

Abstract
Background and Purpose
The clinical use of doxorubicin, an effective anticancer drug, is severely hampered by its cardiotoxicity. 23‐Hydroxybetulinic acid (23‐HBA), isolated from P ulsatilla chinensis, enhances the anticancer effect of doxorubicin while simultaneously reducing its cardiac toxicity, but does not affect the concentration of doxorubicin in the plasma and heart. As the metabolite doxorubicinol is more potent than doxorubicin at inducing cardiac toxicity, in the present study we aimed to clarify the role of doxorubicinol in the protective effect of 23‐HBA.
Experimental Approach
Doxorubicin was administered to mice for two weeks in the presence or absence of 23‐HBA. The heart pathology, function, myocardial enzymes and accumulation of doxorubicin and doxorubicinol were then analysed. A cellular pharmacokinetic study of doxorubicin and doxorubicinol, carbonyl reductase 1 (CBR1) interference and molecular docking was performed in vitro.
Key Results
23‐HBA alleviated the doxorubicin‐induced cardiotoxicity in mice, and this was accompanied by inhibition of the metabolism of doxorubicin and reduced accumulation of doxorubicinol selectively in hearts. In H9c2 cells, the protective effect of 23‐HBA was shown to be closely associated with a decreased rate and extent of accumulation of doxorubicinol in mitochondria and nuclei. siRNA and docking analysis demonstrated that CBR1 has a crucial role in doxorubicin‐mediated cardiotoxicity and 23‐HBA inhibits this metabolic pathway.
Conclusions and Implications
Inhibition of CBR‐mediated doxorubicin metabolism might be one of the protective mechanisms of 23‐HBA against doxorubicin‐induced cardiotoxicity. The present study provides a new research strategy guided by pharmacokinetic theory to elucidate the mechanism of drugs with unknown targets.
Linked Articles
This article is part of a themed section on Chinese Innovation in Cardiovascular Drug Discovery. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2015.172.issue-23
Abbreviations
- 23‐HBA
23‐hydroxybetulinic acid
- AST
aspartate aminotransferase
- CBR1
carbonyl reductase 1
- CMC
carboxymethyl cellulose
- CK
creatine kinase
- CK‐MD
creatine kinase isoenzyme
- EF
ejection fraction
- FS
fractional shortening
- i.g
intragastrically
- LV
left ventricle
- MDR
multidrug resistance
- TCM
traditional Chinese medicine
Tables of Links
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Doxorubicin has been widely used to treat a variety of cancers, including breast cancer, acute leukaemia and malignant lymphoma, since 1970 (Blanco et al., 2008). However, its clinical application is severely limited because of two critical reasons. One is the development of multidrug resistance (MDR) after long‐term treatment, resulting in the failure of chemotherapy. The other is the occurrence of various adverse toxic side effects, the most serious of which is cardiotoxicity or even congestive heart failure (Menna et al., 2007; Takemura and Fujiwara, 2007; Salvatorelli et al., 2012). To date, only one drug, dexrazoxane, has been approved, in 1995, by the Food and Drug Administration to prevent the cardiac toxicity of anthracycline. It can effectively reduce the occurrence of heart failure in patients, but may cause inhibition of bone marrow, dysfunction of liver and kidney, and other adverse reactions (Stěrba et al., 2013). In addition, it was reported that this drug significantly antagonized the inhibitory effects of camptothecin‐ and doxorubicin on growth in vitro (Hasinoff et al., 1996; 1999). Therefore, cardiac safety without reducing the anticancer effect of doxorubicin is an important issue that urgently needs to be solved for safe and effective doxorubicin‐based clinical chemotherapeutic strategies.
In China, the combination of anticancer drugs with traditional Chinese medicines (TCMs) has a long history in clinical cancer therapy, and has been shown to have good efficacy and low toxicity (Qi et al., 2010). TCMs are indispensable sources for the development of modern drugs. In recent years, investigating the active ingredients of TCMs used clinically has led to the development of potential new drugs. For example, 20(S)‐ginsenoside Rh2, a trace active ingredient of ginseng, has been shown to have a marked synergistic effect with an anticancer agent in vitro and in vivo (Xie et al., 2006; Zhang et al., 2010), and it can reverse the doxorubicin‐induced MDR by improving its cellular pharmacokinetic behaviours (Zhang et al., 2012a, 2012b). Although the effectiveness of most natural compounds has been proven in the clinic, their targets are still unknown. Based on our previous studies (Kang et al., 2011; Zhang et al., 2012a, 2012b), our team proposed the concept of ‘reverse pharmacokinetics’ and its importance and convenience for elucidating the mechanisms and discovering the targets of natural active ingredients from TCMs (Hao et al., 2014).
23‐Hydroxybetulinic acid (23‐HBA), an isolated pentacyclic triterpene and major active constituent of Pulsatilla chinensis (Bunge) Regel, is cytotoxic against a variety of tumour cell lines (Ji et al., 2002; Zhou et al., 2007; Zhang et al., 2012a, 2012b). In our previous study, we demonstrated that 23‐HBA enhances the cytotoxicity of doxorubicin by increasing its accumulation in tumours. However, the pharmacokinetic studies revealed that the concentration of doxorubicin in the plasma and heart was not influenced by 23‐HBA, which makes it difficult to explain the protective of 23‐HBA against doxorubicin‐induced cardiotoxicity (Zheng et al., 2010). Doxorubicin can be catalysed into a secondary alcohol metabolite doxorubicinol through two‐electron carbonyl reduction. Doxorubicinol is less potent as an anticancer agent than its parent drug doxorubicin, but it is poorly cleared from the heart and accumulates there to form a long‐lived toxicant to cardiac tissues (Salvatorelli et al., 2012). Therefore, the present study was conducted to investigate whether the protective effect of 23‐HBA on doxorubicin‐induced cardiotoxicity is related to its ability to alter doxorubicin metabolism and doxorubicinol behaviour in the heart. This study was performed from a pharmacokinetic point of view to clarify the underlying mechanism of 23‐HBA from the tissue to the subcellular level.
Methods
Cell culture
Rat H9c2 cells were purchased from American Type Culture Collection (Rockville, MD, USA), and cultured in DMEM supplemented with 10% FBS, and 100 U·mL−1 penicillin and streptomycin (Invitrogen, Carlsbad, CA, USA) at 37°C with 5% CO2.
Animal welfare, ethical statements and treatment
All procedures were approved by the Animal Ethics Committee of China Pharmaceutical University and in compliance with the National Institute of Health Guide for the Care and Use of Laboratory Animals. The in vivo efficacy of 23‐HBA was evaluated using male Balb/c mice (18–22 g) from Military Medical Science Academy (Beijing, Chinese). Mice were housed in a temperature‐ and humidity‐controlled room on a 12 h light/dark cycle and given free access to water and food. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Mice were weighed and randomly divided into eight groups (n = 10, day 0): (1) vehicle control carboxymethyl cellulose (CMC)‐Na [0.2 mL·10 g−1day−1, intragastrically (i.g.)]; (2) 23‐HBA 20 mg·kg−1 day−1 (i.g.); (3) 23‐HBA 40 mg·kg−1 day−1 (i.g.); (4) 23‐HBA 80 mg·kg−1 day−1 (i.g.; (5) doxorubicin 2 mg·kg−1 (i.g., every other day); (6) doxorubicin 2 mg·kg−1 (i.g., every other day) + 23‐HBA 20 mg·kg−1 day−1 (i.g.); (7) doxorubicin 2 mg·kg−1 (i.g., every other day) + 23‐HBA 40 mg·kg−1 day−1 (i.g.); (8) doxorubicin 2 mg·kg−1 (i.g., every other day) + 23‐HBA 80 mg·kg−1 day−1 (i.g.). 23‐HBA or vehicle (0.5% CMC‐Na) was given 1 h before doxorubicin. The body weight, feeding behaviour and motor activity of each animal were monitored as indicators of general health and toxicity.
On day 15, mice were weighed and killed 2 h after doxorubicin administration. The heart, liver, kidney and intestine were collected and stored at −80°C for quantification of doxorubicin and doxorubicinol. Specimens from each heart were fixed in formaldehyde for haematoxylin–eosin (HE) staining, which was detected and semi‐quantified by the Pathology Department, Affiliated Hospital of Southeast University (Nanjing, China) according to the severity of vacuolar degeneration; ‘−’, ‘±’, ‘+’, ‘++’, ‘+++’, respectively, represented none, slight, mild, moderate and severe injury, and the scores were 0, 1, 2, 3 and 4 respectively. The pathology of the cardiac specimens from the eight mice in each group was analysed. Plasma samples were sent to the Traditional Chinese Medicine Hospital of Jiangsu Province (Nanjing, China) to detect the content of myocardial enzymes in the samples.
Echocardiographical analysis
At day 15, echocardiography (Vevo 770 system, VisualSonics, Toronto, Canada) was performed, in mice anaesthetized with 2% isoflurane delivered via a nose cone, to analyse cardiac function. The heart was first imaged in B‐mode in the parasternal long‐axis view to examine the left ventricle (LV). In order to acquire accurate measurements of cardiac dimensions, M‐mode images were obtained from long‐axis and short‐axis B‐mode images by placing the M‐mode sample gate perpendicular to the interventricular septum and LV walls, respectively, at the level of papillary muscles. All M‐mode measurements were performed during end‐diastole (−d) and end‐systole (−s) according to the leading‐edge method of the American Society of Echocardiography (Vinhas et al., 2013). The LV structural parameters measured from the short‐axis view in M‐mode were used in the calculation of LV ejection fraction (EF) and LV fractional shortening (FS).
Quantification of doxorubicin and its metabolite doxorubicinol by LC‐MS/MS
Briefly, samples were protein‐precipitated with methanol (containing azithromycin as an internal standard). After centrifugation, the supernatant was injected into Finnigan TSQ LC‐MS/MS (Thermo Electron, San Jose, CA, USA) for analysis with a Luna C18 column (150 × 2.0 mm, 5 mm, Phenomenex, Lakeforest, CA, USA). The column and autosampler tray temperatures were at 40 and 4°C respectively. The mobile phase consisted of solvent A (0.1% formic acid) and solvent B (methanol) with the following gradient: 0 min; 20% B → 0.5 min; 20% B → 0.8 min; 80% B → 4 min; 80% B → 5.5 min; 20% B → 8 min; 20% B. The flow rate was 0.2 mL·min−1. The MS was operated in positive electrospray ionization mode. MS parameters were as follows: spray voltage, 4.8 kV; sheath gas/auxiliary gas, nitrogen; sheath gas pressure, 42 × 105 Pa; auxiliary gas pressure, 6 × 105 Pa; ion transfer capillary temperature, 285°C. Quantification was performed using selective reaction monitoring mode: m/z 544.7 → 361.0 (doxorubicin); m/z 545.6 → 362.9 (doxorubicinol); m/z749.7→591.4 (azithromycin, internal standard).
Preparation of microsomes and cytosol and the detection of metabolites
Heart and liver tissues were homogenized and centrifuged at 9000 g for 20 min at 4°C. Then the supernatants were centrifuged at 100 000 g for 60 min at 4°C. The supernatant is the cytoplasmic protein (cytosol), and the precipitation is the microsomal protein (microsome).
The microsome and cytosol samples were then incubated with doxorubicin. The samples contained: 148 μL PBS, 10 μL G‐6‐P, 10 μL NADP+, 10 μL G6PDH, 10 μL MgCl2, 10 μL mouse microsome or cytosol (protein concentration was 0.5 mg·mL−1,) and 2 μL doxorubicin (final concentrations were 5, 10, 20, 50, 100, 200 μM). After incubation at 37°C for 60 min, the samples were centrifuged at 40 900× g for 10 min and the supernatant collected for determining the concentration of doxorubicin and doxorubicinol by LC‐MS/MS.
Cell growth inhibition assay
The effects of doxorubicin, doxorubicinol and 23‐HBA on cell viability were determined by the 3‐(4,5‐dimethyl‐2‐thiazolyl)‐2,5‐diphenyl‐2Htetrazolium bromide (MTT) colorimetric assay after the cells had been incubated with various concentrations of doxorubicin in the absence or presence of 23‐HBA (0.2, 2, 20 μM) at 37°C for 24 h. The concentrations required to inhibit growth by 50% (IC50) were calculated from survival curves using the Bliss method.
Apoptosis assays
H9c2 cell extracts were collected to determine the concentration of caspase‐3 using a caspase‐3 (active) ELISA Kit from Invitrogen according to the manufacturer's instructions. The ELISA method is based on the fixation of cleaved caspase‐3 at Asp175/Ser176 between two antibodies. The optical density (O.D.) of this coloured product was measured at 450 nm with an ELISA Reader (Biotek, USA), and this O.D. is directly proportional to the concentration of standard active caspase‐3 present in the kit.
Cells were incubated with Hoechst 33342 (at 10 μg·mL−1) for 30 min after being fixed with 4% formaldehyde. Nuclear morphology was observed by Leica DMI 3000B (Wetzlar, Germany). Representative images were captured and semi‐quantified by Leica QWin V3 software (Leica Microsystems, Wetzlar, Germany).
Quantification of subcellular distribution kinetics of doxorubicin and doxorubicinol
When 90% confluent, cultured cells were treated with 5 μM doxorubicin. After incubation for a designated time (1, 2, 4, 8, 12, 18 h), the nuclei and mitochondria of the cells were isolated according to KeyGen Mitochondria/Nuclei Isolation Kit (Nanjing Keygen Biotech. Co., Ltd., Nanjing, China). The relative purity of the mitochondria and nuclei was calculated and verified by Janus green B and Giemsa's staining (purity > 90%). The concentration of doxorubicin and doxorubicinol in each subcellular compartment was determined by LC‐MS/MS, and further adjusted to the concentrations based on initial dosing volume. All experiments were conducted in triplicate. The intracellular pharmacokinetic parameters were calculated by the method described previously (Zhang et al., 2012b).
RNA interference assay
H9c2 cells were seeded on six‐well cell culture plates, incubated at 37°C, and transfected 24 h later at 70% confluence. CBR1 was transiently knocked down in H9c2 cells by CBR1 siRNA (Santa Cruz Biotechnology, Inc.), which targets CBR1 mRNA. Control siRNA (Santa Cruz Biotechnology, Inc.), a non‐targeting siRNA, was used as a negative control. The transfections were performed for 48 h according to the manufacturer's instructions for the Fugene HD Transfection Reagent. Then the cells were treated with doxorubicin and 23‐HBA for 12 h and collected for the LC‐MS/MS analysis of intracellular doxorubicinol concentration. And the cells treated with doxorubicin and doxorubicinol for 24 h were collected for MTT analysis.
Molecular docking of CBR1
We employed the crystal structure of human CBR1 reported previously (PDB entry code: 3BHI) to complete this docking experiment (Carlquist et al., 2008; El‐Hawari et al., 2009). The CBR1 inhibitors quercetin, epigallocatechin gallate (EGCG), monoHER, hydroxy‐PP and 23‐HBA were docked into the binding site of CBR1. The three‐dimensional structures of the five compounds were built using the Sketch Molecule module by SYBYL 8.1 program package of Tripos, Inc. (St. Louis, MO, USA). These compounds entered the active pocket of CBR1 and mimicked docking by AutoDock software. Then they were scored by Chemscore function.
Data analysis
Data are expressed as mean ± SEM. Statistical analysis included Student's two‐tailed t‐test, one‐way anova and non‐parametric Kruskal–Wallis test. The difference was considered to be statistically significant if the probability value was less than 0.05 (P < 0.05).
Chemicals and reagents
23‐HBA (purity > 99.8%) was a gift from professor W. C. Ye (Jinan University, Guangzhou, China). Doxorubicin was purchased from Sigma‐Aldrich (St. Louis, MO, USA). Doxorubicinol and CBR1 siRNA were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Fugene HD was purchased from Promega (Promega Corporation, Madison, USA). CBR1 antibody was purchased from Abcam (San Francisco, USA). Azithromycin was purchased from the Chinese National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Hoechst 33342 was purchased from Beyotime Institute of Biotechnology (Nantong, China). Caspase‐3 activity kit was purchased from Invitrogen (Life Technologies, Carlsbad, USA). HPLC‐grade methanol was purchased from Sigma‐Aldrich. Deionized water was prepared by Milli‐Q system (Millipore Corporation, Billerica, MA, USA) and was used throughout. All other reagents and solvents were commercially available and of analytical grade.
Results
23‐HBA alleviated myocardial pathological changes and improved cardiac function in mice with doxorubicin‐induced subacute myocardial injury
As shown in Figure 1A, compared with the control group (the score of vacuolar degeneration: 0), mice treated with various doses of 23‐HBA (20, 40 and 80 mg·kg−1) had no obvious pathological damage in cardiac tissue (all groups scored 0). In the doxorubicin group, the staining of myocardial cells had faded and vacuolar degeneration occurred (scored 3.75 ± 0.46), suggesting doxorubicin‐induced cardiac injury. Combined treatment of 20, 40 and 80 mg·kg−1 23‐HBA with doxorubicin dose‐dependently reduced doxorubicin‐induced changes in HE staining and the extent of vacuolar degeneration (scored 3.00 ± 0.76, 2.00 ± 0.53 and 1.50 ± 0.76 respectively).
Figure 1.
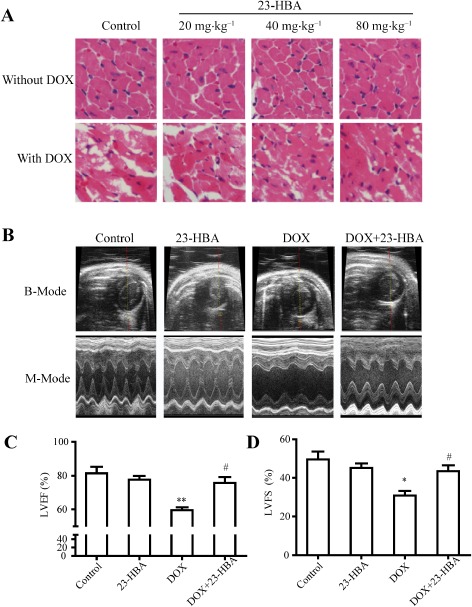
Effects of 23‐HBA on doxorubicin(DOX)‐induced pathological changes in the myocardium and cardiac dysfunction. (A) Representative images of heart tissue sections stained with HE from mice treated with doxorubicin (2 mg·kg−1) and/or 23‐HBA (20, 40, 80 mg·kg−1). Magnification: 400×. (B) Representative B‐mode and M‐mode echocardiography images of left ventricle. Doxorubicin: 2 mg·kg−1; 23‐HBA: 80 mg·kg−1. (C) Echocardiographic measurement of LVEF. (D) Echocardiographic measurement of LVFS. Data are presented as mean ± SEM (n = 5). *P < 0.05 and **P < 0.01 versus control group; # P < 0.05 versus doxorubicin group.
Echocardiography images (Figure 1A) and quantitative analysis (Figure 1C and D) of the doxorubicin group demonstrated a significant impairment of LV function expressed as LVEF (59.50 ± 1.77%) and LVFS (30.81 ± 2.44%) compared with the control group (LVEF: 81.43 ± 3.83%; LVFS: 49.53 ± 4.16%). Combined treatment of 80 mg·kg−1 23‐HBA with doxorubicin resulted in a remarkable improvement in LVEF (75.68 ± 3.52%) and LVFS (43.43 ± 3.12%). However, treatment with 80 mg·kg−1 23‐HBA alone did not obviously change LVEF (77.63 ± 2.31%) and LVFS (45.14 ± 2.38%).
23‐HBA reduced doxorubicin‐induced elevation of myocardial enzymes in mice plasma
Myocardial enzymes, including aspartate aminotransferase (AST), creatine kinase (CK), CK isoenzyme (CK‐MD) and LDH, are important clinical biochemical markers reflecting heart function (Cecen et al., 2011; Ahmed and Urooj, 2012). After a 2 week treatment, 23‐HBA (20, 40 and 80 mg·kg−1 every day, i.g.) did not affect the plasma content of AST, CK, CK‐MD and LDH. I.p. injection of doxorubicin (2 mg·kg−1 every other day) significantly increased the plasma content of AST, CK, CK‐MD and LDH to 2.16, 3.73, 2.11, 1.48 times of that of the control group respectively. Combined treatment with 23‐HBA (20, 40 and 80 mg·kg−1day−1, i.g.) dose‐dependently decreased the doxorubicin‐induced up‐regulation of these four enzymes. In the 80 mg·kg−1 23‐HBA + doxorubicin group, the content of myocardial enzymes recovered to close to the normal level (Figure 2), indicating 23‐HBA could restore the cardiac function.
Figure 2.
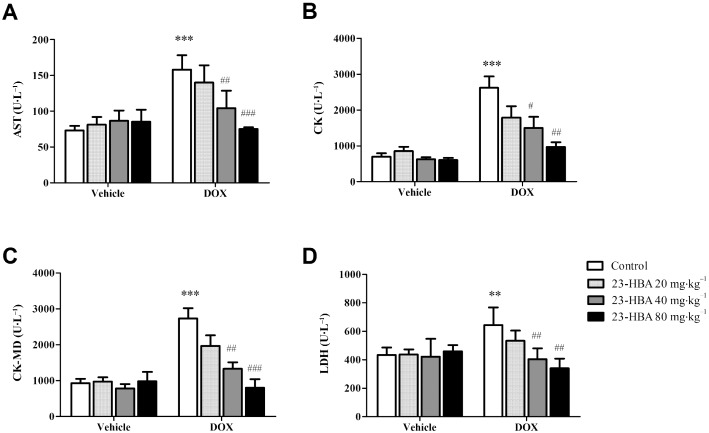
The concentrations of AST, CK, CK‐MD and LDH in the plasma of mice treated with doxorubicin (DOX, 2 mg·kg−1) and/or 23‐HBA (20, 40, 80 mg·kg−1). Data are presented as mean ± SEM (n = 6). **P < 0.01, ***P < 0.001 versus control group without doxorubicin treatment; # P < 0.05, ## P < 0.01, ### P < 0.001 versus control group with doxorubicin treatment.
23‐HBA decreased the accumulation of the metabolite doxorubicinol in heart, but failed to influence the distribution of doxorubicin
After multi‐dose administration of doxorubicin, the concentration of doxorubicin in liver, heart, kidney and intestine showed no obvious difference from that of the single‐dose group (Figure 3A). Doxorubicinol concentration in heart increased to 2.27‐fold that of the single‐dose group, while doxorubicinol concentrations in the other tissues (liver, kidney and intestine) were almost equal to the level of the single‐dose group (Figure 3B), indicating that the metabolite doxorubicinol only accumulated in the heart.
Figure 3.
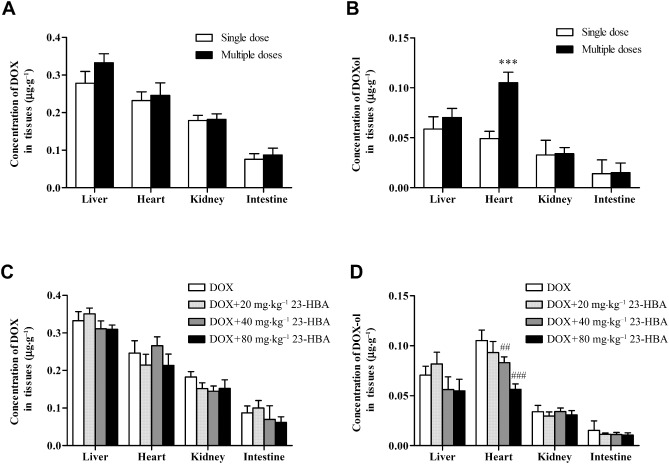
Concentrations of doxorubicin or doxorubicinol in the livers, hearts, kidneys and intestines of mice treated with doxorubicin (DOX) alone or combined with 23‐HBA. (A) Doxorubicin distribution in mice after a single‐ or multi‐dose administration of 2 mg·kg−1 doxorubicin (i.p.). (B) doxorubicinol distribution in mice after single‐ or multi‐dose administration of 2 mg·kg−1 doxorubicin (i.p.). (C) Doxorubicin distribution in mice after multi‐dose administration of 2 mg·kg−1 doxorubicin (i.p.) with or without 20, 40 and 80 mg·kg−1 23‐HBA (i.g.). (D) Doxorubicinol distribution in mice after multi‐dose administration of 2 mg·kg−1 doxorubicin (i.p.) with or without 20, 40 and 80 mg·kg−1 23‐HBA (i.g.). Values are presented as mean ± SEM (n = 6). ***P < 0.001 versus single‐dose administration of doxorubicin; ## P < 0.01, ### P < 0.001 versus doxorubicin alone treatment.
Combined treatment with doxorubicin (2 mg·kg−1 every other day, i.p.) and 23‐HBA (20, 40 and 80 mg·kg−1day−1, i.g.) did not significantly affect the concentration of doxorubicin in mice liver, heart, kidney and intestine when compared with the doxorubicin group (Figure 3C). However, 23‐HBA dose‐dependently reduced the doxorubicinol concentration in the heart, and the combination of 80 mg·kg−1 23‐HBA plus doxorubicin markedly decreased the heart doxorubicinol concentration by 46.52% of the doxorubicin group (Figure 3D). 23‐HBA failed to influence the doxorubicinol concentrations in the other tissues (liver, kidney and intestine).
23‐HBA inhibited the metabolism of doxorubicin in the cytoplasm of mouse live and heart
As carbonyl reductase 1 (CBR1) plays crucial role in the metabolism of doxorubicin (Kassner et al., 2008; Bains et al., 2009), microsomes and cytosol of liver and heart were separated and the expression of CBR1 was investigated. As shown in Figure 4A, CBR1 was highly expressed in both liver and heart and the amount of CBR1 in cytosol was much more than that in microsomes. So the cytosol of liver and heart was separated to investigate the influence of 23‐HBA on the activity of this metabolic enzyme. Intragastric administration of 80 mg·kg−1 23‐HBA significantly inhibited the metabolism of doxorubicin in both liver and heart cytoplasm compared with the control group (Figure 4B and C). Michaelis–Menten kinetic parameters of doxorubicin metabolism were calculated. In liver cytoplasm, the K m of metabolism curve increased from 142.73 to 173.24 μM, Vmax was reduced from 12.03 to 9.61 nM·mg−1·min−1, and intrinsic clearance (Clint, also means Vmax·km−1) decreased from 8.43 × 10−5 to 5.65 × 10−5 mg·min−1. In heart cytoplasm, the K m increased from 139 to 241 μM, Vmax was reduced from 5.02 to 2.87 nM·mg−1·min−1, and Clint decreased from 3.61 × 10−5 to × 10−5 mg·min−1.
Figure 4.
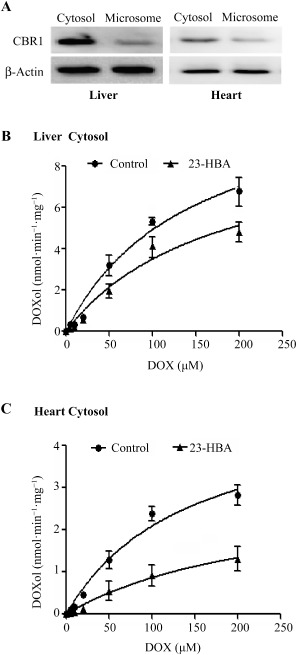
Metabolic characteristics of doxorubicin in mouse liver and heart cytosol. (A) Expression of CBR1 protein in mouse liver and heart cytosol. (B) Curve for the metabolism of doxorubicin in mouse liver cytosol. (C) Curve for the metabolism of doxorubicin in mouse heart cytosol. Values are presented as mean ± SEM (n = 6).
23‐HBA reduced the doxorubicin‐induced cardiac cytotoxicity in H9c2 cells
The results from MTT detection assay showed that doxorubicin (1, 2, 5, 10, 20, 50, 100 μM) concentration‐dependently reduced the survival rate of H9c2 cells. Combined treatment with 23‐HBA (0.2, 2, 20 μM) improved the cell viability in a concentration‐dependent manner, and the IC50 increased from 11.65 μM (IC50 of doxorubicin alone) to12.94, 17.67, 26.55 μM respectively (Figure 5A).
Figure 5.
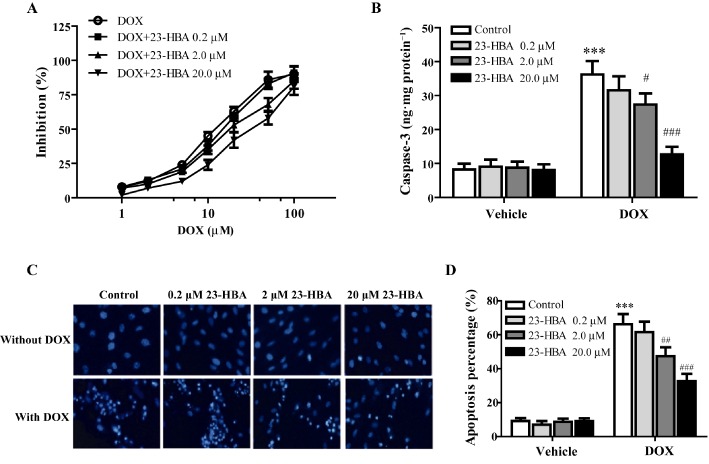
The protective effects of 23‐HBA on doxorubicin‐induced cytotoxicity in H9c2 cells. (A) Logarithmic plots of the inhibition curves obtained after treating the cells with doxorubicin (5 μM) in the presence or absence of 23‐HBA (0.2, 2, 20 μM). (B) Effect of 23‐HBA on caspase‐3 activity induced by doxorubicin in H9c2 cells. (C) Representative fluorescence images of Hoechst 33342 staining of H9c2 cells after different treatments. Magnification: 400×. (D) Quantification analysis of Hoechst staining. Values are expressed as mean ± SEM from six independent experiments. ***P < 0.001 versus control group without doxorubicin, # P < 0.05, ## P < 0.01, ### P < 0.001 versus control group with doxorubicin.
As shown in Figure 5B, 5 μM doxorubicin significantly increased the activity of caspase‐3 in H9c2 cells, which was 4.39 times that of the control group. 23‐HBA (0.2, 2, 20 μM) alone had no obvious effect. When combined with 23‐HBA (0.2, 2.0, 20.0 μM), doxorubicin decreased the caspase‐3 activity by 12.79, 24.44 and 64.91% respectively.
Furthermore, Hoechst 33342 staining was performed to investigate the morphological changes of cells subjected to doxorubicin‐induced apoptosis (Figure 5C) and a semi‐quantification analysis of apoptosis is shown in Figure 5D. The nuclei in the control group were evenly blue, oval with a sharp edge, indicating that the cells grew well. Cells treated with 23‐HBA alone had no obvious changes. But after 5 μM doxorubicin treatment, many cells exhibited pyknosis and nuclear fragmentation, the fluorescence intensity of which was higher (apoptosis: 66.21%) than that of control cells (apoptosis: 9.23%). Combined treatment of 23‐HBA (0.2, 2, 20 μM) significantly alleviated the doxorubicin‐induced morphological changes (apoptosis: 61.58, 47.36 and 32.71%, respectively).
23‐HBA altered the dynamic subcellular distribution of doxorubicin and its metabolite doxorubicinol in H9c2 cells
To quantitatively analyse the time course of the subcellular distribution of doxorubicin and doxorubicinol in the absence and presence of 23‐HBA (0.2, 2.0, 20.0 μM), cytosol, nuclei and mitochondria were separated from H9c2 cells. As shown in Figure 6, doxorubicin mainly accumulated in nuclei, and 23‐HBA did not affect the accumulation curves of doxorubicin in cytosol, nuclei and mitochondria. However, the metabolite doxorubicinol mainly accumulated in mitochondria and nuclei, and 23‐HBA treatment made the concentration–time curve an obvious upward transition in mitochondria and nuclei, indicating a reduction in the amount, distribution and rate of formation of doxorubicinol. The calculated intracellular parameters showed that the rate constant of doxorubicinol for entering mitochondria (Km) was reduced from 0.90 ± 0.11 to 0.63 ± 0.14 h−1 in the presence of 23‐HBA, whereas the rate constant for it entering nuclei (Kn) decreased from 0.92 ± 0.09 to 0.65 ± 0.12 h−1.
Figure 6.
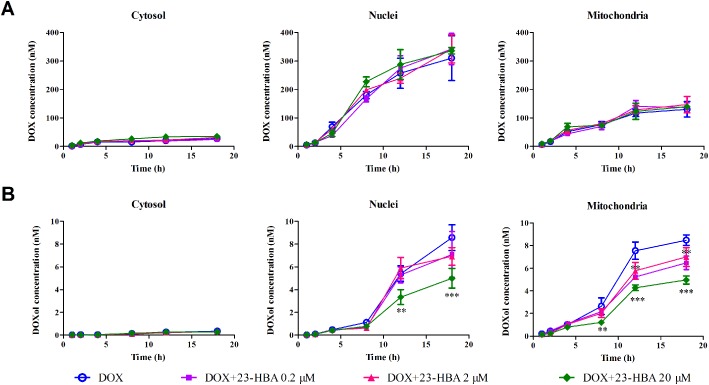
Time–concentration curves of the subcellular distribution of doxorubicin (A) and doxorubicinol (DOXol) (B) in H9c2 cells obtained with the cell fraction approach. H9c2 cells were incubated with 5 μM doxorubicin in the absence or presence of 0.2, 2.0 and 20.0 μM 23‐HBA for 1, 2, 4, 8, 12 and 18 h. The nuclei, mitochondria and cytosol were separated, and doxorubicin or doxorubicinol concentrations in these subcellular organelles were quantified by LC‐MS/MS. Data are expressed as mean ± SEM. of five independent experiments. **P < 0.01, ***P < 0.001 versus doxorubicin group at the same time.
RNA interference of CBR1 reversed the inhibitory effect of 23‐HBA on the production of doxorubicinol in H9c2 cells
As shown in Figure 7A, after gene silence of CBR1 by siRNA, the expression of CBR1 protein in H9c2 cells was significantly reduced. And the intracellular doxorubicinol concentration in the cells of siRNA group was significantly reduced by 45.3% when compared with the control group, indicating CBR1 mediated the metabolism of doxorubicin in H9c2 cells. However, after silencing the CBR1 by siRNA, the 20 μM 23‐HBA combined treatment group exhibited no obvious difference from the group treated with doxorubicin alone (Figure 7B). As shown in Figure 7C and D, the data for cell viability after CBR1 RNA interference showed that the IC50 of doxorubicin (17.54 μM) was increased to 1.85‐fold that of the control group (9.48 μM), while the IC50 of doxorubicinol (1.96 μM) remained at the same level as the control group (2.47 μM).
Figure 7.
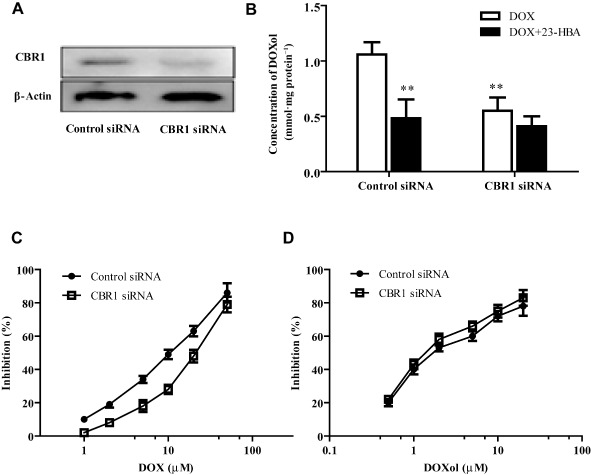
Effects of CBR1 silencing by siRNA on H9c2 cells. (A) Representive images of CBR1 expression detected by Western blotting. (B) Intracellular doxorubicinol concentration of H9c2 cells treated with 5 μM doxorubicin in the presence or absence of 20 μM 23‐HBA after CBR1 interference by siRNA. Values are presented as mean ± SEM (n = 6). **P < 0.01 versus control group treated with doxorubicin. (C) Logarithmic plots of the inhibition curves after doxorubicin treatment in control or CBR1 silenced H9c2 cells. (D) Logarithmic plots of the inhibition curves after doxorubicinol treatment in control or CBR1 silenced H9c2 cells. Values are presented as mean ± SEM (n = 5).
23‐HBA bound to the active centre of CBR1 molecular
Molecular docking analysis was performed with five different CBR1 inhibitors MonoHER, hydroxy‐PP, EGCG, 23‐HBA and quercetin (Carlquist et al., 2008; Gonzalez‐Covarrubias et al., 2008; El‐Hawari et al., 2009). The results from the docking study show that the five compounds bound to the same domain of CBR1 crystal structure, which is the active centre of CBR1 (Figure 8A). The amino acid residues that interacted with the five compounds in a 4–5 Å distance are shown in Figure 8B–F and Table 1. The 23‐hydroxyl and 4‐hydroxy methyl groups of 23‐HBA interacted with the Trp229 and Glu270 amino acid of CBR1. Trp229 residue was also one of the active sites of monoHER where an aromatic–aromatic interaction was likely to occur. The docking affinity of these five compounds with CBR1 protein is shown in Table 1. Higher scores mean a stronger interaction. Generally, a score of >6 is considered as strong inhibition, a score of 6> to >4 is moderate intensity, and a score of <4 means a weak inhibitory effect The results show that the five compounds all exhibited a moderate inhibitory effect on CBR1. The scoring value for 23‐HBA was 4.1734. Among these five compounds, quercetin got the highest score 5.2695, whereas EGCG had the lowest score 3.9295.
Figure 8.
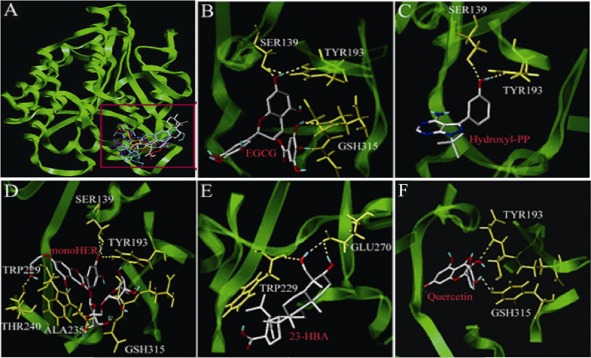
Molecular docking analysis of 23‐HBA and four other CBR1 inhibitors towards CBR1 protein. (A) Three‐dimensional model of the active binding pocket of polymorphic human CBR1 with 23‐HBA and other inhibitors. CBR1 (green) is shown in ribbon form. 23‐HBA (grey), quercetin (red), MonoHER (blue), hydroxy‐PP (yellow) and EGCG (purple) are represented in stick form. (B) Active site of human CBR1 with EGCG. (C) Active site of human CBR1 with hydroxyl‐PP. (D) Active site of human CBR1 with monoHER. (E) Active site of human CBR1 with 23‐HBA. (F) Active site of human CBR1 with quercetin.
Table 1.
Binding residues within ∼4–5 Å of different CBR1 inhibitors
| Ligands | Total score | Crash | Polar | Binding residues within ∼4–5 Å |
|---|---|---|---|---|
| EGCG | 3.9295 | −2.0846 | 2.0532 | Ser139, Tyr193,Gsh315 |
| Hydroxyl‐PP | 5.1335 | −0.7367 | 1.043 | Ser139, Tyr193 |
| MonoHER | 5.1376 | −1.8142 | 3.8614 | Ser139, Tyr193, Gsh315, Thr240, Ala235, Trp229 |
| 23‐HBA | 4.1734 | −0.9207 | 3.5045 | Trp229, Glu270 |
| Quercetin | 5.2695 | −0.9159 | 1.3912 | Tyr193, Gsh315 |
Discussion and conclusions
Cardiac toxicity is the main side effect of doxorubicin, which limits its clinical application (Menna et al., 2007; Takemura and Fujiwara, 2007; Salvatorelli et al., 2012). In our previous study, 23‐HBA increased the antitumour effect of doxorubicin while simultaneously alleviating its ability to induce myocardial injury (Zheng et al., 2010). In support of these findings, in the present study we also showed that 23‐HBA effectively alleviates doxorubicin‐induced myocardial toxicity in a mouse model of subacute myocardial injury; this was assessed by HE pathological staining, echocardiography and myocardial enzyme examination. To date, dexrazoxane is the only licensed antidote for the treatment of anthracycline cardiotoxicity, which can effectively reduce the risk of chronic heart failure after doxorubicin treatment (Stěrba et al., 2013), but may cause bone marrow suppression or adverse gastrointestinal reactions. In addition, it was reported that this drug reduced the anticancer effect of camptothecin and doxorubicin (Hasinoff et al., 1996; 1999). In contrast, 23‐HBA reduced the myocardial toxicity when it increased the antitumour effect of doxorubicin. It exhibited the advantages of natural products with good efficacy and low toxicity (Zheng et al., 2010). It can be considered as a promising candidate for use as an adjuvant drug in clinical tumour chemotherapy.
It is generally believed that pharmacological effects of drugs are often positively correlated with the drug concentrations in plasma or the target tissues when the drug distribution reaches dynamic balance in vivo. However, the present study showed that the concentration of doxorubicin in the heart was not the highest in the main organs, and multiple‐dose treatment did not lead to a cumulative effect in these tissues. It is difficult to explain why the heart is prone to develop myocardial injury after doxorubicin treatment. The plasma protein binding rate of doxorubicin is very low. It can be rapidly distributed in the liver, heart, kidney and intestine after i.v. or i.p. injection. It is mainly metabolized in the liver and excreted in bile, 50% of which are parent drugs, and 23% are active metabolites such as doxorubicinol (Lal et al., 2010). The cytotoxicity of doxorubicinol in tumour tissues is lower than the parent drug doxorubicin, but doxorubicinol exhibits much higher myocardial toxicity (Minotti et al., 2001; Mordente et al., 2003). Doxorubicinol is more polar than its parent drug doxorubicin. So it has a high affinity with mitochondrial cardiolipin, and the myocardial cells are rich in mitochondria, so doxorubicinol prefers to stay in myocardial cells longer (Zucchi and Danesi, 2003; Salvatorelli et al., 2012). Although doxorubicinol cannot directly produce free radicals, it can contribute to Fe2+ release and thus aggravate cardiac oxidative damage (Minotti et al., 2001). The present study confirmed that the concentration of doxorubicinol in the multi‐dose doxorubicin‐treated group accumulated to 2.27 times that of the single‐dose group in myocardial tissue, but no accumulation occurred in the liver, kidney and intestine. This is probably the main reason for the myocardial toxicity of doxorubicin after long‐term administration. Based on the data mentioned earlier, we further investigated whether the mechanisms underlying the protection of 23‐HBA on doxorubicin‐induced myocardial injury was related to an alteration in the concentration of doxorubicinol in the heart. The results supported our hypothesis that 23‐HBA reduces the distribution of doxorubicinol in the heart, but does not affect the distribution of doxorubicin and its metabolite doxorubicinol in the liver, kidney and intestine. The results suggest that the protective effect of 23‐HBA against doxorubicin‐induced cardiotoxicity is closely associated with its ability to inhibit the metabolism of doxorubicin and to changes in the pharmacokinetic behaviour of its metabolite doxorubicinol.
Doxorubicin can be transformed to a semiquinone structure through single‐electron reduction, and it also can form a stable metabolite doxorubicinol through C‐13 hydroxylation by CBR in cytoplasm. CBR1, which is an important enzyme in the catalytic reaction of anthraquinone metabolism, is generally expressed in human and murine liver, heart and other tissues (Kassner et al., 2008; Bains et al., 2009). Forrest et al. (2002) revealed that CBR1 overexpression in the heart advances the development of doxorubicin‐induced cardiotoxicity in transgenic mice, which is accompanied by an increased doxorubicinol concentration in the heart. Similarly, Olson et al. (2003) demonstrated that the metabolism of doxorubicin to doxorubicinol was decreased in Cbr1+/− knockout mice; correspondingly, myocardial toxicity in knockout mice was significantly lower than that of control mice.
The result of Western blotting in the present study showed that CBR1 was highly expressed in the cytoplasm of the liver and heart; however, there was only a small amount of CBR1 in microsomes of the liver and heart, which is in agreement with previous findings (Forrest and Gonzalez, 2000; Mindnich and Penning, 2009). In addition, it was reported that aldo/keto reductases were also involved in the two‐electron reduction of doxorubicin (Bains et al., 2010). But we failed to detect the expression of aldo/keto reductase, because its bands were extremely weak. In fact, there are important variations in the levels of expression and activity of the reductases between human hearts and those of laboratory animals, so doxorubicin could be transformed by different enzymes in humans than in animals. In our present mouse models, CBR1 is the main enzyme responsible for the reduction of doxorubicin to doxorubicinol.
The significant accumulation of doxorubicinol in the heart may be due to two reasons. The first one is the rich blood flow into the heart, which can rapidly deliver the doxorubicinol generated in the liver into the heart. The second reason is the abundant expression of CBR1 in the heart, which can also metabolize doxorubicin in myocardial tissues. 23‐HBA could inhibit the catalytic processes of doxorubicin into doxorubicinol in the cytoplasm of the heart or liver, which may be one of the mechanisms underlying the protective effect of 23‐HBA against doxorubicin‐induced cardiotoxicity.
Based on the in vivo pharmacodynamic and pharmacokinetic data, myocardial H9c2 cells were used to investigate the molecular mechanism underlying the protective effect of 23‐HBA against doxorubicin‐induced cardiotoxicity. The results of MTT, caspase‐3 activity and Hoechst 33342 staining confirmed the myocardial‐protective effects of 23‐HBA in vitro. In order to investigate whether 23‐HBA exerted its protective effect by inhibiting the transformation of doxorubicin to doxorubicinol in the target cells, the progression and characteristics of the accumulation and subcellular distribution of doxorubicin and its metabolite doxorubicinol were studied using the cellular pharmacokinetic methods (Zhang et al., 2012b). Our data showed that doxorubicin was mainly distributed in the nuclei of the target organelles, while most of the doxorubicinol accumulated in the mitochondria and nuclei. The concentration of doxorubicinol in mitochondria was higher than that in the nuclei, which further demonstrated that the place where doxorubicinol exerted myocardial toxicity was the mitochondria. Although the doxorubicin metabolism in H9c2 cells occurred mainly in the cytoplasm (data not shown), doxorubicinol quickly spread to the nuclei and mitochondria after its formation in the cytoplasm because it was more polar and showed a high affinity with the mitochondria and subcellular organelles than doxorubicin (Salvatorelli et al., 2012). As a result, the concentration of doxorubicinol in the cytoplasm is far lower than that in the mitochondria and nucleus. The present study used cellular pharmacokinetic methods to demonstrate that 23‐HBA significantly decreased the extent and velocity of doxorubicinol transported into the nucleus and mitochondria, but failed to affect the dynamic distribution of doxorubicin to the subcellular organelles.
Gene overexpression and knockout studies have already shown that CBR1 is an important enzyme in the catalytic reaction of doxorubicin metabolism, and it is closely associated with doxorubicin‐induced cardiotoxicity (Forrest et al., 2002; Olson et al., 2003; Bains et al., 2009). Therefore, siRNA was used to examine whether the protective effect of 23‐HBA was mediated by CBR1. After the interference of CBR1, the production of doxorubicinol in H9C2 cells was markedly decreased and 23‐HBA failed to affect the intracellular doxorubicinol concentration. From this result, it can be inferred that the inhibitory effect of 23‐HBA on doxorubicin metabolism is the result of an inhibitory effect on CBR1 activity. After CBR1 interference, doxorubicin‐induced cardiotoxicity decreased significantly, while the cytotoxicity of doxorubicinol exhibited no obvious change. This confirmed that the cardiotoxicity induced by doxorubicin depended on the catalytic activity of CBR1. The molecular docking data also showed that 23‐HBA bound to the same region of CBR1 protein (the active centre of the enzyme) as the CBR1 inhibitors quercetin, EGCG, monoHER and Hydroxy‐PP with an equivalent affinity (Carlquist et al., 2008; El‐Hawari et al., 2009). Docking site analysis showed 23‐HBA and monoHER both interacted with the Trp229 of CBR1, suggesting that 23‐HBA and monoHER may have the same mechanism.
In conclusion, inhibition of CBR‐mediated metabolism might be one of the protective mechanisms by which 23‐HBA alleviates doxorubicin‐induced cardiotoxicity. The present study clarified the relationship between the alterations in the behaviour of doxorubicin's metabolite doxorubicinol and the cardiac protective effect of 23‐HBA from a tissue to a subcellular level. This provides a new design strategy for using reverse pharmacokinetic theory to elucidate the mechanism of drugs with unknown targets, especially active constituents isolated from TCMs.
Author contributions
The authors’ responsibilities were as follows: F. Z. and G. W. conceived and designed the study, and gained research‐governance and ethical approval; F. Z., G. H., J. Z. and Y. Z. performed most experiments in the study; F. Z. and G. H. analysed the raw data, and prepared the first draft of the paper; J. Z. established the methods of HPLC‐MS/MS detection and cellular pharmacokinetic; Y. Z. and X. W. assisted with the in vitro experiments; K. H. performed the calculation of pharmacokinetic parameters; F. N., D. L. and Y. S. assisted with sample collection and processing; L. W. performed the molecular docking; W. Y. provided the isolated 23‐HBA; G.W. had primary responsibility for the final content of the paper; and all authors agreed on the final version.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
The work is supported by the Program for New Century Excellent Talents in University (NCET‐11‐0740), China National Nature Science Foundation (No. 30801411, No. 81202591), Jiangsu Province Key Lab of Drug Metabolism and Pharmacokinetics Projects (BM2012012), Jiangsu Province Nature Science Foundation (BK20131308, BK2012354), and the Open Research Fund of State Key Laboratory of Bioelectronics, Southeast University, China ‘Creation of New Drugs’ Key Technology Projects (2015ZX09501001).
References
- Ahmed F, Urooj A (2012). Cardioprotective activity of standardized extract of Ficus racemosa stem bark against doxorubicin‐induced toxicity. Pharm Biol 50: 468–473. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013). The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bains OS, Karkling MJ, Grigliatti TA, Reid RE, Riggs KW (2009). Two nonsynonymous single nucleotide polymorphisms of human carbonyl reductase 1 demonstrate reduced in vitro metabolism of daunorubicin and doxorubicin. Drug Metab Dispos 37: 1107–1114. [DOI] [PubMed] [Google Scholar]
- Bains OS, Grigliatti TA, Reid RE, Riggs KW (2010). Naturally occurring variants of human aldo‐keto reductases with reduced in vitro metabolism of daunorubicin and doxorubicin. J Pharmacol Exp Ther 335: 533–545. [DOI] [PubMed] [Google Scholar]
- Blanco JG, Leisenring WM, Gonzalez‐Covarrubias VM, Kawashima TI, Davies SM, Relling MV et al (2008). Genetic polymorphisms in the carbonyl reductase 3 gene CBR3 and the NAD(P)H:quinine oxidoreductase 1 gene NQO1 in patients who developed anthracycline‐related congestive heart failure after childhood cancer. Cancer 112: 2789–2795. [DOI] [PubMed] [Google Scholar]
- Carlquist M1, Frejd T, Gorwa‐Grauslund MF (2008). Flavonoids as inhibitors of human carbonyl reductase 1. Chem Biol Interact 174: 98–108. [DOI] [PubMed] [Google Scholar]
- Cecen E, Dost T, Culhaci N, Karul A, Ergur B, Birincioglu M (2011). Protective effects of silymarin against doxorubicin‐induced toxicity. Asian Pac J Cancer Prev 12: 2697–2704. [PubMed] [Google Scholar]
- El‐Hawari Y1, Favia AD, Pilka ES, Kisiela M, Oppermann U, Martin HJ et al (2009). Analysis of the substrate‐binding site of human carbonyl reductases CBR1 and CBR3 by site‐directed mutagenesis. Chem Biol Interact 178: 234–241. [DOI] [PubMed] [Google Scholar]
- Forrest GL, Gonzalez B (2000). Carbonyl reductase. Chem Biol Interact 129: 21–40. [DOI] [PubMed] [Google Scholar]
- Forrest GL, Gonzalez B, Tseng W, Li X, Mann J (2002). Human carbonyl reductase overexpression in the heart advances the development of doxorubicin‐induced cardiotoxicity in transgenic mice. Cancer Res 60: 5158–5164. [PubMed] [Google Scholar]
- Gonzalez‐Covarrubias V1, Kalabus JL, Blanco JG (2008). Inhibition of polymorphic human carbonyl reductase 1 (CBR1) by the cardioprotectant flavonoid 7‐monohydroxyethyl rutoside (monoHER). Pharm Res 25: 1730–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao H, Zheng X, Wang G (2014). Insights into drug discovery from natural medicines using reverse pharmacokinetics. Trends Pharmacol Sci 35: 168–177. [DOI] [PubMed] [Google Scholar]
- Hasinoff BB, Yalowich JC, Ling Y, Buss JL (1996). The effect of dexrazoxane (ICRF‐187) on doxorubicin‐ and daunorubicin‐mediated growth inhibition of Chinese hamster ovary cells. Anticancer Drugs 7: 558–567. [DOI] [PubMed] [Google Scholar]
- Hasinoff BB, Chee GL, Thampatty P, Allan WP, Yalowich JC (1999). The cardioprotective and DNA topoisomerase II inhibitory agent dexrazoxane (ICRF‐187) antagonizes camptothecin‐mediated growth inhibition of Chinese hamster ovary cells by inhibition of DNA synthesis. Anticancer Drugs 10: 47–54. [DOI] [PubMed] [Google Scholar]
- Ji ZN, Ye WC, Liu GG, Hsiao W (2002). 23‐Hydroxybetulinic acid‐mediated apoptosis is accompanied by decreases in bcl‐2 expression and telomerase activity in HL‐60 Cells. Life Sci 72: 1–9. [DOI] [PubMed] [Google Scholar]
- Kang A, Hao H, Zheng X, Liang Y, Xie Y, Xie T et al (2011). Peripheral anti‐inflammatory effects explain the ginsenosides paradox between poor brain distribution and anti‐depression efficacy. J Neuroinflammation 8: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassner N, Huse K, Martin HJ, Gödtel‐Armbrust U, Metzger A, Meineke I et al (2008). Carbonyl reductase 1 is a predominant doxorubicin reductase in the human liver. Drug Metab Dispos 36: 2113–2120. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal S, Mahajan A, Chen WN, Chowbay B (2010). Pharmacogenetics of target genes across doxorubicin disposition pathway: a review. Curr Drug Metab 11: 115–128. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menna P, Recalcati S, Cairo G, Minotti G (2007). An introduction to the metabolic determinants of anthracycline cardiotoxicity. Cardiovasc Toxicol 7: 80–85. [DOI] [PubMed] [Google Scholar]
- Mindnich RD, Penning TM (2009). Aldo‐keto reductase (AKR) superfamily: genomics and annotation. Hum Genomics 3: 362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minotti G, Ronchi R, Salvatorelli E, Menna P, Cairo G (2001). Doxorubicin irreversibly inactivates iron regulatory proteins 1 and 2 in cardiomyocytes: evidence for distinct metabolic pathways and implications for iron‐mediated cardiotoxicity of antitumor therapy. Cancer Res 61: 8422–8488. [PubMed] [Google Scholar]
- Mordente A, Minotti G, Martorana GE, Silvestrini A, Giardina B, Meucci E (2003). Anthracycline secondary alcohol metabolite formation in human or rabbit heart: biochemical aspects and pharmacologic implications. Biochem Pharmacol 66: 989–998. [DOI] [PubMed] [Google Scholar]
- Olson LE, Bedja D, Alvey SJ, Cardounel AJ, Gabrielson KL, Reeves RH (2003). Protection from doxorubicin‐induced cardiac toxicity in mice with a null allele of carbonyl reductase 1. Cancer Res 63: 6602–6606. [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al; NC‐IUPHAR (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi F, Li A, Inagaki Y, Gao J, Li J, Kokudo N et al (2010). Chinese herbal medicines as adjuvant treatment during chemo‐ or radio‐therapy for cancer. Biosci Trends 4: 297–307. [PubMed] [Google Scholar]
- Salvatorelli E, Menna P, Gonzalez Paz O, Surapaneni S, Aukerman SL, Chello M et al (2012). Pharmacokinetic characterization of amrubicin cardiac safety in an ex vivo human myocardial strip model. II. Amrubicin shows metabolic advantages over doxorubicin and epirubicin. J Pharmacol Exp Ther 341: 474–483. [DOI] [PubMed] [Google Scholar]
- Stěrba M, Popelová O, Vávrová A, Jirkovský E, Kovaříková P, Geršl V et al (2013). Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxid Redox Signal 18: 899–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemura G, Fujiwara H (2007). Doxorubicin‐induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog Cardiovasc Dis 49: 330–352. [DOI] [PubMed] [Google Scholar]
- Vinhas M, Araújo AC, Ribeiro S, Rosário LB, Belo JA (2013). Transthoracic echocardiography reference values in juvenile and adult 129/Sv mice. Cardiovasc Ultrasound 11: 12. doi: 10.1186/1476‐7120‐11‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Eberding A, Madera C, Fazli L, Jia W, Goldenberg L et al (2006). Rh2 synergistically enhances paclitaxel or mitoxantrone in prostate cancer models. J Urol 175: 1926–1931. [DOI] [PubMed] [Google Scholar]
- Zhang DM, Shu C, Chen JJ, Sodani K, Wang J, Bhatnagar J et al (2012a). BBA, a derivative of 23‐hydroxybetulinic acid, potently reverses ABCB1‐mediated drug resistance in vitro and in vivo . Mol Pharm 9: 3147–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhou F, Wu X, Gu Y, Ai H, Zheng Y et al (2010). 20(S)‐ginsenoside rh2 noncompetitively inhibits p‐glycoprotein in vitro and in vivo: a case for herb‐drug interactions. Drug Metab Dispos 38: 2179–2187. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhou F, Wu X, Zhang X, Chen Y, Zha BS et al (2012b). Cellular pharmacokinetic mechanisms of adriamycin resistance and its modulation by 20(S)‐ginsenoside Rh2 in MCF‐7/Adr cells. Br J Pharmacol 165: 120–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Zhou F, Wu X, Wen X, Li Y, Yan B et al (2010). 23‐Hydroxybetulinic acid from Pulsatilla chinensis (Bunge) Regel synergizes the antitumor activities of doxorubicin in vitro and in vivo . J Ethnopharmacol 128: 615–622. [DOI] [PubMed] [Google Scholar]
- Zhou JP, Li D, Wu XM, Ye WC, Zhang LY (2007). Synthesis and antitumor activity of derivatives of 23‐hydroxybetulinic acid. Chin Chem Lett 18: 1195–1198. [Google Scholar]
- Zucchi R, Danesi R (2003). Cardiac toxicity of antineoplastic anthracyclines. Curr Med Chem Anticancer Agents 3: 151–171. [DOI] [PubMed] [Google Scholar]