

Abstract
Background and Purpose
Although aspirin (acetylsalicylic acid) is commonly used to prevent ischaemic events in patients with coronary artery disease, many patients fail to respond to aspirin treatment. Dietary fish oil (FO), containing ω3 polyunsaturated fatty acids (PUFAs), has anti‐inflammatory and cardio‐protective properties, such as lowering cholesterol and modulating platelet activity. The objective of the present study was to investigate the potential additional effects of aspirin and FO on platelet activity and vascular response to injury.
Experimental Approach
Femoral arterial remodelling was induced by wire injury in mice. Platelet aggregation, and photochemical‐ and ferric chloride‐induced carotid artery thrombosis were employed to evaluate platelet function.
Key Results
FO treatment increased membrane ω3 PUFA incorporation, lowered plasma triglyceride and cholesterol levels, and reduced systolic BP in mice. FO or aspirin alone inhibited platelet aggregation; however, when combined, they exhibited synergistic suppression of platelet activity in mice, independent of COX‐1 inhibition. FO alone, but not aspirin, attenuated arterial neointimal growth in response to injury. Strikingly, a combination of FO and aspirin synergistically inhibited injury‐induced neointimal hyperplasia and reduced perivascular inflammatory reactions. Moreover, co‐administration of FO and aspirin decreased the expression of pro‐inflammatory cytokines and adhesion molecules in inflammatory cells. Consistently, a pro‐resolution lipid mediator‐Resolvin E1, was significantly elevated in plasma in FO/aspirin‐treated mice.
Conclusions and Implications
Co‐administration of FO and low‐dose aspirin may act synergistically to protect against thrombosis and injury‐induced vascular remodelling in mice. Our results support further investigation of adjuvant FO supplementation for patients with stable coronary artery disease.
Linked Articles
This article is part of a themed section on Chinese Innovation in Cardiovascular Drug Discovery. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2015.172.issue-23
Abbreviations
- AA
arachidonic acid
- CO
coconut oil
- CVD
cardiovascular disease
- DHA
docosahexaenoic acid
- EPA
eicosapentaenoic acid
- FO
fish oil
- HDLC
high‐density lipoprotein cholesterol
- LDLC
low‐density lipoprotein cholesterol
- PCI
percutaneous coronary intervention
- PUFA
polyunsaturated fatty acid; TG, triglyceride
Tables of Links
| TARGETS | |
|---|---|
| Catalytic receptors a | Enzymes b |
| CD11b (integrin, alpha M subunit) | COX‐1 |
| LFA‐1 (integrin, beta 2 subunit) | |
| PDGFRβ | |
| VLA‐4 (integrin α4β1) |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (
Introduction
Low‐dose aspirin (acetylsalicylic acid, ASA) is effective for the secondary prevention of heart attack and stroke, and for the primary prevention of nonfatal myocardial infarction (Marangoni and Poli, 2013; Gouya et al., 2014). However, patient responses to this therapy are extremely variable (Floyd and Ferro, 2014). The anti‐platelet effects of aspirin in patients can vary depending on the assay used, genetic background and dosage of aspirin (Mijajlovic et al., 2013). Poor patient compliance is one of the main causes of aspirin resistance. Additionally, high platelet turnover in some inflammatory diseases, such as atherosclerosis and its complications, can lead to a reduced efficacy of aspirin because of a faster regeneration of platelets (Grove et al., 2011). Thus, resistance to aspirin could be avoided by increasing the daily dosage of aspirin (Capodanno et al., 2011; Dillinger et al., 2012) or by administering other agents, such as fish oil (FO) supplements (Lev et al., 2010).
FO has been studied extensively for its role in the attenuation of cardiovascular disease (CVD), induced by lowering total cholesterol (Astrup, 2014), by modulating platelet function (Marangoni and Poli, 2013) and by reducing vascular inflammation (Harris et al., 2013). The cardiovascular benefits of FO are attributed to its ω3 polyunsaturated fatty acid (PUFA) content, particularly eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA). Indeed, some of the protective effects of EPA and DHA are believed to occur through a reduction in pro‐inflammatory prostaglandins (PGs) induced by its ability to inhibit arachidonic acid (AA) metabolic pathways (Calder, 2014). Low‐dose aspirin directly inhibits AA metabolism to PGs, including the powerful platelet agonist TxA2, through the acetylation of COX‐1 on Ser529 (Yu et al., 2005). Recently, compared with aspirin alone, a combination treatment with ω3 PUFAs and low‐dose aspirin was shown to significantly reduce platelet function and potentiate the platelet response to clopidogrel (Gajos et al., 2010; Lev et al., 2010; Block et al., 2012). However, the underlying mechanism for the synergistic effect of ω3 PUFAs and low‐dose aspirin on platelet activity needs to be further investigated.
Arterial restenosis after angioplasty and stent deployment continues to be problematic in coronary intervention treatment, although application of drug‐eluting stents has dramatically increased the success rate compared with that of bare metal stents (Inoue and Node, 2009). Aspirin and clopidogrel are commonly used to prevent thrombotic events after percutaneous coronary intervention (PCI). Surprisingly, aspirin does not reduce restenosis post‐PCI (Schwartz et al., 1988). Also, aspirin was found to have no effect on vascular neointima formation after denudation injury in rat carotid arteries (Yang et al., 2004). In contrast, ω3 PUFAs were shown to attenuate balloon injury‐induced vascular neointima hyperplasia (Faggin et al., 2000), probably through their inhibition of vascular smooth muscle cell proliferation (Pakala and Benedict, 1999). Thus, we hypothesized that co‐administration of FO and aspirin could protect against vascular remodelling in response to injury, as well as potently inhibiting platelet function.
Methods
Experimental animals
All animal care and experimental procedures were performed in accordance with the guidelines of the Animal Care and Use Committee of the Institute for Nutritional Sciences, Chinese Academy of Sciences. The total number of mice used in this study was 327. The COX‐1neo mice – a neomycin (Neo) resistance cassette inserted in COX‐1 intron 10 to ensure hypomorphic expression of COX‐1 gene (Yu et al., 2005), and wild‐type (WT) littermates were initially produced on a mixed C57BL/6/Sv129 genetic background (50%:50%), and maintained on this hybrid background for over 20 generations. Mice were housed in a temperature‐controlled (22 ± 1°C) environment with a 12:12 h light–dark cycle. Mice were fed with a special diet containing 19% FO, 19% coconut oil (CO), with or without aspirin via drinking water (30 mg·L−1). Special diets were provided by Teklad Diets (Harlan Laboratories, Madison, WI, USA) and aspirin was purchased from Sigma (A5376, Saint Louis, MO, USA). After 2 weeks of feeding, BP measurement and thrombotic experiments were conducted and blood, urine and peritoneal macrophages were collected for examination of cell membrane fatty acid and serum lipid, urinary PG metabolites and inflammatory gene expression respectively. For vascular injury experiments, mice were fed specific diets for 2 weeks before surgery and an additional 4 weeks after surgery until the injured arteries were harvested. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Fatty acid analysis
Blood was drawn from the inferior vena cava of 12‐ to 14‐week old mice anaesthetized with chloral hydrate (5 μL g−1, i.p.). The blood was then incubated in a 37°C water bath for 30 min, followed by centrifugation at 900 g for 15 min to remove serum. The blood cells collected were stored at −80°C. The fatty acids from blood cells were extracted as described previously (Zhang et al., 2012). First, saturated fatty acid internal standard (21:0, 50 μg) was added to 0.4 mL blood. Next, hexane (6 mL) and isopropanol (4.5 mL) were added, and the sample was mixed. After centrifugation at 400× g for 5 min, the upper layer containing fatty acids was removed and dried with nitrogen. The fatty acids were then methylated with methylating reagent (methanol : sulphuric acid = 25:1, 4 mL) and extracted with hexane (2 mL). After being washed twice with water (1 mL), the fatty acids were dried with nitrogen, dissolved in isooctane (2 mL), and prepared for GC‐MS analysis.
Serum lipid measurement
Peripheral blood was collected from the retro‐orbital plexus of 10‐ to 12‐week‐old mice. After incubation in a 37°C water bath for 30 min, blood was centrifuged at 900× g for 15 min. Serum was separated and stored at −20°C for later use. Measurement of serum triglycerides (TG), total cholesterol, high‐density lipoprotein cholesterol (HDLC) and low‐density lipoprotein cholesterol (LDLC) levels was performed with commercial kits, following the manufacturer's protocol (BJKT, Beijing, China) (Yan et al., 2014).
BP measurement
Resting systolic BP was measured in conscious mice (10 to 12 weeks old) using a computerized non‐invasive tail cuff system (Visitech Systems, Inc., Apex, NC, USA) as described previously (Cheng et al., 2006). Mice were adapted to the system for 14 days (one 25 min measurement session per day). BP was then recorded daily for 3 consecutive days in the same way. Data were collected and analysed using BP‐2000 software (Visitech Systems, Inc., http://www.visitechsystems.com/).
Platelet aggregation
Blood was isolated from the inferior vena cava of anaesthetized 10‐ to 12‐week‐old mice using a heparin‐containing syringe (15 U·mL−1 blood), and then diluted 1:1 with HEPES‐Tyrode's buffer. Samples were centrifuged at 150× g for 10 min to remove red blood cells. Blood from individual mice in each group was used for aggregation experiments, with the final platelet count adjusted to 2 × 108 cells mL=1 with platelet‐poor plasma from the same mouse. Aggregation was initiated by adding 2.5 μL of ADP with final concentration of 10 μM or 0.5 μL of collagen with final concentration of 2 μg·mL−1 to 250 μL of platelet‐rich plasma (PRP). Platelet aggregation was measured using a lumi‐aggregometer (Chrono‐log Corp., Havertown, PA, USA) and was monitored for 8 min. The area under the aggregation curve was calculated by Image‐Pro Plus software (Media Cybernetics, Silver Spring, MD, USA) for statistical analysis as previously described (Hanke et al., 2010).
Models of thrombogenesis
The photochemical‐induced carotid artery thrombosis model has been described previously (Yu et al., 2005). Briefly, mice (12–14 weeks old) were anaesthetized with 6% chloral hydrate (5 μL·g−1, i.p.); the depth of anaesthesia was assessed by evaluating rear foot reflexes. A midline cervical incision was made, and the right common carotid artery was isolated. A Doppler flow probe (Model 0.5 VB; Transonic Systems, Inc., Ithaca, NY, USA) connected to a flowmeter (Model T105; Transonic Systems, Inc.) was applied to the artery and measurements were obtained using a computerized data acquisition program (PowerLab; AD Instruments, Castle Hill, New South Wales, Australia). In the photochemical arterial thrombosis model, Rose Bengal (11 mg mL−1; 60 mg kg−1) was diluted in PBS and injected into the jugular vein. Before the injection, a 1.5 mW green light laser (540 nm) (Melles Griot, Carlsbad, CA, USA) was applied to the desired site of carotid artery injury from a distance of 10 cm, and blood flow was monitored for 150 min or until stable occlusion occurred. Those mice still showing blood flow after the allotted time were assigned a value of 150 min for the purpose of statistical analysis (Yu et al., 2005). Stable occlusion was defined as a blood flow of 0 mL min−1 for 3 min. In the ferric chloride‐induced model of thrombosis (Petrich et al., 2007; Owens et al., 2011), a 1.2 × 1.2 mm piece of filter paper soaked in 7% ferric chloride was applied to the right common carotid artery for 90s and then the vessel was washed thoroughly with saline. Blood flow was monitored for 30 min until stable occlusion occurred. Mice with an occlusion time longer than 30 min were assigned a value of 30 min for the purpose of statistical analyses. The decrease in blood flow rate (BFR) was determined as the initial BFR minus the BFR at 30 min after ferric chloride treatment.
Thromboxane metabolite (Tx‐M) analysis
Tx‐M was measured in urine collected over the course of 24 h in a metabolic cage and quantified utilizing LC/MS/MS analyses as described previously (Zhang et al., 2013).
Femoral artery injury model
The wire‐induced femoral artery injury model was produced as described previously (Zhang et al., 2013). In brief, 10‐ to 12‐week‐old male WT mice were anaesthetized with isoflurane (2%, delivered in room air) using an induction chamber during the surgery. Bilateral femoral arteries were exposed and wire placement was carried out using a dissecting microscope (S4E, Leica, Wetzlar, Germany). The distal artery and vein were looped with a 6‐0 silk suture and a guide wire (0.38 mm diameter; Cook, Inc., Bloomington, IN, USA) was inserted into the femoral artery. The wire was left in place for 3 min to denude the artery. After this time, the wire was removed and the silk suture was released to restore blood flow. The skin incision was closed with a 5‐0 silk suture.
Histopathological analysis
Femoral arteries were harvested 4 weeks after the injury and fixed in 4% formalin overnight. The samples were processed routinely, embedded in paraffin and stained with haematoxylin and eosin and Ponceaou S‐Picric acid‐Victoria blue routinely. The intimal area was calculated as the area encircled by internal elastic lamina (IEL) minus the lumen area. The medial area was calculated as the area encircled by the external elastic lamina minus intima area. The intima‐to‐media (I/M) ratio was calculated as the intimal area divided by the medial area. The restenosis index (%) was calculated as intimal area divided by the area encircled by IEL (Zhang et al., 2013). All specimens were analysed by an investigator blinded to the study design.
Immunofluorescence staining
For immunohistochemistry, the distribution of macrophages and neutrophils was analysed using an anti‐CD68 antibody (Serotec, Oxford, UK) and an anti‐CD11b monoclonal antibody (BD PharmingenTM, Franklin Lakes, NJ, USA), respectively, followed by a secondary antibody. After being washed three times with PBS, samples underwent diaminobenzidine staining. Sections were counterstained with haematoxylin. For immunofluorescence staining, tissue sections were incubated with primary antibodies against CD68, CD11b and CD3 (Alexa Fluor 488 anti‐mouse, Biolegend, San Diego, CA, USA). After incubation with Alexa Fluor‐conjugated secondary antibodies (1:1000; Invitrogen, Carlsbad, CA, USA), DAPI staining was performed. The samples were mounted on glass slides (ProLong® Gold Antifade Reagent; Invitrogen) and visualized using an inverted fluorescent microscope (Carl Zeiss, Oberkochen, Germany). Subsequent image processing was performed using Photoshop CS2 (Adobe Systems, San Jose, CA, USA). Primary antibodies were used at the following dilutions: CD68, 1:200; CD11b, 1:50; and CD3, 1:200 to identify the inflammatory cells. Six vessel zones (at ×200 view) were selected randomly on four coordinate axes of each stained femoral arterial cross section and three randomly selected sections per mouse were calculated.
RNA extraction and real‐time PCR
Total RNA from circulating white blood cells and vascular tissues was extracted using Trizol reagent (Invitrogen, San Diego, CA, USA), according to the manufacturer's instructions. Briefly, total RNA was reverse‐transcribed to cDNA by use of a reverse transcription reagent kit (TaKaRa Biotechnology, Co., Ltd. Dalian, China), according to the manufacturer's protocol. Real‐time PCR was performed using SYBR Green mix (TaKaRa Biotechnology, Co., Ltd.). Each sample was analysed in triplicate and was normalized to the level of β‐actin mRNA. The PCR protocol was as follows: 5 min at 95°C for one cycle followed by 40 cycles at 95°C for 30 s, 60°C for 30 s and 72°C for 30 s, with a final extension at 72°C for 5 min. A dissociation curve was obtained for each PCR product. Relative gene expression was determined using the ΔΔCT method (Livak and Schmittgen, 2001). All the original data were divided by the mean of WT/CO group. The primer sequences are summarized in Table 1.
Table 1.
Primers used in this study
| Gene | Sense | Anti‐sense |
|---|---|---|
| IL‐6 | ACTCACCTCTTCAGAACGAATTG | CCATCTTTGGAAGGTTCAGGTTG |
| TNF‐α | CCTCTCTCTAATCAGCCCTCTG | GAGGACCTGGGAGTAGATGAG |
| Clec10a | GCTCCGCATACACCTGGATG | GCCGGTCGCATAGTCTGTTC |
| IL‐10 | GACTTTAAGGGTTACCTGGGTTG | TCACATGCGCCTTGATGTCTG |
| MMR | CTACAAGGGATCGGGTTTATGGA | TTGGCATTGCCTAGTAGCGTA |
| Ym‐1 | CAGGTCTGGCAATTCTTCTGAA | GTCTTGCTCATGTGTGTAAGTGA |
| LFA‐1 | AAGTGACGCTTTACCTGCGAC | AAGCATGGAGTAGGAGAGGTC |
| VLA‐4 | CACAACACGCTGTTCGGCTA | CGATCCTGCATCTGTAAATCGC |
| PSGL‐1 | CCTGAGTCTACCACTGTGGAG | GCTGCTGAATCCGTGGACA |
| ICAM‐1 | AACCGCCAGAGAAAGATCAG | TGTGACAGCCAGAGGAAGTG |
| VCAM‐1 | AGTTGGGGATTCGGTTGTTCT | CCCCTCATTCCTTACCACCC |
| P‐selectin | CATCTGGTTCAGTGCTTTGATCT | TGTGCTGTAGTTATAGGTCCACG |
| β‐actin | GGCTGTATTCCCCTCCATCG | CCAGTTGGTAACAATGCCATGT |
Resolvin E1 extraction and detection
Resolvin E1 (5S,12R,18R‐trihydroxy‐6Z,8E,10E,14Z,16E‐EPA) and d5‐Resolvin E1 was prepared by total organic synthesis (Ogawa and Kobayashi, 2009). The structure was confirmed by mass spectral analysis and was consistent with reported properties. The detection of Resolvin E1 generated from dietary FO and aspirin in plasma was performed using a standard LC‐MS/MS protocol with minor modifications (Serhan et al., 2000). Briefly, mouse plasma samples were collected after 2 weeks of FO and aspirin administration. Plasma samples were exacted by ethyl acetate with d5‐Resolvin E1 as internal standard. After centrifugation, the supernatants were analysed by TSQ Vantage LC‐MS/MS (Thermo Scientific, Waltham, MA, USA) using a UPLC column (Acquity UPLC CSH C8, 100 × 2.1 mm, i.d. 1.7 μm). Mobile phase A was (H2O : CH3CN : FA = 63:37:0.1, v v−1v−1) and mobile phase B was (MeOH, with the addition of 2 mM ammonium acetate).
Statistical analysis
Results are expressed as mean ± SEM. The number of samples in each group is shown in the figure legends. Statistical significance was determined by use of Student's unpaired t‐test or one‐way anova followed by post hoc test. A value of P < 0.05 was considered statistically significant.
Results
Dietary FO, but not aspirin, increases the proportion of ω3 PUFAs in erythrocyte membranes and reduces blood TGs in mice
To determine whether FO exerts additional cardiovascular benefits when combined with aspirin, both FO and CO diets (ω6 PUFA‐enriched diet as control, Table 2) were fed to WT, WT treated with low‐dose aspirin (WT/aspirin), and COX‐1neo mice (Yu et al., 2005). As anticipated, FO treatment significantly reduced the proportion of ω6 PUFAs, including linoleic acid (LA) and AA, while increasing levels of ω3 PUFAs, including EPA, docosapentaenoic acid (DPA) and DHA in erythrocyte membranes of all three groups, as compared with CO feeding (Table 3). Among the ω3 PUFAs in the cell membranes, EPA levels in FO‐treated mice were increased approximately 9‐ to 11‐fold, as compared with CO‐treated mice. Interestingly, the ratio of AA to total EPA, DHA and DPA was completely reversed by FO treatment (Table 3). Importantly, aspirin pretreatment and genetic knockdown of COX‐1 alone had no detectable effects on cell membrane fatty acid composition. Thus, FO supplementation could lead to marked increase in membrane incorporation of ω3 PUFAs, particularly EPA.
Table 2.
Contents of experimental diets
| Ingredient | FO diet (g·kg−1) | CO diet (g·kg−1) |
|---|---|---|
| Casein | 240 | 240 |
| L‐cystine | 3.6 | 3.6 |
| Corn starch | 189.208 | 189.208 |
| Sucrose | 150 | 150 |
| Maltodextrin | 100 | 100 |
| Fish oil | 190 | – |
| Coconut oil | – | 190 |
| Soybean oil | 20 | 20 |
| Cellulose | 50 | 50 |
| Mineral mix, AIN‐93G‐MX (94046) | 42 | 42 |
| Calcium phosphate, dibasic | 3 | 3 |
| Vitamin mix, teklad (40060) | 12 | 12 |
| TBHQ (antioxidant) | 0.042 | 0.042 |
| Yellow food colour | 0.15 | – |
| Blue food colour | – | 0.15 |
| Protein (%) | 18.9 | 18.9 |
| Carbohydrate (%) | 38.4 | 38.4 |
| Fat (%) | 42.6 | 42.6 |
Table 3.
Effect of FO on fatty acid composition of blood cell membranes
| PUFAs | WT (%) | WT + aspirin (%) | COX‐1neo (%) | |||
|---|---|---|---|---|---|---|
| CO | FO | CO | FO | CO | FO | |
| ω‐6 | ||||||
| 18:2 ω‐6 (LA) | 15.15 ± 0.84 | 6.49 ± 0.60*** | 14.49 ± 0.49 | 8.85 ± 0.64*** | 14.09 ± 0.56 | 6.63 ± 0.45*** |
| 20:2 ω‐6 (EDA) | 0.31 ± 0.06 | 0.69 ± 0.34 | 0.45 ± 0.20 | 0.15 ± 0.01 | 0.59 ± 0.32 | 0.58 ± 0.35 |
| 20:3 ω‐6 (DGLA) | 2.15 ± 0.27 | 1.96 ± 0.44 | 2.16 ± 0.23 | 1.62 ± 0.38 | 1.77 ± 0.16 | 1.23 ± 0.24 |
| 20:4 ω‐6 (AA) | 13.00 ± 0.58 | 6.80 ± 0.21*** | 12.97 ± 0.54 | 6.66 ± 0.66*** | 13.38 ± 0.89 | 7.47 ± 0.61*** |
| 22:4 ω‐6 (DTA) | 1.16 ± 0.12 | 0.71 ± 0.11 | 1.25 ± 0.06 | 0.89 ± 0.12 | 1.42 ± 0.09 | 0.80 ± 0.13*** |
| 22:5 ω‐6 (DPA) | 0.62 ± 0.06 | 0.61 ± 0.06 | 0.63 ± 0.07 | 0.56 ± 0.06 | 0.61 ± 0.07 | 0.56 ± 0.06 |
| Total | 32.39 ± 0.62 | 17.26 ± 0.66*** | 31.95 ± 0.71 | 18.73 ± 0.87*** | 31.87 ± 1.03 | 17.27 ± 0.98*** |
| ω‐3 | ||||||
| 18:3 ω‐3 (ALA) | 0.24 ± 0.02 | 0.28 ± 0.03 | 0.31 ± 0.06 | 0.39 ± 0.11 | 0.30 ± 0.06 | 0.22 ± 0.02 |
| 20:5 ω‐3 (EPA) | 0.76 ± 0.08 | 8.70 ± 0.37*** | 0.89 ± 0.15 | 7.44 ± 0.42*** | 0.76 ± 0.06 | 7.90 ± 0.29*** |
| 22:5 ω‐3 (DPA) | 0.98 ± 0.15 | 1.95 ± 0.16*** | 1.08 ± 0.08 | 1.83 ± 0.07*** | 1.02 ± 0.10 | 2.07 ± 0.09*** |
| 22:6 ω‐3 (DHA) | 7.82 ± 0.60 | 13.82 ± 0.91*** | 7.86 ± 0.27 | 11.98 ± 0.66*** | 7.53 ± 0.64 | 13.59 ± 0.82*** |
| Total | 9.80 ± 0.79 | 24.75 ± 1.32*** | 10.15 ± 0.24 | 21.65 ± 0.99*** | 9.61 ± 0.72 | 23.79 ± 1.10*** |
| Total PUFAs | 42.19 ± 0.67 | 42.01 ± 0.92 | 42.10 ± 0.85 | 40.37 ± 1.48 | 41.48 ± 1.62 | 41.06 ± 1.42 |
| SFA | 42.17 ± 0.53 | 45.17 ± 1.59 | 42.89 ± 1.09 | 44.74 ± 1.02 | 42.96 ± 0.88 | 45.39 ± 1.38 |
| MFA | 15.59 ± 0.45 | 12.76 ± 0.78 | 14.94 ± 0.49 | 13.86 ± 0.51 | 15.44 ± 0.04 | 13.45 ± 0.38 |
| ω‐6/ω‐3 (of total fractions) | 3.40 ± 0.30 | 0.71 ± 0.06*** | 3.15 ± 0.08 | 0.87 ± 0.05*** | 3.39 ± 0.12 | 0.73 ± 0.06*** |
| AA/EPA + DHA + DPA | 1.38 ± 0.07 | 0.28 ± 0.01*** | 1.32 ± 0.05 | 0.31 ± 0.02*** | 1.45 ± 0.07 | 0.32 ± 0.02*** |
ALA, α‐linolenic acid; DGLA, dihomo‐γ‐linolenic acid; DTA, docosatetraenoic acid; EDA, eicosadienoic acid; LA, linoleic acid; MFA(16:1 + 18:1 + 20:1 + 24:1), monounsaturated fatty acids; SFA(14:0 + 16:0 + 18:0 + 20:0 + 22:0 + 24:0), saturated fatty acids.
Data are given as mean ± SEM (percentage of total fatty acids).
**P < 0.001, ***P < 0.0001 versus CO group; n = 5−6.
We next examined the effects of FO intake on lipid metabolism and BP in mice. As shown in Figure 1, the FO diet significantly reduced plasma levels of TG (Figure 1A) and cholesterol (Figure 1B), including HDLC (Figure 1C) and LDLC (Figure 1D), in WT, aspirin‐treated and COX‐1neo mice, as compared with the CO diet. Moreover, the mice fed with FO diet displayed lower systolic BP than the CO‐fed mice (Figure 2). In contrast, inhibition or genetic knockdown of COX‐1 had no overt effects on lipid metabolism and BP (Figures 1 and 2).
Figure 1.
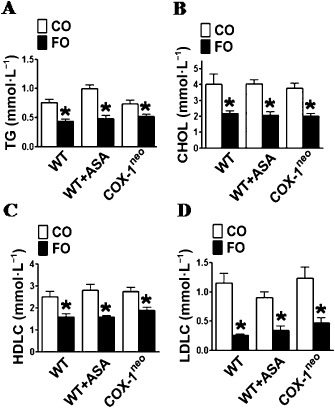
Effect of dietary FO and aspirin (ASA) treatment on serum lipids in mice. WT and COX‐1neo mice were fed 19% FO or CO diet for 2 weeks, and aspirin (30 mg·L −1) was administered in drinking water, as indicated. Serum levels of TG (A), total cholesterol (B), HDLC (C) and LDLC (D)were measured. *P < 0.05 versus CO, n = 10–15.
Figure 2.
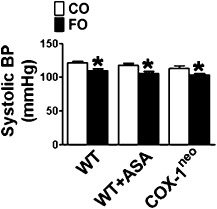
Effect of FO plus aspirin (ASA) on BP. Systolic BP was recorded by tail‐cuff measurement. *P < 0.05 versus CO, n = 9–13.
Aspirin augments the inhibitory effect of FO on platelet activity
To examine the effects of combining FO and low‐dose aspirin on platelet function, PRP from treated mice was prepared, and platelet aggregation was examined in response to ADP and collagen. Platelet aggregation could be induced by either ADP or collagen in a dose‐dependent manner (data not shown). FO‐fed mice displayed reduced platelet aggregation in response to the two agonists, as compared with CO‐fed mice (Figure 3A). Similarly, ADP‐ or collagen‐stimulated platelet activation was markedly diminished upon treatment with aspirin (Figure 3B). Platelet aggregation was further suppressed in mice treated with FO and aspirin, as compared with FO or aspirin treatment alone (Figure 3C and D). Therefore, the combination of FO and aspirin enhanced the inhibition of platelet function.
Figure 3.

Effect of dietary FO and aspirin (ASA) on inhibition of platelet aggregation. (A) Representative tracings of FO‐treated mouse platelet aggregation induced by ADP and collagen; (B) representative tracings of aspirin‐treated mouse platelet aggregation induced by ADP and collagen; (C) representative tracings of FO‐ and aspirin co‐treated mouse platelet aggregation induced by ADP and collagen; (D) quantification of the area under platelet aggregation curve for (A–C). *P < 0.05 versus CO, #P < 0.05 as indicated, n = 5–7. a.u. stands for arbitrary unit. (E) Effect of genetic knock‐down (COX‐1neo) and inhibition (aspirin) of COX‐1 on platelet aggregation from FO‐treated mice. (F) Quantification of the area under aggregation curve for €. *P < 0.05 as indicated, n = 5–7.
Aspirin inhibits platelet activation through the acetylation and deactivation of platelet COX‐1, and the effect of low‐dose aspirin is mimicked in COX‐1neo mice (Yu et al., 2005). We therefore tested whether the synergistic effect of FO and aspirin is dependent on COX‐1 activity. As shown in Figure 3E and F, the aggregation of platelets from FO and aspirin‐treated WT mice was much weaker than that of FO‐fed COX‐1neo mice (genetic COX‐1 knockdown), indicating that the additional effect of FO and aspirin on platelet function is not dependent on COX‐1 inhibition.
Enhanced protective effect of FO and aspirin against thrombosis in vivo
As platelets from mice treated with both FO and aspirin exhibited reduced aggregation in vitro, we next investigated their response to thrombotic stimuli (Rose Bengal or ferric chloride) in vivo. The formation of an occlusive thrombus was monitored by measuring blood flow in the exposed carotid artery with a Doppler probe. In the photochemical‐induced carotid artery thrombosis model, as expected, both FO and aspirin treatment prolonged the time to complete occlusion (Figure 4A) in mice. Strikingly, the combination of FO and aspirin significantly delayed photochemical‐induced thrombus formation, as compared with FO or aspirin treatment alone (FO + aspirin, 114.7 ± 12.8 min; FO alone, 47.50 ± 7.21 min or aspirin alone, 40.67 ± 4.85 min; P < 0.05; Figure 4A). FO‐mediated inhibition of photochemical‐induced thrombosis was also detected in COX‐1neo mice (Figure 4A). However, the FO‐fed COX‐1neo mice exhibited a significantly faster occlusion time than WT mice co‐treated with FO and aspirin (Figure 4A), suggesting that the enhanced inhibitory effect of FO and aspirin on thrombogenesis is not dependent on COX‐1 activity. In the ferric chloride‐induced carotid arterial thrombosis mouse model, both FO intake and COX‐1 inhibition (aspirin and COX‐1neo) prolonged occlusion time (Figure 4B), and co‐administration of FO and aspirin significantly improved BFR decrease induced by ferric chloride as compared with each alone (Figure 4C)
Figure 4.
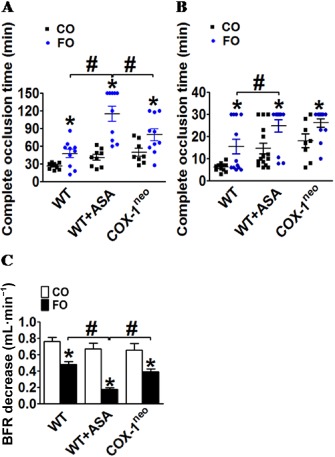
Protective effect of dietary FO plus aspirin against thrombus formation in vivo. (A) Effect of FO and aspirin on complete occlusion time in a photochemical‐induced thrombosis mouse model. *P < 0.05 versus CO, #P < 0.05 as indicated, n = 8–11; effect of FO and aspirin on complete occlusion time (B) and the decrease in blood flow rate (BFR) in a 30 min period (C) in the ferric chloride‐induced thrombosis mouse model. *P < 0.05 versus CO, #P < 0.05 as indicated, n = 8–15.
We also tested the combined effect of FO and aspirin on TxA2 synthesis. As previously described, low‐dose aspirin and COX‐1 knockdown markedly diminished urinary TxA2 metabolite content (∼90% suppression) (Yu et al., 2005). Combined treatment with FO and aspirin resulted in a similar suppression of Tx metabolites as aspirin alone, indicating that the additional effect of FO and aspirin on platelet function did not rely solely on TxA2 production (Figure 5).
Figure 5.

Effect of dietary FO and aspirin (ASA) on TxA2 metabolite (Tx‐M). Urinary Tx‐M was measured in a 24 h urine specimen collected in a metabolic cage. *P < 0.05, n = 10–15.
The combination of FO and aspirin confers additional protection against vascular neointima formation in response to injury
Given the beneficial effect of the combined treatment with FO and aspirin on platelet function, we examined the effect of FO and aspirin on mechanical injury‐induced arterial restenosis in mice. De‐endothelialization of femoral arteries was induced by insertion of a wire into the lumen. Arterial neointima hyperplasia was examined at 28 days post‐injury. As shown in Figure 6, wire injury triggered severe arterial neointima formation and vessel narrowing. Dietary FO intake resulted in a significant reduction in the I/M ratio and in the luminal narrowing, as compared with WT mice fed a CO diet, aspirin‐treated WT mice and COX‐1neo mice. In contrast, aspirin treatment and COX‐1 knockdown had no obvious effect on injury‐induced vascular remodelling of the neointima and luminal narrowing (Figure 6C and D), indicating that constitutively expressed COX‐1 is not involved in the pathogenesis of mechanical injury‐induced vascular remodelling. Interestingly, co‐administration of FO and aspirin attenuated vascular neointima formation more than FO intake alone, significantly reducing the I/M ratio (P < 0.05; Figure 6C) and luminal narrowing (P < 0.05; Figure 6D). In addition, the reductions observed in mice treated with both FO and aspirin were significantly greater than those of FO‐treated COX‐1neo mice (Figure 6C and D), suggesting that the synergistic protective effect of FO and aspirin against injury‐induced vascular remodelling is not dependent on COX‐1 activity.
Figure 6.
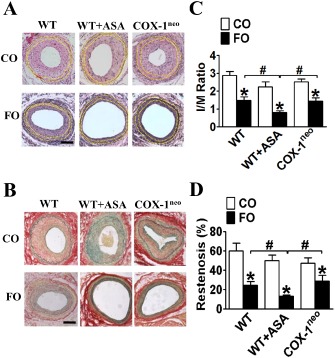
Effect of dietary FO plus aspirin (ASA) on vascular remodelling in response to mechanical injury. Representative haematoxylin and eosin (A) and Ponceaou S‐Picric acid‐Victoria blue (B) staining of cross sections of wire‐injured arteries from WT, aspirin‐treated andCOX‐1neo mice fed with a FO or CO diet. Scale bar, 50 μm; yellow dashed lines indicate both IEL and eternal elastic lamina. Quantification of I/M ratio (C) and restenosis (D) in arteries post‐injury from WT, aspirin‐treated and COX‐1neo mice fed a FO or CO diet. * P < 0.05 versus CO; #P < 0.05 as indicated, n = 7–11.
Dietary FO and aspirin intake suppress inflammatory reaction in response to injury
FO supplementation has been shown to reduce local inflammatory reactions and enhance resolution (Tomasdottir et al., 2013); and low‐dose aspirin exhibits anti‐inflammatory properties to a lesser extent (Morris et al., 2009). We further explored the perivascular inflammatory response to wire injury at day 28. As expected, ingestion of FO markedly suppressed both macrophage (CD68+, Figure 7A and B) and neutrophil (CD11b+, Figure 7C and D) infiltration in WT, WT/aspirin and COX‐1neo mice. Low‐dose aspirin intake tended to decrease inflammatory cell infiltration; however, these reductions did not reach significance (Figure 7B and D). In line with their effects on arterial neointima formation, the combined treatment with FO and aspirin led to a further, significant reduction in the perivascular infiltration of inflammatory cells‐CD68+ and CD11b+ cells (Figure 7B and D ). In addition, we detected less infiltration of inflammatory cells in WT mice treated with FO and aspirin than in FO‐fed COX‐1neo mice (Figure 7B and D). At the early stages of acute inflammation (3 days after injury), we observed a similar suppressive pattern of inflammatory cell infiltration, including CD68+ macrophages, CD11b+ neutrophil and CD3+ T lymphocytes, in mice fed FO alone and those treated with FO and aspirin (Figure 8A–F).
Figure 7.

Synergistic effect of dietary FO and aspirin (ASA) on perivascular inflammatory cell infiltration following injury. (A) Representative immunostaining of CD68 in injured artery cross sections from WT, aspirin‐treated and COX‐1neo mice fed a FO or CO diet. Scale bar, 50 μm; (B) Quantification of CD68+ cell for (A). *P < 0.05 versus CO control; #P < 0.05 as indicated, n = 6–12; (C) Representative immunostaining of CD11b in injured artery cross sections from WT, aspirin‐treated and COX‐1neo mice fed a FO or CO diet. Scale bar, 50 μm; (D) Quantification of CD11b+ cells for (C). * P < 0.05 versus CO; #P < 0.05 as indicated, n = 7–11.
Figure 8.
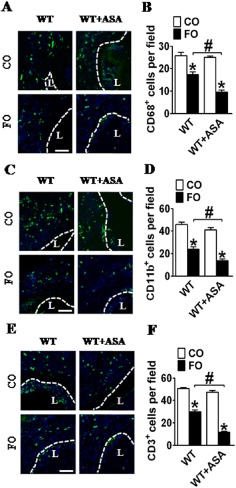
Effect of dietary FO and aspirin (ASA) on early recruitment of inflammatory cells in injured femoral arteries. Representative immunofluorescent staining of CD68(A), CD11b (C) and CD3 (E) in injured femoral artery sections from aspirin alone, FO alone, or FO‐ and aspirin‐treated mice. L, lumen of femoral artery; (B, D, F) Quantification of CD68+, CD11b+ and CD3+ cells in injured femoral artery sections from aspirin alone, FO alone, or FO‐ and aspirin‐treated mice. * P < 0.05 versus CO; #P < 0.05 as indicated, n = 6–7.
Perivascular infiltration by inflammatory cells such as macrophages may contribute to neointimal thickening through the generation of harmful reactive oxygen intermediates and secretion of growth and chemotatic factors (Inoue and Node, 2009). Consistent with previous reports (Oh et al., 2010), FO intake decreased M1 pro‐inflammatory gene expression, including IL‐6, TNF‐α and Clec10a (Figure 9A) in macrophages, and increased M2 anti‐inflammatory gene expression, such as IL‐10, macrophage mannose receptor (MMR), and Ym‐1 (Figure 9B). Aspirin treatment also reduced the expression of some pro‐inflammatory markers, such as TNF‐α and Clec10a (Figure 9A), but had no effects on M2 anti‐inflammatory genes (Figure 9B). Indeed, co‐treatment of FO and aspirin further inhibited M1 pro‐inflammatory gene expression and elevated M2 anti‐inflammatory gene expression, indicating the combination of FO and aspirin had a synergistic inhibitory effect on inflammatory reactions, induced partially by influencing macrophage polarization.
Figure 9.
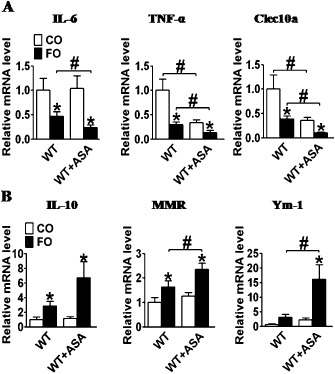
Effect of dietary FO and aspirin (ASA) on mRNA expression of anti‐ and pro‐inflammatory genes in macrophages. (A) IL‐6, TNF‐α and Clec10a gene expression in macrophages from aspirin alone, FO alone, or FO and aspirin‐treated mice; (B) IL‐10, MMR and Ym‐1 gene expression in macrophages from aspirin alone, FO alone, or FO and aspirin‐treated mice. *P < 0.05 versus CO; # P < 0.05 as indicated, n = 6–12.
Effect of dietary FO and aspirin intake on expression of endothelium markers and surface adhesion molecules on leukocytes
Leukocyte infiltration into inflamed areas, such as injured vessels, requires a precise sequence of events that involves the interaction of leukocytes with activated endothelial cells through modulated expression of surface adhesion molecules (Shah, 2003). The first and most important step for leukocyte recruitment is contact with endothelial cells, rolling and temporal arrest of leukocytes (Butcher, 1991). We examined the expression of surface adhesion molecules on leukocytes, such as lymphocyte function‐associated antigen 1 (LFA‐1), very late antigen‐4 (VLA‐4) and P‐selectin glycoprotein ligand‐1 (PSGL‐1). An FO‐supplemented diet reduced the expression of LFA‐1, VLA‐4 and PSGL‐1 on leukocytes as compared with a CO‐supplemented diet. In contrast, low‐dose aspirin had no effect on the expression of these molecules (Figure 10A). Interestingly, we observed that the combination of FO and aspirin significantly diminished the expression of LFA‐1, VLA‐4 and PSGL‐1 on leukocytes, as compared with FO alone.
Figure 10.
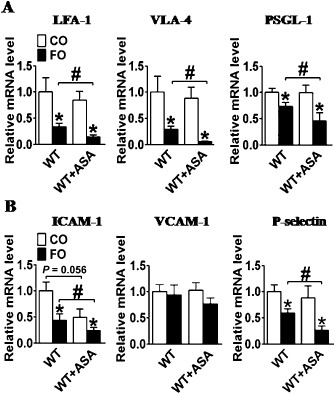
Effect of dietary FO and aspirin (ASA) on the expression of adhesion molecules on leukocytes and endothelium. (A) LFA‐1, VLA‐4, PSGL‐1 gene expression on leukocytes from aspirin alone, FO alone, or FO and aspirin‐treated mice;(B) ICAM‐1, VCAM‐1, and P‐selectin gene expression in aortas from aspirin alone, FO alone, or FO and aspirin‐treated mice.*P < 0.05 versus CO; #P < 0.05 as indicated, n = 5–8.
We next assessed the expression of the corresponding ligands in blood vessels. We observed a similar synergistic inhibition of the vascular expression of LFA‐1 ligand‐intercellular adhesion molecule (ICAM) and PSGL‐1 ligand‐P‐selectin in mice treated with FO and aspirin (Figure 10B).
Resolvin E1 generation in vivo
Resolvins are a group of anti‐inflammatory lipid mediators derived from ω3 PUFAs such as EPA and DHA, and can be generated in the presence of aspirin (Serhan et al., 2008). Hence, we determined plasma resolvin E1 levels in FO‐ or CO‐treated mice by LC‐MS/MS (Figure 11A). The formation of resolvin E1 was confirmed by the MS/MS spectrum consistent with synthetic resolvin E1 (data not shown). Indeed, FO or aspirin alone promoted the production of resolvin E1 in mice (Figure 11B). Strikingly, co‐treatment with FO and aspirin significantly enhanced resolvin E1 biosynthesis in plasma (P < 0.05; Figure 11B).
Figure 11.
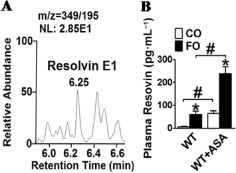
Detection of resolvin E1 in plasma of aspirin (ASA), FO, or FO and aspirin‐treated mice. (A) Selected ion monitoring chromatogram of resolvin E1 isolated from freshly collected plasma; (B) quantification of plasma resolvin E1 production in aspirin, FO, or FO and aspirin‐treated mice. *P < 0.05 versus CO; #P < 0.05 as indicated, n = 8–11.
Discussion and conclusions
In this study, we have shown that dietary FO has broad cardio‐protective effects, such as reducing TG and lowering BP. When combined with low‐dose aspirin, FO ingestion exerted additional protection against platelet aggregation and thrombosis in mouse models; this effect was not dependent on COX‐1 inhibition. Furthermore, we demonstrated that co‐treatment with FO and aspirin significantly reduced arterial neointima formation in response to injury, as compared with either FO or aspirin alone, through the suppression of vascular inflammatory reactions and the induction of M2 macrophage polarization. These findings indicate that FO supplementation could be a favourable adjuvant therapy for patients with stable coronary artery disease (CAD).
ω3 PUFAs, either in FO supplements or as prescription ethyl esters, are an accepted therapy for reducing TG levels (Kris‐Etherton et al., 2002). These substances, which include EPA and DHA, are believed to reduce TG levels primarily by promoting fatty‐acid degradation via peroxisomal β‐oxidation, inhibiting hepatic lipogenesis and accelerating the clearance of TG from the plasma (Harris et al., 2008). Both EPA and DHA have similar TG‐lowering properties (Grimsgaard et al., 1997). Compared with the well‐established TG‐lowering effect of ω3 PUFAs, the effects of FO on HDLC and LDLC appear to be weaker (Harris, 1997). Consistently, our data showed dietary FO intake reduced blood levels of TG, cholesterol, HDLC and LDLC in all three groups tested. However, we did not observe any effect of aspirin on lipid profiles in mice. Despite the fact that high levels of LDLC increase the risk for CVD, the Japan EPA Lipid Intervention Study involving 18 645 patients treated with statins showed that the beneficial effects of EPA on CVD were not associated with reductions in LDLC levels (Yokoyama et al., 2007; Sasaki et al., 2012), indicating that other factors may contribute to the cardio‐protective effect of EPA, including a lowering of BP and inhibitory effect on platelet activity. Indeed, we observed a mild reduction in BP and suppression of platelet aggregation following FO ingestion, consistent with previous reports (Larson et al., 2013; Miller et al., 2014).
The feeding of FO led to the incorporation of ω3 PUFAs (both EPA and DHA) into cell membranes in mice. The incorporated EPA and DHA can inhibit two‐series prostanoid production, including Tx, by competing with AA at the COX enzyme active site. As anticipated, we detected a marked reduction in Tx production in FO‐fed mice. This was similar to mice treated with aspirin, which acetylates COX and blocks the metabolism of AA to Tx in platelets. Furthermore, we also observed a suppression of platelet aggregation in response to either ADP or collagen challenge and impaired photochemical‐ and ferric chloride‐induced arterial thrombosis in FO‐fed mice. Thus, the combination of aspirin and FO may work in parallel to augment the inhibition of platelet aggregation and thrombosis, partially through shifting membrane phospholipid metabolism towards three‐series prostanoid production (Smith, 2005). However, the synergistic effect of aspirin and FO on platelet function is independent of COX‐1 inhibition and TxA2 production. One possible explanation for this is the trans‐endothelial generation of EPA‐derived resolvins, through the interaction of EPA with aspirin, which has previously been shown to block both Tx‐and ADP‐induced platelet activation (Dona et al., 2008; Fredman et al., 2010). Another possible explanation is that the combination of FO and aspirin affects the conversion of lysophosphatidic acid to lysophosphatidylcholine, which is strongly correlated with impaired platelet activation induced by AA (Abdolahi et al., 2014). In healthy humans, co‐administration of EPA, DHA and low‐dose aspirin was recently reported to reduce platelet function when combined, although the individual components did not (Block et al., 2012). A similar synergistic effect of FO and aspirin was also observed in patients with diabetes mellitus (Block et al., 2012; Abdolahi et al., 2014).
Importantly, aspirin resistance or a poor response to aspirin is more common in diabetic patients than in healthy individuals (Pignone et al., 2010). Therefore, the combination of FO and aspirin could be more effective than aspirin alone in reducing platelet aggregation in subjects with cardiovascular risks, particularly those with diabetes mellitus. Despite effectively reducing myocardial infarction and other thrombotic events after PCI, aspirin treatment has failed to inhibit the progression of arterial restenosis (Steinhubl and Berger, 2009). Similar to balloon‐induced carotid artery injury (Yang et al., 2004), aspirin had no effect on vascular neointima growth in our femoral artery injury model. In contrast, co‐treatment with FO and aspirin markedly suppressed neointima formation as compared with either treatment alone; this effect was independent of COX‐1 inhibition, as we did not observe the synergistic effect in COX‐1 knockdown mice. Again, these results suggest that aspirin and FO interact biologically, for example, in producing the mediators E‐ and D‐series resolvins, which are generated from EPA and DHA in the presence of aspirin (Spite and Serhan, 2010). Resolvin E1 was detected in the plasma of human subjects, and its levels were negatively correlated with symptomatic peripheral artery disease (Ho et al., 2010). In vitro, resolvin E1 blocked PDGF‐stimulated migration of human VSMCs by decreasing the phosphorylation of the PDGFRβ (Ho et al., 2010). Indeed, we detected the generation of resolvin E1 in plasma of FO‐ and aspirin‐treated mice (Figure 11). However, the exact roles of EPA‐derived resolvin E1 in injury‐induced vascular remodelling remain to be determined. Restenosis after stent deployment is considered to be a wound‐healing response to mechanical injury. The inflammatory response plays an essential role in transducing signals from the site of vascular injury to ultimately promote neointimal growth (Welt and Rogers, 2002). It has been long known that FO has beneficial anti‐inflammatory properties (Calder and Grimble, 2002), which may contribute to its protective effects against vascular remodelling in response to injury (Faggin et al., 2000; de Roos et al., 2009). In line with the synergistic suppression of injury‐induced neointima growth, the combination of FO and aspirin dramatically attenuated perivascular infiltration of inflammatory cells (macrophages and neutrophils), as compared with FO alone. Moreover, treatment with FO and aspirin markedly reduced the expression of pro‐inflammatory genes (IL‐6, TNF‐α and Celc10a) and enhanced the expression of anti‐inflammatory genes (MMR, Ym‐1) in macrophages, as well as significantly inhibiting the expression of adhesion molecules on leukocytes, as compared with aspirin or FO alone. D‐series resolvins protect against vascular neointima hyperplasia in response to injury (Miyahara et al., 2013). Recently, resolvin E1 has attracted much attention (Buckley et al., 2014); it has been found to ameliorate inflammatory reactions in many conditions such as atopic dermatitis (Kim et al., 2012), allergic airway inflammation (Haworth et al., 2011), colitis (Arita et al., 2005) and acute lung injury (El Kebir et al., 2012). Therefore, the synergistic effect of FO and aspirin on the suppression of injury‐induced neointima growth may be attributed to, at least partially, the anti‐inflammatory effects of pro‐resolving lipid mediators.
In summary, we found that co‐administration of FO and aspirin synergistically inhibited platelet aggregation and vascular neointima formation in response to mechanical injury. Our results indicate that FO supplementation could be used as adjuvant therapy in patients with stable CAD, including those undergoing PCI.
Author contributions
Y. G., M. L., C. D. F. and Y. Y. designed the research associated with the project. Y. G., M. L., L. P., X. L., F. Y., J. Z. and B. X. performed experiments. Q. Z., W. S., H. Y. and L. Z. provided important technical support. Y. G., C. D. F. and Y. Y. wrote the paper.
Conflicts of interest
None.
Acknowledgements
This work was supported by grants from the Ministry of Science and Technology of China (2012CB945100, 2011CB503906), the National Natural Science Foundation of China (81030004), National Science Foundation of China – Canadian Institutes of Health Research joint grant (81161120538 and CIHR‐CCI117951), the One Hundred Talents Program of the Chinese Academy of Sciences (2010OHTP10) and a grant from the Jiangsu Province's Key Discipline of Medicine (XK201118). C. D. F. is a Canada Research Chair holder and recipient of an Ontario Heart and Stroke Foundation Career Investigator award. Y. Y. is a Fellow at the Jiangsu Collaborative Innovation Center for Cardiovascular Disease Translational Medicine.
References
- Abdolahi A, Georas SN, Brenna JT, Cai X, Thevenet‐Morrison K, Phipps RP et al (2014). The effects of aspirin and fish oil consumption on lysophosphatidylcholines and lysophosphatidic acids and their correlates with platelet aggregation in adults with diabetes mellitus. Prostaglandins Leukot Essent Fatty Acids 90: 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013a). The Concise Guide to PHARMACOLOGY 2013/14: Catalytic receptors. Br J Pharmacol 170: 1676–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013b). The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arita M, Yoshida M, Hong S, Tjonahen E, Glickman JN, Petasis NA et al (2005). Resolvin E1, an endogenous lipid mediator derived from omega‐3 eicosapentaenoic acid, protects against 2,4,6‐trinitrobenzene sulfonic acid‐induced colitis. Proc Natl Acad Sci U S A 102: 7671–7676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astrup A (2014). Yogurt and dairy product consumption to prevent cardiometabolic diseases: epidemiologic and experimental studies. Am J Clin Nutr 99 (5 Suppl.): 1235S–1242S. [DOI] [PubMed] [Google Scholar]
- Block RC, Kakinami L, Jonovich M, Antonetti I, Lawrence P, Meednu N et al (2012). The combination of EPA + DHA and low‐dose aspirin ingestion reduces platelet function acutely whereas each alone may not in healthy humans. Prostaglandins Leukot Essent Fatty Acids 87: 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley CD, Gilroy DW, Serhan CN (2014). Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity 40: 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher EC (1991). Leukocyte‐endothelial cell recognition: three (or more) steps to specificity and diversity. Cell 67: 1033–1036. [DOI] [PubMed] [Google Scholar]
- Calder PC (2014). Marine omega‐3 fatty acids and inflammatory processes: effects, mechanisms and clinical relevance. Biochim Biophys Acta. doi: 10.1016/j.bbalip.2014.08.010 [DOI] [PubMed] [Google Scholar]
- Calder PC, Grimble RF (2002). Polyunsaturated fatty acids, inflammation and immunity. Eur J Clin Nutr 56: S14–S19. [DOI] [PubMed] [Google Scholar]
- Capodanno D, Patel A, Dharmashankar K, Ferreiro JL, Ueno M, Kodali M et al (2011). Pharmacodynamic effects of different aspirin dosing regimens in type 2 diabetes mellitus patients with coronary artery disease. Circ Cardiovasc Intervent 4: 180–187. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, FitzGerald GA (2006). Cyclooxygenases, microsomal prostaglandin E synthase‐1, and cardiovascular function. J Clin Invest 116: 1391–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillinger JG, Drissa A, Sideris G, Bal dit Sollier C, Voicu S, Manzo Silberman S et al (2012). Biological efficacy of twice daily aspirin in type 2 diabetic patients with coronary artery disease. Am Heart J 164: 600–606 e601. [DOI] [PubMed] [Google Scholar]
- Dona M, Fredman G, Schwab JM, Chiang N, Arita M, Goodarzi A et al (2008). Resolvin E1, an EPA‐derived mediator in whole blood, selectively counterregulates leukocytes and platelets. Blood 112: 848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Kebir D, Gjorstrup P, Filep JG (2012). Resolvin E1 promotes phagocytosis‐induced neutrophil apoptosis and accelerates resolution of pulmonary inflammation. Proc Natl Acad Sci U S A 109: 14983–14988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faggin E, Puato M, Chiavegato A, Franch R, Pauletto P, Sartore S (2000). Fish oil supplementation prevents neointima formation in nonhypercholesterolemic balloon‐injured rabbit carotid artery by reducing medial and adventitial cell activation. Arterioscler Thromb Vasc Biol 20: 152–163. [DOI] [PubMed] [Google Scholar]
- Floyd CN, Ferro A (2014). Mechanisms of aspirin resistance. Pharmacol Ther 141: 69–78. [DOI] [PubMed] [Google Scholar]
- Fredman G, Van Dyke TE, Serhan CN (2010). Resolvin E1 regulates adenosine diphosphate activation of human platelets. Arterioscl Throm Vas 30: 2005–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajos G, Rostoff P, Undas A, Piwowarska W (2010). Effects of polyunsaturated omega‐3 fatty acids on responsiveness to dual antiplatelet therapy in patients undergoing percutaneous coronary intervention: the OMEGA‐PCI (OMEGA‐3 fatty acids after PCI to modify responsiveness to dual antiplatelet therapy) study. J Am Coll Cardiol 55: 1671–1678. [DOI] [PubMed] [Google Scholar]
- Gouya G, Arrich J, Wolzt M, Huber K, Verheugt FW, Gurbel PA et al (2014). Antiplatelet treatment for prevention of cerebrovascular events in patients with vascular diseases: a systematic review and meta‐analysis. Stroke 45: 492–503. [DOI] [PubMed] [Google Scholar]
- Grimsgaard S, Bonaa KH, Hansen JB, Nordoy A (1997). Highly purified eicosapentaenoic acid and docosahexaenoic acid in humans have similar triacylglycerol‐lowering effects but divergent effects on serum fatty acids. Am J Clin Nutr 66: 649–659. [DOI] [PubMed] [Google Scholar]
- Grove EL, Hvas AM, Larsen SB, Mortensen SB, Kristensen SD (2011). Effect of platelet turnover on whole blood platelet aggregation in patients with coronary artery disease: reply to a rebuttal. J Thromb Haemost 9: 889–890. [DOI] [PubMed] [Google Scholar]
- Hanke AA, Roberg K, Monaca E, Sellmann T, Weber CF, Rahe‐Meyer N et al (2010). Impact of platelet count on results obtained from multiple electrode platelet aggregometry (multiplate). Eur J Med Res 15: 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris WS (1997). n‐3 fatty acids and serum lipoproteins: human studies. Am J Clin Nutr 65 (5 Suppl.): 1645S–1654S. [DOI] [PubMed] [Google Scholar]
- Harris WS, Miller M, Tighe AP, Davidson MH, Schaefer EJ (2008). Omega‐3 fatty acids and coronary heart disease risk: clinical and mechanistic perspectives. Atherosclerosis 197: 12–24. [DOI] [PubMed] [Google Scholar]
- Harris WS, Dayspring TD, Moran TJ (2013). Omega‐3 fatty acids and cardiovascular disease: new developments and applications. Postgrad Med 125: 100–113. [DOI] [PubMed] [Google Scholar]
- Haworth O, Cernadas M, Levy BD (2011). NK cells are effectors for resolvin E1 in the timely resolution of allergic airway inflammation. J Immunol 186: 6129–6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho KJ, Spite M, Owens CD, Lancero H, Kroemer AH, Pande R et al (2010). Aspirin‐triggered lipoxin and Resolvin E1 modulate vascular smooth muscle phenotype and correlate with peripheral atherosclerosis. Am J Pathol 177: 2116–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Node K (2009). Molecular basis of restenosis and novel issues of drug‐eluting stents. Circ J 73: 615–621. [DOI] [PubMed] [Google Scholar]
- Kim TH, Kim GD, Jin YH, Park YS, Park CS (2012). Omega‐3 fatty acid‐derived mediator, Resolvin E1, ameliorates 2,4‐dinitrofluorobenzene‐induced atopic dermatitis in NC/Nga mice. Int Immunopharmacol 14: 384–391. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kris‐Etherton PM, Harris WS, Appel LJ (2002). Fish consumption, fish oil, omega‐3 fatty acids, and cardiovascular disease. Circulation 106: 2747–2757. [DOI] [PubMed] [Google Scholar]
- Larson MK, Tormoen GW, Weaver LJ, Luepke KJ, Patel IA, Hjelmen CE et al (2013). Exogenous modification of platelet membranes with the omega‐3 fatty acids EPA and DHA reduces platelet procoagulant activity and thrombus formation. Am J Physiol Cell Physiol 304: C273–C279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev EI, Solodky A, Harel N, Mager A, Brosh D, Assali A et al (2010). Treatment of aspirin‐resistant patients with omega‐3 fatty acids versus aspirin dose escalation. J Am Coll Cardiol 55: 114–121. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001). Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- Marangoni F, Poli A (2013). Clinical pharmacology of n‐3 polyunsaturated fatty acids: non‐lipidic metabolic and hemodynamic effects in human patients. Atheroscler Suppl 14: 230–236. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijajlovic MD, Shulga O, Bloch S, Covickovic‐Sternic N, Aleksic V, Bornstein NM (2013). Clinical consequences of aspirin and clopidogrel resistance: an overview. Acta Neurol Scand 128: 213–219. [DOI] [PubMed] [Google Scholar]
- Miller PE, Van Elswyk M, Alexander DD (2014). Long‐chain omega‐3 fatty acids eicosapentaenoic acid and docosahexaenoic acid and blood pressure: a meta‐analysis of randomized controlled trials. Am J Hypertens 27: 885–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyahara T, Runge S, Chatterjee A, Chen M, Mottola G, Fitzgerald JM et al (2013). D‐series resolvin attenuates vascular smooth muscle cell activation and neointimal hyperplasia following vascular injury. FASEB J 27: 2220–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris T, Stables M, Hobbs A, de Souza P, Colville‐Nash P, Warner T et al (2009). Effects of low‐dose aspirin on acute inflammatory responses in humans. J Immunol 183: 2089–2096. [DOI] [PubMed] [Google Scholar]
- Ogawa N, Kobayashi Y (2009). Total synthesis of Resolvin E1. Tetrahedron Lett 50: 6079–6082. [Google Scholar]
- Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W et al (2010). GPR120 is an omega‐3 fatty acid receptor mediating potent anti‐inflammatory and insulin‐sensitizing effects. Cell 142: 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens AP, Lu Y, Whinna HC, Gachet C, Fay WP, Mackman N (2011). Towards a standardization of the murine ferric chloride‐induced carotid arterial thrombosis model. J Thromb Haemost 9: 1862–1863. [DOI] [PubMed] [Google Scholar]
- Pakala R, Benedict C (1999). Eicosapentaenoic acid and docosahexaenoic acid selectively attenuate U46619‐induced smooth muscle cell proliferation. Lipids 34: 915–920. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al; NC‐IUPHAR (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrich BG, Fogelstrand P, Partridge AW, Yousefi N, Ablooglu AJ, Shattil SJ et al (2007). The antithrombotic potential of selective blockade of talin‐dependent integrin alpha IIb beta 3 (platelet GPIIb‐IIIa) activation. J Clin Invest 117: 2250–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignone M, Alberts MJ, Colwell JA, Cushman M, Inzucchi SE, Mukherjee D et al (2010). Aspirin for primary prevention of cardiovascular events in people with diabetes: a position statement of the American Diabetes Association, a scientific statement of the American Heart Association, and an expert consensus document of the American College of Cardiology Foundation (vol 33, pg 1395, 2010). Diabetes Care 33: 2129–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Roos B, Mavrommatis Y, Brouwer IA (2009). Long‐chain n‐3 polyunsaturated fatty acids: new insights into mechanisms relating to inflammation and coronary heart disease. Br J Pharmacol 158: 413–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki J, Yokoyama M, Matsuzaki M, Saito Y, Origasa H, Ishikawa Y et al (2012). Relationship between coronary artery disease and non‐HDL‐C, and effect of highly purified EPA on the risk of coronary artery disease in hypercholesterolemic patients treated with statins: sub‐analysis of the Japan EPA Lipid Intervention Study (JELIS). J Atheroscler Thromb 19: 194–204. [DOI] [PubMed] [Google Scholar]
- Schwartz L, Bourassa MG, Lesperance J, Aldridge HE, Kazim F, Salvatori VA et al (1988). Aspirin and dipyridamole in the prevention of restenosis after percutaneous transluminal coronary angioplasty. N Engl J Med 318: 1714–1719. [DOI] [PubMed] [Google Scholar]
- Serhan CN, Clish CB, Brannon J, Colgan SP, Chiang N, Gronert K (2000). Novel functional sets of lipid‐derived mediators with antiinflammatory actions generated from omega‐3 fatty acids via cyclooxygenase 2‐nonsteroidal antiinflammatory drugs and transcellular processing. J Exp Med 192: 1197–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Chiang N, Van Dyke TE (2008). Resolving inflammation: dual anti‐inflammatory and pro‐resolution lipid mediators. Nat Rev Immunol 8: 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah PK (2003). Inflammation, neointimal hyperplasia, and restenosis: as the leukocytes roll, the arteries thicken. Circulation 107: 2175–2177. [DOI] [PubMed] [Google Scholar]
- Smith WL (2005). Cyclooxygenases, peroxide tone and the allure of fish oil. Curr Opin Cell Biol 17: 174–182. [DOI] [PubMed] [Google Scholar]
- Spite M, Serhan CN (2010). Novel lipid mediators promote resolution of acute inflammation: impact of aspirin and statins. Circ Res 107: 1170–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhubl SR, Berger PB (2009). Aspirin following PCI: too much of a good thing? Eur Heart J 30: 882–884. [DOI] [PubMed] [Google Scholar]
- Tomasdottir V, Vikingsson A, Freysdottir J, Hardardottir I (2013). Dietary fish oil reduces the acute inflammatory response and enhances resolution of antigen‐induced peritonitis. J Nutr Biochem 24: 1758–1765. [DOI] [PubMed] [Google Scholar]
- Welt FG, Rogers C (2002). Inflammation and restenosis in the stent era. Arterioscler Thromb Vasc Biol 22: 1769–1776. [DOI] [PubMed] [Google Scholar]
- Yan S, Zhang Q, Zhong X, Tang J, Wang Y, Yu J et al (2014). I Prostanoid Receptor‐mediated inflammatory pathway promotes hepatic gluconeogenesis through activation of PKA and inhibition of AKT. Diabetes 63: 2911–2923. [DOI] [PubMed] [Google Scholar]
- Yang HM, Kim HS, Park KW, You HJ, Jeon SI, Youn SW et al (2004). Celecoxib, a cyclooxygenase‐2 inhibitor, reduces neointimal hyperplasia through inhibition of Akt signaling. Circulation 110: 301–308. [DOI] [PubMed] [Google Scholar]
- Yokoyama M, Origasa H, Matsuzaki M, Matsuzawa Y, Saito Y, Ishikawa Y et al (2007). Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open‐label, blinded endpoint analysis. Lancet 369: 1090–1098. [DOI] [PubMed] [Google Scholar]
- Yu Y, Cheng Y, Fan JJ, Chen XS, Klein‐Szanto A, Fitzgerald GA et al (2005). Differential impact of prostaglandin H synthase 1 knockdown on platelets and parturition. J Clin Invest 115: 986–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Sun Q, Hu FB, Ye XW, Yu ZJ, Zong G et al (2012). Erythrocyte n‐3 fatty acids and metabolic syndrome in middle‐aged and older Chinese. J Clin Endocr Metab 97: E973–E977. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zou FF, Tang J, Zhang QQ, Gong YJ, Wang QS et al (2013). Cyclooxygenase‐2‐derived prostaglandin e‐2 promotes injury‐induced vascular neointimal hyperplasia through the e‐prostanoid 3 receptor. Circ Res 113: 104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]