

Abstract
Background and Purpose
The epithelial sodium channel (ENaC) is expressed in vascular endothelial cells and is a negative modulator of vasodilation. However, the role of endothelial ENaCs in salt‐sensitive hypertension remains unclear. Here, we have investigated how endothelial ENaCs in Sprague‐Dawley (SD) rats respond to high‐salt (HS) challenge.
Experimental Approach
BP and plasma aldosterone levels were measured. We used patch‐clamp technique to record ENaC activity in split‐open mesenteric arteries (MAs). Western blot and Griess assay were used to detect expression of α‐ENaCs, eNOS and NO. Vasorelaxation in second‐order MAs was measured with wire myograph assays.
Key Results
Functional ENaCs were observed in endothelial cells and their activity was significantly decreased after 1 week of HS diet. After 3 weeks of HS diet, ENaC expression was also reduced. When either ENaC activity or expression was reduced, endothelium‐dependent relaxation (EDR) of MAs, in response to ACh, was enhanced. This enhancement of EDR was mimicked by amiloride, a blocker of ENaCs. By contrast, HS diet significantly increased contractility of MAs, accompanied by decreased eNOS activity and NO levels. However, ACh‐induced release of NO was much higher in MAs isolated from HS rats than those from NS rats.
Conclusions and Implications
HS intake increased the BP of SD rats, but simultaneously enhanced EDR by reducing ENaC activity and expression due to feedback inhibition. Therefore, ENaCs may play an important role in endothelial cells allowing the vasculature to adapt to HS conditions.
Linked Articles
This article is part of a themed section on Chinese Innovation in Cardiovascular Drug Discovery. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2015.172.issue-23
Abbreviations
- ENaC
epithelial sodium channel
- HS
high‐salt
- NS
normal‐salt
- SD rat
Sprague‐Dawley rat
- PO
open probability
- MAECs
mesenteric artery endothelial cells
- EDR
endothelium‐dependent relaxation
- SMC
smooth muscle cell
Tables of Links
| TARGETS |
|---|
| Ion channels |
| ENaC, epithelial sodium channel |
| ENaC‐α |
| LIGANDS | |
|---|---|
| ACh | NO |
| Aldosterone | NTG, nitroglycerin |
| Amiloride |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Numerous studies have demonstrated that high salt intake plays a critical role in the pathogenesis of hypertension, but the mechanism(s) by which high salt intake elevates BP is complex. Abnormalities in the renin‐angiotensin‐aldosterone system, the sympathetic nervous system, transmembrane sodium transport and even the vascular endothelium are all involved in the pathogenesis of salt‐sensitive hypertension. A decrease in dietary salt improved endothelial function, as assessed by flow‐mediated dilatation (Dickinson et al., 2009). However, the mechanism by which high salt causes endothelial dysfunction remains unclear.
The amiloride‐sensitive epithelial sodium channel (ENaC) has been most extensively described in the apical plasma membrane of epithelia where it mediates sodium transport in kidney, colon, lung, and sweat glands (Garty and Palmer, 1997). In these tissues, ENaCs mediate Na+ transport across epithelial membranes to regulate salt balance or to maintain an appropriate level of hydration. Recently, ENaCs have been identified in human endothelial cells and their function affects cell volume (Chen et al., 2004; Oberleithner et al., 2004) and stiffness of these cells (Oberleithner et al., 2006; 2007). Interestingly, inhibition of ENaCs in endothelium increased release of NO (Hashikabe et al., 2006; Oberleithner et al., 2007). The regulation of ENaCs is tissue‐specific and is mediated by the mineralocorticoid hormone aldosterone and aldosterone‐induced proteins, for example, the serum‐ and glucocorticoid‐regulated kinase 1 (sgk1; Vallon et al., 2005). In the presence of aldosterone, increasing extracellular sodium by 5 mM could stiffen endothelial cells by 25% and reduced NO release and increased vascular tone in vitro. These effects were prevented by a blocker of ENaCs, amiloride (Oberleithner et al., 2007). A later study showed that inhibition of ENaCs could activate endothelial NOS (eNOS) through the PI3K/Akt pathway, leading to an increase in NO release (Perez et al., 2009). A more recent study in vivo, using atomic force microscopy, revealed that the stiffness of endothelial cells was strongly related to the expression levels of ENaCs. Recent studies, in mice with Liddle's syndrome, revealed that in addition to their role in effects on the kidney, ENaCs in the vascular endothelium may participate in regulation of vascular function, as well as in developing hypertension by increasing the stiffness of endothelial cells (Jeggle et al., 2013).
Earlier studies have mostly focused on the crucial role of ENaCs in regulating the sodium homeostasis in the kidney, especially the functional role in regulating BP (Pratt, 2005; Sun et al., 2011). Excessive salt intake activates ENaCs in the principal cells of the cortical collecting duct, enhances Na+ reabsorption and water retention, and eventually causes salt‐sensitive hypertension (Aoi et al., 2007; Kakizoe et al., 2009). However, to the best of our knowledge, it remains unknown whether dietary salt could target endothelial ENaCs. The results presented here suggest that HS diet increased BP, but the effects of HS on endothelial ENaC activity in Sprague‐Dawley (SD) rats also induced a feedback inhibition of the development of salt‐sensitive hypertension.
Methods
Animals
All animal care and experimental procedures were approved by the Harbin Medical University Animal Supervision Committee. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
A total of 220 male SD rats (220–240 g) were used in the experiments described here. One hundred and forty rats were randomly assigned into seven groups (20 rats in each group) and were fed with either normal salt diet (NS, 0.3% NaCl, w/w for 0, 7, 14 and 21 days) or HS diet (HS, 8% NaCl, w/w for 7, 14 and 21 days). At the end of these treatment periods, the systolic BP (SBP) was measured in conscious SD rats, by the tail‐cuff method (BP 98A, Softon, Tokyo, Japan). Then the rats were killed and blood was collected from main abdominal artery for the measurement of plasma aldosterone. The heart and whole kidney were quickly excised and weighed in cold (4°C) buffer. Samples from six rats of each assigned group were used to provide primary cultures of mesenteric artery endothelial cells (MAECs). The second‐order MAs isolated from 14 rats of each assigned group were prepared to test the vasodilation and to record single‐channel activity of ENaCs. Eighty rats were used to assess additional Western blot, patch‐clamp analysis and Griess assays (14 rats were used to examine the expression levels of α‐ENaC in MAs with or without endothelial cells and ten rats were used to demonstrate the specificity of α‐ENaC antibody in primary cultured MAECs; eight rats were used to characterize the single‐channel conductance, and the remaining 48 rats were used to examine eNOS activity and NO production).
Primary culture of rat MAECs
The MAECs from rat MAs were isolated as previously described (Ashley et al., 2002), with minor modifications. Briefly, SD rats were given heparin and anesthetized with 10% chloral hydrate (4 mL·kg−1). The abdomen was opened, and the heart was perfused with sterile and chilled physiological saline solution (PSS; composition, in mM: 137 NaCl, 5.4 KCl, 0.05 CaCl2, 0.4 KH2PO4, 0.4 Na2HPO4, 4.4 NaHCO3 and 10 HEPES; pH 7.4 with HCl) to remove circulating blood from the vasculature. The mesenteric vascular bed was dissected out and all the venous branches of the mesenteric bed were rapidly excised, under a dissecting microscope. The remaining arterial branches were digested with 0.2 mg·mL−1 collagenase I for 1 h at 37°C with mild shaking. After centrifugation at 1000× g for 5 min, the pelleted cells were resuspended in DMEM supplemented with 20% FBS (v/v), 50 μg·mL−1 heparin, and were then plated in gelatin‐coated Petri dish. Two hours later, non‐adherent cells were removed, and the adherent endothelial cells were cultured at 37°C with 5% CO2 for 3–5 days. Endothelial cells were identified by their cobblestone morphology and immunostaining with an antibody against CD31. These cells were used for experiments without further passage.
In situ patch‐clamp recording
As described by Climent et al., (2011), in situ patch‐clamp recording of ENaC single‐channel current were performed using intact vascular endothelium. Dissected mesenteric vascular bed was placed in a Petri dish containing cold PSS. Second‐order branches of MA were dissected. A U‐shape was cut out on the dorsal wall of the arterial segment. They were then placed on a 5 × 5‐mm cover glass coated with L‐polylysine and transferred to a chamber mounted on an inverted Nikon microscope (Nikon, Tokyo, Japan), allowing direct access to the endothelial cell layer. Single‐channel ENaC currents were recorded in cell‐attached configuration with an Axon Multi‐clamp 200B amplifier (Axon Instruments, Foster City, CA, USA) at room temperature (22–24°C). Patch pipettes were pulled from borosilicate glass with a Sutter P‐97 horizontal puller, and resistance of the pipettes was ranged between 6∼10 MΩ when filled with pipette solution (composition, in mM: 115 NaCl, 4.5 KCl, 0.1 EGTA, 5 HEPES and 5 Na‐HEPES; pH 7.2 with NaOH). The bath solution (composition, in mM: 115 NaCl, 4.5 KCl, 1 MgCl2, 1 CaCl2, 5 HEPES and 5 Na‐HEPES; pH 7.2 with NaOH) was stationary while the chamber volume was ∼0.8 mL. The bath solution contained (in mM): The single‐channel currents were recorded at least over 15 min immediately after gigaseal formation. For most experiments, the data were acquired by application of 0 mV to patch pipettes and were sampled at 5 kHz and low‐pass filtered at 1 kHz with Clampex 10.2 software (Molecular Devices, Sunnyvale, CA, USA). Prior to analysis, the single‐channel traces were further filtered at 30 Hz. Open probability (PO) was calculated as follow: PO = NPO/N, where N (N was estimated by the current amplitude histogram) represents the apparent number of active channels in the patch. The current–voltage (i–V) relationship was constructed using the single‐channel amplitude measured at the indicated pipette voltages, as a function of voltages. (Vpipette means the voltages applied to the electrodes during recordings, therefore, −Vpipette indicates that the intracellular potential deviated from the resting potential of the apical membrane of the MAECs). The slope conductance was fit by linear regression by SigmaPlot software (Jandel Scientific, San Diego, CA, USA).
Wire myograph studies
The relaxtion of rings of isolated MAs was measured using an isometric myograph (Danish Myo Technology, Aarhus, Denmark), as previously described (Yang et al., 2010). The second‐order MAs were prepared as described above. Arterial segments (100–200 μm, lumen diameter, 1.8–2 mm long) were mounted in the myograph, equilibrated in PSS at 37°C and bubbled with a mixture of 5% CO2 in 95% O2. In some rings, the endothelium was removed by gentle rubbing of the intimal surface with a hair (Dora et al., 2000). After measurement of the passive‐tension internal circumference characteristics, the tension was set to an estimated in vivo internal circumference. After a 60 min stabilization period, KPSS (containing 60 mM K+) was added to the chambers and washed out with PSS until a reproducible maximal contraction was achieved. Vessels were pre‐contracted with phenylephrine (10 μM). Endothelium‐dependent and independent relaxation was assessed by measuring dilatory responses to cumulative concentrations of ACh (0.1 nM to 10 μM) and nitroglycerin (NTG, 0.1 nM to 10 μM) respectively. To evaluate the possible role of ENaCs in such relaxation, the artery segments were pre‐incubated with amiloride (0.5 μM) for 10 min before assessing their relaxation response to ACh and NTG. The half maximal effective agonist concentration (EC50) and maximum response (E max) were calculated from each agonist concentration–response curve using a logistic function from OriginPro 8.5 (OriginLab Corporation, Northampton, MA, USA).
Immunofluorescence and Western blotting
MAECs grown to full differentiation were fixed with 4% paraformaldehyde. For the MAs, tissues were dehydrated with 20% sucrose at 4°C overnight, embedded into optimal cutting temperature solution (TissueTek, Sakura Finetek, Torrance, CA, USA), and cut at 6 μm thickness with freezing microtome (Leica Biosystem, Heidelberger, Germany). Cells and artery sections were then permeated with 0.25% Trixiton X‐100 and blocked with 1% BSA 30 min prior to incubation with primary antibody. For double staining, we double‐labelled the cells with antibodies against CD31, an endothelial cell marker (Abcam, Cambridge, MA, USA, ab24590, 1:100) and α‐ENaC (Abcam, ab65710, 1:200 ) for 2 h at room temperature (22–24°C), followed by corresponding secondary fluorescence antibodies. Hoechst 33342 (10 μM) was used to stain nuclei.
For Western blot analysis, the samples were homogenized with CelLyticM lysis reagent and protease inhibitor cocktail (Sigma, Poole, Dorset, UK). Protein concentrations were determined by the Bradford assay method. Equal amounts of total protein were loaded into SDS‐PAGE gel wells. GAPDH of β‐actin was used as the internal standard for protein quantity. Samples prepared with 5× loading buffer were separated on 10% SDS‐PAGE gels and then transferred to nitrocellulose membranes, which were rinsed with Tris‐buffered saline Tween‐20 (TBS‐T) and then blocked in TBS‐T containing 5% milk for 1 h at room temperature (22–24°C). The membrane was incubated with the primary antibodies against α‐ENaC (Novus Biologicals, Littleton, CO, USA, NB100‐74357), phospho‐eNOS (Ser1177; Abcam, ab75639), eNOS (Abcam, ab66127), β‐actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA, sc‐81178) or GAPDH (Santa Cruz Biotechnology, sc‐20357) overnight at 4°C, followed by washing in TBS‐T and incubation with the secondary antibody (1:5000) for another 1 h at room temperature (22–24°C). Membranes were washed with TBS‐T and blots were detected by ECL kit (Invitrogen, Carlsbad, CA, USA) and scanned densitometry (Bio‐Rad, Richmond, CA, USA). Before detecting eNOS activity, MAs isolated from either NS or HS (21 days) group were incubated with or without 1 μM ACh for 10 min. The ratio of phosphorylated eNOS to total eNOS protein expression were used to evaluate eNOS activity.
NO measurement
Isolated MAs were separated longitudinally into two parts with microsurgical scissors, and one half was treated with 1 μM ACh for 10 min while the other half provided the control (no Ach). Total NO production in MA sections was determined by measuring the concentration of nitrate and nitrite, the stable metabolite of NO, with Griess reagent using the NO assay kit (Beyotime Company, Haimen, China) according to the manufacturer's instructions (Liu et al., 2012). The percent change of NO was calculated as follows: (NOACh − NOCtrl)/NOCtrl × 100%, where NOACh represents NO values obtained in the presence of ACh and NOCtrl indicates NO values measured under control conditions.
Data analysis
All data are presented as mean ± SEM. Statistical analysis was performed with SigmaPlot and SigmaStat Software (Jandel Scientific). One‐way anova (followed by Student–Newman–Keuls post hoc test), or Student's t‐test was used to where appropriate for statistical analysis. Differences were considered statistically significant for P < 0.05.
Materials
Unless otherwise noted, all reagents and drugs were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Bioreagents for Western blotting and immunofluorescence were purchased from Promega Corporation (Madison, WI, USA).
Results
HS diet increased BP, but reduced plasma aldosterone in SD rats
Several studies have shown that HS diet induces hypertension especially in salt‐sensitive species. Here we showed that HS diet also increased the SBP of SD rats, measured by tail cuff in conscious animals (n = 20; P < 0.05, Figure 1A). In contrast, the tail SBP of NS rats remained at normal levels (n = 20; P > 0.05). Feeding SD rats with HS diet for 14 and 21 days led to a significant decrease in the plasma aldosterone levels (Figure 1B). As shown in Figure 1C, the ratio of heart: body weights (HW/BW) was not altered by HS diet for 21 days (Figure 1C). In comparison, the ratio of kidney to body weight (KW/BW) was slightly, but significantly higher in HS diet‐treated SD rats than those in SD rats given the NS diet, at day 14 (Figure 1D).
Figure 1.
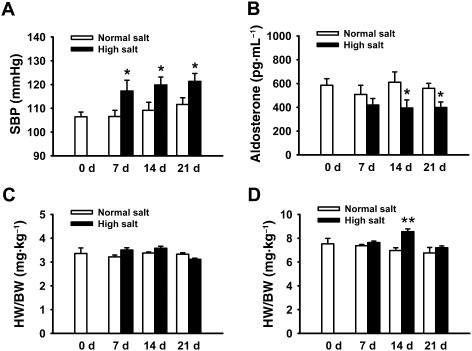
HS diet increased BP, but reduced plasma aldosterone in SD rats. (A) HS intake significantly increases SBP. The BP was measured after 7, 14 or 21 days on the specific diet. *P < 0.05 versus NS diet group at the same day (n = 20). (B) HS intake significantly decreased the plasma aldosterone level in SD rats. The plasma aldosterone levels were respectively measured after 7, 14 or 21 days on the diets. *P < 0.05 versus NS diet group at the same day (n = 20). (C) HS intake has no effect on heart weight as demonstrated by the heart : body weight (HW/BW) ratio, compared with NS intake (n = 10). (D) HS intake in SD rats significantly increased the kidney weights at day 14, as demonstrated by the kidney : body weight (KW/BW) ratio (n = 10). **P < 0.01 versus NS diet group.
HS diet decreased ENaC Po in endothelium of mesenteric arteries
Endothelial cells were isolated from MAs of SD rats and cultured for 3–5 days to confluence in order to characterize the expression profile of ENaCs. The expression of α‐ENaC protein was detected by Western blot in MAECs (Figure 2A). We also examined the expression of ENaCs in isolated MAs, with or without the endothelial layer. The data demonstrate that ENaCs were predominantly expressed in endothelial cells, although these channels were also expressed in smooth muscle cells (SMC), albeit at a very low level (Figure 2B, a and 2B, b). To examine the specificity of the anti‐α‐ENaC antibody, we used A6 cells as a positive control (derived from the distal nephron of Xenopus kidney, an established system for the study of ENaCs) and cardiomyocytes isolated from neonatal rats as a negative control. The data show that the anti α‐ENaC antibody detects products near the expected molecular weight (∼95 kDa) of the ENaC in A6 cells and MAECs, but not in the cardiomyocytes (Supporting Information Figure S1). Immunofluorescence staining images shown in Figure 2C and 2D also demonstrated the expression and distribution of α‐ENaC in MAECs.
Figure 2.
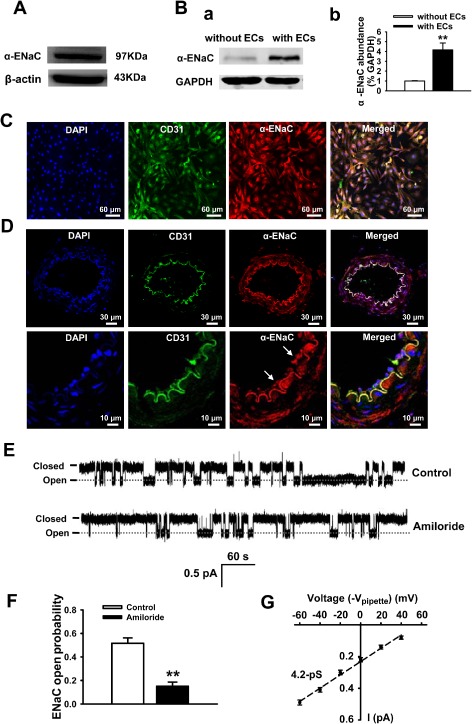
MAs express functional ENaCs. (A, B) The α‐subunit of the ENaC protein was detected in MAECs and in SMCs (n = 6 for each group). (C, D) Representative images showing immunofluorescent staining for α‐ENaC expression in endothelial cells isolated from rat MAs (C) or α‐ENaC expression in intact rat MAs; (E) representative single‐channel traces of ENaC current detected in intact vascular endothelium using split‐opened MAs technique, in the absence of (upper) or presence of 0.5 μM amiloride (lower). (F) Summarized PO of ENaC with and without amiloride incubation (n = 6 paired experiments) **P < 0.01 versus control. (G) The single‐channel conductance was determined by linear regression as indicated by the black dashed line (n = 6–8 for each data point).
Here, for the first time, we used split‐open arteries to directly investigate ENaC activity in SD rats. MAs were isolated and manually split open to access the apical membrane of MAECs. In situ cell‐attached patch‐clamp recording of ENaC single‐channel currents in intact vascular endothelium was performed. As shown in Figure 2E and 2F, the signal‐channel current with the slow kinetics and a very low amplitude was detected in endothelial cells of MAs and this current was significantly blocked by amiloride, a potent blocker of ENaCs (P < 0.01; n = 6). A similar result was seen in MAs isolated from the HS group (data not shown). To further characterize the currents recorded from intact MAs, the I–V relationship was constructed and fitted with linear regression (Figure 2G; n = 6–8 for each data point). The data showed that the single‐channel conductance of the currents was ∼4.2 pS (Figure 2G), which is very comparable with the reported ENaC single‐channel conductance (Ma, 2011). These biophysical features and the pharmacological profile of the current are consistent with the ENaCs previously reported in human dermal microvascular endothelial cells (Wang et al., 2009).
Recent studies suggest that activity of ENaCs in the distal nephron is abnormally up‐regulated by dietary sodium in hypertensive salt‐sensitive rats, and this enhanced activity is one of the major factors causing salt‐sensitive hypertension (Pavlov et al., 2013). In the present study, in situ cell‐attached patch‐clamp experiments were performed to determine whether the HS diet would influence ENaC activity in endothelial cells of split‐open MAs. Our results show that ENaC activity was significantly inhibited in SD rats fed with HS diet for 7 (n = 7; P < 0.01) or for 14 days (n = 7; P < 0.05; Figure 3A‐C and 3E). Interestingly, after 21 days on the HS diet, the ENaC activity returned to normal levels (n = 7; P > 0.05; Figure 3D and 3E). These data together suggest that functional ENaCs were present in the endothelial cells of intact MAs and that activity of ENaCs was reduced by HS challenge, at least for up to 14 days.
Figure 3.
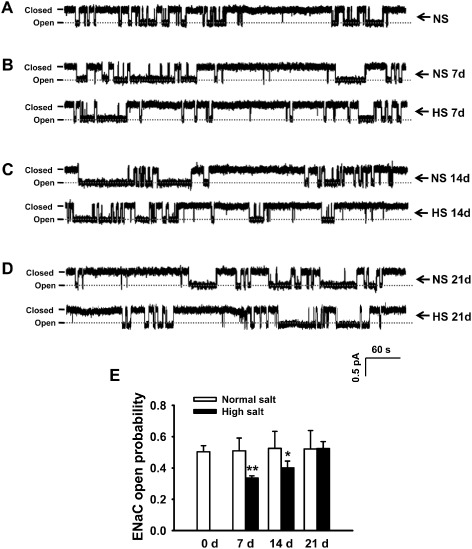
HS intake reduces the activity of ENaCs in MAECs. (A–D) Representative traces of ENaC single‐channel current recorded from rat MAECs in both the HS and NS diet groups, respectively, at days 7, 14 and 21. (E) Summarized PO obtained from the single‐channel recordings as shown in A–D. The data show that PO calculated from HS diet group at days 7 and 14 significantly decreased compared with that from the corresponding NS diet group (n = 7). *P < 0.05, **P < 0.01 versus NS group.
HS diet reduced ENaC expression in MAECs of SD rats
As the HS diet inhibited ENaC single‐channel activity, this diet might also inhibit expression of ENaC proteins. We therefore measured α‐ENaC protein expression in freshly isolated MAECs and found that α‐ENaC expression levels in MAECs were not altered after 2 weeks of HS diet, although ENaC activities were reduced at that time. However, its expression was significantly decreased after 21 days of the HS diet (Figure 4). Because recent studies have indicated that the α‐subunit of ENaC alone can form the pore structure and conduct amiloride‐sensitive Na+ currents (Canessa et al., 1994), it would be interesting to investigate whether the endothelial ENaCs are formed by α‐ENaC only or with other ENaC subunits.
Figure 4.
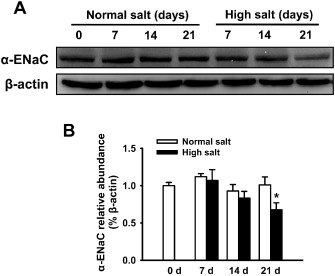
HS intake decreases the expression levels of α‐ENaC protein in MAECs. (A) The Western blots demonstrate the expression levels of α‐ENaC in MAECs under indicated different experimental conditions and days. (B) Summaries of normalized α‐ENaC expression levels. The data show that HS intake up to 14 days did not affect the expression levels of α‐ENaC, and that the expression levels of α‐ENaC were significantly decreased by HS intake at day 21 (n = 6). *P < 0.05 versus NS group.
Endothelial ENaCs play a role in endothelium‐dependent vascular relaxation
Compared with those MAs isolated from rats in a NS diet, MAs isolated from rats in a HS diet had an enhanced endothelium‐dependent relaxation (EDR), as shown by the decrease in EC50 for ACh (Figure 5A and 5B, a; Table 1; n = 6; P < 0.01). However, the EC50 for NTG did not differ between the two groups, suggesting that the HS diet did not affect the endothelium‐independent relaxation caused by the NO donor, NTG (Figure 5F–J; Table 1; n = 6; P > 0.05). To determine the possible involvement of ENaCs, we pre‐incubated the MA segments with the potent ENaC blocker, amiloride (0.5 μM), for 10 min before measuring the ACh‐induced relaxation. The data show that blocking ENaCs in MAs significantly reduced the EC50 of ACh in the NS group, but not in those from the HS group, suggesting that blockade of ENaCs enhanced the EDR in the group on the NS diet (Figure 5A, 5B, b and 5B, c; Table 1; n = 6; P < 0.05). Moreover, there was no ACh‐induced EDR observed in MAs that were lacking endothelium, and removing the endothelial cell layer did not affect the pattern of NTG‐induced vascular relaxation. More importantly, amiloride affected neither ACh‐ nor NTG‐induced relaxation in these MAs (Figure 5C–D). In addition, the HS diet also increased the contractility of MAs in response to 10 μM phenylephrine (Figure 5E; n = 15, P < 0.01), which coincides with increased BP under this condition (Figure 1A) and is also consistent with previously reported results (Sofola et al., 2002). We also performed experiments to examine ACh‐ and NTG‐induced vascular relaxation of MAs isolated from rats, after 14 days on the HS diet. Consistent with the reduction of ENaC activity by this duration of HS diet, the EDR was also enhanced (as suggested by the significantly reduced EC50), and was mimicked by amiloride in MAs isolated from the NS group (Supporting Information Figure S2; Table S1). Finally, the maximum response (E max) of MAs to ACh or NTG was not affected by either HS diet or amiloride (Table 1; Supporting Information Table S1). These results together suggest that dietary HS may enhance EDR by inhibiting the function of ENaCs.
Figure 5.
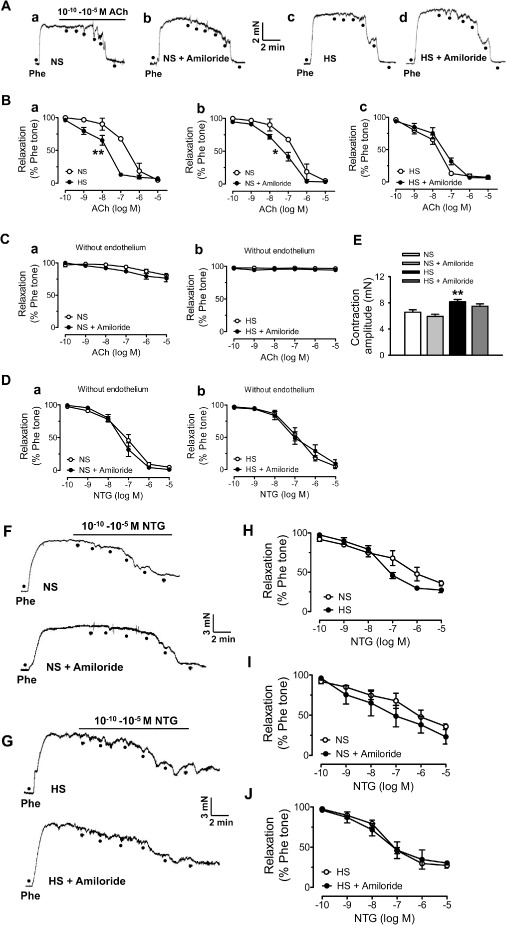
ENaCs regulate the EDR in MAs. (A) Representative traces of ACh‐induced relaxation in MAs from NS rats or HS rats (21 days) in the absence (A, a or A, c) of or in the presence of 0.5 μM amiloride (A, b or A, d). Rings were pre‐contracted with phenylephrine (Phe; 10μM). (B) Summaries of relaxation in response to different doses of ACh, in the absence of or in the presence of 0.5 μM amiloride, from NS or HS groups; the data points were obtained from the experiments shown in A. *P < 0.05, **P < 0.01: EC 50 significantly different between two groups (n = 6, for each data point). (C, a and C, b) Summaries of relaxation in response to different doses of ACh in MAs lacking endothelium, with or without 0.5 μM amiloride, from NS and HS groups (n = 7 for each data point). (D,a and D,b) Summaries of relaxation in response to different doses of NTG in MAs lacking endothelium, with or without amiloride (0.5 μM), in NS and HS groups (n = 7 for each data point). (E) Summarized contractile response to Phe (10 μM) of MAs in NS and HS (21 days) groups (n = 15; **P < 0.01 versus NS). (F‐G) HS intake has no significant effect on endothelium‐independent relaxation. (F) Representative traces of NTG‐induced relaxation in MAs isolated from NS group, in the absence of (upper) and in the presence of 0.5 μM amiloride (lower). (G) Representative traces of NTG‐induced relaxation in MAs in HS group, in the absence of (upper) and in the presence of 0.5 μM amiloride (lower). (H–J) Summaries of relaxation in response to different doses of NTG from NS and HS groups; the data points were obtained under indicated experimental conditions shown in F and G (n = 6).
Table 1.
E max and EC 50 values for ACh‐ and NTG‐induced relaxations in mesenteric arteries
| Normal salt | High salt | ||||
|---|---|---|---|---|---|
| Control | + Amiloride | Control | + Amiloride | ||
| ACh | EC50 (nM) | 174 ± 5 | 63 ± 13* | 50 ± 12** | 50 ± 14 |
| E max (%) | 90.2 ± 2.2 | 90.1 ± 3.5 | 90.7 ± 2.2 | 91.8 ± 3.0 | |
| NTG | EC50 (nM) | 60 ± 26 | 38 ± 24 | 47 ± 17 | 59 ± 29 |
| E max (%) | 46.7 ± 4.9 | 53.7 ± 8.3 | 53.1 ± 5.0 | 55.1 ± 6.5 | |
Data are expressed as the mean ± SEM from six rats per group.
E max and EC50 were calculated from each agonist concentration–response curve using a logistic function from OriginPro 8.5. *P < 0.05, **P < 0.01 versus control in the normal salt diet group.
HS intake attenuated eNOS activity and reduced NO production in MAs
We examined eNOS activity and NO levels in MAs isolated from NS or HS group at day 21, in the presence or in the absence of 1 μM ACh. Compared with the results from the NS group, the ratio of p‐eNOS (Ser1177) to total eNOS was significantly decreased in the HS diet group; however, this HS diet‐induced reduction of eNOS activity was reversed by ACh (Figure 6A and 6B). Consistent with reduced eNOS activity, NO levels in MAs were also significantly decreased in HS group compared with NS group and this HS‐induced reduction of NO was markedly increased by additional ACh (Figure 6C). Interestingly and importantly, the relative NO production induced by ACh was significantly higher in the HS group, compared with that in the NS group (Figure 6D). We suggest that the HS diet reduced eNOS activity and NO production, which could account for the increased contractility of MAs and that the increased stimulated release of NO may be responsible for the ‘enhanced’ EDR in MAs isolated from HS group.
Figure 6.
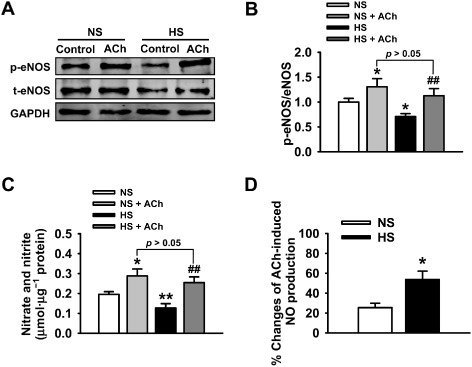
Effects of HS intake on eNOS activity and NO production. (A) The Western blots demonstrate the expression levels of eNOS in MAs under the indicated experimental conditions. (B) Summaries of eNOS activity in response to 1 μM ACh in NS or HS group (n = 6). *P < 0.05 versus NS group; ##P < 0.01 versus HS group. (C) NO production was significantly reduced in MAs isolated from HS group and was significantly increased by adding ACh (n = 6). *P < 0.05, **P < 0.01 versus NS group; ##P < 0.01 versus HS group. (D) The differential percent changes of ACh‐induced NO production, before and after ACh, in MAs isolated from NS or HS group (n = 6; *P < 0.05 vs. NS group).
Discussion
Several lines of evidence have shown that HS diet causes hypertension in Dahl salt‐sensitive rats (Farjah et al., 2003; Aoi et al., 2007; Kakizoe et al., 2009) However, HS intake either only slightly increases (Thierry‐Palmer et al., 2010) or does not alter the systolic BP of SD rats (Farjah et al., 2003). Further, the HS diet increased plasma aldosterone levels in Dahl salt‐sensitive rats but completely suppressed plasma aldosterone in Dahl salt‐resistant rats (Morizane et al., 2012). The differences in the regulation of plasma aldosterone levels in SD and Dahl salt‐sensitive rats may account for salt‐adaptation and salt‐sensitive hypertension. Therefore, we argue that in salt‐sensitive species, following an increase of extracellular sodium, aldosterone is locally elevated in endothelial tissues (our unpublished data) and stimulates the rapid membrane insertion of ENaC molecules, which increase the stiffness of the cells (Jeggle et al., 2013). If aldosterone is raised over a prolonged time, the stiff endothelial cells release less NO, which is perhaps one of the abnormalities causing progressive vascular dysfunction. However, in salt‐insensitive species, a negative feedback mechanism can be initiated to lower the plasma aldosterone levels. In the present study, our data demonstrate that the HS diet reduced plasma aldosterone levels in SD rats. This is not surprising, but for the first time, our patch‐clamp data from split‐open arteries suggest that it was the endothelial ENaC activity which was reduced in SD rats. Consistent with this finding, our Western blot data showed that the HS diet could also reduce α‐ENaC expression. These results together suggest that, rather than being a causative factor in the pathogenesis of hypertension, the reduced ENaC activity may reflect an adaptive response of the vasculature to HS challenge, in SD rats. These suggestions are also supported by our unpublished observations, where HS challenge in salt‐sensitive rats induced more severe hypertension with an increased expression and activity of ENaCs in MAs (data not shown).
We found that ENaCs were not only expressed in endothelial cells, but also in SMC, which was consistent with the previous study (Jernigan et al., 2008). There are several studies suggesting that ENaCs in SMC play an important role in the pressure‐induced constriction, called “myogenic constriction”, in mouse renal arteries (Jernigan and Drummond, 2005; 2006), cerebral vessels (VanLandingham et al., 2009) and rat MAs (Jernigan et al., 2008). Furthermore, we found that, following ENaC inhibition, the MAs were still able to constrict to phenylephrine, suggesting ENaC inhibition specifically blocks pressure‐induced constriction and not the contractility of the vessels (Jernigan and Drummond, 2005). Other studies have demonstrated that ENaCs in SMCs play a vital role in vascular SMC migration (Grifoni et al., 2006). However, our data show that blocking ENaCs with amiloride in MAs (either with or without endothelium) did not affect NTG‐induced vascular relaxation. Therefore, we propose that the ENaCs in SMCs may not be a major player in the regulation of vascular tone, at least in this period of three weeks of HS challenge, especially in the experimental model used for this study.
ENaCs are regulated by a variety of extrinsic and intrinsic factors, including hormone activation, mechanical stretch, various kinases, sodium and metabolic substrates (Bhalla and Hallows, 2008). We found that HS diet for 2 weeks inhibited ENaC activity without affecting its expression and that a reduced ENaC expression was not associated with a decreased ENaC PO , after 3 weeks of HS diet. This disparity between its function and expression profile may be very complex. First, it has been reported that ENaC activity is regulated by changes in both extracellular and intracellular [Na+]. A fast change (over seconds) in channel activity as a result of changes in extracellular [Na+] is known as Na+ self‐inhibition, and a slower change (over hours) in channel activity caused by increased intracellular [Na+] is known as feedback inhibition. Both self‐inhibition and feedback inhibition are physiologically important in limiting Na+ absorption under conditions of HS delivery, thereby mitigating large changes in intracellular [Na+] and cell volume (Turnheim, 1991; Palmer et al., 1998). It is very likely that the reduced ENaC PO in MAECs following the HS diet could be due to feedback inhibition because an elevated intracellular [Na+] indirectly down‐regulates ENaC activity (Abriel and Horisberger, 1999). Second, as in renal epithelial tissue, vascular endothelial cells also express ENaCs and mineralocorticoid receptors (Golestaneh et al., 2001; Oberleithner et al., 2006; Wang et al., 2009). Endothelial ENaC surface abundance can be regulated by aldosterone through activating its gene transcription via the genomic MR‐dependent mechanism (May et al., 1997; Alvarez de la Rosa et al., 2002) and through promoting the trafficking of preformed channels to the plasma membrane via the non‐genomic pathway (McEneaney et al., 2008). Therefore, we speculate that the down‐regulation of ENaC expression at 3 weeks after HS challenge may be due to the reduced plasma aldosterone levels, which is consistent with previously reported results (Oberleithner, 2007). Finally, HS intake may induce increased oxidative stress in the cardiovascular system, which might be associated with the disparity between ENaC function and its expression profile because reactive oxygen species regulates, at least, the PO of ENaCs (Zhang et al., 2013).
We noted that the effect of salt loading on vasodilatation responses is still a matter of debate. Shultz (Shultz and Tolins, 1993) and Linder (Linder et al., 1990) show that the plasma level of NO can be significantly increased in SD rats in response to HS challenge. However, Tolins and Shultz (1994) suggest that the salt sensitivity is determined by the level of endogenous NO; Sofola et al. (2002) and Lenda et al. (2000) show that HS diet either does not affect or decreases ACh‐induced relaxation of vasculature. We found that HS challenge significantly decreased eNOS activity in MAs, as well as the production of NO. These results may explain, at least in part, why HS increases BP and contractility of MAs. Our data also demonstrate that the relatively stronger EDR seen in MAs isolated from the HS rats may not reflect real vascular function, but rather suggest that the HS diet (at least for 3 weeks) in SD rats did not impair its ability to activate eNOS and/or to generate NO in response to ACh. In addition, either in rats on a long‐term HS diet or in hypertensive patients, HS intake appears to worsen vasculature relaxation (Tolins and Shultz, 1994; Lenda et al., 2000).
Nevertheless, our data show that HS intake induced a relatively stronger EDR induced by ACh, at least after 3 weeks of the HS diet. We speculate that this enhanced relaxation would be correlated with endothelial ENaC activity because blockade of ENaCs by amiloride can enhance the EDR, but not to endothelium‐independent relaxation, as induced by NTG. How HS diet affects endothelial ENaC activity in salt‐sensitive rats may lead us to a clearer conclusion about the role of the vasculature in salt‐sensitive hypertension.
Author contributions
Z‐R. Zhang, H‐B. Liu and H‐P. Ma conceived and designed the experiments; J. Zhang, Y‐Y. Sun, X‐Y Li, S. Jiang, M‐Y. Liu, J. Shi, B‐L. Song and D. Zhao collected, analysed and interpreted the data; H‐B. Liu and J. Zhang drafted the manuscript; Z‐R. Zhang and H‐P. Ma revised the manuscript; and all authors approved the final version of the manuscript.
Conflict of interest
None declared.
Supporting information
Figure S1 The anti α‐ENaC antibody specifically detects ENaC proteins near the predicted size (95 kDa) in mesenteric artery endothelial cells (MAECs) and A6 cells (A6), but not in neonatal mouse cardiomyocytes (CMs).
Figure S2 (A, B) Summaries of relaxation in response to different doses of ACh in MAs isolated from rats 14 days after NS or HS diet, in the presence or in the absence of 0.5 μM amiloride (n = 7 for each data point; **represents that EC50 was significantly different between two groups; P < 0.01 vs. NS group). (C) Summaries of NTG‐induced relaxation in MAs isolated from rats 14 days after NS or HS diet, in the absence or in the presence of 0.5 μM amiloride (n = 7 for each data point). (D) Summarized contractile response to phenylephrine (10 μM) of MAs 14 days NS or HS diet (n = 15; **indicates P < 0.01).
Table S1 E max and EC50 values for ACh‐ and NTG‐induced relaxations in mesenteric arteries
Acknowledgements
This study was supported by Key Project of Chinese National Program for Fundamental Research and Development (973 Program 2014CB542401, 2012CB517803 to Z. Z.), National Natural Science Foundation of China (81270340 and 81320108002 to Z. Z. and 81300191 to H. L.), Department of Health and Human Services (National Institute of Health Grant R01‐DK100582) to H. M. and China Postdoctoral Science Foundation (2012M520775 to H. L.) and Research Project of Health and Family Planning Commission of Heilongjiang Province (2014‐330 to BL. Song).
References
- Abriel H, Horisberger JD (1999). Feedback inhibition of rat amiloride‐sensitive epithelial sodium channels expressed in Xenopus laevis oocytes. J Physiol 516 (Pt 1): 31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA et al (2013). The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol 170: 1607–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez de la Rosa D, Li H, Canessa CM (2002). Effects of aldosterone on biosynthesis, traffic, and functional expression of epithelial sodium channels in A6 cells. J Gen Physiol 119: 427–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoi W, Niisato N, Sawabe Y, Miyazaki H, Tokuda S, Nishio K et al (2007). Abnormal expression of ENaC and SGK1 mRNA induced by dietary sodium in Dahl salt‐sensitively hypertensive rats. Cell Biol Int 31: 1288–1291. [DOI] [PubMed] [Google Scholar]
- Ashley RA, Dubuque SH, Dvorak B, Woodward SS, Williams SK, Kling PJ (2002). Erythropoietin stimulates vasculogenesis in neonatal rat mesenteric microvascular endothelial cells. Pediatr Res 51: 472–478. [DOI] [PubMed] [Google Scholar]
- Bhalla V, Hallows KR (2008). Mechanisms of ENaC regulation and clinical implications. J Am Soc Nephrol 19: 1845–1854. [DOI] [PubMed] [Google Scholar]
- Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD et al (1994). Amiloride‐sensitive epithelial Na+ channel is made of three homologous subunits. Nature 367: 463–467. [DOI] [PubMed] [Google Scholar]
- Chen W, Valamanesh F, Mirshahi T, Soria J, Tang R, Agarwal MK et al (2004). Aldosterone signaling modifies capillary formation by human bone marrow endothelial cells. Vascul Pharmacol 40: 269–277. [DOI] [PubMed] [Google Scholar]
- Climent B, Zsiros E, Stankevicius E, de la Villa P, Panyi G, Simonsen U et al (2011). Intact rat superior mesenteric artery endothelium is an electrical syncytium and expresses strong inward rectifier K+ conductance. Biochem Biophys Res Commun 410: 501–507. [DOI] [PubMed] [Google Scholar]
- Dickinson KM, Keogh JB, Clifton PM (2009). Effects of a low‐salt diet on flow‐mediated dilatation in humans. Am J Clin Nutr 89: 485–490. [DOI] [PubMed] [Google Scholar]
- Dora KA, Hinton JM, Walker SD, Garland CJ (2000). An indirect influence of phenylephrine on the release of endothelium‐derived vasodilators in rat small mesenteric artery. Br J Pharmacol 129: 381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farjah M, Roxas BP, Geenen DL, Danziger RS (2003). Dietary salt regulates renal SGK1 abundance: relevance to salt sensitivity in the Dahl rat. Hypertension 41: 874–878. [DOI] [PubMed] [Google Scholar]
- Garty H, Palmer LG (1997). Epithelial sodium channels: function, structure, and regulation. Physiol Rev 77: 359–396. [DOI] [PubMed] [Google Scholar]
- Golestaneh N, Klein C, Valamanesh F, Suarez G, Agarwal MK, Mirshahi M (2001). Mineralocorticoid receptor‐mediated signaling regulates the ion gated sodium channel in vascular endothelial cells and requires an intact cytoskeleton. Biochem Biophys Res Commun 280: 1300–1306. [DOI] [PubMed] [Google Scholar]
- Grifoni SC, Gannon KP, Stec DE, Drummond HA (2006). ENaC proteins contribute to VSMC migration. Am J Physiol Heart Circ Physiol 291: H3076–H3086. [DOI] [PubMed] [Google Scholar]
- Hashikabe Y, Suzuki K, Jojima T, Uchida K, Hattori Y (2006). Aldosterone impairs vascular endothelial cell function. J Cardiovasc Pharmacol 47: 609–613. [DOI] [PubMed] [Google Scholar]
- Jeggle P, Callies C, Tarjus A, Fassot C, Fels J, Oberleithner H et al (2013). Epithelial sodium channel stiffens the vascular endothelium in vitro and in Liddle mice. Hypertension 61: 1053–1059. [DOI] [PubMed] [Google Scholar]
- Jernigan NL, Drummond HA (2005). Vascular ENaC proteins are required for renal myogenic constriction. Am J Physiol Renal Physiol 289: F891–F901. [DOI] [PubMed] [Google Scholar]
- Jernigan NL, Drummond HA (2006). Myogenic vasoconstriction in mouse renal interlobar arteries: role of endogenous beta and gammaENaC. Am J Physiol Renal Physiol 291: F1184–F1191. [DOI] [PubMed] [Google Scholar]
- Jernigan NL, LaMarca B, Speed J, Galmiche L, Granger JP, Drummond HA (2008). Dietary salt enhances benzamil‐sensitive component of myogenic constriction in mesenteric arteries. Am J Physiol Heart Circ Physiol 294: H409–H420. [DOI] [PubMed] [Google Scholar]
- Kakizoe Y, Kitamura K, Ko T, Wakida N, Maekawa A, Miyoshi T et al (2009). Aberrant ENaC activation in Dahl salt‐sensitive rats. J Hypertens 27: 1679–1689. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenda DM, Sauls BA, Boegehold MA (2000). Reactive oxygen species may contribute to reduced endothelium‐dependent dilation in rats fed high salt. Am J Physiol Heart Circ Physiol 279: H7–H14. [DOI] [PubMed] [Google Scholar]
- Linder L, Kiowski W, Buhler FR, Luscher TF (1990). Indirect evidence for release of endothelium‐derived relaxing factor in human forearm circulation in vivo. Blunted response in essential hypertension. Circulation 81: 1762–1767. [DOI] [PubMed] [Google Scholar]
- Liu L, Liu J, Wong WT, Tian XY, Lau CW, Wang YX et al (2012). Dipeptidyl peptidase 4 inhibitor sitagliptin protects endothelial function in hypertension through a glucagon‐like peptide 1‐dependent mechanism. Hypertension 60: 833–841. [DOI] [PubMed] [Google Scholar]
- Ma HP (2011). Hydrogen peroxide stimulates the epithelial sodium channel through a phosphatidylinositide 3‐kinase‐dependent pathway. J Biol Chem 286: 32444–32453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May A, Puoti A, Gaeggeler HP, Horisberger JD, Rossier BC (1997). Early effect of aldosterone on the rate of synthesis of the epithelial sodium channel alpha subunit in A6 renal cells. J Am Soc Nephrol 8: 1813–1822. [DOI] [PubMed] [Google Scholar]
- McEneaney V, Harvey BJ, Thomas W (2008). Aldosterone regulates rapid trafficking of epithelial sodium channel subunits in renal cortical collecting duct cells via protein kinase D activation. Mol Endocrinol 22: 881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C (2010). Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160: 1573–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morizane S, Mitani F, Ozawa K, Ito K, Matsuhashi T, Katsumata Y et al (2012). Biphasic time course of the changes in aldosterone biosynthesis under high‐salt conditions in Dahl salt‐sensitive rats. Arterioscler Thromb Vasc Biol 32: 1194–1203. [DOI] [PubMed] [Google Scholar]
- Oberleithner H (2007). Is the vascular endothelium under the control of aldosterone? Facts and hypothesis. Pflugers Arch 454: 187–193. [DOI] [PubMed] [Google Scholar]
- Oberleithner H, Ludwig T, Riethmuller C, Hillebrand U, Albermann L, Schafer C et al (2004). Human endothelium: target for aldosterone. Hypertension 43: 952–956. [DOI] [PubMed] [Google Scholar]
- Oberleithner H, Riethmuller C, Ludwig T, Shahin V, Stock C, Schwab A et al (2006). Differential action of steroid hormones on human endothelium. J Cell Sci 119: 1926–1932. [DOI] [PubMed] [Google Scholar]
- Oberleithner H, Riethmuller C, Schillers H, MacGregor GA, de Wardener HE, Hausberg M (2007). Plasma sodium stiffens vascular endothelium and reduces nitric oxide release. Proc Natl Acad Sci U S A 104: 16281–16286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LG, Sackin H, Frindt G (1998). Regulation of Na+ channels by luminal Na+ in rat cortical collecting tubule. J Physiol 509 (Pt 1): 151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov TS, Levchenko V, O'Connor PM, Ilatovskaya DV, Palygin O, Mori T et al (2013). Deficiency of renal cortical EGF increases ENaC activity and contributes to salt‐sensitive hypertension. J Am Soc Nephrol 24: 1053–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al; NC‐IUPHAR (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl. Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez FR, Venegas F, Gonzalez M, Andres S, Vallejos C, Riquelme G et al (2009). Endothelial epithelial sodium channel inhibition activates endothelial nitric oxide synthase via phosphoinositide 3‐kinase/Akt in small‐diameter mesenteric arteries. Hypertension 53: 1000–1007. [DOI] [PubMed] [Google Scholar]
- Pratt JH (2005). Central role for ENaC in development of hypertension. J Am Soc Nephrol 16: 3154–3159. [DOI] [PubMed] [Google Scholar]
- Shultz PJ, Tolins JP (1993). Adaptation to increased dietary salt intake in the rat. Role of endogenous nitric oxide. J Clin Invest 91: 642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofola OA, Knill A, Hainsworth R, Drinkhill M (2002). Change in endothelial function in mesenteric arteries of Sprague‐Dawley rats fed a high salt diet. J Physiol 543: 255–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Zhang JN, Zhao D, Wang QS, Gu YC, Ma HP et al (2011). Role of the epithelial sodium channel in salt‐sensitive hypertension. Acta Pharmacol Sin 32: 789–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierry‐Palmer M, Tewolde TK, Emmett NL, Bayorh MA (2010). High dietary salt does not significantly affect plasma 25‐hydroxyvitamin D concentrations of Sprague Dawley rats. BMC Res Notes 3: 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolins JP, Shultz PJ (1994). Endogenous nitric oxide synthesis determines sensitivity to the pressor effect of salt. Kidney Int 46: 230–236. [DOI] [PubMed] [Google Scholar]
- Turnheim K (1991). Intrinsic regulation of apical sodium entry in epithelia. Physiol Rev 71: 429–445. [DOI] [PubMed] [Google Scholar]
- Vallon V, Wulff P, Huang DY, Loffing J, Volkl H, Kuhl D et al (2005). Role of Sgk1 in salt and potassium homeostasis. Am J Physiol Regul Integr Comp Physiol 288: R4–R10. [DOI] [PubMed] [Google Scholar]
- VanLandingham LG, Gannon KP, Drummond HA (2009). Pressure‐induced constriction is inhibited in a mouse model of reduced betaENaC. Am J Physiol Regul Integr Comp Physiol 297: R723–R728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Meng F, Mohan S, Champaneri B, Gu Y (2009). Functional ENaC channels expressed in endothelial cells: a new candidate for mediating shear force. Microcirculation 16: 276–287. [DOI] [PubMed] [Google Scholar]
- Yang D, Luo Z, Ma S, Wong WT, Ma L, Zhong J et al (2010). Activation of TRPV1 by dietary capsaicin improves endothelium‐dependent vasorelaxation and prevents hypertension. Cell Metab 12: 130–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Chen S, Liu H, Zhang B, Zhao Y, Ma K et al (2013). Hydrogen sulfide prevents hydrogen peroxide‐induced activation of epithelial sodium channel through a PTEN/PI(3,4,5)P3 dependent pathway. PLoS ONE 8: e64304. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 The anti α‐ENaC antibody specifically detects ENaC proteins near the predicted size (95 kDa) in mesenteric artery endothelial cells (MAECs) and A6 cells (A6), but not in neonatal mouse cardiomyocytes (CMs).
Figure S2 (A, B) Summaries of relaxation in response to different doses of ACh in MAs isolated from rats 14 days after NS or HS diet, in the presence or in the absence of 0.5 μM amiloride (n = 7 for each data point; **represents that EC50 was significantly different between two groups; P < 0.01 vs. NS group). (C) Summaries of NTG‐induced relaxation in MAs isolated from rats 14 days after NS or HS diet, in the absence or in the presence of 0.5 μM amiloride (n = 7 for each data point). (D) Summarized contractile response to phenylephrine (10 μM) of MAs 14 days NS or HS diet (n = 15; **indicates P < 0.01).
Table S1 E max and EC50 values for ACh‐ and NTG‐induced relaxations in mesenteric arteries