

Abstract
Prostate cancer (PCa) is the second leading cause of cancer death in American men, and curing metastatic disease remains a significant challenge. Nearly all patients with disseminated PCa initially respond to androgen deprivation therapy (ADT), but virtually all patient will relapse and develop incurable castration-resistant prostate cancer (CRPC). A high-throughput RNAi screen to identify signaling pathways regulating PCa cell growth led to our discovery that Checkpoint Kinase 2 (CHK2) knockdown dramatically increased PCa growth and hypersensitized cells to low androgen levels. Mechanistic investigations revealed that the effects of CHK2 were dependent on the downstream signaling proteins CDC25C and CDK1. Moreover, CHK2 depletion increased androgen receptor (AR) transcriptional activity on androgen-regulated genes, substantiating the finding that CHK2 affects PCa proliferation, partly, through the AR. Remarkably, we further show that CHK2 is a novel AR-repressed gene, suggestive of a negative feedback loop between CHK2 and AR. Additionally, we provide evidence that CHK2 physically associates with the AR, and that cell cycle inhibition increased this association. Finally, immunohistochemical analysis of CHK2 in prostate cancer patient samples demonstrated a decrease in CHK2 expression in high-grade tumors. In conclusion, we propose that CHK2 is a negative regulator of androgen sensitivity and PCa growth, and that CHK2 signaling is lost during prostate cancer progression to castration resistance. Thus, perturbing CHK2 signaling may offer a new therapeutic approach for sensitizing CRPC to ADT and radiation.
Keywords: Checkpoint Kinase 2, Androgen Receptor, Prostate Cancer
Introduction
Prostate cancer (PCa) is the most common non-cutaneous malignancy in American men. With approximately 30,000 deaths yearly, PCa is the second leading cause of cancer mortality, following lung carcinoma. The standard treatment for advanced PCa is radiation and androgen deprivation therapy (ADT). Despite the high response rate to ADT, the outgrowth of castration-resistant cells will invariably occur with progression to a lethal disease and a relative 5-year survival rate of 28% [www.cancer.org]. Even with the recent advances in ADT that have led to improvements in overall survival, castration-resistant prostate cancer (CRPC) is still incurable. Therefore, insights into the mechanisms by which androgens stimulate PCa growth will lead to new therapies or the improvement of current treatments for the management of CRPC.
Current research has demonstrated that androgen receptor (AR) signaling continues to play a crucial role in CRPC progression. AR overexpression is sufficient to drive androgen-dependent PCa cells to castration resistance and facilitate proliferation in low androgen levels (1). In addition to its cognate steroid hormone, AR can be regulated by interactions with a constellation of co-regulatory and signaling molecules (2). Several mechanisms, including extensive networks between androgen and peptide growth factor signaling pathways, multiple genetic mutations, and the genetic plasticity of cancer, contribute to the inherent and acquired resistance to ADT (3). Previous studies have shown that signal transduction pathways can stimulate AR activation. These studies illustrate that modulating signaling pathways in PCa models regulates castration-resistant growth, AR transcription, AR protein stability, AR DNA binding, and AR phosphorylation (4–6). Together, these reports suggest that the ability of signaling cascades to influence AR function may have a significant role in CRPC progression, and that CRPC may not be effectively treated by ligand-directed therapy alone.
Comparative genomic hybridization studies of cancer have shown that loss of genetic material is much more common than gains or amplifications, suggesting that tumor suppressor genes play a crucial role in tumor development (7). Individuals with defects in DNA damage response (DDR) signaling lose the natural protection against tumorigenesis and are more susceptible to cell transformation and cancer. Checkpoint kinase 2 (CHK2), a serine/threonine kinase and candidate tumor suppressor gene, is essential in the cell-cycle checkpoint activated in response to radiation-induced DNA double-strand breaks (DSBs) (8,9). Activated CHK2 phosphorylates multiple downstream effectors involved in regulating cell-cycle arrest, apoptosis, DNA repair, mitotic spindle assembly, and chromosomal stability (9,10). Rare CHK2 germline mutations have been identified in several familial cancers and rare somatic mutations have been reported in various carcinomas, including those arising in the prostate, breast, and colon (11–13). These mutations affect the stability or kinase activity of CHK2, suggesting that they may be involved in the pathogenesis of these neoplasms. Loss of heterozygosity studies suggest that CHK2 mutations may contribute to tumor development through haploinsufficiency (14). While loss of the q arm on chromosome 22 where CHK2 is located has been observed in breast, colorectal, and ovarian cancers, mutation of the remaining allele was not associated with this loss (15). However, the function of the remaining normal allele can be disrupted (16). CHK2 splice variants lacking kinase activity or mislocalized to the cytoplasm have been detected in breast cancer (17). Reduced expression or total loss of CHK2 protein that was associated with mutation or promoter methylation has been found in many cancers including those of the breast, urinary bladder, colon, ovary, and lung (15,18,19). Reports of other tumors lacking CHK2 expression without any detectable mutation or promoter methylation suggest that additional epigenetic, post-transcriptional, or post-translational mechanisms can decrease CHK2 protein levels (20). Thus, these studies provide support that CHK2 behaves as a tumor suppressor gene.
In this study, we demonstrated that, in addition to dramatically increasing PCa proliferation, CHK2 depletion hypersensitized PCa cells to castrate androgen levels. Conversely, CHK2 overexpression decreased cell growth. The CHK2-mediated effects on growth required the downstream signaling proteins CDC25C and CDK1. We also found that reduced CHK2 protein levels correlated with increasing Gleason score in patient samples. CHK2 knockdown increased AR transcriptional activity on both androgen-activated and androgen-repressed genes, providing evidence that CHK2 affected PCa proliferation, at least in part, through increasing AR activity. Finally, we show for the first time that CHK2 is a novel AR-regulated gene that physically associates with the AR. Cellular perturbations that halt the cell cycle increase this interaction. Based on these findings, we propose that CHK2 is a negative regulator of androgen sensitivity and PCa cell growth, and that CHK2 signaling is lost during progression to castration resistance.
Materials and Methods
Cell Culture, Reagents, and Immunoblotting
LNCaP and C4-2 cells (a gift from Dr. L. W. K. Chung) were grown in T-Medium (Invitrogen) with 5% Non-Heat Inactivated serum (Gemini) as previously described (21). CWR22Rv1 (Rv1), VCaP, and PC3 cells (gifts from Drs. Steven Balk, Karen Knudsen, and L. W. K. Chung, respectively) were grown in Dulbecco's Modified Eagle's Media (Invitrogen) with 10% Heat Inactivated serum. DU-145 cells (a gift from Dr. Chung) were grown in RPMI-1640 (Invitrogen) with 10% Heat Inactivated serum. LHS cells (a gift from Dr. William Hahn) were grown in ProstaLife Epithelial Cell Medium (Lifeline). For growth and RNA experiments, phenol-red free RPMI-1640 media with 5% Charcoal-Stripped Serum (CSS) (Gemini) was used. Commercial DNA fingerprinting kits (DDC Medical) verified cell lines. The following STR markers were tested: CSF1PO, TPOX, TH01, Amelogenin, vWA, D16S539, D7S820, D13S317 and D5S818. Allelic score data revealed a pattern related to the scores reported by the ATCC, and consistent with their presumptive identity.
Antibodies
CHK2, CDC25C, CDK1, ERK1/2, and HA (Cell Signaling); AR21 (in-house). Western blotting performed as previously described (21,22).
CHK2 primers were based on the human CHEK2 sequence obtained from Genbank (BC004207.2) (Supplemental Fig.S1A). PCR was performed using iProof High-Fidelity DNA Polymerase (Bio-Rad) and MGC Human CHEK2 Sequence-Verified cDNA as the template (Thermo Scientific). Amino-terminal triple HA-tagged wild-type (wt) CHK2 was ligated into AgeI and EcoRI sites of the lentiviral expression vector pLJM1 (Addgene plasmid 19319) using T4 DNA ligase (Promega), transformed into Stbl3 bacteria (Life Technologies), and clones were sequenced for verification.
TRC lentiviral human shRNAs targeting CHK2, CDC25C, and CDK1, and pLKO vector control (Thermo Scientific)
CyQuant Growth Assays
Assay was performed as previously described (22). Briefly, shRNA or pLKO control virus was added to fibronectin-coated (1μg/ml) 96well plates. Cells were plated in RPMI-1640 plus 5%CSS with vehicle, 0-0.05nM R1881, and/or 10μM MDV3100 (Selleck). Quantification was performed on Day 7 using a BioTek Synergy 2 plate reader.
RNA Isolation and qPCR
RNA isolation and quantitative real-time PCR (qPCR) was performed as previously described (21,22). RNA concentrations were determined using a NanoDrop 2000 UVVis Spectrophotometer (Thermo Scientific). Primer sequences and annealing temperatures are described in Supplemental Fig.S1B.
Chromatin Immunoprecipitation (ChIP)
ChIP analyses and qPCR were performed as previously described (23) using primers described in Supplemental Fig.S1C.
Immunohistochemistry
Approval from the Institutional Review Board at the University of Virginia was obtained for studies involving human specimens. The avidin-biotin immunoperoxidase technique was used on zinc formalin-fixed, paraffin-embedded tissues. After slides had been placed in a citrate buffer pH 6.0, they were subjected to microwave heat followed by application of the CHK2 antibody (1:400 dilution) for 1 hour. DAB was used as the chromogen.
Results
CHK2 regulates androgen-dependent and castration resistant prostate cancer cell growth
We recently screened a panel of shRNAs that target the human kinome against LNCaP prostate cancer cells grown in the presence or absence of androgen (22) The screen identified CHK2 as a potential negative regulator of growth, while CHK1 knockdown had no effect on cell growth. We evaluated the effect of CHK2 knockdown on growth using the CyQuant Assay, which measures DNA content as a surrogate for cell number. Androgen-sensitive LNCaP or castration-resistant C42 or Rv1 prostate cancer cells were transduced with lentiviral particles expressing two independent shRNAs specific for CHK2 or pLKO empty vector control in the presence or absence of synthetic androgen (R1881). Each CHK2 shRNA generated a reproducible decrease of approximately 70% in protein expression compared to pLKO (Fig.1A). Growth was increased 2-5-fold in the absence of androgen when CHK2 was knocked down. As expected, androgen stimulated proliferation. Remarkably, CHK2 depletion from all three cell lines increased growth in the presence of R1881 compared to pLKO control cells expressing CHK2 under similar conditions. Since these three AR-positive cell lines have mutated AR, we also assessed the effect of CHK2 down-regulation on cell growth in a PCa cell line with wtAR, VCaP. CHK2 knockdown in these cells significantly increased growth in the absence and presence of androgen (Fig.1A). We confirmed that the CHK2 effects on growth was not due to the inhibition of a regulator of cell cycle progression, as cell cycle analysis in response to CHK2 knockdown by flow cytometry showed no marked differences in the proportion of cells in each phase of the cell cycle between CHK2-expressing and CHK2-depleted cells (data not shown). These findings suggest that CHK2 may be a negative regulator of PCa cell growth.
Figure 1.

CHK2 regulates androgen-dependent and androgen-independent prostate cancer cell growth. CyQuant Assay measured DNA content as a surrogate for cell number 7 days after shRNA transduction. Growth was compared to untreated empty vector control and the values were averaged across biological replicates. Error bars represent standard error of the mean. (A) Cell lysate was blotted for CHK2 and ERK1/2. Plotted is the CHK2 signal normalized to total ERK1/2. Quantitation was performed on Odyssey LICOR imaging system, n=3. The effect of two independent shRNAs on cell growth in LNCaP, VCaP, Rv1, and C42 cells in the absence or presence of 0.05nM R1881, n=5. (B) Knockdown of CHK2 enhances sensitivity to castration levels of androgen. The effect of two independent shRNAs on cell growth in LNCaP and Rv1 cells in the absence or presence of 0-0.05nM R1881, n=3. (C) The effect of two independent CHK2 shRNAs on cell growth in LNCaP and Rv1 cells in the presence of R1881 (0.05nM) and/or the anti-androgen MDV3100 (10μM). (D) Cell lysate was blotted for CHK2 and ERK1/2. Quantitation represents the CHK2 signal normalized to total ERK1/2, n=3. The effect of two independent shRNAs on cell growth in PC3 and DU145 cells in the absence or presence of 0.05nM R1881, n=3. Statistical analysis was performed using two-way ANOVA. * p<0.0001. (E) CHK2 knockdown has no effect on cell growth or AR transcription in non-AR expressing cells. The effect of two independent shRNAs on cell growth in LHS cells, n=3. Cell lysate was blotted for CHK2 and ERK1/2. Quantitation on Odyssey LICOR imaging system represents the CHK2 signal normalized to total ERK1/2, n=3.
Androgen hypersensitivity, a condition where cells can tolerate and continue to proliferate in low androgen concentrations, was reported as one mechanism of resistance to ADT (24). Since CHK2 knockdown dramatically increased growth in the presence of 0.05nM R1881, we hypothesized that CHK2 depletion would enhance sensitivity to lower levels of androgen. To test this, LNCaP and Rv1 cells were transduced with two independent CHK2 shRNAs or empty vector, exposed to multiple R1881 doses, and growth was determined (Fig.1B). Consistent with our previous results, CHK2 knockdown increased growth in the absence of androgen. Notably, a comparison of the slope of the lines from the CHK2-depleted and CHK2-expressing cells showed that there was a significant concentration-dependent augmentation in R1881 sensitivity. Upon exposure to 0.006nM R1881, LNCaP cells expressing CHK2 shRNAs exhibited a 5-fold growth advantage over pLKO control cells. The growth benefit gained from CHK2 knockdown was most evident at 0.025nM R1881, where the proliferative difference between control and CHK2-depleted cells was up to 7-fold. Similar to these results, CHK2 depletion in Rv1 cells heightened androgen sensitivity. Thus, these results indicate that CHK2 knockdown hypersensitizes PCa cells to castrate androgen levels.
To investigate whether the AR plays a role in CHK2-regulated growth, cell growth was assessed using the CyQuant Assay in LNCaP and Rv1 cells expressing two independent CHK2 shRNAs or pLKO and treated with R1881 and/or the anti-androgen MDV3100. We determined that the increase in PCa proliferation following CHK2 knockdown could be blocked by MDV3100, indicating a dependence on AR activity for growth, even when growth is enhanced due to CHK2 loss (Fig.1C). However, the inhibitory effect of MDV3100 was less in CHK2-depleted cells compared to pLKO-expressing cells under similar culture conditions. It is not surprising that the effect of MDV3100 is not as great in Rv1 cells, since this PCa line represents castration resistant disease. Moreover, we measured the effect of CHK2 knockdown on growth in AR-null PC3 and DU145 cells and observed that there was no difference in cell number in the presence or absence of androgen in cells expressing CHK2 compared to CHK2-depleted cells (Fig.1D). Finally, in order to determine if the growth effects mediated by CHK2 were specific to PCa cells, we measured cell number in response to two independent CHK2 shRNAs in LHS cells, which are non-tumorigenic immortalized human prostate epithelial cells. Down-regulation of CHK2 in normal prostate cells had no significant effects on cell growth (Fig.1E). Taken together, these results indicate that the CHK2-mediated growth effects in PCa are regulated in both an androgen-dependent and androgen-independent manner.
CHK2 is required for PCa cell growth in response to hormone
We hypothesized that, if CHK2 knockdown increased growth in PCa cells, then CHK2 overexpression would have the opposite effect. Rv1 or VCaP cells were transduced with lentiviral particles expressing wtCHK2 or pLJM1 vector in the presence or absence of R1881. Overexpression of CHK2 in Rv1 cells (Fig.2A) decreased growth by 55% in the absence of androgen compared to cells expressing endogenous CHK2. While androgen supplementation increased growth in cells expressing the empty vector, CHK2 overexpression inhibited the R1881-induced growth by 65%. Similar results were seen in VCaP cells, where the overexpression of CHK2 suppressed growth by 27% and 51% in the absence and presence of R1881, respectively. These data indicate that CHK2 negatively regulates PCa growth and response to hormone.
Figure 2.

CHK2 is required for prostate cancer cell growth in response to hormone. CyQuant Assay measured cell number 7 days after transduction. (A) CHK2 overexpression decreases cell growth. Cell lysate was blotted for HA (exogenous CHK2), CHK2 (endogenous CHK2), and ERK1/2. The effect of empty vector (pLMJ1) or wild-type CHK2 (pLJM1-wtCHK2) overexpression on growth in Rv1 and VCaP cells in the absence or presence of 0.05nM R1881, n=3. (B) Overexpression of CHK2 in cells depleted of CHK2 abrogated the growth effect of CHK2 knockdown alone. Cell lysate was blotted for HA, CHK2, and ERK1/2. The effect of empty vector (pLKO), CHK2 shRNA, or CHK2 shRNA/wtCHK2 overexpression on growth in LNCaP, Rv1, and C42 cells in the absence or presence of 0.05nM R1881, n=3. Statistical analysis was performed using two-way ANOVA. * p<0.01.
To confirm that CHK2 was required for the growth changes in response to hormone, we performed rescue experiments that took advantage of an shRNA which targeted the 3′-UTR of CHK2, and therefore, only knocked down endogenous protein but not exogenous CHK2 (Fig.2B). LNCaP, Rv1, and C42 cells were transduced with an empty vector, shCHK2-3, or shCHK2-3/wtCHK2, exposed to R1881, and growth was determined. Consistent with our previous data, CHK2 depletion increased growth in the absence and presence of androgen. Notably, reconstitution of wtCHK2 in cells depleted of CHK2 repressed the growth augmentation resulting from CHK2 knockdown and androgen stimulation by at least 68% in these cell lines. Thus, these data show that CHK2 is necessary for the regulation of PCa cell growth and hormone response.
CHK2 protein is inversely correlated with PCa grade
We analyzed CHK2 staining in primary PCa samples from radical prostatectomies of 85 patients and found a significant decrease in CHK2 expression with increasing Gleason score (Fig.3). There was a significant correlation between patient samples with higher Gleason scores [scores 7(4+3), and 8-10] and low CHK2 expression. Furthermore, patient samples with higher CHK2 expression were significantly associated with lower Gleason scores [scores 6 and 7(3+4)]. These data suggest that a reduction in CHK2 expression may contribute to PCa progression.
Figure 3.
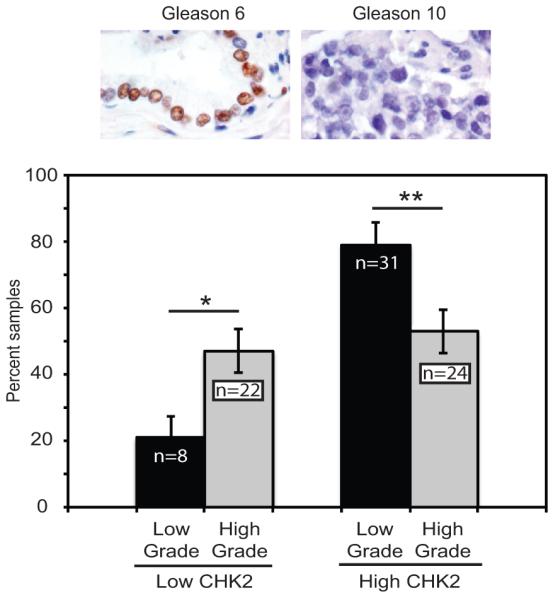
CHK2 protein levels are inversely correlated with prostate cancer grade. CHK2 decreases with increasing Gleason score. Immunohistochemistry for CHK2 in 85 zinc formalin-fixed, paraffin-embedded primary prostate cancers consisting of tissue microarrays, radical prostatectomies, and biopsies evaluated by a pathologist (HFF). High-grade tumors consisted of 3 Gleason 7(4+3), 11 Gleason 8, 14 Gleason 9, and 18 Gleason 10; and low-grade tumors included 15 Gleason 6 and 24 Gleason 7(3+4). The extent of staining was scored on a percentage basis. The number of cells staining for CHK2 was scored as 0; 1 (<5% positive); 2 (5-25%); 3 (26-50%), and 4 (>50%). Plotted is CHK2 level of staining versus percent samples. The CHK2 antibody was validated prior to staining the patients samples by evaluating the specificity of the antibody on zinc formalin-fixed, paraffin-embedded CHK2-depleted and CHK2-expressing (20% vs 90% positive staining) LNCaP cells using varying concentrations of anti-CHK2 antibody. Statistical analysis was performed using the chi-squared test. * p<0.05 and ** p<0.02.
CHK2 regulates AR transcriptional activity
Since the AR is a major regulator of PCa growth, we determined whether CHK2 was affecting PCa growth through modulation of AR transcriptional activity. To examine the effect of CHK2 knockdown on AR target gene transcription, qPCR was used to determine transcript levels of AR-induced genes in LNCaP cells transduced with two independent CHK2 shRNAs. We quantitated gene transcription at 2 hours following androgen exposure to separate the effect of cell proliferation from the CHK2 knockdown effect. As expected, androgen induced transcription of AR target immediate-early genes (NKX3.1, SGK1, STAG1, TMPRSS2) in the pLKO control cells (Fig.4A). CHK2 knockdown significantly increased the induction of NKX3.1 transcription in the absence and presence of 1nM dihydrotestosterone (DHT). The magnitude of the transcriptional amplification of the three other AR-induced genes (TMPRSS2, SGK1, STAG1) was dependent on the target gene and shCHK2 construct. The shCHK2-1 and shCHK2-3 viruses consistently elicited greater than 60% knockdown of CHK2 mRNA compared to pLKO (Fig.4A), whereas another shCHK2 construct (shCHK2-4) produced approximately 30% knockdown (data not shown). We also measured transcript levels of an AR-induced gene (FKBP51) in VCaP cells transduced with two independent CHK2 shRNAs (Fig.4B). In response to androgen, FKBP51 transcript levels were amplified 2-5-fold further in CHK2-depleted cells compared to control cells. Moreover, we quantitated gene transcription of six AR-responsive genes at 24 hours to evaluate the effect of CHK2 knockdown on steady-state levels of AR transcriptional activity (Fig.4C). PSA was the most affected androgen-induced gene tested, with a significant 3-fold induction in transcription upon CHK2 knockdown in the absence and presence of androgen. TMPRSS2 and KLK2 message levels were dramatically increased in the absence of R1881, whereas SGK1 and ORM1 transcripts were amplified in the presence of androgen. CHK2 knockdown decreased FST transcript levels by 50-60% in the absence of androgen. We did not see any changes in FKBP51 transcript levels in response to CHK2 knockdown in AR-null PC3 cells (Fig.4D). Together, these results suggest that the growth increase following CHK2 knockdown may be due to the regulation of AR transcriptional activity.
Figure 4.

CHK2 regulates AR transcriptional activity. Transcript levels of AR target genes in LNCaP, VCaP, or PC3 cells transduced with two independent shRNAs and pLKO control were measured by qPCR. RNA was isolated 2 hours following 1nM DHT exposure and 24 hours after addition of 1nM R1881. Transcript levels were normalized to the housekeeping gene, PSMB6, and compared to pLKO. Values were averaged across biological replicates +/− standard error of the mean, n=3. Shown are the histograms for (A) four immediate-early androgen-activated genes (NKX3.1, TMPRSS2, SGK1, and STAG1) and CHK2 in LNCaP cells. Statistical analysis was performed using two-way ANOVA. * p<0.05. (B) one androgen-activated genes (FKBP51) and CHK2 in VCaP cells. Statistical analysis was performed using two-way ANOVA. * p<0.05. (C) Table showing the fold changes in mRNA for six androgen-responsive genes (PSA, TMPRSS2, KLK2, SGK, ORM1, FST) in response to CHK2 knockdown and hormone treatment in LNCaP cells. Statistical analysis was performed using two-way ANOVA. * p<0.05 (vs pLKO, vehicle), ∧ p<0.05 (vs pLKO, 1nM R1881). (D) CHK2 knockdown in AR-null PC3 cells has no effect on AR transcriptional activity. Shown are the histograms for FKBP51 and CHK2.
CHK2 regulates cell growth through a signaling pathway involving CDC25C, CDK1, and AR
Cell division cycle 25C (CDC25C) is a dual specificity phosphatase downstream of CHK2 and overexpressed in PCa (9,25). CHK2 negatively regulates CDC25C through S216 phosphorylation, which creates a binding site for 14-3-3 and results in cytoplasmic sequestration and proteasome degradation (26). Studies show the importance of CDC25C in the regulation of the cell cycle during the G2/M transition and DDR (27,28). To determine whether CDC25C is necessary for the CHK2 effects on growth, we performed epistasis experiments where CDC25C expression was decreased in CHK2-depleted LNCaP cells with two independent CDC25C shRNAs (Fig.5A). CDC25C was analyzed since it was the highest expressed CDC25 isoform in LNCaP cells (data not shown). Transduced cells were treated with vehicle or R1881, and cell number was assessed. If the increase in growth resulting from CHK2 knockdown is dependent upon CHK2 signaling to CDC25C, then CDC25C depletion will block the growth increase resulting from CHK2 knockdown. This is what we observed; the growth increase induced by R1881 in CHK2-depleted cells was completely blocked when CDC25C and CHK2 were diminished together under similar culture conditions (Fig.5B). Thus, these results suggest that CHK2 mediates effects on cell growth through CDC25C.
Figure 5.

CHK2 regulates cell growth through a signaling pathway involving CDC25C, CDK1, and AR. (A) Cell lysate was blotted for CHK2, CDC25C, and ERK1/2. The effect of CHK2 shRNA and two independent (B) CDC25C or (D) CDK1 shRNAs on cell growth in LNCaP cells. CyQuant Assay was performed 7 days after shRNA transduction. The experiment was done in the absence and presence of 0.05nM R1881. Cell growth was compared to untreated pLKO control and the values were averaged across biological replicates. Error bars represent standard error of the mean, n=3. (C) Cell lysate was blotted for CHK2, CDK1, and ERK1/2. The relative effect of CHK2 and two independent (E) CDC25C or (F) CDK1 shRNAs on cell growth in LNCaP cells in the presence of R1881 (0.05nM) and/or the anti-androgen MDV3100 (10μM). Cell growth was compared to untreated pLKO control and the values were averaged across biological replicates. Error bars represent standard error of the mean, n=3. Statistical analysis was performed using two-way ANOVA. * p<0.001.
CDC25C facilitates cell cycle progression by removing inhibitory phosphorylations on cyclin-dependent kinase 1 (CDK1), which is also downstream of CHK2 and upregulated in PCa (29). To determine whether CDK1 is required for the CHK2 effects on growth, epistasis experiments were performed in LNCaP cells where CDK1 protein was decreased in combination with CHK2 depletion (Fig.5C). Cells expressing shRNAs for CHK2, CDK1, or pLKO were exposed to vehicle or R1881, and the CyQuant Assay was used to quantitate cell growth. Consistent with reports that CDK1 is downstream of CDC25C and CHK2, knockdown of CDK1 in combination with CHK2 depletion abrogated the androgen-induced increase in growth resulting from CHK2 knockdown alone (Fig.5D). These data indicate that CDK1 is necessary for the CHK2 effects on proliferation.
We did not see any growth inhibition when CDC25C or CDK1 alone were knocked down, even in the presence of CHK2. This may be due to the nature of RNAi experiments, which do not completely abrogate protein expression and activity. Thus, the shRNA knockdown functions similar to a hypomorphic allele. The residual CDC25C or CDK1 expression allowed cells to continue to cycle during the course of the experiment. Moreover, previous studies showed that there were no differences in growth when comparing pLKO empty vector to a non-target control (22). Taken together, our findings show that CHK2 regulates cell growth through CDC25C and CDK1.
Since CHK2 regulated growth through CDC25C and CDK1, we evaluated the impact of AR inhibition on the CHK2 growth effects in epistasis experiments where CDC25C (Fig.5E) or CDK1 (Fig.5F) expression was decreased in CHK2-depleted LNCaP cells. Transduced cells were treated with R1881 and/or MDV3100, and cell growth was measured. As expected, MDV3100 blocked the growth increase induced by R1881 in CHK2-depleted PCa cells, and the decrease in growth resulting from AR inhibition was lower in cells depleted of CHK2 than in cells expressing pLKO. Importantly, CDC25C or CDK1 knockdown in CHK2-depleted cells significantly augmented the inhibitory effect of MDV3100 on growth. The influence of CDK1 knockdown on the growth inhibitory effect of MDV3100 was greater than that of CDC25C knockdown suggesting that CDK1 repression may cooperate more with AR inhibition to suppress PCa growth. Together, these data show that a CHK2-CDC25C-CDK1-AR signaling pathway regulates PCa cell growth.
AR negatively regulates CHK2 expression
MatInspector [www.genomatix.de], a program that locates transcription factor binding sites in DNA sequences, predicted that five out of eight reported CHK2 transcripts possessed candidate androgen response elements (AREs) in the promoter region, suggesting direct regulation of CHK2 mRNA by the AR (Fig.6A). Four transcripts contained half ARE sites, whereas one transcript possessed the full ARE site. To determine whether AR regulated CHK2 expression, we used qPCR to quantitate CHK2 transcript levels in LNCaP cells 24 hours after androgen exposure (Fig.6B). Consistent with the MatInspector analysis, androgen regulated transcription of CHK2 mRNA in LNCaP cells repressing mRNA by 45%. Next, we determined whether AR was recruited to the candidate ARE in the CHK2 gene by ChIP-qPCR. While androgen deprivation coincided with low AR occupancy at the candidate ARE, the addition of androgen significantly increased AR occupancy by 4-fold, suggesting that androgen promoted AR binding and AR-dependent repression of CHK2 transcription (Fig.6C). We observed that AR only occupied the CHK2 transcript that contained the full ARE site in the presence of androgen. Finally, we verified the presence of AREs in the CHK2 promoter by evaluating the expression of CHK2 protein in LNCaP and Rv1 cells exposed to R1881 by western blot. There was a significant androgen-induced decrease of 71% and 42% in CHK2 protein in LNCaP and Rv1 cells, respectively (Fig.6D). The repression of CHK2 protein in LNCaP and Rv1 cells suggested that the effect of androgen was not specific to one cell line. To confirm that AR regulated CHK2 protein expression, we examined CHK2 levels in AR-null DU145 cells in the absence and presence of hormone. R1881 had no effect on CHK2 expression in DU-145 cells, strengthening the idea that AR may be required for the modulation of CHK2 protein levels. These data indicate that CHK2 is a novel AR-regulated gene down-regulated by androgen.
Figure 6.

AR negatively regulates CHK2 expression. (A) Model of a candidate ARE in the promoter of the CHK2 gene. (B) Transcript levels of CHK2 in LNCaP cells were measured by qPCR. RNA was isolated 24 hours after addition of 1nM R1881. Transcript levels were compared to LNCaP cells in the absence of hormone and normalized to the housekeeping gene, PSMB6. Values were averaged across biological replicates +/− standard error of the mean, n=3. Statistical analysis was performed using two-way ANOVA. * p<0.05. (C) LNCaP cells were cultured in hormone-depleted media for 48 hours and then treated with 10nM DHT for 2 hours. Samples were harvested for ChIP analysis and AR was immunoprecipitated with AR21 antibody and analyzed using primers targeting the candidate ARE in the CHK2 promoter, n=5. Statistical analysis was performed using two-way ANOVA. * p<0.02. (D) Cell lysate was blotted for CHK2 and ERK1/2. Representative blots are shown. LNCaP, Rv1, and DU145 cells were grown in phenol-red free RPMI media with 5% CSS in the absence or presence of 1nM R1881 for 48 hours. Plotted is the CHK2 signal normalized to total ERK1/2. Quantitation was performed on Odyssey LICOR imaging system. Error bars represent standard error of the mean, n=3. Statistical analysis was performed using two-way ANOVA. * p<0.05.
CHK2 physically associates with AR and CDK1 in PCa cells
To determine if the CHK2 effects on growth and AR transcription are mediated through associations between CHK2 and AR, we measured CHK2/AR interactions in LNCaP and Rv1 cells (Fig.7A). Surprisingly, CHK2 immune complexes revealed that AR co-immunoprecipitated with CHK2 in both PCa cell lines. Since CDK1 was required for the CHK2 effects on growth, we assessed whether CDK1 was also present in CHK2/AR complexes (Fig.7B). CDK1 immune complexes showed that both CHK2 and AR interacted with CDK1. These data suggest that the impact of CHK2 on PCa growth and AR transcriptional activity may be mediated through physical interactions with AR and CDK1. Since CHK2 is a cell cycle regulator, we assessed whether perturbations to the cell cycle affected CHK2/AR interactions. We measured CHK2/AR immune complexes in LNCaP, Rv1, and C42 cells grown in the presence of CSS or complete growth media (Fig.7C). CHK2/AR interactions were significantly increased under conditions of serum starvation, as seen in CHK2 immune complexes where more AR co-precipitated with equal amount of CHK2. Thus, our results show for the first time that CHK2 physically binds to AR, and that these interactions increase when there are halts in the cell cycle.
Figure 7.

CHK2 physically associates with AR and CDK1. (A) Physical association of CHK2 and the AR in prostate cancer cells. CHK2 immune complexes were generated from 1mg cell extract from LNCaP and Rv1 cells grown in the appropriate media supplemented with 5% FBS, separated by 7.5% SDS-PAGE, and immunoblotted with AR21 and CHK2 antibodies. (B) The interaction of CHK2, CDK1, and the AR in prostate cancer cells. CDK1 immune complexes were generated from 1mg cell extract from LNCaP and Rv1 cells cultured in the appropriate growth media, separated by 10% SDS-PAGE, and immunoblotted with AR21, CHK2, and CDK1 antibodies. For comparison, 100μg cell extract was immunoblotted with the same antibodies. (C) CHK2/AR interactions increase in response to serum starvation. CHK2 immune complexes were generated from 1mg cell extract from LNCaP, Rv1, and C42 cells grown in complete growth media or RPMI-1640 plus 5%CSS, separated by 7.5% SDS-PAGE, and immunoblotted with AR21 and CHK2 antibodies. Plotted is the AR signal normalized to total CHK2 and compared to cells growth in complete growth media. Quantitation was performed on Odyssey LICOR imaging system. Error bars represent standard error of the mean, n=4. Statistical analysis was performed using two-way ANOVA. * p<0.05. (D) Model of the CHK2-CDC25C-CDK1-AR signaling axis.
Discussion
Signaling networks governing PCa growth and AR activity have been recognized as key contributors in the progression of androgen-dependent PCa to CRPC (2,6). Our study identified a CHK2-CDC25C-CDK1-AR signaling pathway as a novel regulator of PCa cell growth and AR transcriptional activity (Fig.7D). We utilized loss-of-function and gain-of-function studies to demonstrate a critical role for CHK2 in the negative modulation of PCa growth and androgen sensitivity, which was not compensated for by the presence of the related kinase CHK1. Genetic and protein expression studies in tumors from germline CHK2 mutation carriers and non-carriers suggest that a reduced level rather than total lack of CHK2 protein is necessary for tumorigenesis (7,11–14). CHK2 was the most common gene lost in gliomas and copy number loss was significantly associated with lower gene expression (30). Our analysis of human prostate cancers showed that there was a significant correlation between low CHK2 expression and advanced Gleason score. Cell lines from prostate cancer, lung cancer, osteosarcomas, and myelodysplastic syndromes have documented CHK2 alterations. In addition to genetic variations that generate mutated CHK2 forms, changes in protein expression impair its kinase activity (9). These modified proteins may impair other tumor suppressive pathways in a dominant-negative manner (31).
Genomic instability is believed to be crucial for tumorigenesis because the mutations required for malignancy are difficult to accumulate in a normal cell (32). Mutations in genes required for the preservation of genomic integrity could be a mechanism for genome instability. Individuals carrying certain CHK2 mutations suffer from a statistically significant increase in incidence of prostate, breast, and other cancers (11,33,34). In addition, germline CHK2 mutations are found in 63% of familial PCa (8), while sporadic CHK2 mutations are observed in preliminary reports of prostate tumor data from The Cancer Genome Atlas (cBio). Our data, when considered in the context of the CHK2 mutations in PCa and the role of CHK2 in the DDR, strongly suggest that CHK2 may act as a tumor suppressor gene and CHK2 loss may be causal in PCa progression.
Several mechanisms responsible for the regulation of CHK2 expression have been reported in various cancers. In addition to inactivating mutations, a dominant-negative effect exerted by CHK2 splice variants in breast cancer reduces expression of the wild-type protein (31). Promoter methylation results in the down-regulation of CHK2 in non-small cell lung carcinomas (19), while decreased CHK2 expression in aggressive, non-Hodgkin lymphomas was due to post-transcriptional regulation, as no mutations, deletions, or hypermethylation were present in the promoter region (20). Functional p53 negatively regulated CHK2 expression in colon cancer cell lines under physiologic conditions and following X-ray and ultraviolet radiation exposure (35). Protein phosphatase 2A and Wip1 decreased CHK2 kinase activity and protein stability through T68 dephosphorylation (36,37). We showed for the first time that AR repressed CHK2 expression in response to hormone. Since CHK2 regulated androgen-dependent growth and transcription, these results indicate that the relationship between CHK2 and AR is reciprocal. While the increase in AR transcriptional activity resulting from CHK2 depletion suggests that CHK2 may represent a novel AR corepressor, the AR-CHK2 negative feedback loop may serve as a mechanism to halt checkpoint signaling once DNA damage is repaired. It is also possible that cancer cells may hijack this feedback loop to perturb CHK2 levels to optimize their survival and resistance to therapy.
The CDC25 family, which consists of three isoforms (CDC25 A, B, and C), activate CDKs which regulate cell cycle progression. As key components of checkpoint signaling pathways activated by DNA damage, the activity and expression of CDC25s are negatively regulated by CHK2. All three CDC25 proteins are overexpressed in prostate tumors (25,38,39). CHK2 loss in PCa could contribute to the reduced rate of degradation and subsequent CDC25C upregulation. Failure to inactivate CDC25C in response to DNA damage may promote genomic instability and cancer predisposition described in syndromes where CHK2 is affected (40). The literature supports a role for CDC25 in regulating AR activity, where CDC25A acts as a repressor and CDC25B as an activator of AR transcriptional activity (38,39). We determined that CDC25C may activate androgen-dependent PCa growth since the dual knockdown of CDC25C and CHK2 repressed growth.
The progression through the cell cycle must be strictly regulated in space and time to maintain genomic stability. It is possible that CDC25C overexpression, accompanied by increased phosphatase activity (25), could lead to CDK1 activation, eventually pushing the cell through the checkpoint barrier and acquiring genetic mutations. CDK1 was identified as a mitosis-phase gene that was upregulated in CRPC (29). CDK1 activity promotes AR S81 phosphorylation and protein stabilization, thereby increasing AR activity (41). Recently, we reported that CDK1 phosphorylates AR on S308, and that this phosphorylation may regulate AR localization during mitosis (42). CDK1 activity increases as PCa progresses (29,43). Elevated CDK1 activity could be a mechanism for increasing AR expression and stability in response to low androgen levels (41). Inappropriate cell cycle progression also requires the escape from checkpoint mechanisms, which is consistent with loss of CHK2 activity in CRPC.
CHK2 was epistatic with CDC25C and CDK1, consistent with these proteins functioning in the same pathway. Decreases in CHK2 could contribute to PCa by relieving regulation of CDC25C and CDK1. Interestingly, we noticed that CDC25C and CDK1 protein levels increased in shCHK2-expressing LNCaP cells. This could suggest that CHK1 does not compensate for CHK2 since under some conditions CHK1 also phosphorylates CDC25C leading to cytoplasmic sequestration and/or degradation (26). Furthermore, these results support the possibility that CDK1 activity is upregulated due to elevated CDC25C levels following CHK2 loss, ultimately leading to increased growth.
Proper regulation of the DDR is essential for the cell to prevent the propagation of mutations that lead to cancer development. CHK2 is a cell-cycle regulator and a significant component of the DDR pathway initiated by radiation-induced DSBs (44). Our data showed that CHK2 knockdown increased NKX3.1 transcription following androgen stimulation. NKX3.1 has been shown to increase PCa cell survival and clonogenicity in response to DNA damage by enhancing DSB repair (45). It was recently demonstrated that AR was activated by radiation-induced DNA damage and initiated a transcriptional program regulating DNA repair, thereby providing a potential mechanism for the synergy of ADT with ionizing radiation (46,47). Deficiencies in DNA repair resulting from CHK2 loss may contribute to the pronounced genomic instability seen in advanced PCa.
We presented the first evidence that CHK2 physically associated with AR. Using mass spectroscopy analysis or AR immunoprecipitates, CDK1 was found to interact with AR (21,41). CDC25C and CHK2 co-immunoprecipitated in Daudi and T47D cells (48). AR recruited DNA repair proteins (KU70, ATM, DNA-PK) to AR target sites upon transcriptional activation (49). A small pool of CHK2 associated with chromatin (50). Thus, it is conceivable that these proteins form a complex that might function as a molecular node to assimilate mitogenic and checkpoint signals. CHK2 loss may result in the inability to properly regulate these interactions, rendering them active during the wrong phase of the cell cycle.
In this study, we present data to show that the loss of CHK2 signaling increases AR activity and facilitates cell proliferation. Our data substantiates a new role for CHK2 in the regulation of androgen sensitivity and PCa growth, and directly links a critical member of the DDR with AR-mediated transcription and proliferation in PCa. These findings are clinically relevant since several CHK and second-generation CDK1 inhibitors are in clinical trials and the CHK2 signaling pathway is activated in response to radiation-induced DNA damage (43). These data may assist in the rational application of existing therapies and lead to the development of novel PCa therapeutics. Based on our findings, we would predict that modulation of the CHK2-CDC25C-CDK1-AR signaling pathway would cooperate with ADT and thereby sensitize prostate tumor cells with CHK2 loss to radiation therapy.
Supplementary Material
Acknowledgements
We thank the laboratory members and Drs. Michael Weber, Michael Harding, Stephan Culp, Mark Jameson, Kevin Janes, and Mark Axelrod for their critical comments.
Grant Support: National Cancer Institute (R01 CA124706 and CA178338, DG), and the Paul Mellon Urologic Cancer Institute (DG)
Footnotes
Conflict of interest disclosure: The authors have no conflicting financial interests
References
- 1.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004 Jan;10(1):33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 2.Gioeli D. Signal transduction in prostate cancer progression. Clin Sci. 2005 Apr;108(4):293–308. doi: 10.1042/CS20040329. [DOI] [PubMed] [Google Scholar]
- 3.Gioeli DG. The promise of novel androgen receptor antagonists. Cell Cycle. 2010 Feb;9(3):440–1. doi: 10.4161/cc.9.3.11045. [DOI] [PubMed] [Google Scholar]
- 4.Ikonen T, Palvimo JJ, Kallio PJ, Reinikainen P, Jänne OA. Stimulation of androgen-regulated transactivation by modulators of protein phosphorylation. Endocrinology. 1994 Oct;135(4):1359–66. doi: 10.1210/endo.135.4.7925097. [DOI] [PubMed] [Google Scholar]
- 5.Culig Z, Hobisch A, Cronauer MV, Radmayr C, Hittmair A, Zhang J, et al. Regulation of prostatic growth and function by peptide growth factors. Prostate. 1996 Jun;28(6):392–405. doi: 10.1002/(SICI)1097-0045(199606)28:6<392::AID-PROS9>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 6.Ta HQ, Gioeli D. The convergence of DNA damage checkpoint pathways and androgen receptor signaling in prostate cancer. Endocr Relat Cancer. 2014 Aug; doi: 10.1530/ERC-14-0217. ERC – 14–0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Visakorpi T, Kallioniemi AH, Syvänen AC, Hyytinen ER, Karhu R, Tammela T, et al. Genetic changes in primary and recurrent prostate cancer by comparative genomic hybridization. Cancer Res. 1995 Jan;55(2):342–7. [PubMed] [Google Scholar]
- 8.Dong X, Wang L, Taniguchi K, Wang X, Cunningham JM, McDonnell SK, et al. Mutations in CHEK2 associated with prostate cancer risk. Am J Hum Genet. 2003 Feb;72(2):270–80. doi: 10.1086/346094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Antoni L, Sodha N, Collins I, Garrett MD. CHK2 kinase: cancer susceptibility and cancer therapy - two sides of the same coin? Nat Rev Cancer. 2007 Dec;7(12):925–36. doi: 10.1038/nrc2251. [DOI] [PubMed] [Google Scholar]
- 10.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003 May;3(5):421–9. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 11.Cybulski C, Huzarski T, Górski B, Masojć B, Mierzejewski M, Debniak T, et al. A novel founder CHEK2 mutation is associated with increased prostate cancer risk. Cancer Res. 2004 Apr;64(8):2677–9. doi: 10.1158/0008-5472.can-04-0341. [DOI] [PubMed] [Google Scholar]
- 12.Van Puijenbroek M, van Asperen CJ, van Mil A, Devilee P, van Wezel T, Morreau H. Homozygosity for a CHEK2*1100delC mutation identified in familial colorectal cancer does not lead to a severe clinical phenotype. J Pathol. 2005 Jun;206(2):198–204. doi: 10.1002/path.1764. [DOI] [PubMed] [Google Scholar]
- 13.Nevanlinna H, Bartek J. The CHEK2 gene and inherited breast cancer susceptibility. Oncogene. 2006;25(43):5912–9. doi: 10.1038/sj.onc.1209877. [DOI] [PubMed] [Google Scholar]
- 14.Sodha N, Mantoni TS, Tavtigian SV, Eeles R, Garrett MD. Rare germ line CHEK2 variants identified in breast cancer families encode proteins that show impaired activation. Cancer Res. 2006 Sep;66(18):8966–70. doi: 10.1158/0008-5472.CAN-06-1990. [DOI] [PubMed] [Google Scholar]
- 15.Williams LH, Choong D, Johnson SA, Campbell IG. Genetic and Epigenetic Analysis of CHEK2 in Sporadic Breast, Colon, and Ovarian Cancers. Clin Cancer Res. 2006 Dec;12(23):6967–72. doi: 10.1158/1078-0432.CCR-06-1770. [DOI] [PubMed] [Google Scholar]
- 16.Bartek J, Falck J, Lukas J. CHK2 kinase--a busy messenger. Nat Rev Mol Cell Biol. 2001 Dec;2(12):877–86. doi: 10.1038/35103059. [DOI] [PubMed] [Google Scholar]
- 17.Staalesen V, Falck J, Geisler S, Bartkova J, Børresen-Dale A-L, Lukas J, et al. Alternative splicing and mutation status of CHEK2 in stage III breast cancer. Oncogene. 2004 Nov;23(52):8535–44. doi: 10.1038/sj.onc.1207928. [DOI] [PubMed] [Google Scholar]
- 18.Bartkova J, Guldberg P, Grønbaek K, Koed K, Primdahl H, Møller K, et al. Aberrations of the Chk2 tumour suppressor in advanced urinary bladder cancer. Oncogene. 2004 Nov;23(52):8545–51. doi: 10.1038/sj.onc.1207878. [DOI] [PubMed] [Google Scholar]
- 19.Zhang P, Wang J, Gao W, Yuan B-Z, Rogers J, Reed E. CHK2 kinase expression is down-regulated due to promoter methylation in non-small cell lung cancer. Mol Cancer. 2004 May;3:14. doi: 10.1186/1476-4598-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tort F, Hernàndez S, Beà S, Martínez A, Esteller M, Herman JG, et al. CHK2-decreased protein expression and infrequent genetic alterations mainly occur in aggressive types of non-Hodgkin lymphomas. Blood. 2002 Dec;100(13):4602–8. doi: 10.1182/blood-2002-04-1078. [DOI] [PubMed] [Google Scholar]
- 21.Gordon V, Bhadel S, Wunderlich W, Zhang J, Ficarro SB, Mollah SA, et al. CDK9 regulates AR promoter selectivity and cell growth through serine 81 phosphorylation. Mol Endocrinol. 2010 Dec;24(12):2267–80. doi: 10.1210/me.2010-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whitworth H, Bhadel S, Ivey M, Conaway M, Spencer A, Hernan R, et al. Identification of Kinases Regulating Prostate Cancer Cell Growth Using an RNAi Phenotypic Screen. PLoS One. 2012 Jun;7(6) doi: 10.1371/journal.pone.0038950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu M, Rao M, Wu K, Wang C, Zhang X, Hessien M, et al. The Androgen Receptor Acetylation Site Regulates cAMP and AKT but Not ERK-induced Activity. J Biol Chem. 2004 Jul;279(28):29436–49. doi: 10.1074/jbc.M313466200. [DOI] [PubMed] [Google Scholar]
- 24.Fujimoto N, Miyamoto H, Mizokami A, Harada S, Nomura M, Ueta Y, et al. Prostate Cancer Cells Increase Androgen Sensitivity by Increase in Nuclear Androgen Receptor and Androgen Receptor Coactivators; A Possible Mechanism of Hormone-Resistance of Prostate Cancer Cells. Cancer Invest. 2007 Jan;25(1):32–7. doi: 10.1080/07357900601130698. [DOI] [PubMed] [Google Scholar]
- 25.Ozen M, Ittmann M. Increased expression and activity of CDC25C phosphatase and an alternatively spliced variant in prostate cancer. Clin cancer Res an Off J Am Assoc Cancer Res. 2005 Jul;11(13):4701–6. doi: 10.1158/1078-0432.CCR-04-2551. [DOI] [PubMed] [Google Scholar]
- 26.Thanasoula M, Escandell JM, Suwaki N, Tarsounas M. ATM/ATR checkpoint activation downregulates CDC25C to prevent mitotic entry with uncapped telomeres. EMBO J. 2012 Aug;31(16):3398–410. doi: 10.1038/emboj.2012.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawabe T, Suganuma M, Ando T, Kimura M, Hori H, Okamoto T. Cdc25C interacts with PCNA at G2/M transition. Oncogene. 2002 Mar;21(11):1717–26. doi: 10.1038/sj.onc.1205229. [DOI] [PubMed] [Google Scholar]
- 28.Matsuoka S, Huang M, Elledge SJ. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science. 1998 Dec;282(5395):1893–7. doi: 10.1126/science.282.5395.1893. [DOI] [PubMed] [Google Scholar]
- 29.Balk SP, Knudsen KE. AR, the cell cycle, and prostate cancer. Nucl Recept Signal. 2008;6:e001. doi: 10.1621/nrs.06001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Squatrito M, Brennan CW, Helmy K, Huse JT, Petrini JH, Holland EC. Loss of ATM/Chk2/p53 pathway components accelerates tumor development and contributes to radiation resistance in gliomas. Cancer Cell. 2010 Dec;18(6):619–29. doi: 10.1016/j.ccr.2010.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berge EO, Staalesen V, Straume AH, Lillehaug JR, Lønning PE. Chk2 splice variants express a dominant-negative effect on the wild-type Chk2 kinase activity. Biochim Biophys Acta. 2010 Mar;1803(3):386–95. doi: 10.1016/j.bbamcr.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 32.Beckman RA, Loeb LA. Efficiency of carcinogenesis with and without a mutator mutation. Proc Natl Acad Sci. 2006 Sep;103(38):14140–5. doi: 10.1073/pnas.0606271103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walsh T, Casadei S, Coats KH, Swisher E, Stray SM, Higgins J, et al. Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA J Am Med Assoc. 2006 Mar;295(12):1379–88. doi: 10.1001/jama.295.12.1379. [DOI] [PubMed] [Google Scholar]
- 34.Cybulski C, Wokołorczyk D, Kładny J, Kurzawski G, Kurzwaski G, Suchy J, et al. Germline CHEK2 mutations and colorectal cancer risk: different effects of a missense and truncating mutations? Eur J Hum Genet EJHG. 2007 Feb;15(2):237–41. doi: 10.1038/sj.ejhg.5201734. [DOI] [PubMed] [Google Scholar]
- 35.Matsui T, Katsuno Y, Inoue T, Fujita F, Joh T, Niida H, et al. Negative Regulation of Chk2 Expression by p53 Is Dependent on the CCAAT-binding Transcription Factor NF-Y. J Biol Chem. 2004 Jun;279(24):25093–100. doi: 10.1074/jbc.M403232200. [DOI] [PubMed] [Google Scholar]
- 36.Dozier C, Bonyadi M, Baricault L, Tonasso L, Darbon J-M. Regulation of Chk2 phosphorylation by interaction with protein phosphatase 2A via its B’ regulatory subunit. Biol Cell. 2004 Sep;96(7):509–17. doi: 10.1016/j.biolcel.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 37.Fujimoto H, Onishi N, Kato N, Takekawa M, Xu XZ, Kosugi A, et al. Regulation of the antioncogenic Chk2 kinase by the oncogenic Wip1 phosphatase. Cell Death Differ. 2006 Jul;13(7):1170–80. doi: 10.1038/sj.cdd.4401801. [DOI] [PubMed] [Google Scholar]
- 38.Ngan ESW, Hashimoto Y, Ma Z-Q, Tsai M-J, Tsai SY. Overexpression of Cdc25B, an androgen receptor coactivator, in prostate cancer. Oncogene. 2003 Feb;22(5):734–9. doi: 10.1038/sj.onc.1206121. [DOI] [PubMed] [Google Scholar]
- 39.Chiu Y-T, Han H-Y, Leung SC-L, Yuen H-F, Chau C-W, Guo Z, et al. CDC25A Functions as a Novel Ar Corepressor in Prostate Cancer Cells. J Mol Biol. 2009 Jan;385(2):446–56. doi: 10.1016/j.jmb.2008.10.070. [DOI] [PubMed] [Google Scholar]
- 40.Lee SB, Kim SH, Bell DW, Wahrer DC, Schiripo TA, Jorczak MM, et al. Destabilization of CHK2 by a missense mutation associated with Li-Fraumeni Syndrome. Cancer Res. 2001 Nov;61(22):8062–7. [PubMed] [Google Scholar]
- 41.Chen S, Xu Y, Yuan X, Bubley GJ, Balk SP. Androgen receptor phosphorylation and stabilization in prostate cancer by cyclin-dependent kinase 1. Proc Natl Acad Sci U S A. 2006 Oct;103(43):15969–74. doi: 10.1073/pnas.0604193103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koryakina Y, Knudsen KE, Gioeli D. Cell-cycle-dependent regulation of androgen receptor function. Endocr Relat Cancer. 2015 Apr;22(2):249–64. doi: 10.1530/ERC-14-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov. 2009 Jul;8(7):547–66. doi: 10.1038/nrd2907. [DOI] [PubMed] [Google Scholar]
- 44.Tan Y, Raychaudhuri P, Costa RH. Chk2 mediates stabilization of the FoxM1 transcription factor to stimulate expression of DNA repair genes. Mol Cell Biol. 2007 Feb;27(3):1007–16. doi: 10.1128/MCB.01068-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bowen C, Gelmann EP. NKX3.1 activates cellular response to DNA damage. Cancer Res. 2010 Apr 15;70(8):3089–97. doi: 10.1158/0008-5472.CAN-09-3138. [DOI] [PubMed] [Google Scholar]
- 46.Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013 Sep; doi: 10.1158/2159-8290.CD-13-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goodwin JF, Schiewer MJ, Dean JL, Schrecengost RS, Leeuw R de, Han S, et al. A Hormone–DNA Repair Circuit Governs the Response to Genotoxic Insult. Cancer Discov. 2013 Sep; doi: 10.1158/2159-8290.CD-13-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bahassi EM, Myer DL, McKenney RJ, Hennigan RF, Stambrook PJ. Priming phosphorylation of Chk2 by polo-like kinase 3 (Plk3) mediates its full activation by ATM and a downstream checkpoint in response to DNA damage. Mutat Res. 2006 Apr;596(1-2):166–76. doi: 10.1016/j.mrfmmm.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 49.Haffner MC, Aryee MJ, Toubaji A, Esopi DM, Albadine R, Gurel B, et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat Genet. 2010 Aug;42(8):668–75. doi: 10.1038/ng.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li J, Stern DF. DNA damage regulates Chk2 association with chromatin. J Biol Chem. 2005 Nov;280(45):37948–56. doi: 10.1074/jbc.M509299200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.