

Abstract
Pharmacologic augmentation of γ-globin expression sufficient to reduce anemia and clinical severity in patients with diverse hemoglobinopathies has been challenging. In studies here, representative molecules from four chemical classes, representing several distinct primary mechanisms of action, were investigated for effects on γ-globin transcriptional repressors, including components of the NuRD complex (LSD1 and HDACs 2-3), and the downstream repressor BCL11A, in erythroid progenitors from hemoglobinopathy patients. Two HDAC inhibitors (MS-275 and SB939), a short-chain fatty acid derivative (sodium dimethylbutyrate [SDMB]), and an agent identified in high-throughput screening, Benserazide, were studied. These therapeutics induced γ globin mRNA in progenitors above same subject controls up to 20-fold, and increased F-reticulocytes up to 20%. Cellular protein levels of BCL11A, LSD-1, and KLF1 were suppressed by the compounds. Chromatin immunoprecipitation assays demonstrated a 3.6-fold reduction in LSD1 and HDAC3 occupancy in the γ-globin gene promoter with Benserazide exposure, 3-fold reduction in LSD-1 and HDAC2 occupancy in the γ-globin gene promoter with SDMB exposure, while markers of gene activation (histone H3K9 acetylation and H3K4 demethylation), were enriched 5.7-fold. These findings identify clinical-stage oral therapeutics which inhibit or displace major co-repressors of γ-globin gene transcription and may suggest a rationale for combination therapy to produce enhanced efficacy.
Introduction
Sickle cell disease and beta thalassemia syndromes, classified as a global health burden, are caused by mutations which produce mutant or deficient beta-globin protein [1-3]. It is well established that the clinical severity of both conditions is reduced in individuals who produce significantly elevated HbF levels, generally from 20-30%, with HbF expression in a significant proportion of their red blood cells considered a major determinant of clinical severity [4-9]. Hydroxyurea (HU) has provided a major advance in sickle cell disease, but many adult sickle cell patients and most β-thalassemia subjects still require additional agents to achieve ameliorating levels of HbF [7-10]. Identification of additional inducers of fetal globin expression, multiple and differing mechanisms of action, could offer therapeutic options and potential for combination therapy [3-5, 11-28].
In-depth understanding of the molecular basis for adult-stage γ-globin gene silencing has identified several repressors of γ-globin expression which act at the gene promoter or interactions which disrupt binding of the LCR (locus control region) to the gene promoter [3-4, 29-43]. The transcription factor BCL11A, encoding a zinc finger transcription factor, has also been shown to function as a negative regulator of fetal globin expression in several model systems, and its absence strongly induces γ-globin in knock-out mice [34-36]. Down-regulation of BCL11A expression in adult human erythroid cells leads to robust induction of HbF [33]. BCL11A interacts with the Mi-2/NuRD chromatin remodeling complexes, as well as the erythroid transcription factors GATA1, FOG1, SOX6, and LSD1, in erythroid progenitors to repress γ-globin gene transcription [36-38]. LSD1, a demethylase, strongly represses γ-globin gene expression by binding to the promoter and altering histone methylation, and its inhibition or suppression de-represses and activates γ-globin transcription [31-32]. KLF1 typically enhances β-globin synthesis, in part through interaction with the BCL11A gene [40-43], but is also recruited to the γ-promoter, coincident with induction of γ-globin transcription, by certain SCFADs capable of inducing HbF expression [35,43]. Available evidence indicates that the collaborative action of multiple complex transcriptional repressors are required for γ-globin gene silencing [4;7; 15-20; 36-37].
Many reports have identified therapeutic candidates which induce the fetal globin gene promoter in reporter assays through unknown mechanisms, or inhibit acetylation of different histones. In studies here, we investigated potential mechanisms of action of four orally active, clinical-stage γ-globin-inducing therapeutics which represent four chemical classes of therapeutics and have favorable safety profiles. One objective was to determine if multiple molecular actions could be identified which could be considered for future application in combinations, for potentially greater efficacy in patients than agents operating through one mechanism alone [5, 63]. The agents tested included MS275 (Etinostat), a class I HDAC inhibitor of the benzamide family; SB939 (Pracinostat), a pan-HDAC inhibitor of the hydroxamic acid family; sodium dimethylbutyrate (SDMB), a short chain fatty acid derivative which induces the fetal globin promoter but is not a pan-HDAC inhibitor; and Benserazide, a therapeutic approved for another medical condition for activity is as a dopa decarboxylase inhibitor, recently identified as a potent inducer of fetal globin (Perrine, submitted). SDMB has the additional activity of prolonging STAT-5 phosphorylation/activation, acting through a signaling pathway also utilized by erythropoietin, which stimulates erythroid cell proliferation [21]. We found that these candidates induce γ-globin expression from 2 to 20-fold over subject control cells cultured from hemoglobinopathy patients or cord blood, and reduce binding of multiple known co-repressors from the γ-globin gene promoter. Further, enhancement of histone transcriptional activation marks H3K4me2 and H3K9Ac were detected at the γ-globin gene promoter following exposure to two agents. These studies therefore identify multiple molecular actions of orally active therapeutic candidates, which act on established mediators of γ-globin silencing through different components, suggesting potential to combine agents with different mechanisms to induce higher level γ-globin expression in the hemoglobinopathies as needed for many patients.
Materials and Methods
Erythroid progenitor cultures with candidate drugs
De-identified peripheral blood samples from patients with HbE-β0-thalassemia or sickle cell disease, or from normal cord blood, were collected in heparin and studied with approval of the Institutional Review Board of the Boston University School of Medicine and the Thalassemia Research Center at Mahidol University. Erythroid progenitors were cultured in a two-phase system, as previously described [28]. The first phase media utilized HyClone™ Iscove's Modified Dulbecco's Medium (IMDM, Hyclone, Logan, Utah) containing 30% charcoal-treated fetal bovine serum (Atlanta Biologicals, Miami, FL) and rHu IL-3 (25 ng/ml), rHuSCF (50 ng/ml) (STEMCELL Technologies Inc., Vancouver, BC), and rHuEPO (0.1u/ml) (Amgen, Thousand Oaks, CA) for the first 7 days, followed by replacement with new differentiation media, with 30% charcoal-absorbed fetal bovine serum (Atlanta Biologicals), rHuEPO at 3 U/ml, and rHuIL-3 (0.1 ng/ml) (Stem Cell Technologies). RhuSCF was not utilized during the erythroid phase (Phase 2) to avoid its confounding effects on induction of fetal globin expression and rapid differentiation. Test compounds were added on day 7 of the second phase (Phase 2) erythroid differentiation cultures. Erythroid progenitors were confirmed by Wright-Giemsa stain, and FACS analysis (FACScan, BD Biosciences, San Jose, CA) utilizing antibodies to glycophorin A and CD71 (BD Biosciences, San Jose, CA) to identify erythroid progenitors. Benserazide HCl (Enzo, Farmingdale, NY), MS-275 (Selleckchem, Houston, TX), SB939 (Selleckchem, Houston, TX) and sodium dimethylbutyrate (SDMB) (Frontage Laboratories, Exton, PA) were tested at concentrations which were first established to not inhibit cell proliferation, as follows: Benserazide 0.2 μM, SB939 0.1 μM, MS275 0.5 μM, and SDMB 400 μM. Viable cells were enumerated by hemacytometry and harvested on day 12-13 of the erythroid phase for mRNA and ChIP analysis, and on day 14 for analysis of F-reticulocytes by flow cytometry, or for immunoblot analyses, as described previously [28, 47]. Each compound was tested in at least 6 different hemoglobinopathy subjects' progenitors; Benserazide was studied in cells from 30 patient sources.
K562 cells were purchased from ATCC (American Type Culture Collection, Manassas, VA), and cultured in RPMI Medium 1640 (Life Technologies, Grand Island, NY) with 10% FBS (Atlanta Biologicals, Miami, FL). Concentrations of the test agents required for induction of γ-globin mRNA were one log higher than was required for induction in primary erythroid progenitor cells, consistent with prior studies.
RNA preparation, cDNA synthesis, and RT-PCR
RNA was extracted and quantitative real time (RT)-PCR was performed as previously described [28]. Briefly, total RNA was extracted from the cells using STAT-60 reagent (TEL-TEST, Inc., Friendswood, TX) following directions of the manufacturer. First-strand cDNA was synthesized using TaqMAN Universal PCR Master MIX (Applied Biosystems, Branchburg, New Jersey). Relative quantification PCR was performed using the ABI 7500 Real-Time PCR system (Applied Biosystems, Foster City, CA) by the ΔΔCt method, as described previously [28, 46]. Relative abundance of γ-globin-mRNA was determined as compared to α-globin mRNA or 18S as internal controls. γ-globin mRNA was analyzed in progenitors cultured from >60 different subjects. Relative mRNA abundance for LSD1, BCL11A, and KLF1 was similarly determined, using 18S rRNA as an internal control. TaqMan® Gene Expression primers were utilized from Life Technology (Grand Island, NY). Primer information is as follows: human γ-globin (Hs00361131_g1), human β-globin (Hs00758889_s1); human α-globin (Hs00361191_g1), 18S (Hs9999901_s1); LSD1 (Hs01002741 m1); BCL11A (Hs01093199_m1); EKLF1 (Hs00610592_M1) [28, 31].
F-reticulocyte analysis by flow cytometry
The proportion of reticulocytes expressing HbF (% F-reticulocytes) in erythroid progenitor cells were analyzed by immunofluorescent staining and flow cytometry, as previously described [21-23, 28, 35]. Briefly, pelleted cells were fixed with 3% formaldehyde in PBS/0.1% BSA for 20 minutes at room temperature, permeabilized with 0.5% Saponin in PBS/0.1% BSA for 10 minutes. Cells were washed, pelleted by centrifugation, and stained for 30 minutes with a PerCP-conjugated mouse anti-human fetal hemoglobin antibody, or PerCP mouse Isotype Control (BD Biosciences, San Jose, CA). The cells were then washed once with permeabilization buffer and washed twice with PBS/0.1% BSA, and stained with Thiazole Orange (BD Biosciences, San Jose, CA) to detect reticulum. The samples were analyzed by flow cytometery (BD FACScan) using CellQuest software (BD Biosciences, San Jose, CA). The fraction of F-reticulocytes were determined by setting gates based on unstained isotype controls, and compared between untreated control cells and the cultured cells from the same subjects, treated with the test compounds.
Immunoblotting
Erythroid progenitor cells were lysed in Laemmli sample buffer and subjected to 8% or 12% SDS-PAGE, as previously described [47]. Proteins were transferred to a nitrocellulose membrane and probed with antibodies to LSD1 (Abcam, Cambridge, Massachusetts), BCL11A (Novus Biological, Littleton, CO), KLF-1 (Santa Cruz Biotechnology, Dallas, Texas), β-actin (Sigma A, St. Louis, MO), or HbF (sc-21756, Santa Cruz Biotechnology, Dallas, Texas) and β-actin (Sigma A, St. Louis, MO). Proteins were visualized with the GE Imaging System (Image Quant, LSD4010 (GE Healthcare, Piscataway, NJ) and quantified with ImageJ software (NIH, Bethesda, MD).
Chromatin immunoprecipitation assays
Chromatin immunoprecipitation (ChIP) assays were performed as previously described [47] using at least 4 × 106 cells for each assay from patient or cord blood erythroid progenitors cultured alone or with test compounds Benserazide (0.2 uM) and sodium dimethylbutyrate (SDMB) (400 uM). DNA was extracted from sonicated whole cells (with DNA fragment lengths of 150-500 bp) and incubated with antibodies to LSD1, HDAC2, HDAC3, H3K4me2 (Abcam, Cambridge, Massachusetts), or H3K9Ac (Millipore, Billerica, MA) overnight at 4°C. Immunocomplexes were precipitated using protein A Sepharose beads (Sigma A, St. Louis, MO) for 4 hours at 4°C. The precipitated DNA was quantified by real-time quantitative PCR assay with primer pairs for human γ-globin promoter sequences as follows: γ-globin promoter Forward: AACGGTCCCTGGCTAAACTC; γ-globin promoter Reverse: GCTGAAGGGTGCTTCC-TTTT. The qRT-PCR data from chromatin immunoprecipitation was normalized to input DNA. Triplicate PCR reactions for each sample were performed and each ChIP condition was performed in at least 6 independent experiments.
Statistical analyses
Statistical analyses were performed using paired Student's t-tests, a level of ≤ 0.05 was considered significant.
Results
Therapeutic effects on γ-globin mRNA and F-reticulocytes by multiple therapeutic agents
As shown in Figure 1, the percentage of erythroid progenitors produced in this two-phase differentiation system was > 95% at the time that cells were collected for analysis. The candidate therapeutic agents induced γ-globin mRNA expression up to 20-fold over untreated control progenitors from the same patient to a statistically significant degree, with positive responses found in at least 70% of progenitors, as shown in Figure 2A. MS275 exposure induced a mean increase of 1.8 fold γ-globin mRNA above controls, (p<0.001); SDMB induced a 2.2 fold increase,(p<0.001), and SB939 induced 3.1 fold mean increase, (p<0.001); Benserazide induced a mean increase in γ-globin mRNA of 12-fold over subject control, (range 1.5 to 20 fold, p<0.001). Increases in proportions of erythroid cells expressing HbF protein (% F-reticulocytes) were observed with therapeutic treatment compared to control cells from the same subject, as shown in Figure 2B. SDMB induced a mean absolute increase of 5%, range 2% to 9%, (p<0.05); MS275 induced a mean increase in F-reticulocytes of 8% above control, (range 3% to 11%, p<0.01). Benserazide induced a mean increase of 10 %, (range 5% to 21.5 %, p<0.001). The pan-HDAC inhibitor SB939 induced a mean increase of 11.6 %, (range 2.5% to 22%, p<0.001). All changes from controls were statistically significant. To additionally confirm the changes at the protein level, Western analysis was performed with cells treated with Benserazide; increases were 2.6-fold and 1.6-fold above same subject controls, in thalassemic and cord blood samples respectively, shown in Figure 2C.
Figure 1. Differentiation of erythroid progenitors.
Photomicrographs of Wright-Giemsa stained erythroid progenitors in Phase 2 of culture, showing glycophorin-positive cells on day 5 (panel A), day 10 (panel B), and day 15 (panel C). Scale, 10 μm.
Figure 2. γ-globin mRNA and F-reticulocytes induced by therapeutic candidates.
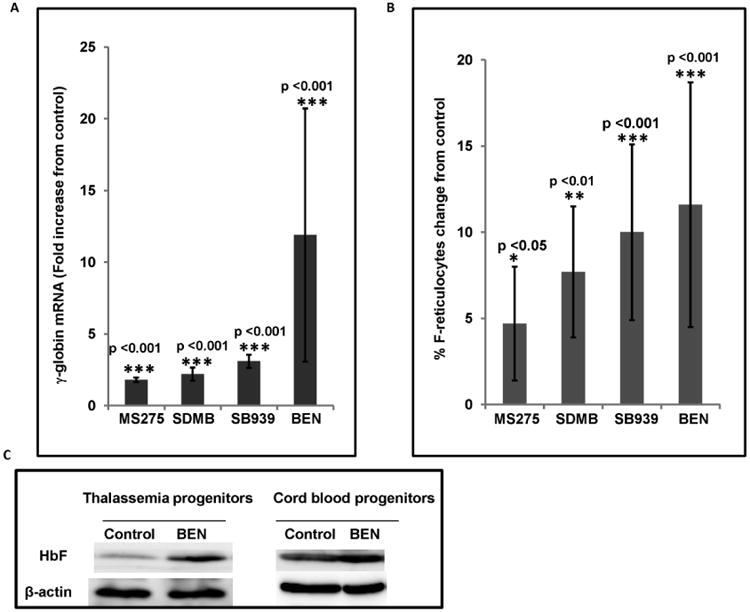
A: Mean fold change in γ-globin mRNA in erythroid progenitor cells treated with the designated therapeutic candidates (MS-275, SDMB, SB939, or Benserazide), compared to vehicle-treated cells from the same subject. All changes are significant, p <0.001. Error bars indicate SD.
B. Mean change in proportions of cells expressing HbF protein (F-reticulocytes) in erythroid progenitor cells treated with therapeutic candidates (SDMB, Benserazide, MS275, SB939), compared to control cells from the same subject. Error bars indicate SD. * Asterisks indicate statistically significant differences (* p<0.05, ** p<0.01. *** p<0.001)
C. Western blot demonstrating increased total HbF in thalassemic or cord blood erythroid progenitors cultured with Benserazide, compared to control cells from the same sources. Fetal globin protein increased by 2.6-fold and 1.6 fold above control levels.
Reduction of the mRNA levels of co-repressors of γ-globin gene expression
Binding of repressor proteins to the proximal γ-globin gene promoter, altering interactions with the LCR, is established as an important mechanism of γ-globin gene silencing [3-5, 29-31, 64]. To elucidate potential mechanisms by which these agents induce γ-globin expression, we first determined whether the cellular transcript levels of the established γ-globin gene co-repressor proteins LSD1, BCL11A, and EKLF1, were affected by the test agents. Exposure to MS275 and Benserazide resulted in significantly reduced LSD1 mRNA levels, by 3.6 and 4.7-fold respectively (p<0.05), SB939 and SDMB treatment produced smaller changes in LSD1 transcript levels, and were not significant (shown in Figure 3A). LSD1 is a potent silencer of γ-globin gene expression; knockdown or chemical inhibition of LSD1 has been shown to significantly increase γ-globin gene expression [31-32]. These findings indicate that reduced LSD1 transcription may be a mechanism of action through which Benserazide and MS275, and to a lesser degree SDMB, and SB939, (which are not LSD1 enzyme inhibitors), de-repress γ-globin gene silencing. Exposure to all of the candidate compounds resulted in significant reductions in BCL11A mRNA; exposure to MS275, SDMB, Benserazide, and SB939, reduced BCL11A transcripts by 1.7, 2.9, 3.1, and 3.6- fold respectively (Figure 3B). KLF1 transcripts, an activator of β-globin expression, was down-regulated by MS275 exposure, by 3.3-fold, but not by other compounds (Figure 3C).
Figure 3. Mean mRNA levels of repressor components in erythroid cells treated with therapeutic candidates.
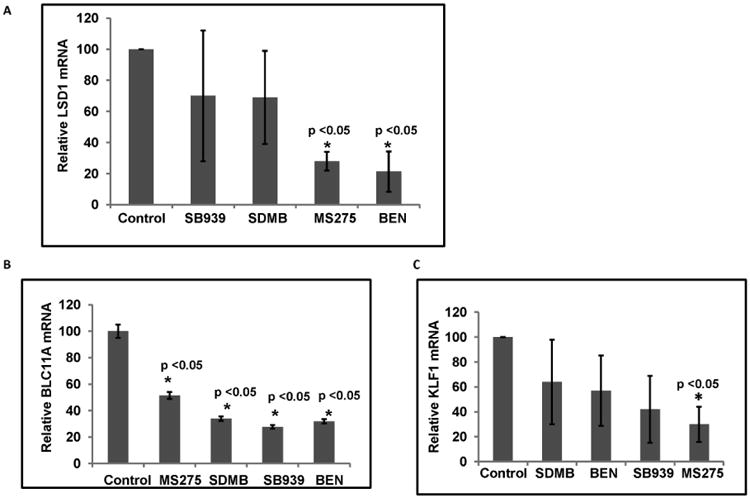
3A) Mean transcript levels of LSD1, 3B) BCL11A and 3C) KLF1 assayed by quantitative RT-PCR relative to levels in control cells are shown; 18S transcripts were used as internal controls. The mean value represents data from at least three independent experiments. The error bars represent the SD. Asterisks indicate significant differences between the treatment relative to the controls (* p<0.05).
Treatment with therapeutic candidates reduces the protein levels of specific co-repressors of γ-globin expression
To evaluate effects of these therapeutic agents on co-repressor expression at the protein level, immunoblot analyses of LSD1, BCL11A, and KLF1 levels were performed in erythroid progenitors cultured from subjects with HbE β-thalassemia or sickle cell disease, or normal cord blood. Consistent with the qRT-PCR data showing decreased LSD1 transcripts in cells exposed to MS275 or Benserazide, and to a lesser extent SDMB, (Figure 3A), LSD1 protein levels in HbE β-thalassemia cells were significantly reduced (5-fold) by exposure to Benserazide and MS275 (Figure 4A), while SDMB had lesser effects on LSD1 protein expression (Figure 4A). BCL11A protein levels were suppressed by all therapeutic candidates (Figure 4B). Protein levels of KLF1 were reduced to the greatest degree by MS275 by 3.3 fold (Figure 4C), and to a lesser degree by the other candidates (data not shown). The findings demonstrate suppression of BCL11A and LSD1 protein levels by three therapeutic agents, and suppression of KLF1 with exposure to MS275.
Figure 4. Effects of therapeutic candidates on LSD1, BCL11A and KLF1 protein levels in hemoglobinopathy erythroid cells from hemoglobinopathy patients.

A. LSD1 protein in HbE β-thalassemia cells treated with Benserazide, MS275 or SDMB. The ratio of LSD1:β-actin protein is shown below each panel. Each panel designates an immunoblot performed from at least two different patients. Control designates protein in untreated cells from the same subject.
B. BCL11A protein in sickle cell progenitors treated with SB939, in HbE β-thalassemia cells treated with Benserazide or SDMB (left panel). BCL11A protein in cord blood erythroid progenitor cells treated with MS275, SB939, or Benserazide (right panel).
C. KLF-1 protein in erythroid progenitor cells treated with MS275.
Co-repressor complexes at the γ-globin gene promoter are disrupted by candidate inducers
Several co-repressor complexes have been identified that suppress γ-globin gene transcription with binding in the γ-globin promoter or LCR region [27]. The first complex identified was the nucleosome remodeling complex (NuRD), an ATP-dependent chromatin remodeler of which HDACs 1-3 are major components [29-30]. Recently, LSD1 was demonstrated as important in the NuRD complex binding the γ-globin promoter which is a major silencing mechanism of γ-globin gene expression [31-32]. Our finding that certain of these diverse γ-globin inducing agents repress LSD1 cellular protein levels suggested that they may thereby affect LDS1 occupancy of the promoter. We therefore used chromatin-immunoprecipitation (ChIP) to quantitate LSD1 promoter occupancy, with HDAC2 or -3 occupancy as additional markers of the NuRD complex. Benserazide exposure reduced LSD1 and HDAC3 occupancy at the γ-globin gene promoter by 3.4-fold and 3.6-fold respectively; it did not alter HDAC2 occupancy (Figure 5A and 5B), in patients' and cord blood erythroid progenitors. SDMB exposure decreased HDAC2 and LSD1 binding at the γ-globin promoter by 6.4- and 1.7-fold respectively; it did not affect HDAC3 (Figure 5C). These data demonstrate potent effects of Benserazide and SDMB on disrupting binding of the co-repressors LSD1, and either HDAC2 or HDAC3 respectively, at the γ-globin promoter, coincident with suppression of LSD1 protein levels in the cells and with γ-globin gene induction.
Figure 5. Chromatin immunoprecipitation assays of binding of co-repressors in the γ-globin gene promoter.
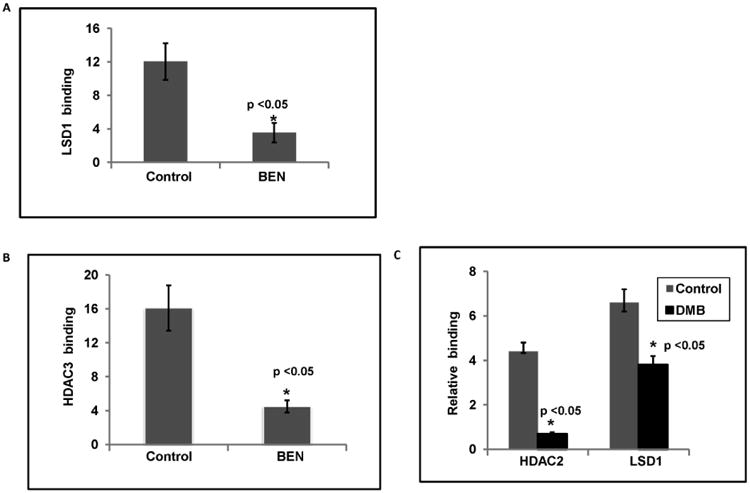
A) LSD1 protein binding in γ-globin gene promoter in HbE β-thalassemia progenitor cells treated with Benserazide. The bars represent the standard deviation (* p< 0.05).
B) HDAC3 protein binding in the γ-globin gene promoter in HbE β-thalassemia cells progenitors treated with Benserazide. The bars represent the standard deviation (* p< 0.05).
C) LSD1 and HDAC2 protein binding in γ-globin gene promoter in K562 cells treated with SDMB. The error bars represent the standard deviation. Asterisks indicate significant differences between the treated and vehicle-treated cells (* p<0.05).
Histone marks associated with gene activation are enriched at the γ-globin promoter by exposure to Benserazide
A growing body of evidence has shown that histone tail modifications are important in the control of transcriptional activity. In globin loci, histone marks for active chromatin, such as demethylation of lysine 4 and acetylation of lysine 9 on histone H3, are strongly associated with active transcription of globin genes [26]. In contrast, transcriptionally-silenced globin genes lack these activation marks. After demonstrating that Benserazide can regulate LSD1 and HDAC3 binding at the γ-globin gene promoter, we examined histone modification in the promoter during exposure to these agents. H3K4me2 is a target of LSD1, and demethylation of H3K4Me2 results in suppression of γ-globin gene transcription [26]. H3K4me2 was enriched by 5.7-fold at the γ-globin promoter with Benserazide treatment (Figure 6), coincident with decreased occupancy of LSD1 at the promoter (Figure 5A). H3K9Ac is an epigenetic signature implicated in transcriptional activation and targeted (deacetylated and inactivated) by HDACs in repressor complexes. ChIP analysis showed that H3AcK9 is enriched by 3-fold at the γ-globin promoter after Benserazide treatment (Figure 6), coincident with the decreased occupancy of HDAC3 at these promoters (Figure 5B). Benserazide treatment resulted in enhancement of H3K4me2 and H3K9Ac accumulation at the γ-globin promoter within the time-frame during which increases in transcription of the γ-globin gene and decreases in LSD1 and HDAC3 promoter occupancy were detected, suggesting one possible mechanism for γ-globin induction by Benserazide.
Figure 6. Activation markers of histone modification (H3AcK9 & H3K4me2) in the γ-globin gene promoter in HbE β-thalassemia progenitors treated with Benserazide (BEN).
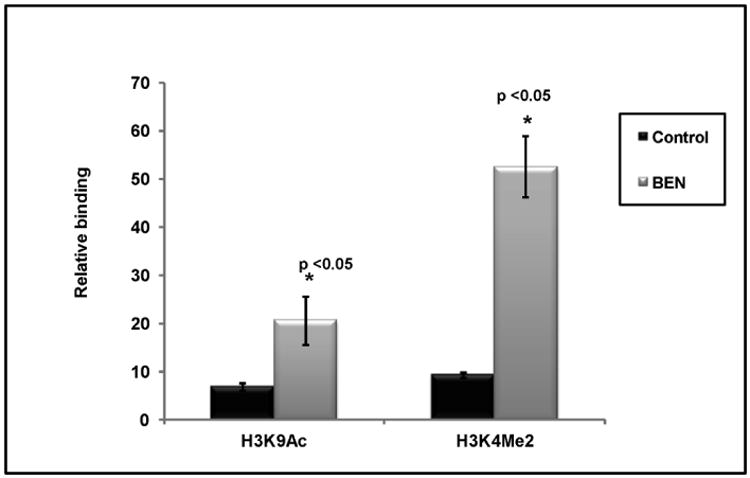
H3K4me2 and H3AcK9 enrichment in the γ-globin gene promoter is shown in Benserazide-treated HbE β-thalassemia progenitors and vehicle-treated control. The error bars represent the SEM. Asterisks indicate significant differences between the vehicle controls and Benserazide-treated cells (*p<0.05).
Discussion
A significant body of biochemical, epidemiologic, clinical, and pharmacologic data has demonstrated that adequately elevated fetal globin protein levels are associated with less severe anemia in β-thalassemia patients and reduced clinical complications in sickle cell disease [2-6, 19, 59, 63, 64]. While any increment in fetal globin was established to be clinically beneficial [6], significant amelioration of anemia or most clinical symptoms requires high-level expression, with 20-30% HbF, and high proportions of F-cells, which are enriched in patients through selective survival [reviews 2, 5, 69]. Hydroxyurea (HU), the single FDA-approved therapeutic has good activity in young patients and beneficial effects in approximately 50% of adults [5,7-10]. Additional non-cytotoxic HbF inducers, which could be used long-term, would be beneficial for treatment of patients with β-hemoglobinopathies [5, 63,67]. Clinical trials have shown significant HbF elevation, reduced anemia, and fewer hospital days in patients treated with arginine butyrate or decitabine, given parenterally [12,14]. More than 70 compounds have been identified which enhance fetal globin expression [5,14-26; 37; 45]. However, oral therapeutics with therapeutic activity similar to, or greater than, these parenteral drugs would be more feasible for long-term therapy for this global population. Therapeutic agents which result in reduced transcriptional repressor complex occupancy on the γ-globin promoter, thereby re-enabling pro-transcriptional interactions of the LCR with the γ-globin gene promoter would provide targeted therapies. The therapeutics investigated here are all orally active, and have shown favorable safety profiles in clinical trials or with long-term treatment in other medical conditions. [68] Increasing proportions of cells expressing HbF was recently proposed to be critically important, which these agents do in vitro. [69] We hypothesize that the two HDAC inhibitors studied here, which increase chromatin accessibility, particularly offer complimentary actions for combination use with an agent with more targeted action. Interestingly, while direct inhibitors of HDAC enzymatic activity, SB939 and MS275, can act within the repressor complex promoter without affecting promoter occupancy by the complex, all of these inducers resulted in reduced binding of at least one HDAC from the γ-globin promoter.
These four therapeutics which induce γ-globin expression in patients' progenitors were found to have differential actions on three potent γ-globin transcriptional repressors: LSD1, BCL11A, and KLF1 (Figure 7). LSD1 inhibition strongly induces γ-globin expression [31], LSD1 and the Co-REST histone demethylase complex interacts with BCL11A; these proteins co-occupy the human β-globin gene cluster and cooperate in silencing γ-globin transcription in adult erythroid progenitors [34]. BCL11A down-regulation in multiple models in mice and in primary adult erythroid cells was reported to be associated with robust HbF expression [27]. BCL11A occupancy reconfigures the β-globin cluster by modulating chromosomal loop formation and altering LCR interactions to activate beta globin expression, functioning in concert with the NuRD-repressor complex, KLF-1, and HDACs. [37,41] KLF1, an erythroid-specific transcription factor, which activates β-globin expression [39,40] has also been demonstrated to activate γ-globin expression when recruited to the promoter after exposure to certain SCFADs or a compound designated RB 7 [45; 52]. Polymorphisms which reduce expression of KLF1 have been shown to enhance γ globin in beta thalassemia [42, 44]. All four γ-globin-inducing compounds reduced BCL11A at the mRNA by 2.5 to 3.7-fold and at the protein levels by 10-fold in patients' progenitors. While the studies presented herein do not yet causally link the inhibitory effects of these candidate therapeutics on repressor proteins with the resulting increases in γ-globin transcription, the well-established ability of inhibition of either LSD1 or BCL11A, by any of a number of molecular or pharmacological means, to induce high level γ-globin expression strongly suggests that these are among the relevant molecular mechanisms by which these candidates act.
Figure 7.
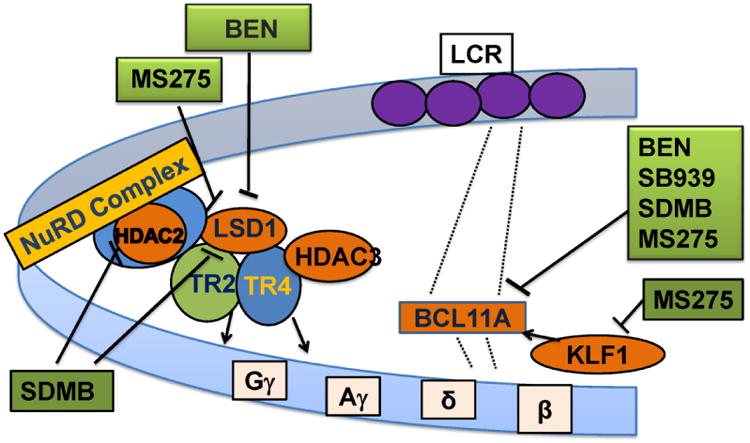
Schematic diagram of sites of molecular mechanisms of γ-globin induction by Benserazide (BEN), MS275 (Etinostat), SDMB (dimethylbutyrate), and SB939 (Pracinostat).
Benserazide, an L-amino acid decarboxylase inhibitor, has been used clinically for more than 40 years to increase plasma levels of L-dopa, an active modality for treatment of Parkinson's disease. [68] Its previously unrecognized ability to also induce fetal globin expression was discovered in a high-throughput screen of an approved or clinically utilized drug library. This agent has demonstrated γ-globin induction in vivo in beta globin YAC transgenic mice and in anemic baboons (manuscript under review). The studies herein suggest probable mechanisms of action. Benserazide exposure virtually abolished BCL11A protein expression in patients' progenitors and downregulated its transcription. Exposure to Benserazide also profoundly reduced LSD1 mRNA and protein levels and binding of LSD1 and HDAC3 in the γ-globin promoter region. SDMB is an oral short chain fatty acid derivative which does not cause general histone deacetylase inhibition, [21, 23] but here was found to displace HDAC2 at the γ-globin promoter; selective HDAC2 inhibition is a demonstrated mechanism of HbF induction [34]. These actions on γ-globin transcriptional repressor complexes were demonstrated in association with locally increased enrichment of histone activation markers at the promoter in ChIP assays, without causing global histone acetylation, which is typically associated with erythroid growth inhibition. Treatment with MS275 or SB939 in combination with Benserazide or SDMB may result in a more accessible chromatin structure in erythroid cells to facilitate γ-globin transcription without affecting multiple genes and should avoid undesirable off-target effects, providing a therapeutic profile recommended by Fathallah and Atweh [63]. Benserazide has been used safely clinically, for the separate action of inhibiting aromatic amino acid decarboxylase, for four decades. Its distinct actions identified here suggest that complementary regimens using two agents, or with pan-HDAC inhibitors, which typically inhibit cell proliferation, in combination with either Benserazide or SDMB, may achieve high–level γ-globin expression, without inhibiting erythroid cell proliferation, which is undesirable in these conditions of anemia.
Identifying dosing regimens to produce sustained high levels of fetal globin with therapeutics that are readily tolerable in patients has been daunting [5, 14, 25]. Many factors, such as individual patients' rates of erythropoiesis and erythroid apoptosis, differential susceptibility to induction dependent upon molecular mutations, genetic modifiers, translation of fetal globin, erythropoietin levels, extent of prior marrow infarcts, iron bioavailability, and other unknown factors all make development of therapeutics for the diverse hemoglobinopathies challenging. [5, 48-65] While concern has been raised regarding prolonged suppression of BCL11A on B-cell differentiation long-term in the context of gene editing approaches, these and other small molecules can be readily administered intermittently, thereby preventing sustained off-target effects. [67, 70] The findings here suggest a basis for combining pharmacologic agents to induce higher HbF expression through complementary molecular mechanisms if one alone is not adequate.
Acknowledgments
Supported by NIH grants R01 DK-52962, R41 HL-108516, and R42-110727. S. Perrine, D. V. Faller, Y. Dai, D. Chui, and S. Fucharoen designed the studies. Y. Dai, Yuan Luo Hong, Jose Sangerman performed assays. S. Perrine, D.V. Faller, and Y. Dai wrote the paper.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115:4331–6. doi: 10.1182/blood-2010-01-251348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steinberg MH, Rodgers GP. Pharmacologic modulation of fetal hemoglobin. Medicine (Baltimore) 2001;80:328–44. doi: 10.1097/00005792-200109000-00007. [DOI] [PubMed] [Google Scholar]
- 3.Wilber A, Nienhuis AW, Persons DA. Transcriptional regulation of fetal to adult hemoglobin switching: new therapeutic opportunities. Blood. 2011;117:3945–53. doi: 10.1182/blood-2010-11-316893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bauer DE, Kamran SC, Orkin SH. Reawakening fetal hemoglobin: prospects for new therapies for the beta-globin disorders. Blood. 2012;120:2945–53. doi: 10.1182/blood-2012-06-292078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perrine SP, Pace BS, Faller DV. Targeted fetal hemoglobin induction for treatment of beta hemoglobinopathies. Hematol Oncol Clin North Am. 2014;28:233–48. doi: 10.1016/j.hoc.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 6.Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–44. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 7.Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: A 17.5 year follow-up. Am J Hematol. 2010;85:403–8. doi: 10.1002/ajh.21699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med. 1995;332:1317–22. doi: 10.1056/NEJM199505183322001. [DOI] [PubMed] [Google Scholar]
- 9.Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG) Lancet. 2011;377:1663–72. doi: 10.1016/S0140-6736(11)60355-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fucharoen S, Siritanaratkul N, Winichagoon P, et al. Hydroxyurea increases hemoglobin F levels and improves the effectiveness of erythropoiesis in beta-thalassemia/hemoglobin E disease. Blood. 1996;87:887–92. [PubMed] [Google Scholar]
- 11.Ley TJ, DeSimone J, Anagnou NP, et al. 5-azacytidine selectively increases gamma-globin synthesis in a patient with beta+ thalassemia. N Engl J Med. 1982;307:1469–1475. doi: 10.1056/NEJM198212093072401. [DOI] [PubMed] [Google Scholar]
- 12.Saunthararajah Y, Hillery CA, Lavelle D, et al. Effects of 5-aza-2′-deoxycytidine on fetal hemoglobin levels, red cell adhesion, and hematopoietic differentiation in patients with sickle cell disease. Blood. 2003;102:3865–3870. doi: 10.1182/blood-2003-05-1738. [DOI] [PubMed] [Google Scholar]
- 13.Perrine SP, Ginder GD, Faller DV, et al. A short-term trial of butyrate to stimulate fetal-globin-gene expression in the beta-globin disorders. N Engl J Med. 1993;328:81–6. doi: 10.1056/NEJM199301143280202. [DOI] [PubMed] [Google Scholar]
- 14.Atweh GF, Sutton M, Nassif I, et al. Sustained induction of fetal hemoglobin by pulse butyrate therapy in sickle cell disease. Blood. 1999;93:790–797. [PMC free article] [PubMed] [Google Scholar]
- 15.Ikuta T, Kan YW, Swerdlow PS, et al. Alterations in protein-DNA interactions in the gamma globin gene promoter in response to butyrate therapy. Blood. 1998;92:2924–2933. [PubMed] [Google Scholar]
- 16.Collins AF, Pearson HA, Giardina P, et al. Oral sodium phenylbutyrate therapy in homozygous beta thalassemia: a clinical trial. Blood. 1995;85:43–49. [PubMed] [Google Scholar]
- 17.Perrine SP, Dover GH, Daftari P, T C, et al. Isobutyramide, an orally bioavailable butyrate analogue, stimulates fetal globin gene expression in vitro and in vivo. Br J Haematol. 1994;88:555–61. doi: 10.1111/j.1365-2141.1994.tb05073.x. [DOI] [PubMed] [Google Scholar]
- 18.Cappellini D, Graziadei MG, Ciceri L, et al. Oral isobutyramide therapy in patients with thalassemia intermedia: results of a phase II open study. Blood Cells Mol Dis. 2000;26:105–111. doi: 10.1006/bcmd.2000.0283. [DOI] [PubMed] [Google Scholar]
- 19.Singer ST, Kuypers FA, Olivieri NF, et al. Fetal haemoglobin augmentation in E/beta(0) ia: clinical and haematological outcome. Br J Haematol. 2005;131:378–388. doi: 10.1111/j.1365-2141.2005.05768.x. [DOI] [PubMed] [Google Scholar]
- 20.Constantoulakis P, Knitter G, Stamatoyannopoulos G. On the induction of fetal hemoglobin by butyrates: in vivo and in vitro studies with sodium butyrate and comparison of combination treatments with 5-AzaC and AraC. Blood. 1989;74:1963–71. [PubMed] [Google Scholar]
- 21.Boosalis MS, Bandyopadhyay R, Bresnick EH, et al. Short-chain fatty acid derivatives stimulate cell proliferation and induce STAT-5 activation. Blood. 2001;97:3259–67. doi: 10.1182/blood.v97.10.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pace BS, White GL, Dover GJ, et al. Short-chain fatty acid derivatives induce fetal globin expression and erythropoiesis in vivo. Blood. 2002;100:4640–8. doi: 10.1182/blood-2002-02-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castaneda S, Boosalis MS, Emery D, et al. Enhancement of growth and survival and alterations in Bcl-family proteins in beta-thalassemic erythroid progenitors by novel short-chain fatty acid derivatives. Blood Cells Mol Dis. 2005;35:217–226. doi: 10.1016/j.bcmd.2005.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kutlar A, Ataga K, Reid M, et al. A phase I/II trial of HQK-1001, a fetal globin gene inducer, in sickle cell disease. Am J Hematol. 2012 doi: 10.1002/ajh.23306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuchareon S, Inati A, Siritanaraku N, et al. A randomized Phase I/II trial of HQK-1001, an oral foetal globin gene inducer, in beta thalassemia intermedia and HbE beta thalassemia. Brit J Haematol. 2013 May;161(4):587–93. doi: 10.1111/bjh.12304. doi:10. 1111/bjh.12304. Epub 2013 Mar 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bohacek R, Boosalis MS, McMartin C, et al. Identification of novel small-molecule inducers of fetal hemoglobin using pharmacophore and ‘PSEUDO’ receptor models. Chem Biol Drug Des. 2006;67:318–28. doi: 10.1111/j.1747-0285.2006.00386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Testa U. Fetal hemoglobin chemical inducers for treatment of hemoglobinopathies. Ann Hematol. 2009;88 doi: 10.1007/s00277-008-0637-y. [DOI] [PubMed] [Google Scholar]
- 28.Boosalis MS, Castaneda SA, Trudel M, et al. Novel therapeutic candidates, identified by molecular modeling, induce gamma-globin gene expression in vivo. Blood Cells Mol Dis. 2011;47:107–16. doi: 10.1016/j.bcmd.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xue Y, Wong J, Moreno GT, et al. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell. 1998;2:851–61. doi: 10.1016/s1097-2765(00)80299-3. [DOI] [PubMed] [Google Scholar]
- 30.Tanabe O, McPhee D, Kobayashi S, et al. Embryonic and fetal beta-globin gene repression by the orphan nuclear receptors, TR2 and TR4. EMBO J. 2007;26(9):2295–2306. doi: 10.1038/sj.emboj.7601676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi L, Cui S, Engel JD, et al. Lysine-specific demethylase 1 is a therapeutic target for fetal hemoglobin induction. Nat Med. 2013;19:291–4. doi: 10.1038/nm.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cui S, Kolodziej KE, Obara N, et al. Nuclear receptors TR2 and TR4 recruit multiple epigenetic transcriptional corepressors that associate specifically with the embryonic beta -type globin promoters in differentiated adult erythroid cells. Mol Cell Biol. 2011 Aug;31(16):3298–311. doi: 10.1128/MCB.05310-11. Epub 2011 Jun 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cao H, Stamatoyannopoulos G, Jung M. Induction of human gamma globin gene expression by histone deacetylase inhibitors. Blood. 2004;103:701–709. doi: 10.1182/blood-2003-02-0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bradner JE, Mak R, Tanguturi SK, et al. Chemical genetic strategy identifies histone deacetylase 1 (HDAC1) and HDAC2 as therapeutic targets in sickle cell disease. Proc Natl Acad Sci U S A. 2010;107:12617–22. doi: 10.1073/pnas.1006774107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perrine SP, Mankidy R, Boosalis MS, et al. Erythroid Kruppel-like factor (EKLF) is recruited to the gamma-globin gene promoter as a co-activator and is required for gamma-globin gene induction by short-chain fatty acid derivatives. Eur J Haematol. 2009;82:466–76. doi: 10.1111/j.1600-0609.2009.01234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Z, Luo HY, Steinberg MH, et al. BCL11A represses HBG transcription in K562 cells. Blood Cells Mol Dis. 2009;42:144–149. doi: 10.1016/j.bcmd.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 37.Sankaran VG, Menne TF, Xu J, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322:1839–42. doi: 10.1126/science.1165409. [DOI] [PubMed] [Google Scholar]
- 38.Uda M, Galanello R, Sanna S, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci U S A. 2008;105:1620–5. doi: 10.1073/pnas.0711566105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Siatecka M, Bieker JJ. The multifunctional role of EKLF/KLF1 during erythropoiesis. Blood. 2011;118:2044–54. doi: 10.1182/blood-2011-03-331371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bieker JJ, Southwood CM. The erythroid Kruppel-like factor transactivation domain is a critical component for cell-specific inducibility of a beta-globin promoter. Mol Cell Biol. 1995;15:852–60. doi: 10.1128/mcb.15.2.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou D, Liu K, Sun CW, et al. KLF1 regulates BCL11A expression and gamma- to beta-globin gene switching. Nat Genet. 2010;42(9):742–744. doi: 10.1038/ng.637. [DOI] [PubMed] [Google Scholar]
- 42.Manwani D, Bieker JJ. KLF1: when less is more. Blood. 2014;124:672–3. doi: 10.1182/blood-2014-05-576967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mankidy R, Faller DV, Mabaera R, et al. Short-chain fatty acids induce gamma-globin gene expression by displacement of a HDAC3-NCoR repressor complex. Blood. 2006;108:3179–86. doi: 10.1182/blood-2005-12-010934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Borg J, Papadopoulos P, Georgitsi M, et al. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat Genet. 2010;42:801–5. doi: 10.1038/ng.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sangerman JI, Boosalis MS, Shen L, et al. Identification of new and diverse inducers of fetal hemoglobin with High Throughput Screening (HTS). [abstract] Blood. 2010;116:4277. [Google Scholar]
- 46.TaqMan, Life Technologies TaqMan Gene Expression. Invest Ophthalmol Vis Sci. 2014 May 13;55(8):5382–93. doi: 10.1167/iovs.14-14179. https://www.lifetechnologies.com/order/genome-database/details/gene-expression/Hs00361131_g1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dai Y, Ngo D, Forman LW, et al. Sirtuin 1 is required for antagonist-induced transcriptional repression of androgen-responsive genes by the androgen receptor. Mol Endocrinol. 2007;21:1807–21. doi: 10.1210/me.2006-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mabaera R, West RJ, Conine SJ, et al. A cell stress signaling model of fetal hemoglobin induction: what doesn't kill red blood cells may make them stronger. Exp Hematol. 2008;36:1057–72. doi: 10.1016/j.exphem.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 49.Schaeffer EK, West RJ, Conine SJ, et al. Multiple physical stresses induce gamma-globin gene expression and fetal hemoglobin production in erythroid cells. Blood Cells Mol Dis. 2014;52:214–24. doi: 10.1016/j.bcmd.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 50.Centis F, Tabellini L, Lucarelli G, et al. The importance of erythroid expansion in determining the extent of apoptosis in erythroid precursors in patients with beta-thalassemia major. Blood. 2000;96:3624–3629. [PubMed] [Google Scholar]
- 51.Mathias LA, Fisher TC, Zeng L, et al. Ineffective erythropoiesis in beta-thalassemia major is due to apoptosis at the polychromatophilic normoblast stage. Exp Hematol. 2000;28:1343–1353. doi: 10.1016/s0301-472x(00)00555-5. [DOI] [PubMed] [Google Scholar]
- 52.Pootrakul P, Sirankapracha P, Hemsorach S, et al. A correlation of erythrokinetics, ineffective erythropoiesis, and erythroid precursor apoptosis in Thai patients with thalassemia. Blood. 2000;96:2606–2612. [PubMed] [Google Scholar]
- 53.Garner C, Mitchell J, Hatzis T, et al. Haplotype mapping of a major quantitative-trait locus for fetal hemoglobin production, on chromosome 6q23. Am J Hum Genet. 1998;62:1468–1474. doi: 10.1086/301859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Macari ER, Schaeffer EK, West RJ, et al. Simvastatin and t-butylhydroquinone suppress KLF1 and BCL11A gene expression and additively increase fetal hemoglobin in primary human erythroid cells. Blood. 2013;121:830–9. doi: 10.1182/blood-2012-07-443986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lettre G, Sankaran VG, Bezerra MA, et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta- globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci USA. 2008;105(33):11869–11874. doi: 10.1073/pnas.0804799105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Labie D, Pagnier J, Lapoumeroulie C, et al. Common haplotype dependency of high G gamma-globin gene expression and high Hb F levels in beta-thalassemia and sickle cell anemia patients. Proc Natl Acad Sci USA. 1985;82:2111–2114. doi: 10.1073/pnas.82.7.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thein SL, Menzel S, Lathrop M, et al. Control of fetal hemoglobin: new insights emerging from genomics and clinical implications. Hum Mol Genet. 2009;18:R216–223. doi: 10.1093/hmg/ddp401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nuinoon M, Makarasara W, Mushiroda T, et al. A genome-wide association identified the common genetic variants influence disease severity in beta0-thalassemia/hemoglobin E. Hum Genet. 2010;127:303–314. doi: 10.1007/s00439-009-0770-2. [DOI] [PubMed] [Google Scholar]
- 59.Sheehan VA, Luo Z, Flanagan JM, et al. Genetic modifiers of sickle cell anemia in the Baby HUG cohort: influence on laboratory and clinical phenotypes. Am J Hematol. 2013 Apr 20; doi: 10.1002/ajh.23457. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 60.Borg J, Papadopoulos P, Georgitsi M, et al. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat Genet. 2010;42(9):801–805. doi: 10.1038/ng.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sedgewick AE, Timofeev N, Sebastiani P, et al. BCL11A is a major HbF quantitative trait locus in three different populations with beta-hemoglobinopathies. Blood Cells Mol Dis. 2008;41(3):255–258. doi: 10.1016/j.bcmd.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Menzel S, Garner C, Gut I, et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet. 2007;39(10):1197–1199. doi: 10.1038/ng2108. [DOI] [PubMed] [Google Scholar]
- 63.Fathallah H, Atweh GF. Induction of fetal hemoglobin in the treatment of sickle cell disease. Hematology Am Soc Hematol Educ Program. 2006:58–62. doi: 10.1182/asheducation-2006.1.58. [DOI] [PubMed] [Google Scholar]
- 64.A Bank. Regulation of human fetal hemoglobin: new players, new complexities. Blood. 2006;107:435–43. doi: 10.1182/blood-2005-05-2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen JJ, Perrine S. Stressing HbF synthesis: role of translation? Blood. 2013;122:467–8. doi: 10.1182/blood-2013-06-506139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Krivega I, Byrnes C, de Vasconcellos JF, et al. Inhibition of G9a methyltransferase stimulates fetal hemoglobin production by facilitating LCR/γ-globin looping. Blood. 2015 Jul 30;126(5):665–72. doi: 10.1182/blood-2015-02-629972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Peterson KR, Costa FC, Fedosyuk H, et al. A cell-based high-throughput screen for novel chemical inducers of fetal hemoglobin for treatment of hemoglobinopathies. PLoS One. 2014 Sep 16;9(9):e107006. doi: 10.1371/journal.pone.0107006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hoffman-La Roche Limited. Product Monograph for PROLOPA, levodopa and benserazide combination Capsules 50-12.5, 100-25, 200-50 Pharmaceutical standard: professed Antiparkinson Agent. Submission control No. 128706. [Google Scholar]
- 69.Steinberg MH, Chui DHK, Dover GJ, et al. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood. 2014;123(4):459–600. doi: 10.1182/blood-2013-09-528067. DOI: http://dx.doi/10.1182/blood-2013-09-528067. [DOI] [PubMed] [Google Scholar]
- 70.Pace BS, Liu L, Li B, et al. Cell signaling pathways involved in drug-mediated fetal hemoglobin induction: Strategies to treat sickle cell disease. Experimental Biology and Medicine. 2015;240:1050–1064. doi: 10.1177/1535370215596859. [DOI] [PMC free article] [PubMed] [Google Scholar]