is worth investigating whether a high salt intake can directly jeopardize the function of cardiac mitochondria during cardiac hypertrophy.
In addition to pharmacotherapy for the management of cardiac failure, several dietary factors have been shown to exert protective effects on the heart in animal studies (Baccarelli et al., 2008; Ma et al., 2011; Xu et al., 2011). Baicalein, a flavonoid in the root of Scutellaria baicalensis, was found to prevent cardiac hypertrophy by inhibiting MEK‐ERK1/2 signalling (Zong et al., 2013). We showed that chronic capsaicin administration, the main pungent ingredient in hot pepper, prevented the development of obesity, improved glucose homeostasis and attenuated atherosclerosis, all through its ability to activate TRPV1 (Zhang et al., 2007; Ma et al., 2011; Wang et al., 2012). However, whether TRPV1 activation by dietary capsaicin has a beneficial effect on cardiac hypertrophy induced by high salt intake is still uncertain.
In this study, we sought to examine the hypothesis that TRPV1 activation by capsaicin prevents cardiac hypertrophy induced by high salt intake by improving cardiac mitochondrial function. Our results have shown for the first time that a dysfunction of mitochondrial Complex I has a prominent role in causing the cardiac impairments induced by a high salt intake. TRPV1 activation by chronic dietary capsaicin reversed cardiac hypertrophy by restoring the function of Complex I and by up‐regulating the expression of sirtuin 3.
Methods
Animals and study design
Male C57BL/6J wild‐type (WT) and TRPV1 knockout (TRPV1−/−) mice (strain information: B6.129X1‐Trpv1tm1Jul/J, stock number: 003770) were purchased from Jackson Laboratory (Bar Harbor, Maine, USA). Both types of male mice, aged 6–8 weeks, were randomly grouped and fed with a normal‐salt diet (NS, 0.5% NaCl by weight), high‐salt diet (HS, 8% NaCl by weight) or high‐salt plus capsaicin diet (HC, 8% NaCl and 0.01% capsaicin by weight) for 24 weeks, respectively (Hao et al., 2011; Ma et al., 2011). All of the mice were housed under a 12 h/12 h day/night cycle; chow and water were available ad libitum. The temperature of the room was kept at 20°C. A total of 36 TRPV1−/− mice and 36 age‐matched wild‐type WT mice (C57BL‐6J) were used for this experiment. Body weight, blood pressure, heart rate, running endurance and echocardiographic parameters were measured. All of the experimental procedures were performed in accordance with protocols approved by the Institutional Animal Care and Research Advisory Committee of the Third Military Medical University. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Changes in the heart and the expressions of its associated proteins were examined in cardiac tissues from these mice. Cardiac mitochondrial respiration and ATP production were also measured in mice fed a high salt diet with and without 0.01% capsaicin. Details of the experimental protocols are available in the online Supporting Information.
Cell culture and treatment
Embryonic rat‐heart‐derived H9C2 cells (Cell Bank, Chinese Academy of Sciences, Shanghai, China) were propagated in high‐glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% foetal bovine serum, 100 U ml−1 penicillin and 100 mg ml−1 streptomycin at 37°C. The medium was changed every 2 days. After incubation to near confluence, the H9C2 cells were then deprived of serum and incubated for another 24 h before treatment. The vehicle was ethanol and its final concentration was 0.2%.
These H9C2 cells were randomly divided into the following groups: (1) the control group (CON), in which cells were incubated in normal medium; (2) the HS group, in which cells were incubated in DMEM containing 10 mM sodium chloride; (3) the HC group, in which 1 μM capsaicin was added before exposure to 10 mM sodium chloride; (4) the iRTX+HC group, in which 1 μM 5′‐iodoresiniferatoxin (iRTX) was added before exposure to 10mM sodium chloride plus capsaicin; (5) the RES+HC group, in which 20 μM resveratrol (RES) was added before exposure to 10mM sodium chloride plus capsaicin; (6) the NAM+RES+HC group, in which 20 mM nicotinamide (NAM) was added before exposure to 10mM sodium chloride plus capsaicin and resveratrol; (7) the RES+HS group, in which 20 μM RES was added before exposure to 10mM sodium chloride; and (8) the NAM+RES+HS group, in which 20 mM NAM was added before exposure to 10mM sodium chloride and resveratrol.
Chemicals including capsaicin, iRTX, RES, and NAM were purchased from Sigma‐Aldrich Co. (St Louis, MO, USA). Dulbecco's modified Eagle's medium and foetal bovine serum were purchased from Gibco Co. (Grand Island, NY, USA).
These in vitro studies were used to determine the underlying mechanisms.
Data analysis
All data are expressed as means ± SEM. Experiments were carried out in triplicate. Data were evaluated with the SPSS 12.0 software (SPSS Inc, Chicago, IL, USA). The significance of differences between groups was analysed using Student's t‐test or one‐way anova; Tukey's multiple comparisons test was used as a post hoc test. P values less than 0.05 were considered statistically significant.
Results
Activation of TRPV1 prevented high salt intake‐induced cardiac hypertrophy and dysfunction
WT and TRPV1−/− mice on a HS diet for 24 weeks were found to have an elevated systolic BP and higher heart weight to body weight (HW/BW) ratio (Table 1). Echocardiography confirmed the impairment in cardiac structure and function in mice fed the HS diet (Figure 1A). The thicknesses of the diastolic left ventricular posterior wall (LVPWd) and anterior/interventricular septal wall (IVSd) were significantly increased in WT mice fed the HS diet compared with WT mice fed a normal‐salt diet (ND) (Figure 1B,C). A reduction in the % fractional shortening (FS) of the left ventricle indicated a cardiac dysfunction in mice fed a HS diet (Figure 1D). No differences were found in the left ventricular internal diameter during diastole or the % ejection fraction (EF) between WT mice with or without HS diet, although the EF tended to be smaller in the HS group (Figure 1E,F). Chronic dietary capsaicin for 24 weeks significantly reduced the HW/BW ratio, LVPWd, and IVSd and also increased FS in WT mice on a HS diet but not TRPV1−/− mice (Table 1, Figure 1A–D). Similarly, an increase in EF was also found in WT mice fed a HS diet plus capsaicin, but this effect was not found in TRPV1−/− mice (Figure 1E). The present results indicate that long‐term capsaicin administration prevents HS diet‐induced cardiac dysfunction by activating TRPV1.
Table 1.
Characteristics of WT and TRPV1 −/− mice fed a normal diet or high‐salt diet, with or without capsaicin
| WT | TRPV1−/− | |||||
|---|---|---|---|---|---|---|
| ND | HS | HC | ND | HS | HC | |
| Weight (g) | 27.9 ± 4.2 | 27.5 ± 3.2 | 26.9 ± 3.8 | 27.7 ± 2.0 | 26.1 ± 2.3 | 26.2 ± 2.2 |
| SBP (mmHg) | 108 ± 4 | 125 ± 7** | 114 ± 5# | 108 ± 5 | 122 ± 4** | 122 ± 5** |
| HR (beat·min−1) | 619 ± 24 | 610 ± 26 | 616 ± 21 | 617 ± 31 | 617 ± 23 | 622 ± 25 |
| HW/BW (mg·g−1) | 4.97 ± 0.30 | 6.90 ± 0.55** | 5.49 ± 0.38# | 6.19 ± 0.53 | 7.73 ± 0.94** | 7.60 ± 0.76** |
| Food intake (g·day−1) | 4.3 ± 0.8 | 4.3 ± 0.5 | 3.9 ± 0.8 | 4.1 ± 0.6 | 4.2 ± 0.7 | 4.1 ± 0.6 |
| Water intake (mL·day−1) | 5.4 ± 1.4 | 7.0 ± 1.9 | 7.1 ± 1.8 | 5.6 ± 0.8 | 8.1 ± 1.5** | 7.9 ± 1.7** |
Values are expressed as the means ± SD; n = 9 per group.
*P < 0.05, **P < 0.01, significantly different from ND; # P < 0.05, significantly different from HS.
HC, high‐salt plus capsaicin diet; HS, high‐salt diet; ND, normal‐salt diet.
Figure 1.
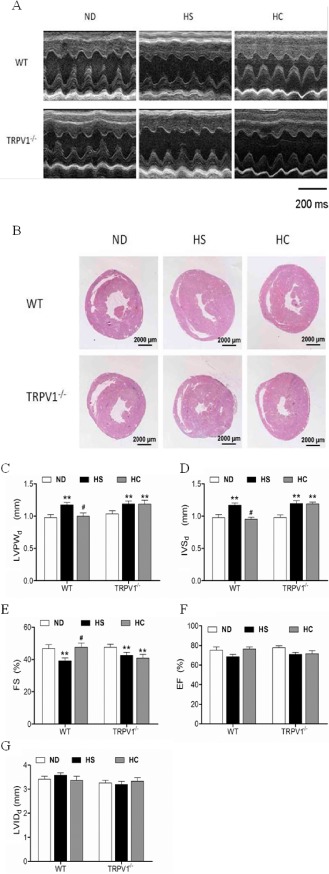
Characteristics of cardiac structure and function in mice on different diets. (A) Representative images of mouse hearts by M‐mode echocardiography. (B) Low magnification images of hematoxylin and eosin‐stained sections at the papillary muscle level. (C–G) Posterior wall thickness of LV (LVPW), interventricular septum thickness (IVS), left ventricular (LV) fractional shortening (%FS), LV ejection fraction (%EF) and LV internal diameter in diastole (LVIDd). Values obtained from echocardiography are expressed as the means ± SEM. (n = 8 for TRPV1−/− mice and WT littermates). **P < 0.01, versus mice on a ND; # P < 0.05, versus mice on a HS diet. HC, high salt plus capsaicin diet.
The amelioration of cardiac dysfunction was associated with an increased endurance capacity, energy expenditure and physical performance in a TRPV1‐dependent manner
To examine the effect of cardiac dysfunction on mouse physical activity in vivo, we monitored mouse endurance capacity, energy expenditure and physical performance. The endurance test protocol using stepwise increasing workloads is illustrated in Figure 2A. Mice on a HS diet performed less well than those mice on ND in the endurance test, with 26.6% and 22.3% reduced endurance capacity for WT on a HS diet and TRPV1−/− mice compared with control mice respectively (Figure 2B). The energy expenditure study showed that mice on a HS diet consumed lower amounts of O2, produced less heat and had a reduced respiratory exchange ratio compared to mice on a ND (Figure 2C–E). Mice on a HS diet showed reduced total locomotor activity compared with mice on a ND (Figure 2F). Interestingly, heat production and total locomotor activity were reduced in TRPV1−/− mice compared with WT mice (Figure 2D,F). The endurance capacity, energy expenditure and physical performance were improved in WT mice fed a HS diet plus capsaicin compared with WT mice fed only a HS diet; these effects of capsaicin were absent in TRPV1−/− mice (Figure 2B–F). These results indicate that dietary capsaicin ameliorated cardiac dysfunction and improved endurance capacity, energy expenditure and physical performance in a TRPV1‐dependent manner.
Figure 2.
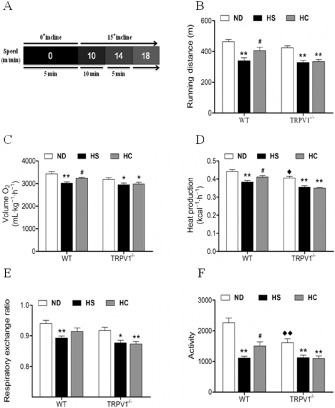
Treadmill endurance capacity and energy expenditure in WT and TRPV1 −/− mice. (A) Endurance capacity test protocol. (B) Changes in endurance capacity. (C) Oxygen consumption of WT and TRPV1 −/− mice on ND, HS and high salt plus capsaicin diet (HC) over 48 h. (D) Heat production of WT and TRPV1 −/− mice on ND, HS and HC over 48 h. (E) Respiratory exchange ratio of WT and TRPV1 −/− mice on ND, HS and HC over 48 h. (F) Locomotor activity of WT and TRPV1−/− mice on ND, HS and HC over 48 h. The results are shown as the means ± SEM (n = 6–8). *P < 0.05, **P < 0.01 vs. mice on a ND; # P < 0.05 versus HS; ◆ P < 0.05, ◆◆ P < 0.01 versus mice with different genotypes.
TRPV1 activation amended HS diet‐induced CI dysfunction
We next examined whether mitochondrial dysfunction contributed to cardiac disturbances caused by a HS diet and determined the role of TRPV1 in this process. First we showed that TRPV1 is expressed in both the mitochondria and cytoplasm (Figure 3A). Furthermore, the presence of mitochondrial TRPV1 was validated by immunofluorescence staining (Figure 3B).
Figure 3.
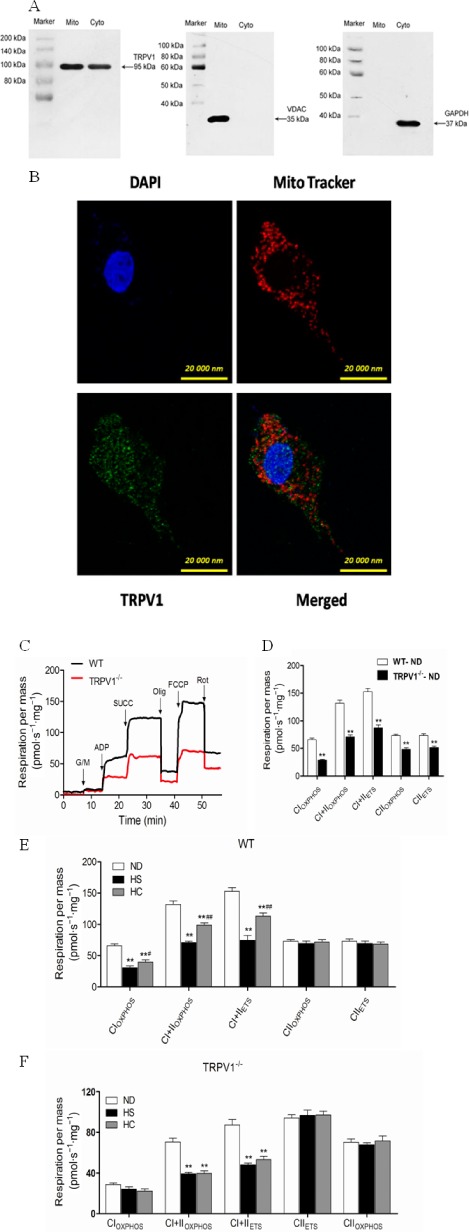
Characterization of TRPV1 in H9C2 cells and heart, and cardiac mitochondrial respiratory chain function in WT and TRPV1 −/− mice fed a HS diet in the presence or absence of capsaicin administration. (A) TRPV1 is localized to the cardiac mitochondria. The purity of the cardiac mitochondrial fractions in WT mice was tested using VADC, a mitochondrial marker, and GAPDH, a cytoplasmic marker. (B) Immunofluorescent staining with TRPV1 antibody in H9C2 cells. Mitochondria were labelled with MitoTracker® Red CMXRos. (C) Representative measurements of oxygen consumption in isolated cardiac mitochondria from WT and TRPV1 −/− mice. (D) The mitochondrial respiratory rate in WT and TRPV1 −/− mice. (E) The mitochondrial respiratory rate was assessed in WT mice on a ND, HS, high salt plus capsaicin (HC) diet. (F) The mitochondrial respiratory rate in TRPV1 −/− mice on a ND, HS, HC diet. The data are presented as the means ± SEM (n = 5–8). *P < 0.05, **P < 0.01 versus mice on a ND; # P < 0.05, ## P < 0.01 versus mice on a HS diet.
We also measured mitochondrial respiration and O2 consumption in isolated cardiac mitochondria in TRPV1−/− and WT mice fed a HS diet (Figure 3C). Compared with WT mice, TRPV1−/− mice showed an overall reduction in mitochondrial respiration, indicating an impaired mitochondrial function (Figure 3D). Furthermore, mitochondrial respiration, which was represented by CI and CI plus Complex II (CII) oxidative phosphorylation (OXPHOS), CI and CII LEAK, as well as CI plus CII electron transfer system (ETS), was markedly reduced in WT mice on a HS diet and in TRPV1−/− mice compared with WT on a ND (Figure 3E,F). No differences were found in CII‐supported OXPHOS and ETS (Figure 3E,F), indicating the high salt intake impaired cardiac mitochondrial CI. Chronic dietary capsaicin significantly improved the reduction in CI function in WT mice and increased CI OXPHOS, CI and CII OXPHOS, CI and CII LEAK, as well as CI and CII ETS by 29.1%, 140.0%, 48.3% and 113.8% respectively (Figure 3E,F). However, the effect of dietary capsaicin on cardiac CI function was not found in TRPV1−/− mice. Altogether, these data suggest that both the TRPV1 knockout and high salt intake induce cardiac mitochondrial dysfunction, which can be improved by dietary capsaicin through TRPV1 activation.
TRPV1 knockout‐ and high salt intake‐mediated Complex I dysfunction is associated with a reduction in its activity
We further investigated the cardiac CI activity in WT mice fed a HS diet and in TRPV1‐/‐ mice. Mitochondrial citrate synthase (CS) activity was determined as an enzymatic measure of mitochondrial mass. In general, TRPV1−/− mice showed an overall reduction in CI activity, ATP production and CS activity compared with WT mice; however, cardiac CII activity was not different between WT and TRPV1−/− mice (Figure 4). Furthermore, cardiac mitochondria had lower CI activity, CS activity, and ATP production in WT mice on a HS diet compared with WT mice on a ND (Figure 4). In addition, the mitochondrial mass was significantly reduced in WT mice fed with HS diet compared with WT mice with ND. Chronic dietary capsaicin significantly increased CI activity, ATP production and CS activity in WT mice on a HS diet (Figure 4). In contrast, these effects of capsaicin were absent in TRPV1−/− mice. Therefore, these data indicate that CI dysfunction caused by TRPV1 knockout and high salt intake is associated with reduction in CI activity.
Figure 4.
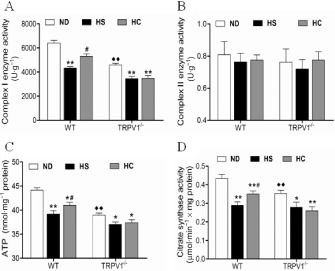
Cardiac mitochondrial enzymatic activities and ATP levels in WT and TRPV1 −/− mice fed a HS diet in the presence or absence of capsaicin administration. (A) Enzymatic activity of Complex I (U·g−1). (B) Enzymatic activity of Complex II (U·g−1). (C) ATP level (nmol·mg−1). (D) Citrate synthase activity (μmol·min−1 × mg protein). The results are shown as the means ± SEM. (n = 5–7), *P < 0.05, **P < 0.01 versus mice on a ND; # P < 0.05 versus mice on a HS diet; ◆◆ P < 0.01 versus mice with different genotypes.
Effect of TRPV1 activation on mitochondrial sirtuin 3 in vitro
Sirtuin 3 is located primarily in the mitochondria and can regulate CI activity, ATP production and mitochondrial content (Ahn et al., 2008). We examined the protein expressions of TRPV1 and sirtuin 3 in cultured H9C2 cells in the presence of high‐sodium with or without TRPV1, a sirtuin 3 agonist and TRPV1 inhibitor (Figure 5A). Compared with high‐sodium concentration stimulation, capsaicin markedly enhanced the expressions of TRPV1 and sirtuin 3. In contrast, 5′‐iodoresiniferatoxin (iRTX), a TRPV1 antagonist, significantly reduced the expression of TRPV1 and sirtuin 3 (Figure 5B,C). Compared with high sodium plus capsaicin, the sirtuin 3 agonist resveratrol (RES) slightly increased the expression of sirtuin 3 but not TRPV1 (Figure 5C). Subunit 9 (NDUFA9) is involved in the CI assembly process and is crucial for CI biogenesis and activity (Stroud et al., 2013). Sirtuin 3 affects the acetylation state of NDUFA9 (Ahn et al., 2008). Compared with high‐sodium stimulation, the expression levels of sirtuin 3 and NDUFA9 were up‐regulated by the sirtuin 3 agonist RES. The effect of RES was abolished by its inhibitor, nicotinamide (Figure 5D–F). Altogether, our results suggest that sirtuin 3 regulates the expression of NDUFA9 under high‐sodium stimulation and benefits mitochondrial function.
Figure 5.
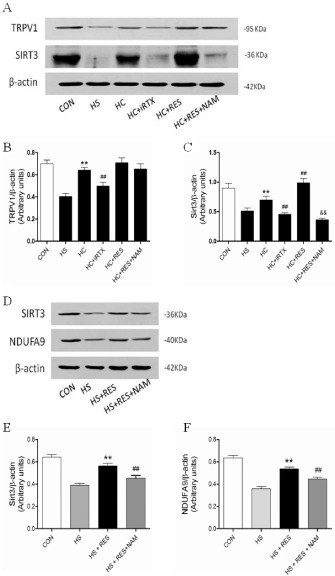
Changes in sirtuin 3 (SIRT3) and NDUFA9 expression in cultured cardiomyoblasts. (A) Representative blots of TRPV1 and sirtuin 3. (B,C) Quantitative analysis of TRPV1 and sirtuin 3 expression in cultured cardiomyoblasts in the presence of high‐sodium medium with or without TRPV1 and sirtuin 3 agonist and inhibitor treatment for 24 h (n = 5). The results are shown as the means ± SEM (n = 5). **P < 0.01 versus mice on a HS diet; ## P < 0.01 versus mice on a HS diet plus capsaicin; && P < 0.01 versus mice on a HS diet with resveratrol (RES). (D) Representative protein blots of sirtuin 3 and NDUFA9. (E,F) Quantitative analysis of sirtuin 3 and NDUFA9 expression in cultured cardiomyoblasts in the presence of high‐sodium medium with or without sirtuin 3 agonist and inhibitor treatment for 24 h (n = 5). The results are shown as the means ± SEM. (n = 5), **P < 0.01 versus mice on a HS diet; ## P < 0.01 versus mice on a HS diet with RES.
TRPV1 activation protected mitochondria from dysfunction by up‐regulating sirtuin 3 in vivo
Finally, the effect of TRPV1 activation by capsaicin on sirtuin 3 and its related target genes was validated in mice in vivo. The expressions of TRPV1, sirtuin 3 and NDUFA9 in isolated cardiac mitochondria were reduced in mice on a HS diet compared with mice on a ND. In contrast, chronic dietary capsaicin significantly increased the expressions of TRPV1, sirtuin 3 and NDUFA9 in isolated cardiac mitochondria from WT mice on a HS diet. Importantly, dietary capsaicin did not affect the expressions of TRPV1, sirtuin 3 and NDUFA9 in isolated cardiac mitochondria from TRPV1−/− mice (Figure 6). These data further validate that TRPV1 activation by capsaicin can prevent cardiac mitochondria dysfunction caused by high salt intake.
Figure 6.
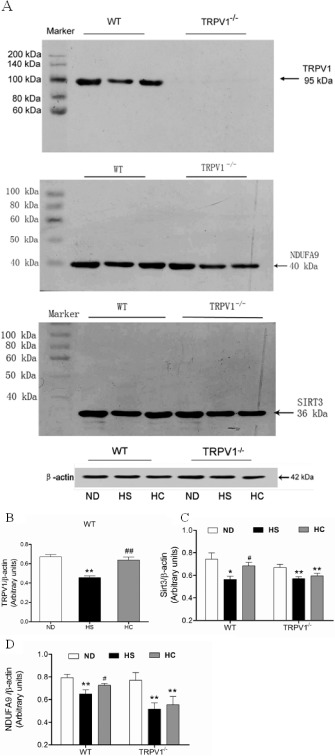
TRPV1, sirtuin 3 (SIRT3) and NDUFA9 expression in freshly isolated mitochondria from the cardiac tissues of WT and TRPV1 −/− mice. (A) Representative blots of TRPV1, sirtuin 3 and NDUFA9. (B–D) Quantitative analysis of TRPV1, sirtuin 3, NDUFA9 expressions in WT and TRPV1−/− mice fed a HS diet with or without capsaicin treatment. The values are presented as the means ± SEM (n = 5). *P < 0.05, **P < 0.01 versus mice on a ND; # P < 0.05, ## P < 0.01 versus HS diet.
Discussion and conclusions
There are several findings from this study. Firstly, high salt intake‐mediated cardiac dysfunction and reduced physical activity are associated with a disturbance in energy metabolism. Secondly, TRPV1 activation by chronic capsaicin administration can improve high salt‐induced cardiac dysfunction in WT but not in TRPV1‐/‐ mice. Thirdly, TRPV1 knockout and high salt intake significantly jeopardized cardiac mitochondrial CI OXPHOS capacity and reduced CI enzyme activity. However, chronic dietary capsaicin increased cardiac mitochondrial function in a TRPV1‐dependent manner. Together, these findings highlight an essential role for TRPV1 as a regulator of cardiac energy expenditure.
High salt consumption leads to cardiac hypertrophy with an elevated systolic BP, cardiac dysfunction and cardiac fibrosis in rats (Takeda et al., 2000; Lal et al., 2003). In this study, we also confirmed that long‐term high salt intake caused cardiac hypertrophy as well as a reduction in physical activity and energy expenditure. However, these mice on a HS diet presented early cardiac disturbance with minor cardiac dysfunction and without cardiac fibrosis.
The mechanism responsible for high salt‐induced cardiac hypertrophy remains unclear. Several mechanisms have been suggested, such as the overstimulation of the sympathetic activity and activation of the renin‐angiotensin system (Le Corvoisier et al., 2010). Antagonizing sympathetic activity or activation of the renin‐angiotensin system can effectively improve cardiac hypertrophy and dysfunction both in human and animal hearts (Lal et al., 2003; Machackova et al., 2010).
ATP is primarily produced in the mitochondrial respiratory chain by OXPHOS. Previous studies showed that a dysfunction of mitochondrial bioenergetics contributes to the cardiac hypertrophy and can lead to heart failure (Wilkins et al., 2004; Osterholt et al., 2013). Mitochondrial changes during cardiac hypertrophy include disrupted mitochondrial substrate metabolism, respiratory chain activity and proteomic remodelling as well as oxidative stress (Marin‐Garcia et al., 2013).
Our results showed that high salt intake reduced cardiac CI OXPHOS capacity without altering CII OXPHOS capacity. Diminished activity of the respiratory chain enzymes may impair mitochondrial respiration, thereby reducing ATP production in the heart (Vyatkina et al., 2004). Several studies have reported TRPC3 and TRPC6 as essential components of the AngII‐induced cardiac hypertrophic signalling pathway (Eder and Molkentin, 2011). We found that TRPV1 activation improved vascular function by reducing vascular ROS production in high salt intake‐induced hypertensive mice (Hao et al., 2011). In the present study, we further showed that TRPV1 is also expressed in the mitochondria in cultured cardiomyoblasts. Compared with WT mice, TRPV1−/− mice had an overall reduction in cardiac mitochondrial respiration.
Mitochondrial CI is the largest component involved in the regulation of cellular energy production (Pagliarini et al., 2008). CI dysfunction is closely associated with cardiac hypertrophy (Dugan et al., 2013). Human CI consists of seven subunits. These subunits are encoded by mitochondrial DNA and 38 nuclear‐encoded subunits are assembled together. Alterations in the CI subunit structure impairs the mitochondrial respiratory process and eventually leads to heart failure. Inactivation of the Ndufs4 gene, a protein critical for CI assembly, accelerates heart failure through its inhibition of sirtuin 3 activity (Karamanlidis et al., 2013).
We showed that mitochondrial CI is dsysfunctional in WT mice fed a HS diet and in TRPV1 knockout mice. Chronic dietary capsaicin significantly improved the reduction in cardiac CI function in WT mice fed a HS diet, but not in TRPV1−/− mice. This result indicates that the TRPV1 mutation exacerbates cardiac mitochondrial dysfunction.
We also examined the relationship between CI and sirtuin 3 in the heart. sirtuin 3 is located primarily in the mitochondria, where it contributes to the regulation of mitochondrial content, CI protein deacetylation and the maintenance of cellular ATP levels in a NAD+‐dependent manner (Ahn et al., 2008). Recent studies showed that sirtuin 3−/− mice had normal cardiac function under normal conditions, but their cardiac function was impaired during certain stress conditions (Ahn et al., 2008). The association of sirtuin 3 with CI can be reversed by the administration of the CI inhibitor rotenone or hydrogen peroxide exposure (Zhang and Brown, 2004). Subunit 9 (NDUFA9) of CI, a mitochondrial protein, is a target of sirtuin 3 in the mitochondrial respiratory chain (Ahn et al., 2008). Our results showed that the sirtuin 3 agonist resveratrol remarkably up‐regulated the sirtuin 3 and NDUFA9 expressions, indicating that maintaining normal sirtuin 3 status is crucial for mitochondrial function.
In the present study, long‐term high salt intake as a type of unhealthy lifestyle led to cardiac disturbance through an impairment of mitochondrial function and reduction in ATP production in the heart. Although antagonizing neurohormonal activation ameliorates mitochondrial dysfunction in overt heart failure, patients in the early stages of cardiac failure are recommended to change their lifestyle. Epidemiological studies have shown that many dietary factors can affect cardiovascular diseases. Over the past two decades, strategies such as limitation of sodium intake, increasing dietary potassium and consuming fruits as well as vegetables, based on the ‘DASH diet’, have been validated as effective antihypertensive measures (Appel et al., 1997; Erkkila and Lichtenstein, 2006). Capsicum species, or hot peppers, are consumed worldwide as vegetables and spices. Capsaicin is the main pungent ingredient in hot pepper and produces a flavour to food. Capsaicin is also a specific agonist for the TRPV1 channel (Caterina et al., 1997). Beneficial effects of capsaicin on the cardiovascular system have been validated by our study and others (Yang et al., 2010; Hao et al., 2011; Ma et al., 2011; Xu et al., 2011; Wang et al., 2012). In this study, chronic dietary capsaicin attenuated high salt intake‐induced cardiac disturbance by improving CI capacity, increasing ATP production and up‐regulating sirtuin 3 and NDUFA9 expression in a TRPV1‐dependent manner, all of which benefited cardiac mitochondrial function.
In conclusion, chronic dietary capsaicin consumption benefits cardiac mitochondrial function in mice on a HS diet. Our mechanistic evidence suggests that this cardiac protection is associated with TRPV1 activation mediated by enhancing CI capacity and ATP production as well as increasing sirtuin 3 and NDUFA9 levels. Our study provides insights into the promising role of the cardiac TRPV1 channel in the regulation of cardiac energy metabolism. Long‐term dietary capsaicin may represent a novel preventive strategy for patients with early stage cardiac failure.
Author contributions
H. L. and Q. L. performed most of the experiments, analysed the data and wrote the manuscript. X. H., P. G. and Q. S. reviewed and edited the manuscript. H. Y., P. L., Z. L., S. X., T. Y., Y. Z. and H. Z. performed some experiments. Z. Z. and D. L. contributed to the discussion and edited the manuscript. Z. Z. designed the experiments and wrote and edited the manuscript. Z. Z. is the guarantor of this work and, as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Conflicts of interest
None.
Supporting information
Appendix S1 Supplementary methods.
Figure S1 Immunoblots of TRPV1 (95 kDa), sirtuin3 (36 kDa) and NDUFA9 (40 kDa) in H9C2 cells.
Acknowledgements
We thank Tingbin Cao and Lijuan Wang (Chongqing Institute of Hypertension, China) for technical assistance. This study was funded by the National Basic Research Program of China (2013CB531205, 2012CB517805, 2013CB531104 and 2011CB503902), the National Natural Science Foundation of China (81370353, 81130006, 91339112 and 91339000) and supported by PCSIRT.
References
- Abel ED, Doenst T (2011). Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc Res 90: 234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn B‐H, Kim H‐S, Song S, Lee IH, Liu J, Vassilopoulos A et al (2008). A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci USA 105: 14447–14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013a). The concise guide to PHARMACOLOGY 2013/14: ion channels. Br J Pharmacol 170: 1607–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013b). The concise guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appel LJ, Moore TJ, Obarzanek E, Vollmer WM, Svetkey LP, Sacks FM et al (1997). A clinical trial of the effects of dietary patterns on blood pressure. DASH Collaborative Research Group. N Engl J Med 336: 1117–1124. [DOI] [PubMed] [Google Scholar]
- Baccarelli A, Cassano PA, Litonjua A, Park SK, Suh H, Sparrow D et al (2008). Cardiac autonomic dysfunction effects from particulate air pollution and protection by dietary methyl nutrients and metabolic polymorphisms. Circulation 117: 1802–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beech DJ (2013). Characteristics of transient receptor potential canonical calcium‐permeable channels and their relevance to vascular physiology and disease. Circ J 77: 570–579. [DOI] [PubMed] [Google Scholar]
- Burnier M, Phan O, Wang Q (2007). High salt intake: a cause of blood pressure‐independent left ventricular hypertrophy? Nephrol Dial Transpl 22: 2426–2429. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997). The capsaicin receptor: a heat‐activated ion channel in the pain pathway Nature 389: 816–824. [DOI] [PubMed] [Google Scholar]
- Dugan LL, You YH, Ali SS, Diamond‐Stanic M, Miyamoto S, DeCleves AE et al (2013). AMPK dysregulation promotes diabetes‐related reduction of superoxide and mitochondrial function. J Clin Invest 123: 4888–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder P, Molkentin JD (2011). TRPC channels as effectors of cardiac hypertrophy. Circ Res 108: 265–272. [DOI] [PubMed] [Google Scholar]
- Erkkila AT, Lichtenstein AH (2006). Fiber and cardiovascular disease risk: how strong is the evidence? J Cardiovasc Nurs 21: 3–8. [DOI] [PubMed] [Google Scholar]
- Frey N, Olson E (2003). Cardiac hypertrophy: the good, the bad, and the ugly. Ann Rev Physiol 65: 45–79. [DOI] [PubMed] [Google Scholar]
- Hao X, Chen J, Luo Z, He H, Yu H, Ma L et al (2011). TRPV1 activation prevents high salt diet‐induced nocturnal hypertension in mice. Pflugers Arch 461: 345–353. [DOI] [PubMed] [Google Scholar]
- Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG et al (2005). ACC/AHA 2005 Guideline Update for the Diagnosis and Management of Chronic Heart Failure in the Adult. Circulation 112: e154–e235. [DOI] [PubMed] [Google Scholar]
- Jula AM, Karanko HM (1994). Effects on left ventricular hypertrophy of long‐term nonpharmacological treatment with sodium restriction in mild‐to‐moderate essential hypertension. Circulation 89: 1023–1031. [DOI] [PubMed] [Google Scholar]
- Karamanlidis G, Lee CF, Garcia‐Menendez L, Kolwicz SC Jr, Suthammarak W, Gong G et al (2013). Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab 18: 239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp CD, Conte JV (2012). The pathophysiology of heart failure. Cardiovasc Pathol 21: 365–371. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal A, Veinot JP, Leenen FHH (2003). Prevention of high salt diet‐induced cardiac hypertrophy and fibrosis by spironolactone. Am J Hypertens 16: 319–323. [DOI] [PubMed] [Google Scholar]
- Le Corvoisier P, Adamy C, Sambin L, Crozatier B, Berdeaux A, Michel JB et al (2010). The cardiac renin‐angiotensin system is responsible for high salt diet‐induced left ventricular hypertrophy in mice. Eur J Heart Fail 12: 1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Zhong J, Zhao Z, Luo Z, Ma S, Sun J et al (2011). Activation of TRPV1 reduces vascular lipid accumulation and attenuates atherosclerosis. Cardiovasc Res 92: 504–513. [DOI] [PubMed] [Google Scholar]
- Machackova J, Sanganalmath SK, Barta J, Dhalla KS, Dhalla NS (2010). Amelioration of cardiac remodeling in congestive heart failure by beta‐adrenoceptor blockade is associated with depression in sympathetic activity. Cardiovasc Toxicol 10: 9–16. [DOI] [PubMed] [Google Scholar]
- Marin‐Garcia J, Akhmedov AT, Moe GW (2013). Mitochondria in heart failure: the emerging role of mitochondrial dynamics. Heart Fail Rev 18: 439–456. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onohara N, Nishida M, Inoue R, Kobayashi H, Sumimoto H, Sato Y et al (2006). TRPC3 and TRPC6 are essential for angiotensin II‐induced cardiac hypertrophy. EMBO J 25: 5305–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterholt M, Nguyen TD, Schwarzer M, Doenst T (2013). Alterations in mitochondrial function in cardiac hypertrophy and heart failure. Heart Fail Rev 18: 645–656. [DOI] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE et al (2008). A mitochondrial protein compendium elucidates complex I disease biology. Cell 134: 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri V, Tracy RP, Roman MJ, Liu JE, Best LG, Bella JN et al (2003). Relation of left ventricular hypertrophy to inflammation and albuminuria in adults with type 2 diabetes the Strong Heart Study. Diabetes Care 26: 2764–2769. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai VB, Sundaresan NR, Jeevanandam V, Gupta MP (2010). Mitochondrial SIRT3 and heart disease. Cardiovasc Res 88: 250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosca MG, Hoppel CL (2010). Mitochondria in heart failure. Cardiovasc Res 88: 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder RE, Hilgers KF, Schlaich MP, Schmidt BM (2007). Renin‐angiotensin system and cardiovascular risk. Lancet 369: 1208–1219. [DOI] [PubMed] [Google Scholar]
- Seddon M, Looi YH, Shah AM (2007). Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart 93: 903–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud DA, Formosa LE, Wijeyeratne XW, Nguyen TN, Ryan MT (2013). Gene knockout using transcription activator‐like effector nucleases (TALENs) reveals that human NDUFA9 protein is essential for stabilizing the junction between membrane and matrix arms of complex I. J Biol Chem 288: 1685–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda Y, Yoneda T, Demura M, Miyamori I, Mabuchi H (2000). Sodium‐induced cardiac aldosterone synthesis causes cardiac hypertrophy. Endocrinology 141: 1901–1904. [DOI] [PubMed] [Google Scholar]
- Takimoto E, Kass DA (2007). Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 49: 241–248. [DOI] [PubMed] [Google Scholar]
- Tao R, Vassilopoulos A, Parisiadou L, Yan Y, Gius D (2014). Regulation of MnSOD enzymatic activity by Sirt3 connects the mitochondrial acetylome signaling networks to aging and carcinogenesis. Antioxid Redox Signal 20: 1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui H, Kinugawa S, Matsushima S (2009). Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc Res 81: 449–456. [DOI] [PubMed] [Google Scholar]
- Vyatkina G, Bhatia V, Gerstner A, Papaconstantinou J, Garg N (2004). Impaired mitochondrial respiratory chain and bioenergetics during chagasic cardiomyopathy development. Biochim Biophys Acta 1689: 162–173. [DOI] [PubMed] [Google Scholar]
- Wang P, Yan Z, Zhong J, Chen J, Ni Y, Li L et al (2012). Transient receptor potential vanilloid 1 activation enhances gut glucagon‐like peptide‐1 secretion and improves glucose homeostasis. Diabetes 61: 2155–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM et al (2004). Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res 94: 110–118. [DOI] [PubMed] [Google Scholar]
- Xu X, Wang P, Zhao Z, Cao T, He H, Luo Z et al (2011). Activation of transient receptor potential vanilloid 1 by dietary capsaicin delays the onset of stroke in stroke‐prone spontaneously hypertensive rats. Stroke 42: 3245–3251. [DOI] [PubMed] [Google Scholar]
- Yang D, Luo Z, Ma S, Wong WT, Ma L, Zhong J et al (2010). Activation of TRPV1 by dietary capsaicin improves endothelium‐dependent vasorelaxation and prevents hypertension. Cell Metab 12: 130–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LL, Liu DY, Ma LQ, Luo ZD, Cao TB, Zhong J et al (2007). Activation of transient receptor potential vanilloid type‐1 channel prevents adipogenesis and obesity. Circ Res 100: 1063–1070. [DOI] [PubMed] [Google Scholar]
- Zhang T, Brown JH (2004). Role of Ca2+/calmodulin‐dependent protein kinase II in cardiac hypertrophy and heart failure. Cardiovasc Res 63: 476–486. [DOI] [PubMed] [Google Scholar]
- Zong J, Zhang D, Zhou H, Bian Z, Deng W, Dai J et al (2013). Baicalein protects against cardiac hypertrophy through blocking MEK‐ERK1/2 signaling. J Cell Biochem 114: 1058–1065. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supplementary methods.
Figure S1 Immunoblots of TRPV1 (95 kDa), sirtuin3 (36 kDa) and NDUFA9 (40 kDa) in H9C2 cells.